

Abstract
Aims: The aim of this study was to develop and demonstrate an approach for describing the diversity of human pathogenic viruses in an environmentally isolated viral metagenome.
Methods and Results: In silico bioinformatic experiments were used to select an optimum annotation strategy for discovering human viruses in virome data sets and applied to annotate a class B biosolid virome. Results from the in silico study indicated that <1% errors in virus identification could be achieved when nucleotide‐based search programs (BLASTn or tBLASTx), viral genome only databases and sequence reads >200 nt were considered. Within the 51 925 annotated sequences, 94 DNA and 19 RNA sequences were identified as human viruses. Virus diversity included environmentally transmitted agents such as parechovirus, coronavirus, adenovirus and aichi virus, as well as viruses associated with chronic human infections such as human herpes and hepatitis C viruses.
Conclusions: This study provided a bioinformatic approach for identifying pathogens in a virome data set and demonstrated the human virus diversity in a relevant environmental sample.
Significance and Impact of the Study: As the costs of next‐generation sequencing decrease, the pathogen diversity described by virus metagenomes will provide an unbiased guide for subsequent cell culture and quantitative pathogen analyses and ensures that highly enriched and relevant pathogens are not neglected in exposure and risk assessments.
Keywords: bioinformatics, biosolids, next‐generation DNA sequencing, pathogen, viral metagenome, virome, virus
Introduction
Next‐generation DNA sequencing has recently been applied to study viral metagenomes (viromes) in varied environmental matrices including fresh water (Djikeng et al. 2009; Lopez‐Bueno et al. 2009), oceans (Angly et al. 2006) and reused wastewater (Rosario et al. 2009). Most of these studies are interested in describing gene diversity, but a potential application within virome studies is to determine which human viral pathogens are most prevalent in a given environmental matrix. The major limitation to method extension towards human viral identifications is that postsequencing bioinformatic protocols for analysing viromes are in nascent stages of development and require careful consideration to produce the high‐quality virome annotations required for pathogen identification. Viruses do not contain ubiquitous genetic elements such as the 16s rRNA encoding genes (Rohwer and Edwards 2002); hence, virome studies must sort and assemble sequences that could come from any location on the viral genome. Unresolved virome construction and annotation concerns include uncertainty about the optimal sequence read length for identification, as well as appropriate use of databases and database search programs. An additional limitation is the unknown sequencing depth required to reach the rare human viruses amidst the ubiquity of bacteriophages in the environment (Breitbart and Rohwer 2005).
The goal of this study was to develop and test a method for describing the diversity of human pathogenic viruses in an environmentally isolated virome. To improve sequence analysis methods, we conducted an in silico study of ten known human viral pathogen genomes with the aim of decreasing errors in annotating next‐generation sequencing reads as pathogenic viral nucleic acids. As a demonstration, viral DNA and RNA (cDNA) extracted from sewage sludge residuals resulting from municipal wastewater treatment (termed biosolids) were sequenced using 454 Life Sciences pyrosequencing technology. We then applied the optimal annotation schemes identified by the in silico study to describe the diversity and abundance of viral pathogens and to determine the sequencing depth required to portray this viral pathogen diversity. Biosolids are ideal for demonstrating virome pathogen recovery as this waste stream originates from the solid residuals of wastewater treatment plants serving up to one million people, their pathogen content is not well documented (Gerba et al. 2002; Viau and Peccia 2009) and growing public opposition to the land application of biosolids as a soil conditioning product has initiated an expressed desire for comprehensive viral pathogen surveys in biosolids (NRC 2002).
Materials and methods
Bioinformatic experiments
An in silico study was conducted by parsing the genomes of ten environmentally relevant viruses into short sequences and determining the sequence length, BLAST program and viral databases that resulted in the highest confidence of correct annotation. Human virus genotypes were chosen to represent common environmental viral diseases caused by inhalation and ingestion exposure routes (Table 1). Artificial reads were produced at every location along the genome, and lengths were set to represent common read lengths produced by next‐generation sequencing platforms: 100 nucleotide (nt) reads from Illumina Genome Analyzer, 200 nt paired‐end reads from Illumina HiSeq2000 and 400 nt reads from 454 Life Sciences GS FLX sequencer with titanium chemistry.
