

Abstract
Glutamate carboxypeptidase II (GCP2) is a membrane-bound cell-surface peptidase which is implicated in several neurological disorders, and is also over-expressed in prostate tumor cells. There is significant interest in the inhibition of GCP2 as a means of neuroprotection, while GCP2 inhibition as a method to treat prostate cancer remains a topic of further investigation. The key zinc-binding functional group of the well characterized classes of GCP2 inhibitors (phophonates and phosphoramidates) is tetrahedral and negatively charged at neutral pH, while glutamyl urea class of inhibitors possess a planar and neutral zinc-binding group. This study introduces a new class of GCP2 inhibitors, N-substituted glutamyl sulfonamides, which possess a neutral tetrahedral zinc-binding motif. A library containing 15 secondary sulfonamides and 4 tertiary (N-methyl) sulfonamides was prepared and evaluated for inhibitory potency against purified GCP2 enzyme activity. While most inhibitors lacked potency at 100 μM, short alkyl sulfonamides exhibited promising low micromolar potency, with the optimal inhibitor in this series being glutamyl N-propylsulfonamide (2g). Lastly, molecular docking was used to develop a model to formulate an explanation for the relative inhibitory potencies employed for this class of inhibitors.
Keywords: glutamate carboxypeptidase II, GCP2, prostate specific membrane antigen, PSMA, sulfonamide, computational docking
Introduction
Neurotoxicity caused by the presence of excess glutamate has been implicated in several neurological disorders including ischaemia, traumatic brain injury (TBI), stroke, amyotrophic lateral sclerosis (ALS).1–3 By their nature, substances that limit the release of glutamate in the nervous system can be considered neuroprotective agents. One source of glutamate in the nervous system is proteolysis of the short neuropeptide N-acetylaspartylglutamate (NAAG), a reaction catalyzed by the membrane-bound cell-surface peptidase glutamate carboxypeptidase II (GCP2). As such, inhibition of GCP2 causes decreased levels of extracellular glutamate,4 as well as also increased levels of NAAG which itself has a neuroprotective role.5
GCP2 is also expressed in the human prostate epithelium, and thus another name for the enzyme is prostate-specific membrane antigen (PSMA).6 PSMA is over-expressed in prostate cancer tissue and non-prostatic tumour-associated neovasculature7,8. There are studies suggesting that the inhibition of PSMA by small molecules9,2 and monoclonal antibodies10,11,2,12 have potential as therapeutic strategies against prostate cancer. However, the most established use of PSMA inhibitors has been in diagnostic imaging of prostate cancer.2,13–17,1,18–21
There are a number of known classes of GCP2 inhibitor scaffolds, typically characterized by a functional group connected to either a glutaryl moiety or the glutamyl amino group, which either terminates the structure or serves as a linker to another molecular fragment.1,2 In most inhibitors, glutarate/glutamate appears to occupy the S1′ pocket of GCP2,22,23 while an alternative motif (if present) occupies the S1 pocket. Key classes of inhibitors include: phosphonates (1a), phosphates (1b), phosphoramidates (1c–1e), and ureas (1f).1,2 In the case of the phosphorous based inhibitors, the functionalities appear to serve as zinc binding group (ZBG) to the catalytic zinc atoms in the active site,22,23. A recent structural investigation shows the oxygen in urea-based inhibitors also interact with the catalytic zinc atoms.24 A problem with the highly potent (yet multiply charged) phosphonate inhibitor 2-PMPA (1a), is poor oral bioavailability25 thereby limiting its practical value as a clinical neuroprotective agent.
One goal of our research program is to explore alternative functional groups that could serve as a ZBG group to interact with the catalytically active zinc centers in GCP2. Alternatively we envision that these functional groups could passively serve as a linker between glutamate and another molecular fragment making favorable contacts in the active site. One attractive scaffold is the glutamyl ureas, which have been explored extensively by Kozikowski et al,2 and have also been co-crystallized with GCP2.24 Another functionality is the tetrahedral sulfonamide, due to its trivial installation into small molecule inhibitors. Sulfonamides are attractive for pharmaceutical development, compared to similar phosphororus-based groups (1a–1e) due to their aqueous stability despite their net neutral charge. One target where sulfonamides have been successfully used as a zinc-binding group is carbonic anhydrase.26–28 Sulfonamide-based inhibitors have also been explored in the context of antiviral and antitumor drug discovery.29–31
An important feature of sulfonamides (as well as ureas) is the ease in which they can be conjugated to chiral and inexpensive protected glutamate building blocks (4) to generate an optically active inhibitor scaffold. In contrast, the highly potent phosphonate inhibitors (e.g. 2-PMPA) are more challenging to produce in optically active form.32
We envisioned a first-generation of simple sulfonamides based on glutamic acid, in which the SAR of various R1 groups on the sulfur atom could be explored (Figure 2). Two classes of inhibitors were considered: secondary sulfonamides (2) and tertiary (N-methyl) sulfonamides (3). The latter class of compounds was considered in order to examine importance of the free NH group for both inhibitory potency against GCP2, and its potenial as a zinc binding group.
