

1. Introduction
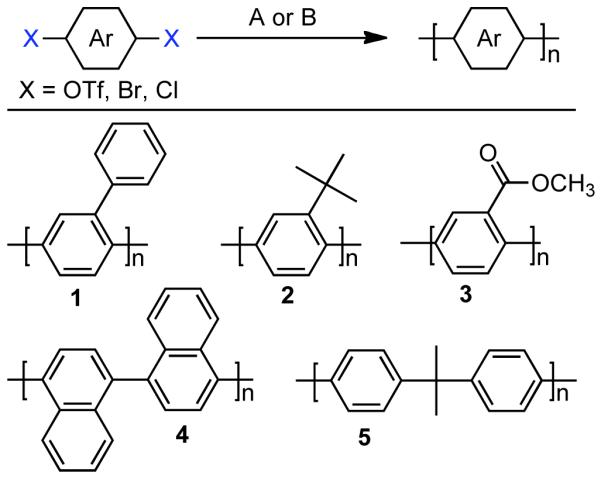
1.1. Overview and Historical Background of Nickel-Catalyzed Cross-Couplings
Synthetic organic chemists are Ångstrom-scale architects who take great pleasure in drafting the blueprints for the edification of intricate molecular topologies and architectures. In order to turn the molecular dreams into a reality, synthetic chemists must ultimately roll up their sleeves and become molecular construction workers building their structure brick by brick. From the dawn of the modern chemistry in the ages of Boyle, Dalton and Lavoisier to the present days the tools for molecular construction have evolved, progressing through notable paradigm shifts from the development of the atomistic theory to the rigorous application of the retrosynthetic method focusing on the enantioselective synthesis of medicinal natural products.1,2,3 More recently academic and industrial focus has moved toward efficient chemistry utilizing low-cost and green reagents in transformations designed with high atom-economy starting for bio-sourced feedstocks as well as beyond the small molecule in strategies to produce designed macromolecular materials4 and supramolecular ensembles.5,6 Numerous strategies have been explored for the synthesis of relatively large organic molecules and polymers, but typically the coupling of pre-fabricated units together is more expeditious.7,8,9,10,11,12,13,14,15
Early in the development of homocoupling, the joining of identical chemical fragments, and cross-coupling, early transition metals such as Ni, were identified as useful reagents and catalysts. However, in the intervening years more attention was invested in the development of later transition metal catalysis, particularly Pd-catalyzed Heck,16,17 Hiyama,18 Kumada,19 Negishi,20,21 Suzuki-Miyaura,22,23 Sonogashira,24 Stille25,26 coupling techniques due to some advantages in terms of the diversity and tenability of preparable catalysts, their oxidative and aqueous stability, and relatively facile isolation and structural analysis of their complexes which aided mechanistic and methodologic developments. These, typically Pd-catalyzed,27 coupling reactions employed a diversity of organometallic transmetalating reagents or unsaturated functionalities for coordination/insertion chemistry, but nearly uniformly required aryl, vinyl, allyl or sometimes alkyl halides as electrophiles. While this manuscript was under revision the pioneering work on Pd-catalyzed cross-coupling was recognized with the 2010 Nobel Prize in Chemistry for Professors Heck, Negishi, and Suzuki. However, there is growing interest in using more available, economical, and environmentally friendly phenol and enol-derived electrophiles.
1.2.The Value of Phenol- and Enol-Derived Electrophiles
Industrial chemists are now able to provide a wide array of aryl, polyaryl, vinyl, allyl, and alkyl halides to the hands of bench chemists. However, such species are far less available from natural sources and are certainly not used as coupling partners in biosynthetic pathways. A key benefit to phenol- and enol-derived electrophiles is the ready accessibility of these substrates. In the case of phenols, such compounds are naturally abundant or can be readily prepared from other easily accessed aromatic species.28 A Scifinder search reveals that over 50,000 phenol and aryl polyol derivatives are commercially available.29 Moreover, for certain heteroaromatic compounds, a phenol-type substituent can often be derived from a lactam precursor. The advantages are similar for enol-derived electrophiles, as cross-coupling partners of this class can be accessed by enolization and subsequent trapping of readily available carbonyl compounds.