Table 1.
Human viral pathogens included in the in silico study
| Virus | Nucleic acid | Genome size (nt) | Accession number |
|---|---|---|---|
| Adenovirus | dsDNA | 34 794 | AC_000019 |
| Astrovirus | ssRNA | 6813 | NC_001943 |
| Coronavirus | ssRNA | 27 317 | NC_002645 |
| Hepatitis A virus | ssRNA | 7478 | NC_001489 |
| Norovirus | ssRNA | 7654 | NC_001959 |
| Parechovirus | ssRNA | 7348 | NC_001897 |
| Polyomavirus JC | dsDNA | 5130 | NC_001699 |
| Respiratory Syncytial virus | ssRNA | 15 225 | NC_001781 |
| Rhinovirus | ssRNA | 7152 | NC_001617 |
| Rotavirus | dsRNA | 17 448 | NC_011507* |
*Segment 1, successive segments also included.
To choose optimal sequence search and alignment programs, the following NCBI BLASTALL programs were compared: BLASTn nucleotide to nucleotide searches, BLASTx translated nucleotide to amino acid searches and tBLASTx translated nucleotide to translated nucleotide searches. Each BLASTALL program was applied to two search databases including the full NCBI database (nt, nonredundant nucleotide database, and nr, nonredundant amino acid database) and the NCBI viral databases (vnt, nucleotide database, and vnr, amino acid database).
When the top hit, as determined by lowest E‐value, matched the human pathogen strain that was searched for and there were no ambiguous classifications (i.e. same virus but different host), the read was listed as correct. In the case of multiple hits with equivalent E‐values, the highest bit score was used for annotation. Reads were classified as missing if they contained no hits at or below the 10−3E‐value threshold. The sum of ambiguous and missing sequences were grouped and reported as total classification errors. Classifications were only made when an E‐value of 10−3 or less was observed. This E‐value threshold was based on precedent set in prior virome studies (Zhang et al. 2005; Lopez‐Bueno et al. 2009; Rosario et al. 2009) and also from an evaluation of annotating 100‐nt adenovirus sequence segments using an E‐value of either 10−3 or 10−5. This evaluation revealed that by excluding a greater number of correct sequence reads, an average 18% increase in error was observed when the E‐value threshold was set at 10−5 instead of 10−3 (Table S1).
Biosolid sample preparation and sequencing
Class B biosolids were sampled from an anonymous US wastewater treatment facility that collected solid residuals by primary sedimentation and secondary activated sludge clarification and treated by mesophilic anaerobic digestion (35–37°C, 15 d solid retention time). Digested sludge was dewatered by belt pressing to 17% solid content. Previous class B biosolid indicator and pathogen monitoring from this plant revealed faecal coliform concentrations of 5·1 × 104 colony forming units per dry g, male‐specific coliphage concentrations of 2·7 × 104 plaque forming units per dry g and adenovirus concentrations of 3 × 106 genomic units per dry g (Viau and Peccia 2009).
Five 100 g grab samples were collected in accordance with US EPA method 1680 (USEPA 2006) and shipped on ice overnight to the laboratory. Within 24 h of collection, biosolid samples were recombined to form a composite sample and viruses were eluted and concentrated following a US EPA method for the recovery of viruses from sludge (USEPA 1999). The concentrated viral solution was passed through a 0·45‐μm filter to remove any remaining bacterial and eukaryotic cells and DNase‐/RNase‐digested with OmniCleave endonuclease (Epicentre Biotechnologies, Madison, WI, USA) to remove any naked nucleic acids. Purified viral extracts were stored at −80°C.
Both DNA and RNA were recovered from the viral concentrate. Three DNA extractions were performed each with 0·6 ml of viral concentrate using the MoBio PowerSoil DNA kit (MoBio Laboratories, Carlsbad, CA, USA) and modifications described elsewhere (Viau and Peccia 2009). Triplicate RNA extractions were performed with 2 ml of the viral concentrate each using the MoBio PowerSoil RNA kit (MoBio) followed by DNA digestion. Viral RNA was converted to cDNA with a Multiscribe high‐capacity cDNA reverse transcription kit (Life Technologies™ AB, Carlsbad, CA, USA).