Figure 2.
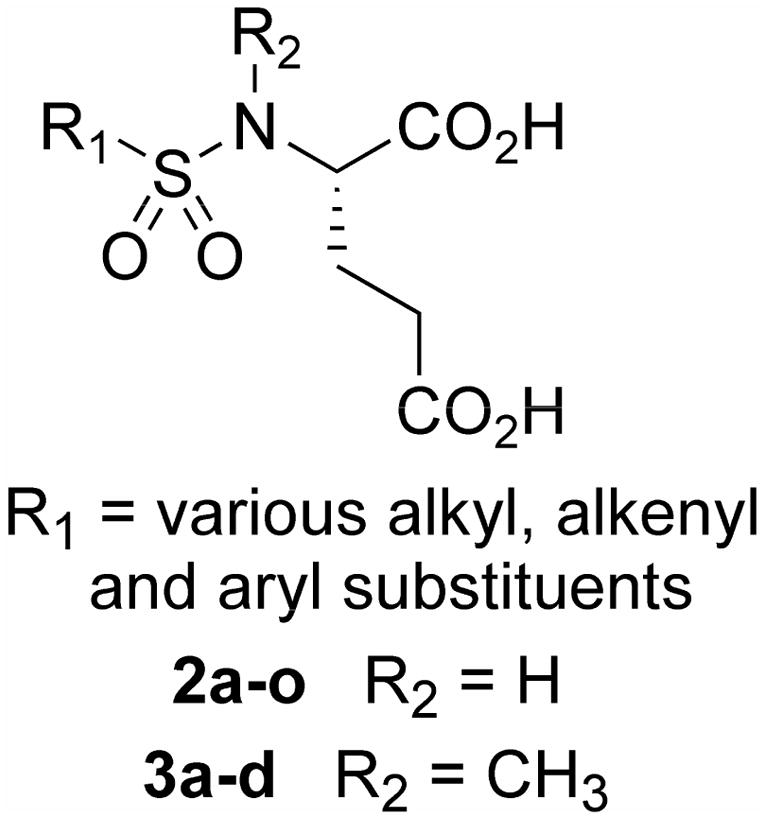
General structure of sulfonamide inhibitors
Results and Discussion
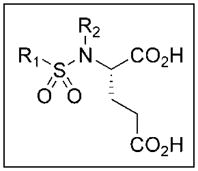
A simple method was employed for the preparation of our library of sulfonamide inhibitors (Scheme 1). Diprotected glutamic acid (4) was directly conjugated with commercially available sulfonyl chlorides to generate sulfonamides 5, and deprotected with TFA to the produce the first library of sulfonamide inhibitors 2. Alternatively, compound 4 was conjugated with sulfonyl chlorides and then treated with methyl sulfate to generate N-methylsulfonamide analogs 6, which were deprotected to generate the final N-methylsulfonamide inhibitor library 3. In general the purity of the final products, as evidenced by 1H and 13C NMR was very high (>95%), and the products did not require additional purification. The composition of the library is outlined in Table 1.
Scheme 1.

Synthesis of substituted glutamyl sulfonamides. Reagents and conditions: (a) sulfonyl chloride or triflic anhydride (1.05 eq), Et3N (3.0 eq), 1h; (b) TFA (20 eq), DCM; (c) CH3I (4.0 eq), K2CO3 (4.0 eq), 16-crown-6 (1.0 eq).
Table 1.
Inhibitory potency of sulfonamide derivatives against GCP2 enzymatic activity (IC50 values with error indicated in parenthesis).