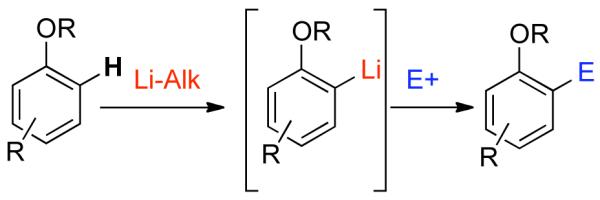
Other advantages to using phenol-derived electrophiles exist. Most notably, oxygenation on the aromatic ring can be used to introduce additional substituents via a number of pathways including electrophilic aromatic substitution.30 Depending on the nature of the O-substituent and the electrophile it is often possible to control the preferred formation of para or ortho substituted products. Ortho substitution of phenol derivatives can also be achieved using directed ortho-metallation (DoM).31 Using this methodology, numerous functional groups, such as phenols, ethers, carbamates, and sulfamates can direct ortho-lithiation (Scheme 1). Subsequent quenching of the resulting organolithium species with an electrophilic species E+ provides the ortho-substituted product, which could potentially be used as a cross-coupling substrate. Recently, aryl pivalates32 and aryl carbamates33,34 have been ortho-arylated directly using palladium-catalyzed functionalization methodologies.
Scheme 1.
The advantage of using phenol-derived electrophiles in coupling reactions is readily apparent. Nevertheless, the implementation of such a strategy is often not as simple as for aryl halides, largely due to a higher C-O bond strength relative to C-Cl, C-Br, and C-I. Often the energy of activation of the C-O bond rivals the energy for deleterious side-reactions and the selectivity of the coupling strategy is diminished. Typically, the C-O bond must be first activated, most often through conversion of the phenols to more reactive sulfonates, ideally through the use of inexpensive methanesulfonyl chloride. However, some recent examples utilizing alternative activation as esters or carbamates, or even directly employing ethers and phenols have been reported.
1.3. Ni, Not Just an Alternative to Pd and Pt
From the perspective of economics, Ni is clearly more desirable than the later elements in the d10 group. Ni is a commodity metal with a cost of roughly $1.20 per mol, whereas Pd and Pt are precious metals, which command a significantly higher price of $1,500 and $10,000 per mol, respectively.35 Thus, unless a process is viable with very low levels of Pd, or Pt can be used and recycled, or very high levels of Ni are required, a Ni-catalyzed approach would be preferred on a cost-basis. In addition to often advantageous economics, Pd-catalysis and Pt-catalysis is not as readily applied to coupling reactions involving phenol-derived electrophiles. Despite cohabitation of group 10, Pt ([Xe]4f145d96s1), is chemically dissimilar from its lower period cousin Pd ([Kr]4d10) and Ni ([Ar]4s23d8), more readily accessing higher oxidation states (+4) and participating in oxygenation and other chemistries outside the scope of this review. Pd is typically observed in the (0 and +2) oxidation states and is very efficient in coupling reactions involving C-X electrophiles. Recent advances in ligand design36,37 mostly from the laboratory of Buchwald have provided improved reactivity toward phenol-derived electrophiles. Nevertheless, the early transition metal Ni (found in 0, +2 as well as +1,+3 oxidation states), is more nucleophilic on account of its smaller size and can harness phenol derived as well as other less reactive electrophiles, employing typically less exotic ligands or even under ligand-free conditions. Therefore, we find Ni to be a privileged reagent for cross-coupling from the standpoints of economics and versatility.
1.4. Scope of the Review
This review will cover the historical development through the present state-of-the art in the Ni-catalyzed homocoupling, cross-coupling, and functionalization of C-O bonds. The functionalization of C-O bonds is not only critical for installing the ultimate functionality of the target molecule but also for providing reactive handles for subsequent cross-coupling reactions. The review is intended to be comprehensive for Ni-mediated chemistry up until late July 2010. Work utilizing other transition metals, notably Pd will be covered less comprehensively and typically for the purpose of historical context and comparison. In some cases extensive discussion of Ni-catalyzed transformations of organohalides was necessary in order to understand the development and the mechanism in the corresponding sulfonates, ethers, or esters.