Samples were combined, and a total of 5 μg of DNA and cDNA each were sent to the Yale Center for Excellence in Genome Science for shotgun pyrosequencing on a 454 GS FLX sequencer using titanium chemistry (Roche Diagnostics Corporation, Indianapolis, IN, USA). One quarter of a microwell plate was used for this analysis. Prior to sequencing, DNA was fragmented by nebulization into 300‐ to 800‐nt sequences.
Virome annotation
To remove artificial replicates, a known artefact of 454 pyrosequencing, the 454 replicate filter with default settings was used (Gomez‐Alvarez et al. 2009). Filtered sequence reads were assembled with the Newbler runAssembly program from the 454 Life Sciences Data Analysis 2.3 package (Branford, CT, USA). Sequence assembly settings utilized a minimum overlap of 40 bp and a minimum identity of 90%, while all other settings were default. Unassembled sequences (singletons) were then extracted and combined with assembled contiguous sequences (contigs) for annotation. The virome data were annotated by tBLASTx searches within the NCBI viral database from January 2010. Annotation used the previously described E‐score selection criteria. Sequences are available from the NCBI Sequence Read Archive under accession SRX016659.
Results
Annotation accuracy
For the in silico experiments, the percentage of erroneous reads (Fig. 1) suggests that the most appropriate annotation strategy for viral pathogen identification will be a nucleotide‐based search (BLASTn or tBLASTx) with a virus only database and read lengths of 200 nt and greater. Average error rate in classification using these methods was 0·1% with a 1·2–0% range among the ten viruses.
Figure 1.
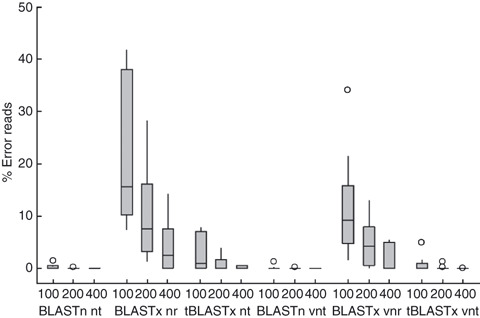
Box plot of total classification errors for ten human viruses according to read length and annotation method. Total errors include both ambiguous and missing identifications. Groupings are by the read length (100, 200, 400 nt), the BLAST search program (BLASTn, BLASTx, tBLASTx), and the database where ‘nt’ represents nucleotide database, ‘nr’ represents amino acid database, and ‘v’ represents virus only database. In each box the centreline represents the median, the top and bottom of the box represent the 25th and 75th error percentiles, and the lines represent the data spread. Outliers, marked by circles, were outside three standard deviations of the median. Outliers for Rotavirus were >80% error for the BLASTx nr and tBLASTx nt cases and were excluded from the graph. Complete results for each individual virus are listed in Table S2.
Four other important trends emerged for annotating human viruses from short virome sequences. First, smaller, more focused viral databases resulted in less incorrect classifications than the larger databases (Fig. 1). Viral databases resulted in less total error in 87 of 90 search scenarios conducted. Secondly, the amino acid‐based search program, BLASTx, produced a greater amount of total errors than the nucleotide‐based search programs, BLASTn and tBLASTx, when comparing both the complete and virus only databases. For example, when using 200‐nt artificial reads and the virus database, BLASTx per cent errors averaged 355 times greater than BLASTn errors and 35 times greater than tBLASTx errors. Results from BLASTn and tBLASTx were statistically indistinguishable for total errors using the viral databases at read lengths of 200 nt (P =0·367). Third, increasing read length either maintained or reduced the number of errors in all scenarios considered. When read length was increased from 100 to 200 nt, the tBLASTx overall error decreased five times to 0·13%, while average error for 400 nt reads was decreased to 0·0015%. Finally, the number of errors was extremely dependent on the type of virus (Table S2). BLASTx nr total errors ranged from 7·4% for norovirus to 94·5% for rotavirus for 100‐nt reads. Total error rates in rotavirus were high‐end outliers to the other genome sequences owing to the high similarity of genome sequences between human and animal rotaviruses.