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Cmpd | R1 | R2 | % Yield initial sulfonamide formation | % Yield sulfonamide N-methylation | % Yield final deprotection | IC50 against purified PSMA μM (error) |
| 1 | 2a | methyl | H | 88 | n/a | quant | >100 |
| 2 | 2b | trifluoromethyl | H | 90 | n/a | quant | >100 |
| 3 | 2c | chloromethyl | H | 94 | n/a | quant | 17.2 (1.5) |
| 4 | 2d | ethyl | H | 65 | n/a | quant | 11.6 (0.4) |
| 5 | 2e | vinyl | H | 90a | n/a | quant | >100 |
| 6 | 2f | methylazido | H | 66b | n/a | quant | >100 |
| 7 | 2g | propyl | H | 73 | n/a | 97 | 5.0 (0.8) |
| 8 | 2h | butyl | H | 98 | n/a | 95 | 55.3 (1.9) |
| 9 | 2i | phenyl | H | 77 | n/a | quant | >100 |
| 10 | 2j | 4-methylphenyl | H | 93 | n/a | quant | >100 |
| 11 | 2k | 2-bromophenyl | H | 94 | n/a | 75 | 34.0 (2.9) |
| 12 | 2L | 2-nitrophenyl | H | 94 | n/a | 85 | 35.3 (2.2) |
| 13 | 2m | 2-pyridyl | H | 94 | n/a | 25 | >100 |
| 14 | 2n | 3-pyridyl | H | 60 | n/a | 30 | >100 |
| 15 | 2o | 2-thiophenyl | H | 82 | n/a | 75 | >100 |
| 16 | 3a | chloromethyl | CH3 | (above) | 82 | 34 | >100 |
| 17 | 3b | ethyl | CH3 | (above) | 73 | quant | >100 |
| 18 | 3c | propyl | CH3 | (above) | 98 | quant | >100 |
| 19 | 3d | butyl | CH3 | (above) | 65 | 33 | >100 |
Vinyl sulfonamide inhibitor 2e and methylazido sulfonamide inhibitor 2f could not be prepared from commercially available sulfonyl chlorides, and were synthesized as described in Scheme 2. Thus, protected amino acid 4 was treated with chloroethanesulfonyl chloride to generate intermediate sulfonamide 7, which underwent elimination in situ to generate vinyl sulfonamide 4e, which was then deprotected as before to 2e. Likewise, protected chloromethyl sulfonamide 4c was treated with sodium azide to generate 4f, which was subsequently deprotected to 2f.33
Scheme 2.

Synthesis of sulfonamides 2e and 2f. Reagents and conditions: (a) chloroethanesulfonyl chloride (1.4 eq), Et3N (2.5 eq); (b) TFA (20 eq), DCM; (c) NaN3 (4.0 eq), 15-crown-5 (0.5 eq), DMF, overnight, 95 °C.
Using the methodology described in Scheme 1, a library of inhibitor candidates was prepared, to explore a variety of simple alkyl (2a–2d, 2f–2h), vinyl (2e) and aryl (2i–2L) fragments at the R1 position, and also to explore the effect of incorporating an N-methyl substituent into the sulfonamide inhibitor structure (3a–3d) (Table 1). In addition to substituted aromatic rings, three heteroaromatic ring systems were also evaluated: 2m–2o, with the rational that at least 2m and 2o contain hydrogen bond acceptor atoms (N and S) which could potentially enhance zinc binding with GCP2.
Once prepared, the sulfonamide inhibitors 2a–2o and 3a–3d were assayed for inhibition against purified GPC2 using conditions that were described previously 34 with results indicated as 50% inhibitory (IC50) values. The majority of the compounds screened were inactive at 100 μM. However, we found that several small alkyl inhibitors in compound series 2 possessed measurable potency, including the chloromethyl (2c), ethyl (2d), and propyl (2g) sulfonamides, with the latter inhibitor being the most potent inthis series. The butyl homolog (2h) obtained relatively weak potency (55 μM) but was still more potent than the majority of the inactive inhibitors that lacked notable potency at 100 μM. Overall, comparing the short N-alkyl sulfonamide inhibitors it appears that propyl (2g) has the optimal carbon chain length (IC50 5 μM). The ethyl (2d) and bioisosteric chloromethyl (2c) sulfonamide obtained reduced potency (IC50 11.6 and 17.2 μM, respectively) while methyl and trifluoromethyl sulfonamides were inactive. In terms of aromatic sulfonamides, the 2-bromophenyl (2k) and 2-nitrophenyl (2L) sulfonamides were more potent than others in this class, but could not compete with the short alkyl sulfonamide inhibitors. The heteroaromatic sulfonamide inhibitors (2m–2o) appeared to be devoid of potency.