The review is organized into sections according to the type of phenol-derived electrophile employed. These sections are further divided between homocoupling, cross-coupling, and functionalization and divided into subchapters according to either the transmetallating agent employed or the type of Ni-catalyst used.
2. Nickel-Catalyzed Reactions of Aryl and Vinyl Sulfonates and Sulfates
2.1. Homocoupling of Aryl and Vinyl Sulfonates and Sulfates
2.1.1. Homocoupling of Aryl Halides to Produce Biaryls
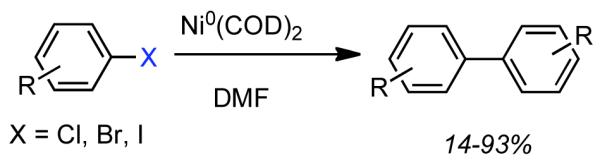
Ni0 complexes first attracted attention as reagents for organic coupling reactions through early work of Wilke on the cyclooligomerization of butadiene.38 A. Yamamoto and Saito elaborated Ni0 mediated reactions involving olefins and established that both linear oligomerization and cyclooligomerization proceeded through a coordination/insertion mechanism.39,40 Interest in the Ni0-catalyzed homocoupling of aryl halides and pseudohalides emerged in 1966, when Saito and A. Yamamoto prepared Et2NiII(bpy), and determined that upon heating it decomposed to butane or ethane and ethylene via free-radical coupling or disproportionation, respectively.41 1971 witnessed the pioneering disclosure by Semmelhack that stoichiometric Ni0(COD)2 could mediate homocoupling of aryl chlorides, bromides, and iodides in DMF at 25-45 °C (Scheme 2).42 This technique was immediately recognized as a very mild, robust and selective approach to the synthesis of biaryls and heterobiaryls. At the time state-of-the-art Ullmann coupling approaches frequently required temperatures exceeding 200 °C, while competitive aryllithium and Grignard techniques were intolerant toward electrophilic substrates. Semmelhack investigated alternative reagents such as Ni0(CO)4, but its use was complicated by competitive CO insertion. It was demonstrated that the rate of homocoupling depended strongly on the nature of the leaving group with -I > -Br > -Cl >> -OSO2R~0. A tentative three-step mechanism was invoked: (1) oxidative addition of Ar-X to Ni0(COD)2 to form NiIIArX(COD)2, (2) further oxidative addition of Ar-X to form NiIVAr2X2, followed by (3) reductive elimination to generate biaryl and NiIIX2. The involvement of a NiIV species was supported by faster consumption of Ar-X than Ni0(COD)2. While clearly a promising discovery, limitations to this early embodiment of Ni0-mediated homocoupling were evident, such as poor reactivity with ortho-substituted aryl halides, decomposition of the catalyst at elevated temperature, reduction of the C-X bond when protic substrates were employed, and limited solvent scope.
Scheme 2.
In subsequent studies, Semmelhack explored the utility of Ni0-mediated homocoupling in the synthesis of natural products, specifically the capability of Ni0 to facilitate the synthesis of cyclic biphenyls.43,44 Ni0(COD)2 alone, nor in conjunction with phosphine ligand additives, was able to produce any form of cyclic biphenyl synthesis. Nevertheless, precomplexed Ni0(PPh3)4 could afford cyclic biaryls of various size from acyclic bis(aryliodides) (Scheme 3). Ni0-mediated annulation was readily applied to the synthesis of dimethyl ether variant of plant derived natural product Anulsone, while the aforementioned incompatibility with ortho-substituents prohibited the elaboration of Stegnone and related compounds through this approach.
Scheme 3.