Human viruses in class B biosolids
After replicate filtering, sequencing provided 123 893 raw sequences. Reads were assembled into 1028 contigs that averaged 874 nt and 46 153 singletons that averaged 260·7 nt. Through tBLASTx comparison with the NCBI viral nt database, 51 925 total sequences were annotated and classified as being of viral origin (215 contigs comprising 48 831 sequences and 3094 singletons) Within these viral classifications, ten different human pathogen viruses (16 strains) representing 113 sequences were identified and included 94 DNA virus sequences and 19 RNA virus sequences (Table 2). Only three sequences identified as human pathogens were ambiguous and were excluded from these results. Through comparisons with the Greengeens core set rDNA database, <0·2% of all sequences were annotated as bacteria (10−30E‐value threshold) (DeSantis et al. 2006).
Table 2.
Human pathogenic viruses identified in the class B biosolid virome
| Virus | Nucleic acid | Genome length (nt) | Number of sequences identified |
|---|---|---|---|
| Human herpesvirus 2 | dsDNA | 154 746 | 46 |
| Human herpesvirus 8 type P | dsDNA | 137 868 | 12 |
| Human herpesvirus 1 | dsDNA | 152 261 | 10 |
| Human herpesvirus 4 | dsDNA | 171 823 | 3 |
| Human herpesvirus 6A | dsDNA | 159 322 | 1 |
| Human coronavirus 229E | ssRNA | 27 317 | 9 |
| Human coronavirus HKU1 | ssRNA | 29 926 | 1 |
| Tanapox virus | dsDNA | 144 565 | 9 |
| Orf virus | dsDNA | 139 962 | 8 |
| Human parechovirus | ssRNA | 7348 | 7 |
| Human adenovirus D | dsDNA | 35 083 | 2 |
| Human adenovirus E | dsDNA | 35 994 | 1 |
| Human adenovirus type 1 | dsDNA | 36 001 | 1 |
| Aichi virus | ssRNA | 8521 | 1 |
| Hepatitis C virus genotype 1 | ssRNA | 9646 | 1 |
| Torque Teno Virus‐like minivirus | ssDNA | 2916 | 1 |
When using shotgun sequencing techniques, it is recognized that the likelihood of a viral fragment being identified is a function of both the virus’ abundance and genome size (Angly et al. 2009). Figure 2 shows the potential number of viral genomes relative to adenovirus content after correction for viral genome size. These results indicate that the RNA viruses parechovirus and coronavirus and the DNA virus herpesvirus were the most abundant human viruses in the biosolid sample tested here. Overall, annotated viral sequences consisted of 33·8% eukaryotic viruses and 66·2% bacteriophages, while human pathogenic viruses comprised <0·1% of total sequences (Fig. 2inset). Table S3 provides a complete list of eukaryotic viruses identified in this study.
Figure 2.
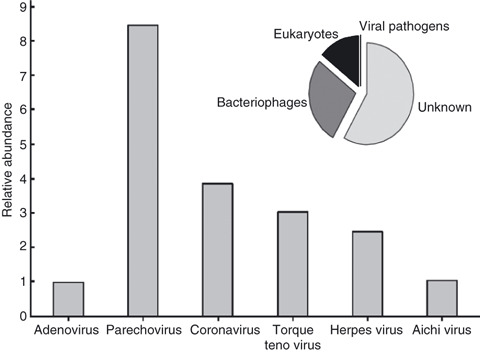
Relative abundance of pathogenic viruses in biosolid virome normalized by genome size and the abundance of adenovirus. Inset: Pie chart of sequence identifications (n = 51 000). Human viral pathogens represent <0·1% of total sequences.