To establish a tentative mode of binding of the most potent set of inhibitors, computational docking was employed. As such, the entire set of sulfonamides (2a–2o and 3a–3f) was docked into a high-resolution X-ray crystal structure (PDB=2C6C)35, which was co-crystallized with the potent phosphonate inhibitor GPI-18431. Additionally, the compounds were docked into a different crystal structure (PDB=3D7H),24 which was co-crystallized with the urea inhibitor DCIBzL. Docking of each inhibitor was performed with FRED2 (OpenEyes) employing a library of ligand conformations generated by OMEGA (OpenEyes) with protonation states determined by the small molecule ionization tool implemented in PIPELINE PILOT (Accelrys).
Upon docking this series of inhibitors, several observations were made:
the N-alkyl sulfonamide inhibitors do not adopt the binding mode of the other well characterized inhibitor classes (phosphonate, phosphoramidate, urea, etc), in which the glutamate clearly fits in the S1′ binding pocket making connections to Lys699 and Asn257 (gamma-carboxyl); and Arg210 (alpha-carboxyl) (Figure 3a). This was concluded by examining not only at the lowest energy binding mode of the inhibitors, but an entire selection of 20 alternate poses for the alkyl inhibitors. The steric features simply exclude this possibility for the alkyl sulfonamide inhibitors, as well as the aromatic and N-methyl inhibitors.
The binding mode of the alkyl sulfonamide inhibitors appears ‘backward’, in which the glutamate and R group on the sulfonamide are configured oppositely when compared to the conventional mode. Notably, there are known inhibitors that lack a glutamate residue, indicating that filling the S1’ pocket with glutamate is not necessary for binding.36,23 The new binding mode was observed consistently with a series of alkyl sulfonamide compounds, docking into two different X-ray crystal structures (2G6C and 3D7H).
-
The feature we observe with our most potent inhibitor (propyl, 2g, IC50=5 μM) is the near filling of a cavity flanked by Asn257 and Lys699 (Figure 3c). The less potent ethyl analog (2d) (IC50 = 10 μM) (Figure 3b) and chloromethyl analog (IC50=17.2 μM) fills this cavity less completely. In these three molecules (among other small alkyl sulfonamides), a key interaction occurs between the glutamate α-carboxyl and the catalytic zinc atoms. One sulfonamide oxygen atom makes an additional putative interaction with zinc, while the other oxygen interacts with Arg210, and the phenols of Tyr700 and Tyr552
The relatively poor butyl sulfonamide inhibitor (IC50=55 μM) does not fit well into this cavity, due to its size, creating a steric clash (Figure 3d). The clash is resolved by forcing the alpha carboxyl group away from the GCP2 catalytic zinc atoms, destroying a potentially valuable interaction. This most likely costs the inhibitor in a significant drop in potency.
Figure 3.

Binding mode of sulfonamide GCP2 inhibitors. (a) conventional mode of binding for phosphonates, phosphoramidates, ureas determined by X-ray crystallography, as demonstrated by inhibitor GPI-18431 (PDB = 2C6C) (b) tentative binding mode for alkyl sulfonamide inhibitors, in this case the ethyl sulfonamide (2d), which has limited hydrophobic contacts with the cavity flanked by Lys699 and Asn257; (c) tentative binding model for propyl sulfonamide (2g), which fills the Asn257 and Lys699 cavity more completely, possibly explaining its improved potency; (d) tentative binding mode for the butyl sulfonamide inhibitor (2h), which has relatively low potency (IC50=55 μM), possibly explained by the extra alkyl CH2 group forcing the α-carboxyl away from GCP2 zinc atoms, eliminating this key proposed interaction.
(4) The N-methyl inhibitors generally dock in an orientation preventing the key interactions from occurring (particularly between the α-carboxyl group and the zinc atoms), and their inactivity is not surprising. The aromatic sulfonamides also adopt a different binding mode, closer to the conventional inhibitor binding mode, although they are still unable to make the key γ-carboxyl interactions with Asn257, Lys699 that the conventional inhibitors make (data not shown).