As first reported by Tolman, Ni0(PPh3)4 is expected to exhibit appreciable dissociation in solution into Ni0(PPh3)3 and free PPh3 ligand, due to the steric bulk of PPh3.45,46 Kende realized that this dissociation phenomenon meant that Ni0(PPh3)3 is likely the active catalyst in Ni0 -mediated homocoupling and sought to develop a convenient method to prepare this highly air sensitive reagent in situ.47 Ultimately, Kende determined that NiIICl2(PPh3)2 could be converted to Ni0(PPh3)3 using Zn0 powder in the presence of excess PPh3 in deoxygenated DMF as solvent. This in situ generation of active Ni0 was harnessed in the homocoupling of diversely substituted aryl bromides and other halides (42-85% yield). Electron deficient aryl halides exhibited higher homocoupling yields than electron-rich examples. Likewise, lower yields were observed for singly ortho-substituted aryl halides, while no conversion was documented for doubly ortho-substituted aryl halides. A similarly powerful steric inhibition by ortho-substituents was noted by Semmelhack in his seminal work.42
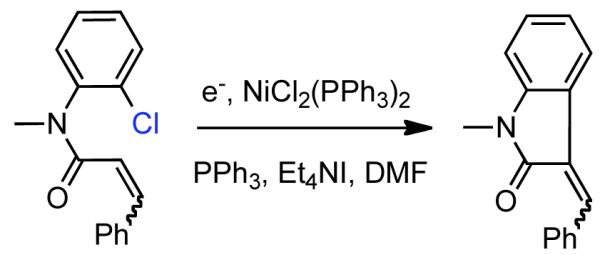
Using electrochemistry,48 Jennings provided early mechanistic insight into Ni0-catalyzed homocoupling of phenyl bromide.49 Here, Ni0 was generated from NiII(acac)2 in situ via electrochemical reduction in DMF in the presence or absence of PPh3 as a stabilizing ligand. In the absence of metal acetylacetonato (acac), no coupling product was formed, confirming the involvement of low-valent Ni complexes generated in situ. Alternatively, Ban reported the electrochemical generation of Ni0(PPh3)3 from NiCl2(PPh3)2, in DMF in the presence of additional PPh3 and Et4NI as an electrolyte.50 Ni0(PPh3)3 generated in this fashion was suitable for the homocoupling of some aryl halides. Of the substrates tested bromobenzene provided the highest yield in the shortest time (80% in 4h). Aryl chlorides or electron-rich aryl bromides reacted slower and often did not achieve complete conversion. Electron deficient aryl bromides seemed to exhibit phenyl transfer from triphenylphosphine ligand. Like Ni0 generated in situ from chemical reduction,51,52 electrochemically generated Ni0(PPh3)3 was synthetically useful and was able to mediate intramolecular Heck-addition to form indoles (Scheme 4). Generation of Ni0 from NiIIX2(PPh3)2 via electrochemical reduction in THF/HMPT and its ability to mediate the homocoupling of aryl bromides and chlorides was also demonstrated. 53
Scheme 4.
In 1977, Kumada realized that a major deficiency of the emerging Ni0 mediated homocoupling reactions was the stoichiometric levels of the Ni0 or NiII precursor employed,54 and demonstrated that the homocoupling reaction could be made catalytic in Ni if stoichiometric levels of Zn0 were used. Scouting experiments revealed that the homocoupling of bromobenzene to generate biphenyl could provide a high yield (89%) when the following ratio of the reactants was employed at 50 °C [PhBr]o:[Zn0]o:[PPh3]o:[NiIICl2(PPh3)2]o = 1 : 1 : 0.4 : 0.5. Lower levels of PPh3, NiIICl2(PPh3), or the use of other solvents than DMF such as MeCN, THF, or EtOH resulted in diminished yields. Under these catalytic conditions the homocoupling reaction was similarly successful for p-bromotoluene, p-bromoanisole, methyl p-bromobenzoate, and p-bromoacetophenone. Lower yields were achieved for ortho-substituted aryl bromides and heterocyclic thienyl bromide. At the time of this work, the prevailing assumption was that Zn0 served as a reducing agent, rather than as a precursor to organozinc intermediates.55 Kumada observed a rate-acceleration of the homocoupling when iodide anions were added. The iodide anions seemed to act as electron-transfer accelerants presumably vis-à-vis a bridging interaction between Ni and Zn. Through the use of stoichiometric KI, the homocoupling yield of bromobenzene after 24 h at room temperature was increased from 24% to 81%.