Discussion
Bioinformatic approaches to improve annotation certainty
Use of the viral database with either BLASTn or tBLASTx search programs and read lengths >200 nt is recommended for annotating human pathogen diversity in environmental virome sequences. This recommendation is, however, specific for the goal of pathogen identification and differs from the common practice of using BLASTx for annotating functional genes and nonpathogenic viruses in previous metagenome studies (Breitbart et al. 2002; Angly et al. 2006; Vega Thurber et al. 2008; Djikeng et al. 2009; Lopez‐Bueno et al. 2009; Coetzee et al. 2010). Here, the BLAST search program tBLASTx was used to annotate human pathogens from the biosolid virome. Higher error rates in the translated nucleotide to amino acid BLASTx searches are likely due to the presence of noncoding viral genome regions in queries. Searches conducted by tBLASTx include non‐protein‐encoding regions that are left out of BLASTx searchers. Although the per cent errors associated with the nucleotide to nucleotide BLASTn searches were statistically indistinguishable to those associated with the translated nucleotide to translated nucleotide tBLASTx searches, the latter offers advantages associated with amino acid conservation that are not included in BLASTn searches. This amino acid conservation advantage is demonstrated in the biosolid virome sequencing effort where a BLASTn search of the viral nucleotide database yielded only 8726 sequence identifications compared to the total of 51 925 sequences identifications using tBLASTx. Finally, and in addition to decreased computational time, advantages of the focused virus database are that it does not include similar sequences from nontarget organisms that may be deposited into full databases and thus results in lower ambiguity than the full NCBI database. Physical separation of virus‐sized particles and destruction of free DNA and RNA during sample preparation ensure that the gene sequences produced were of viral origin and obviate the need for annotation using databases that contain additional, nonviral, nucleic acid sequences.
Viral pathogen diversity in class B biosolids
A major public health concern surrounding the land application of biosolids is the risk of infection from viruses that are aerosolized when spread onto land or viruses that enter into ground or surface water supplies (Westrell et al. 2004; Brooks et al. 2005; Eisenberg et al. 2008). To date, viruses previously found in biosolids by PCR‐based methods or culturing include enterovirus (Gerba et al. 2002; Wong et al. 2010), polyomavirus (Bofill‐Mas et al. 2006), reovirus (Gallagher and Margolin 2007), hepatitis A virus (Straub et al. 1994), norovirus (Wong et al. 2010) and adenovirus (Viau and Peccia 2009; Wong et al. 2010). The resulting data from these different studies suggest that adenoviruses are the most abundant human virus in class B biosolids (Viau and Peccia 2009; Schlindwein et al. 2010; Wong et al. 2010). These previous efforts, however, were limited by a requirement that investigators must choose the viruses that will be searched for. By contrast, the production of a viral metagenome produces a list of viruses that is based on abundance and is independent of researcher bias.
Of the viruses described in Fig. 2, their high prevalence within the general population further improves confidence in their identification in biosolids. Viruses found in this study with known environmental routes of exposure and causing respiratory and gastroenteritis infections include adenovirus (Crabtree et al. 1997), parechovirus (Baumgarte et al. 2008), aichi virus (Le Guyader et al. 2008), torque teno virus (TTV) (Griffin et al. 2008) and coronavirus (Yu et al. 2004). Coronavirus is recognized as a major cause of the common cold (Falsey et al. 2002), and 95% of humans are infected with parechovirus within 2–5 years of age (Joki‐Korpela and Hyypiä 2001). Commonly used enterovirus qPCR primers do not include parechovirus; thus, this virus’s presence has not been reflected in previous qPCR enterovirus monitoring (Wong et al. 2010). Although commonly enumerated in class B biosolids (Gerba et al. 2002), enteroviruses were not detected by this study. TTV also circulates in healthy individuals with an estimated worldwide prevalence of 80%, and researchers have suggested its use as a faecal indicator (Bendinelli et al. 2001; Griffin et al. 2008). Both aichi virus and parechovirus have been identified by a sequencing efforts in reused wastewater (Rosario et al. 2009), providing support for their presence in wastewater residuals. Among viral agents described that do not have environmental exposure routes, herpesvirus may be carried by as much as 90% of the population (Arbuckle et al. 2010).