Conclusion and Future Directions
A small library of N-substituted glutamyl sulfonamide inhibitors of GCP2 was prepared. Inhibition data was determined for the series of compounds. While the majority of the inhibitors were inactive, a small collection of inhibitors containing short alkyl sulfonamide substituents possessed low micromolar activity, with the best compound being the propyl sulfonamide (2g). The relative potencies were rationalized by a model generated through computational docking. Namely the most potent compound (2g) optimally filled a pocket flanked by the residues Asn257 and Lys699. We suggest that sulfonamides that contained fewer or more or more carbon atoms were unable to fill this cavity, and in turn achieved reduced potency. Based on the SAR model determined by modeling, we plan to generate additional compounds which we expect will obtain improved electrostatic interactions (particularly with Lys699), and these results will be reported in due course.
Materials and Methods
IC50 Determinations for PSMA Inhibition
Inhibition studies were performed as described previously with only minor modifications 34. Working solutions of the substrate (N-[4-(phenylazo)-benzoyl]-glutamyl-g-glutamic acid, PABGgG) and inhibitors were made in TRIS buffer (50 mM, pH 7.4 containing 1% Triton X-100). Working solutions of purified PSMA (50 μg/mL) were diluted in TRIS buffer (50 mM, pH 7.4 containing 1% Triton X-100) to provide from 15% to 20% conversion of substrate to product in the absence of inhibitor. A typical incubation mixture (final volume 250 μL) was prepared by the addition of either 25 μL of an inhibitor solution or 25 μL TRIS buffer (50 mM, pH 7.4 containing 1% Triton X-100) to 175 μL TRIS buffer (50 mM, pH 7.4 containing 1% Triton X-100) in a test tube. PABGgG (25 μL, 100 mM) was added to the above solution. The enzymatic reaction was initiated by the addition of 25 μL of the PSMA working solution. In all cases, the final concentration of PABGgG was 10 mM while the enzyme was incubated with five serially diluted inhibitor concentrations providing a range of inhibition from 10% to 90%. The reaction was allowed to proceed for 15 min with constant shaking at 37 °C and was terminated by the addition of 25 μL methanolic TFA (2% trifluoroacetic acid by volume in methanol) followed by vortexing. The quenched incubation mixture was quickly buffered by the addition of 25 μL K2HPO4 (0.1 M), vortexed, and centrifuged (10 min at 7,000g). An 85 μL aliquot of the resulting supernatant was subsequently quantified by HPLC as previously described.37,38 IC50 values were calculated using KaleidaGraph 3.6 (Synergy Software).
Synthesis
All solvents used in reactions were both anhydrous and obtained as such from commercial sources. 1H and 13C NMR spectra were recorded on a Bruker DRX 300 MHz or Bruker Avance 500 MHz spectrometer. 1H NMR chemical shifts are relative to TMS (δ = 0.00 ppm) or CDCl3 (δ = 7.26 ppm). 13C NMR chemical shifts are relative to CDCl3 (δ = 77.23 ppm). High resolution mass spectra were obtained by the University of Notre Dame Mass Spectrometry & Proteomics Facility, Notre Dame, IN 46556–5670 using ESI either by direct infusion on a Bruker micrOTOF-II or by LC elution via an ultra-high pressure Dionex RSLC with C18 column coupled with a Bruker micrOTOF-Q II.
General procedure for sulfonamide formation reactions
2-(Ethane-1-sulfonylamino)-pentanedioic acid di-tert-butyl ester (5e)
To a solution of HCl * Glu(OtBu)-OtBu (4) (100 mg, 0.338 mmol) in DCM (3.3 ml) was added Et3N (0.141 ml, 1.014 mmol, 3.0 eq) and ethanesulfonyl chloride (0.034 ml, 0.355 mmol, 1.05 eq). The reaction was stirred 1–2h, whereupon TLC showed a new spot suspected to be product, and the majority of the amino acid starting material appeared consumed. The product mixture was dissolved in diethyl ether (25 ml), washed with aq. NaHCO3 (5%) (25 ml), HCl (1M) (25 ml), and NaCl (satd. aq) (25 ml). The organic layer was dried over Na2SO4, and concentrated in vacuo to generate the product (79 mg, 65%) as a colorless oil. 1H NMR (500 mHz, CDCl3) δ 1.37 (t, 3H, J = 7 Hz), 1.43 (s, 9H), 1.48 (s, 9H), 1.84–1.89 (m, 1H), 2.08–2.12 (m, 1H), 2.83–2.41 (m, 2H), 2.98–3.03 (m, 2H), 3.96–4.01 (m, 1H), 5.07 (d, 1H, J = 9 Hz). 13C NMR (125 mHz, CDCl3) δ 8.8, 28.6, 28.7, 29.2, 31.8, 48.4, 56.6, 81.5, 83.6, 171.9, 172.6.