Simultaneously, Caubère elaborated chemistry to modulate the basicity of NaH through the formation of so-called “Complex Reducing Reagents” (CRA)s. CRAs were defined as a mixture of NaH, sodium alkoxides and other metal salts and their preparation typically involved the treatment of alcohols with NaH.56 In particular Caubère reported a 4:2:1 mixture of NaH, t-AmONa and NiIIOAc, NiCRA, which was effective at mediating the reduction of aryl iodides, bromides, chlorides, and somewhat surprisingly fluorides under relatively mild conditions.57 NiCRA, also effectively mediated the reduction of gem-dihalocyclopropanes58 as well as alkyl, allyl, benzyl, and vinyl halides.59,60 The stereochemical outcome of the reduction of 7,7-dibromonorcarane by NiCRA, for example, indicated the potential for a radical mechanism.58 Interestingly, treatment of 1-bromonaphthalene with NiCRA in the presence of PPh3 as ligand, afforded 70% 1,1′-binaphthalene and 25% naphthalene.59 The use of 2,2′-bipyridine (bpy) as coligand in conjunction with NiCRA helped to eliminate side reactions such as aryl exchange, allowing for the homocoupling of ortho-, meta-, and para-substituted aryl chlorides and bromides.61 NiCRA could also be employed for the synthesis of bipyridines.62 In the formation of the NiCRA more H2 is generated than would be expected for complete consumption of the t-AmOH in the presence of superstoichiometric NaH. The excess H2 generated was attributed to the hydridic reduction of NiII to Ni0.63 Therefore, it is likely that in the presence of ligand, Ni0(bpy) or Ni0(PPh3)n complexes are formed in solution and the mechanism of NiCRA mediated homocoupling is akin to that of other Ni0 reagents.
Supplementary to Zn0 or hydride mediated reduction of NiII, Rieke demonstrated that reduction of NiIII2 with potassium64 or lithium65 metal in the presence of PEt3 produced a highly active fine metal powder,66 “Rieke Nickel.” Treatment of this powder with bromopentafluorobenzene resulted in the formation of an isolable trans-haloaryl NiII complex, BrNiII(PEt3)2C6F5 in 60% yield.64 In subsequent work, a preliminary demonstration of the ability of Rieke Nickel to mediate the homocoupling of iodobenzene and bromobenzene was provided,67 followed by a full exploration of the scope of Rieke Nickel catalyzed homocoupling of aryl bromides and ioides.65 In the latter study, it was concluded that the nature of the progenitor NiII salt generally did not have a significant effect and that Rieke Nickel prepared from NiIICl2, NiIIBr2, and NiIII2 were similarly effective homocoupling reagents. Nevertheless, treatment of Rieke Nickel derived from NiIIBr2 with p-bromochlorobenzene resulted in the predominately reduction product, while Rieke Ni synthesized from NiIII2 efficiently produced 4,4′-dichlorobiphenyl. It is therefore possible that iodide generate from the preparation of the Rieke Ni affected a halogen exchange to generate a more selectively reactive p-chloroiodobenzene. Though not explicitly mentioned, it is also possible that the iodide could help accelerate electron transfer processes in this reaction. In general, the most significant effects to product outcome were derived from the structure of the aryl halide. Aryl iodides were more reactive than aryl bromides after the same amount of time at comparable temperatures, though good homocoupling yields for aryl bromides could be achieved after longer time at higher temperature. Electron-deficient aryl halides were more readily homocoupled than electron-rich aryl halides. As in other catalytic systems, ortho-substitution reduced homocoupling and favored reduction of the halide, while the presence of a nitro-group completely inhibited the reaction.