The identifications generated by this viral metagenome sequencing effort are intended to direct quantitative pathogen monitoring efforts, not replace them. Obtaining a more unbiased view of virus diversity through virome production is labour‐intensive and costly. Given limited database size and the inherent genetic similarity between host specific viruses, some level of classification error may always be present. An example of this is the unexpected identifications by this sequencing effort of variola virus (Table S3). Because smallpox (variola virus) has been eradicated worldwide since 1980, it is likely that these identifications are from some other members of the family Poxviridae. While the results indicate that some forms of Poxviridae are present, these known ambiguities highlight the need to accompany virus metagenome‐based pathogen identifications with more in‐depth, confirmatory analysis. The ability to distinguish host specificity was demonstrated in our in silico study; however, this may not extend to all viruses. These limitations suggest that rather than applying massively parallel sequencing as the only form of virus detection, a more appropriate approach for using virus metagenome information is to describe the viral pathogen diversity of a class of environmental samples (e.g. class B biosolids) in order to efficiently guide quantitative and confirmatory analysis of selected agents of interest.
Conclusions
Through an in silico study of simulated viral pathogen reads and an initial biosolid virome sequencing effort, this work has demonstrated the utility of next‐generation DNA sequencing for identifying human viruses in environmental samples of concern. An annotation approach specific for pathogen identification is described that delineates appropriate BLAST programs (tBLASTx, BLASTn), databases (virus only database) and required sequence lengths (>200nt) to achieve <1% error in viral pathogen classification. Several viruses not previously identified in biosolids, including coronavirus, herpesvirus, TTV and parechovirus, were identified and ranked as highly abundant compared to adenoviruses. These results indicate the importance of obtaining an unbiased view of viral pathogen diversity as a guide for subsequent cell culture and specific quantitative PCR investigations required to fully understand biosolid pathogen content.
Acknowledgements
This work was supported by the National Science Foundation grant BES0348455. Computing was carried out at the Yale University Center for High Performance Computation in Biology and Biomedicine (HPC) which is supported by NIH grant RR19895. The authors were grateful for the advice and assistance of Nicholas Carriero and Rob Bjornsen at the high performance computation center. K.B. was supported by a fellowship from the Environmental Research and Education Foundation and STAR fellowship Assistance Agreement no. FP917115 awarded by the US Environmental Protection Agency (EPA). This article has not been formally reviewed by the EPA. The views expressed in this article are solely those of the authors, and the EPA does not endorse any products or commercial services mentioned in this article.
Supplementary Material
Table S1 Percent ambiguous and missing sequence errors for 100 nt reads of adenovirus using E‐values of 10−3 and 10−5
Table S2 Annotation error results for individual viruses by BLAST technique, database and type of error. Numbers in columns represent the percentage erroneous reads
Table S3 Eukaryotic virus classification and number of sequences found in class B biosolids sample, E‐value < 10−3