General procedure for TFA deprotection reactions
2-Ethenesulfonylamino-pentanedioic acid (2e)
To a solution of 2-Ethenesulfonylamino-pentanedioic acid di-tert-butyl ester (5e) (0.026 g, 0.076 mmol) was added DCM:TFA (2:1) (0.5 ml). The solution was stirred at RT for 5h, whereupon TLC showed conversion of starting material to product. Solvent was removed in vacuo to generate the product as a colorless film (0.018 g, 100%). NMR and MS data tabulated in supporting information.
General procedure for N-methylation reactions
2-[Methyl-(propane-1-sulfonyl)-amino]-pentanedioic acid di-tert-butyl ester (6c)
To a solution of 2-propanesulfonylamino-pentanedioic acid di-tert-butyl ester (5g) (15 mg, 0.041 mmol) in DMF (3 ml) was added K2CO3 (0.023 g, 0.164 mmol) and 18-crown-6 (11 mg, 0.041 mmol, 1.0 eq), followed by iodomethane (0.010 ml, 0.164 mmol). The solution was heated to 50 °C for 3h, whereupon TLC showed consumption of starting material and formation of product. The reaction mixture was taken up in ether-ethyl acetate (1:1) (20 ml), washed with H2O (3 × 20 ml) followed by NaCl (satd. aq.) (20 ml). Subsequently the organic layer was dried over anhydrous Na2SO4, and concentrated in vacuo to provide the product as a colorless oil (11 mg, 73%). 1H NMR (300 mHz, CDCl3) δ 1.04 (t, 3H, J = 7 Hz), 1.45 (s, 9H), 1.47 (s, 9H), 1.83–1.90 (m, 2H), 2.31–2.36 (t, 2H, J = 7 Hz), 2.81 (s, 3H), 2.97–3.04 (m, 2H), 4.41–4.46 (m, 1H).
2-Azidomethanesulfonylamino-pentanedioic acid di-tert-butyl ester (4f)
To a solution of 2-chloromethanesulfonylamino-pentanedioic acid di-tert-butyl ester (5c) (23 mg, 0.062 mmol) in DMF (1 ml) was added sodium azide (16 mg, 0.246 mmol, 4 eq) and 15-crown-5 (0.006 ml, 0.031 mmol, 0.5 eq). The mixture was heated overnight at 100 °C. The reaction mixture was taken up in H2O (15 ml) and extracted with EtOAc (4 × 20 ml). The organic layer was combined and concentrated in vacuo. The product was isolated after purification by flash column chromatography (4:1 hexanes:ethyl acetate) to yield the product as a colorless oil (15 mg, 66% yield). 1H NMR (300 mHz, CDCl3) δ 1.45 (s, 9H), 1.49 (s, 9H), 1.83–1.93 (m, 1H), 2.08–2.19 (m, 1H), 2.39–2.44 (m, 2H), 4.07–4.11 (m, 1H), 4.25 (d, 1H, J = 15 Hz), 4.34 (d, 1H, J = 15 Hz), 5.35 (d, 1H, J = 6 Hz). 13C NMR (75 mHz, CDCl3) δ 28.1, 28.3, 28.4, 31.3, 56.8, 67.1, 81.3, 83.5, 170.8, 172.3.
Supplementary Material
Figure 1.

Representative inhibitors of GCP2.
Acknowledgments
This work was supported in part by grants from the National Institutes of Health (SC2 GM-084867-01 A1 and 1R01CA140617-01A2). Mass-spectrometry services at University of Notre Dame were supported by grants from the National Science Foundation (CHE-0741793). Lastly, the authors would like to acknowledge OpenEyes Inc. and Accelrys Inc. for providing free site licenses to the academic community.