In earlier work Semmelhack had suggested that the Ni-mediated homocoupling in DMF solvent using stoichiometric Ni0 preceded via sequential oxidative additions involving three distinct oxidation states of Ni: 2ArX +Ni0 ➔ ArX + ArNiIIX ➔ Ar2NiIVX2.42 However, the only evidence for NiIV in this reaction was the observation that Ar-X may be consumed faster by Ni0. On the other hand Kochi suggested that a NiI/NiIII radical-chain pathway was more likely in nonpolar solvents (Scheme 5).68,69 In this mechanism, one equivalent of Ar-X oxidatively adds to Ni0L3 to form the ArNiIIXL2 complex, whilst the second equivalent of Ar-X oxidatively adds to NiIXL3 to form ArNiIIIX2L2. Exchange of the NiII bound aryl group for a NiIII bound halide provides Ar1Ar2NiIIIXL2 and the NiIIX2L2. Reductive elimination of Ar1Ar2NiIIIXL2 regenerates NiIXL3 and produces the homocoupled biaryls. The mechanism by which NiI or NiIII is generated from NiII or Ni0 progenitors is more complex and not as well understood. In fact, it has been suggested that treatment of NiII(PPh3)3 with Ar-I produces barely any biaryls product.70 If Zn0 is present, it is likely only once this catalytic cycle, to convert NiIIX2L2 to Ni0L3. In a much later report T. Yamamoto investigated the mechanism of the Ni0 mediated dehalogenative coupling that he adopted from Semmelhack to develop a robust polymerization technique for haloaryl and haloheteroaryl monomers.71 In Semmelhack’s original disclosure, Ni0(COD)2 was used without the presence of a co-ligand. Yamamoto demonstrated that species such as Ni0(COD)bpy are more active. Of most concern was the observation via kinetic studies that the homocoupling was second-order in the nickel-complex and therefore, that oxidative addition is not the rate-determining step. The observation that the rate of the reaction was independent of aryl halide concentration conflicted Kochi and Semmelhack’s mechanisms.
Scheme 5.

Based on his previous observation of an isolable trans-haloaryl Ni-complex, ArNiIIX(PEt3)264, Rieke concluded that Ar-X + Ni0 + PEt3 ➔ ArNiIIX(PEt3)265 must represent the first step of the reaction. An alternative mechanism to Semmelhack and Kochi was suggested (Scheme 6), wherein two equivalents of ArNiIIX(PEt3) undergo metathesis/disproportionation to provide Ar2NiII(PEt3)2 which produces the homocoupled product via reductive elimination. While more reasonable than the aforementioned mechanism involving a NiII/NiIV redox cycle, there is no substantial evidence that NiII metathesis can actually occur.
Scheme 6.
In addition to Rieke Nickel, Cheng reported an electrochemical method to produce Ni0 amalgam from NiIISO4.72 Cheng’s Nickel could mediate the homocoupling of aryl bromides and iodides including polybrominated compounds to generate useful polyaromatic structures. Somewhat surprising the addition of KI to the reaction mixture allowed for the rapid homocoupling of aryl chlorides as well.
In a 1981 patent73 and in the open literature in 1986, Colon reported other methods to generate the Ni0 complex in situ from simple NiII salts.74 He found that in the presence of PPh3 additive, NiIICl2 and NiIIBr2 could be reduced by other low-valent metals in situ to form Ni0 complexes that are capable to mediate the homocoupling of chlorobenzene in quantitative yield. NiIII2·6H2O, NiII(OAc)2·4H2O, and NiII(acac)2·2H2O were less effective as precatalysts, providing a mixture of homocoupling and hydrodehalogenation under these reaction conditions. On the other hand, NiII(NO3)2·6H2O, NiIIO and NiIIF2 did not generate useful Ni0 complexes via in situ reduction with metal, and were therefore, completely incompatible with the homocoupling methodology. Of the metals investigated by Colon, Zn provided the optimum balance of rate, product selectivity (i.e. homocoupling vs dehalogenation), and yield, with Mn and Mg as close seconds. Al, Ca, and Na were far less effective and Fe provided no reaction at all. It was intriguing to note that under Semmelhack’s conditions,42 which presumably would generate the same active catalyst as the one described by Colon, Ni0(PPh3)3, only 14% yield was achieved for the homocoupling of chlorobenzene, while in the case of Colon’s system 98+% was obtained. Alternatively, in the presence of stoichiometric Ni0(PPh3)4 prepared fresh in a separate flask, but in the absence of reducing metal, only 40% yield was obtained after 24 h. Treatment of this stalled reaction with Zn after this 24 h period restarted the reaction and provided quantitative yield, clearly demonstrating that it is the excess Zn in Colon’s system that allows for the greater catalytic turnover and higher yield. An investigation of precatalysts suggested that those bearing monodentate triarylphosphine ligands were superior to both those bearing bidendate bis(diarylphosphine) ligands and trialkylphospine ligands, as the latter two classes provided for more sluggish reactions.