Contributor Information
K. Bibby, Department of Chemical and Environmental Engineering, Yale University, New Haven, CT, USA
E. Viau, Department of Chemical and Environmental Engineering, Yale University, New Haven, CT, USA
J. Peccia, Department of Chemical and Environmental Engineering, Yale University, New Haven, CT, USA.
References
- Angly,F.E., Felts,B., Breitbart,M., Salamon,P., Edwards,R.A., Carlson,C., Chan,A.M., Haynes,M. et al. (2006) The marine viromes of four oceanic regions. PLoS Biol 4, e368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angly,F.E., Willner,D., Prieto‐Davó,A., Edwards,R.A., Schmieder,R., Vega‐Thurber,R., Antonopoulos,D.A., Barott,K. et al. (2009) The GAAS metagenomic tool and its estimations of viral and microbial average genome size in four major biomes. PLoS Comput Biol 5, e1000593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbuckle,J.H., Medveczky,M.M., Luka,J., Hadley,S.H., Luegmayr,A., Ablashi,D., Lund,T.C., Tolar,J. et al. (2010) The latent human herpesvirus‐6A genome specifically integrates in telomeres of human chromosomes in vivo and in vitro. Proc Natl Acad Sci USA 107, 5563–5568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgarte,S., de Souza Luna,L.K., Grywna,K., Panning,M., Drexler,J.F., Karsten,C., Huppertz,H.I. and Drosten,C. (2008) Prevalence, types, and RNA concentrations of human parechoviruses, including a sixth parechovirus type, in stool samples from patients with acute enteritis. J Clin Microbiol 46, 242–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendinelli,M., Pistello,M., Maggi,F., Fornai,C., Freer,G. and Vatteroni,M.L. (2001) Molecular properties, biology, and clinical implications of TT virus, a recently identified widespread infectious agent of humans. Clin Microbiol Rev 14, 98–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bofill‐Mas,S., Albinana‐Gimenez,N., Clemente‐Casares,P., Hundesa,A., Rodriguez‐Manzano,J., Allard,A., Calvo,M. and Girones,R. (2006) Quantification and stability of human adenoviruses and polyomavirus JCPyV in wastewater matrices. Appl Environ Microbiol 72, 7894–7896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitbart,M. and Rohwer,F. (2005) Here a virus, there a virus, everywhere the same virus? Trends Microbiol 13, 278–284. [DOI] [PubMed] [Google Scholar]
- Breitbart,M., Salamon,P., Andresen,B., Mahaffy,J.M., Segall,A.M., Mead,D., Azam,F. and Rohwer,F. (2002) Genomic analysis of uncultured marine viral communities. Proc Natl Acad Sci USA 99, 14250–14255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks,J.R., Tanner,B.D., Josephson,K.L., Gerba,C., Haas,C.N. and Pepper,I. (2005) A national survey on the residential impact of biological aerosols from the land application of biosolids. J Appl Microbiol 99, 310–322. [DOI] [PubMed] [Google Scholar]
- Coetzee,B., Freeborough,M.‐J., Maree,H.J., Celton,J.‐M., Rees,D.J.G. and Burger,J.T. (2010) Deep sequencing analysis of viruses infecting grapevines: virome of a vineyard. Virology 400, 157–163. [DOI] [PubMed] [Google Scholar]
- Crabtree,K.D., Gerba,C.P., Rose,J.B. and Haas,C.N. (1997) Waterborne adenovirus: a risk assessment. Water Sci Technol 35, 1–6. [Google Scholar]
- DeSantis,T.Z., Hugenholtz,P., Larsen,N., Rojas,M., Brodie,E.L., Keller,K., Huber,T., Dalevi,D. et al. (2006) Greengenes, a Chimera‐checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 72, 5069–5072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djikeng,A., Kuzmickas,R., Anderson,N.G. and Spiro,D.J. (2009) Metagenomic analysis of RNA viruses in a fresh water lake. PLoS ONE 4, e7264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg,J.N.S., Moore,K., Soller,J.A., Eisenberg,D.M. and Colford,J.M. Jr (2008) Microbial risk assessment framework for exposure to amended sludge projects. Environ Health Perspect 116, 727–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falsey,A.R., Walsh,E.E. and Hayden,F.G. (2002) Rhinovirus and coronavirus infection‐associated hospitalizations among older adults. J Infect Dis 185, 1338–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher,E.M. and Margolin,A.B. (2007) Development of an integrated cell culture – Real‐time RT‐PCR assay for detection of reovirus in biosolids. J Virol Methods 139, 195–202. [DOI] [PubMed] [Google Scholar]
- Gerba,C., Pepper,I. and Whitehead,L. (2002) A risk assessment of emerging pathogens of concern in the land application of biosolids. Water Sci Technol 46, 225–230. [PubMed] [Google Scholar]