References and Notes
- 1.Tsukamoto T, Wozniak KM, Slusher BS. Drug Discov Today. 2007;12:767–76. doi: 10.1016/j.drudis.2007.07.010. [DOI] [PubMed] [Google Scholar]
- 2.Zhou J, Neale JH, Pomper MG, Kozikowski AP. Nat Rev Drug Discov. 2005;4:1015–26. doi: 10.1038/nrd1903. [DOI] [PubMed] [Google Scholar]
- 3.Whelan J. Drug Discov Today. 2000;5:171–172. doi: 10.1016/s1359-6446(00)01486-0. [DOI] [PubMed] [Google Scholar]
- 4.Vornov JJ, Wozniak K, Lu M, Jackson P, Tsukamoto T, Wang E, Slusher B. Ann N Y Acad Sci. 1999;890:400–5. doi: 10.1111/j.1749-6632.1999.tb08019.x. [DOI] [PubMed] [Google Scholar]
- 5.Kamiya H, Shinozaki H, Yamamoto C. J Physiol. 1996;493(Pt 2):447–55. doi: 10.1113/jphysiol.1996.sp021395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carter RE, Feldman AR, Coyle JT. Proc Natl Acad Sci U S A. 1996;93:749–53. doi: 10.1073/pnas.93.2.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haffner MC, Kronberger IE, Ross JS, Sheehan CE, Zitt M, Muhlmann G, Ofner D, Zelger B, Ensinger C, Yang XJ, Geley S, Margreiter R, Bander NH. Hum Pathol. 2009;40:1754–61. doi: 10.1016/j.humpath.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 8.Chang SS, O’Keefe DS, Bacich DJ, Reuter VE, Heston WD, Gaudin PB. Clinical cancer research : an official journal of the American Association for Cancer Research. 1999;5(10):2674–81. [PubMed] [Google Scholar]
- 9.Tang H, Brown M, Ye Y, Huang G, Zhang Y, Wang Y, Zhai H, Chen X, Shen TY, Tenniswood M. Biochem Biophys Res Commun. 2003;307:8–14. doi: 10.1016/s0006-291x(03)01119-7. [DOI] [PubMed] [Google Scholar]
- 10.Chang SS. Curr Opin Investig Drugs. 2004;5:611–5. [PubMed] [Google Scholar]
- 11.Chang SS. Rev Urol. 2004;6(Suppl 10):S13–8. [PMC free article] [PubMed] [Google Scholar]
- 12.Elsasser-Beile U, Buhler P, Wolf P. Curr Drug Targets. 2009;10:118–25. doi: 10.2174/138945009787354601. [DOI] [PubMed] [Google Scholar]
- 13.Bander NH, Milowsky MI, Nanus DM, Kostakoglu L, Vallabhajosula S, Goldsmith SJ. J Clin Oncol. 2005;23:4591–601. doi: 10.1200/JCO.2005.05.160. [DOI] [PubMed] [Google Scholar]
- 14.Foss CA, Mease RC, Fan H, Wang Y, Ravert HT, Dannals RF, Olszewski RT, Heston WD, Kozikowski AP, Pomper MG Johns Hopkins University, BMUSA. Clinical cancer research : an official journal of the American Association for Cancer Research. 2005;11(11):4022–8. doi: 10.1158/1078-0432.CCR-04-2690. [DOI] [PubMed] [Google Scholar]
- 15.Pomper MG, Musachio JL, Zhang J, Scheffel U, Zhou Y, Hilton J, Maini A, Dannals RF, Wong DF, Kozikowski AP. Mol Imaging. 2002;1:96–101. doi: 10.1162/15353500200202109. [DOI] [PubMed] [Google Scholar]
- 16.Guilarte TR, McGlothan JL, Foss CA, Zhou J, Heston WD, Kozikowski AP, Pomper MG Department of Environmental Health Sciences, JHSoPHBMDUSA. Neuroscience letters. 2005;387(3):141–4. doi: 10.1016/j.neulet.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 17.Humblet V, Lapidus R, Williams LR, Tsukamoto T, Rojas C, Majer P, Hin B, Ohnishi S, De Grand AM, Zaheer A, Renze JT, Nakayama A, Slusher BS, Frangioni JV. Mol Imaging. 2005;4:448–62. doi: 10.2310/7290.2005.05163. [DOI] [PubMed] [Google Scholar]