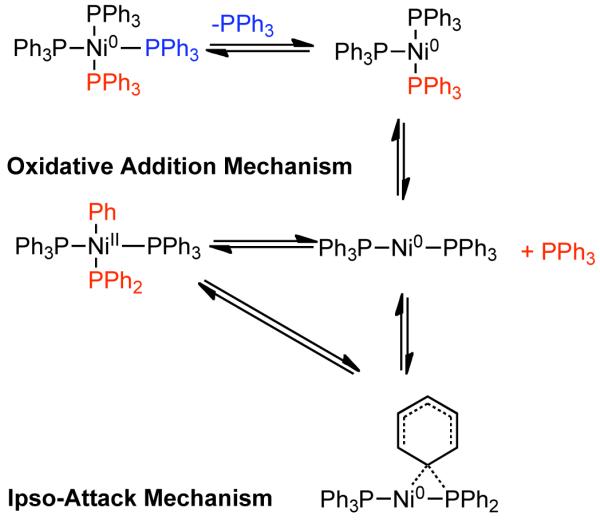
Colon noted similar reactivity trends to those documented previously by others for the various substituted chloroarenes investigated. Additionally, he observed that electron-withdrawing substituents on the arene were most effective for boosting the yield of biaryls, while electron-donating substituents provided more significant levels of monosubstituted biaryl impurities. The formation of the monosubstituted biphenyl adduct appears to be the result of aryl exchange75,76 from the phosphine ligand (Scheme 7). Experimental work demonstrated that the process of aryl-exchange was accelerated by higher reaction temperatures and suppressed by the presence of additional phosphine ligand. At room temperature Ni0(PPh3)3 is the most stable ligation state of Ni0 in solution, while at elevated temperature further ligand dissociation may occur. The resulting, Ni0(PPh3)2 might oxidatively add to PPh3 and provide PhNiIIPPh2(PPh3)2. This trans-phosphinoaryl Ni-complex might undergo subsequent cross-coupling of the Ni-bound phenyl with Ar-X (Scheme 7).76,77 Alternatively, phenyl-migration may occur directly via ipso-substitution on the phenyl ring, where the aryl carbon is transferred from P to Ni (Scheme 7).75,78 As expected, this aryl exchange process which results in monosubstituted biphenyl side products, could be diminished somewhat by the addition of bidentate ligands such as 2,2′-bipyridine, which do not dissociate as readily and provide for a more stable active catalyst. As noted by others, nitro-groups were completely incompatible due to either reductive side-reactions or potentially due to the formation of nitro-Ni0 complexes.93 Protic chlorobenzene derivatives were prone to direct reduction, for example p-chlorophenol to phenol. This reduction process can be exploited through the injection of additional H2O to provide a mild approach for aryl hydrodehalogenation.119 Reduction to the nonhalogenated arene was also more prevalent in aryl bromides and aryl iodides. The mechanism of this hydrodehalogenation may involve the formation of reductive nickel-hydrides in the presence of protic substrate or adventitious moisture. Alternatively, the trans-haloaryl Ni intermediate that results from the oxidative addition of Ni0(PPh3)3 to Ar-X may react with a proton source and decompose to the hydrodehalogenated arene.
Scheme 7.
Colon confirmed an earlier observation of Kumada54 that the addition of salts was shown to accelerate the rate of the homocoupling reaction, and specifically that this acceleration followed the trend F−<SO42−<Cl−<<Br−<I−. This rate enhancement is not thought to be due to halide substitution on the arene, as aryl iodides alone were less effective for homocoupling due to competitive reduction and the fact that no acceleration was observed in the absence of Zn.