- Gomez‐Alvarez,V., Teal,T.K. and Schmidt,T.N. (2009) Systematic artifacts in metagenomes from complex microbial communities. ISME J 3, 1314–1317. [DOI] [PubMed] [Google Scholar]
- Griffin,J., Plummer,J. and Long,S. (2008) Torque teno virus: an improved indicator for viral pathogens in drinking waters. Virol J 5, 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joki‐Korpela,P. and Hyypiä,T. (2001) Parechoviruses, a novel group of human picornaviruses. Ann Med 33, 466–471. [DOI] [PubMed] [Google Scholar]
- Le Guyader,F.S., Le Saux,J.‐C., Ambert‐Balay,K., Krol,J., Serais,O., Parnaudeau,S., Giraudon,H., Delmas,G. et al. (2008) Aichi virus, norovirus, astrovirus, enterovirus, and rotavirus involved in clinical cases from a French oyster‐related gastroenteritis outbreak. J Clin Microbiol 46, 4011–4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez‐Bueno,A., Tamames,J., Velazquez,D., Moya,A., Quesada,A. and Alcami,A. (2009) High diversity of the viral community from an Antarctic Lake. Science 326, 858–861. [DOI] [PubMed] [Google Scholar]
- NRC (2002) Biosolids Applied to Land: Advancing Standards and Practices. Washington DC: National Research Council of the National Academies. [Google Scholar]
- Rohwer,F. and Edwards,R. (2002) The phage proteomic tree: a genome‐based taxonomy for phage. J Bacteriol 184, 4529–4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosario,K., Nilsson,C., Lim,Y.W., Ruan,Y. and Breitbart,M. (2009) Metagenomic analysis of viruses in reclaimed wastewater. Environ Microbiol 11, 2806–2820. [DOI] [PubMed] [Google Scholar]
- Schlindwein,A.D., Rigotto,C., Simoes,C.M.O. and Barardi,C.R.M. (2010) Detection of enteric viruses in sewage sludge and treated wastewater effluent. Water Sci Technol 61, 537–544. [DOI] [PubMed] [Google Scholar]
- Straub,T.M., Pepper,I.L. and Gerba,C.P. (1994) Detection of naturally occurring enteroviruses and hepatitis A virus in undigested and anaerobically digested sludge using the polymerase chain reaction. Can J Microbiol 40, 884–888. [DOI] [PubMed] [Google Scholar]
- USEPA (1999) Environmental Regulations and Technology: Control of Pathogens and Vector Attraction in Sewage Sludge. Washington DC: Office of Research and Development, US Environmental Progection Agency. [Google Scholar]
- USEPA (2006) Method 1680: Fecal Coliforms in Sewage Sludge (Biosolids) by Multiple‐Tube Fermentation using Lauryl Tryptose Broth (LTB) and EC Medium. Washington, DC: USEPA. [Google Scholar]
- Vega Thurber,R.L., Barott,K.L., Hall,D., Liu,H., Rodriguez‐Mueller,B., Desnues,C., Edwards,R.A., Haynes,M. et al. (2008) Metagenomic analysis indicates that stressors induce production of herpes‐like viruses in the coral Porites compressa. Proc Natl Acad Sci USA 105, 18413–18418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viau,E. and Peccia,J. (2009) Survey of wastewater indicators and human pathogen genomes in biosolids produced by class A and class B stabilization treatments. Appl Environ Microbiol 75, 164–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westrell,T., Schonning,C., Stenstrom,T.A. and Ashbolt,N.J. (2004) QMRA (quantitative microbial risk assessment) and HACCP (hazard analysis and critical control points) for management of pathogens in wastewater and sewage sludge treatment and reuse. Water Sci Technol 50, 23–30. [PubMed] [Google Scholar]
- Wong,K., Onan,B.M. and Xagoraraki,I. (2010) Quantification of enteric viruses, pathogen indicators, and salmonella bacteria in class B anaerobically digested biosolids by culture and molecular methods. Appl Environ Microbiol 76, 6441–6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu,I.T.S., Li,Y., Wong,T.W., Tam,W., Chan,A.T., Lee,J.H.W., Leung,D.Y.C. and Ho,T. (2004) Evidence of airborne transmission of the severe acute respiratory syndrome virus. N Engl J Med 350, 1731–1739. [DOI] [PubMed] [Google Scholar]
- Zhang,T., Breitbart,M., Lee,W.H., Run,J.‐Q., Wei,C.L., Soh,S.W.L., Hibberd,M.L., Liu,E.T. et al. (2005) RNA viral community in human feces: prevalence of plant pathogenic viruses. PLoS Biol 4, e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Percent ambiguous and missing sequence errors for 100 nt reads of adenovirus using E‐values of 10−3 and 10−5
Table S2 Annotation error results for individual viruses by BLAST technique, database and type of error. Numbers in columns represent the percentage erroneous reads
Table S3 Eukaryotic virus classification and number of sequences found in class B biosolids sample, E‐value < 10−3