- 18.Banerjee SR, Foss CA, Castanares M, Mease RC, Byun Y, Fox JJ, Hilton J, Lupold SE, Kozikowski AP, Pomper MG. J Med Chem. 2008;51:4504–17. doi: 10.1021/jm800111u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mease RC, Dusich CL, Foss CA, Ravert HT, Dannals RF, Seidel J, Prideaux A, Fox JJ, Sgouros G, Kozikowski AP, Pomper MG. Clin Cancer Res. 2008;14:3036–43. doi: 10.1158/1078-0432.CCR-07-1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lapi SE, Wahnishe H, Pham D, Wu LY, Nedrow-Byers JR, Liu T, Vejdani K, VanBrocklin HF, Berkman CE, Jones EF. J Nucl Med. 2009;50:2042–8. doi: 10.2967/jnumed.109.066589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dijkgraaf I, Boerman OC. Cancer Biother Radiopharm. 2009;24:637–47. doi: 10.1089/cbr.2009.0694. [DOI] [PubMed] [Google Scholar]
- 22.Barinka C, Rovenska M, Mlcochova P, Hlouchova K, Plechanovova A, Majer P, Tsukamoto T, Slusher BS, Konvalinka J, Lubkowski J. J Med Chem. 2007;50:3267–73. doi: 10.1021/jm070133w. [DOI] [PubMed] [Google Scholar]
- 23.Mesters JR, Henning K, Hilgenfeld R. Acta Crystallogr D Biol Crystallogr. 2007;63:508–13. doi: 10.1107/S090744490700902X. [DOI] [PubMed] [Google Scholar]
- 24.Barinka C, Byun Y, Dusich CL, Banerjee SR, Chen Y, Castanares M, Kozikowski AP, Mease RC, Pomper MG, Lubkowski J. J Med Chem. 2008;51:7737–43. doi: 10.1021/jm800765e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peng XQ, Li J, Gardner EL, Ashby CR, Jr, Thomas A, Wozniak K, Slusher BS, Xi ZX. Eur J Pharmacol. 2010;627:156–61. doi: 10.1016/j.ejphar.2009.10.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bonnac L, Innocenti A, Winum J-Y, Casini A, Montero J-L, Scozzafava A, Barragan V, Supuran CT. J Enzyme Inhib Med Chem. 2004:275–278. doi: 10.1080/14756360410001689522. [DOI] [PubMed] [Google Scholar]
- 27.Temperini C, Cecchi A, Scozzafava A, Supuran CT. Org Biomol Chem. 2008;6:2499–506. doi: 10.1039/b800767e. [DOI] [PubMed] [Google Scholar]
- 28.Supuran CT. Nat Rev Drug Discov. 2008;7:168–181. doi: 10.1038/nrd2467. [DOI] [PubMed] [Google Scholar]
- 29.Scozzafava A, Owa T, Mastrolorenzo A, Supuran CT. Curr Med Chem. 2003;10:925–53. doi: 10.2174/0929867033457647. [DOI] [PubMed] [Google Scholar]
- 30.Supuran CT, Casini A, Scozzafava A. Med Res Rev. 2003;23:535–58. doi: 10.1002/med.10047. [DOI] [PubMed] [Google Scholar]
- 31.Supuran CT, Innocenti A, Mastrolorenzo A, Scozzafava A. Mini Rev Med Chem. 2004;4:189–200. doi: 10.2174/1389557043487402. [DOI] [PubMed] [Google Scholar]
- 32.Vitharana D, France JE, Scarpetti D, Bonneville GW, Majer P, Tsukamoto T. Tetrahedron: Asymmetry. 2002;13:1609–1614. [Google Scholar]
- 33.Zhou A, Rayabarapu D, Hanson PR. Organic Letters. 2008;11:531–534. doi: 10.1021/ol802467f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu LY, Anderson MO, Toriyabe Y, Maung J, Campbell TY, Tajon C, Kazak M, Moser J, Berkman CE. Bioorg Med Chem. 2007;15:7434–43. doi: 10.1016/j.bmc.2007.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mesters JR, Barinka C, Li W, Tsukamoto T, Majer P, Slusher BS, Konvalinka J, Hilgenfeld R. Embo J. 2006;25:1375–84. doi: 10.1038/sj.emboj.7600969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang H, Byun Y, Barinka C, Pullambhatla M, Bhang HE, Fox JJ, Lubkowski J, Mease RC, Pomper MG. Bioorg Med Chem Lett. 2010;20:392–7. doi: 10.1016/j.bmcl.2009.10.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maung J, Mallari JP, Girtsman TA, Wu LY, Rowley JA, Santiago NM, Brunelle AN, Berkman CE. Bioorg Med Chem. 2004;12:4969–79. doi: 10.1016/j.bmc.2004.06.031. [DOI] [PubMed] [Google Scholar]
- 38.Anderson MO, Wu LY, Santiago NM, Moser JM, Rowley JA, Bolstad ES, Berkman CE. Bioorg Med Chem. 2007;15:6678–6686. doi: 10.1016/j.bmc.2007.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.