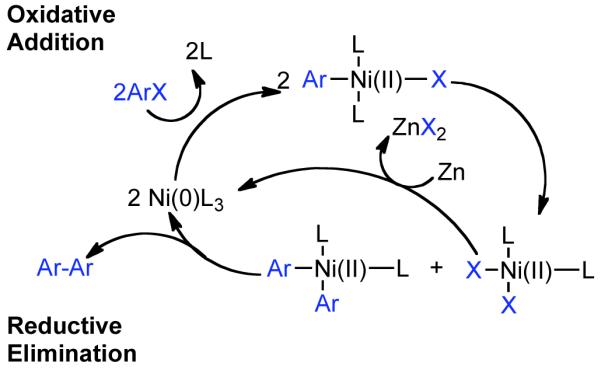
Colon suggested that the great dissimilarity of reaction conditions and associated experimental findings, suggested that various mechanisms for Ni0 catalyzed homocoupling are plausible, but nevertheless that their system was most probably described by the mechanism of Bontempelli, where all electrochemical reductions are replaced with Zn0-mediated electron-transfer (Scheme 8).79 Here, Zn0 reduction of the NiIICl2L2 precatalyst to Ni0L3, where L is a generic phosphine, often monodentate phosphine ligand, PPh3, or solvent. An aryl halide can oxidatively add to Ni0L3 or as will be described later, an aryl sulfonate, to form a σ-bonded ArNiIIXL2 complex. Further reduction with ½ equivalent of Zn0 and ligand addition can provide ArNiIL3. Subsequent oxidative addition with a second equivalent of aryl halide or aryl sulfonate would result in Ar1Ar2NiIIIXL2. Reductive elimination of this complex, would generate the homocoupled biaryls and the NiIXL3 complex. Further, reduction with ½ equivalent of Zn0 would provide Ni0L3, thereby closing the catalytic cycle. Alternatively, oxidative addition of Ar-X to NiIXL3 could furnish the ArNiIIIX2L2 complex, which can productively re-enter the main catalytic cycle by reduction with an equivalent Zn0 to form ArNiIIL3. Amatore demonstrated that this mechanism has been substantiated for aryl halides through detailed electrochemical studies.80
Scheme 8.

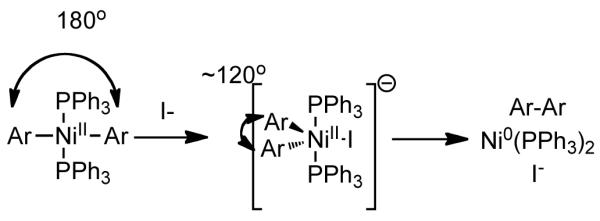
In considering the mechanism of homocoupling, Colon noted the autocatalytic role of aryl halides in the absence of additional halide additives, and the pseudo-zero-order dependence of aryl chloride in the presence of sufficient external halide. The observation of autocatalysis strongly suggests that oxidative addition is not rate-limiting. However, reductive elimination is certainly a plausible rate-limiting step. If the reaction rates are compared under conditions of suppressed auto-catalysis, it was found that the rates increased according to I<Br<Cl for iodobenzene, bromobenzene, and chlorobenzene homocoupling, respectively. These observations are the inverse of those made by Semmelhack in regard to Ni0(PPh3)4 mediated homocoupling in the absence of Zn, and therefore provide some evidence that the Zn-mediated and Zn-free reactions follow different mechanisms. Colon proposed that the rate acceleration offered by halide ion, was due to its complexation with square planar Ar2NiL2 which generates a trigonal bypyramidal structure that would enhance reductive elimination by bringing the aryl substituents closer together (Scheme 9). The only observation that contradicts reductive elimination as being the rate-determining step was that the reaction rate was dependant on the level of Zn provided, which suggests that one of the electron transfer steps may be rate determining. In this case, the role of the halide additive may be as a bridging ligand54 or to enhance the dielectric properties of the solvent, either of which should accelerate the electron transfer. From electrochemical data and other observations Colon speculated that the reduction of ArNiIICl(PPh3)2 to ArNiIL3 by Zn was the least favored and the slowest step. The role of additive halide in accelerating this step, may once again be due to the formation of a penta-coordinated nickel intermediate that could allow for more facile bridging with the Zn surface for subsequent reduction. However, the rate-determining step in any homocoupling reaction will be dependent upon the substrate and progress of the reaction. For example, arenes bearing electron-withdrawing groups or high conversion may cause oxidative addition to become rate determining.
Scheme 9.
In the same report, Colon also disclosed preliminary investigations of Ni0–catalyzed cross-coupling. While, addition of two aryl halides to the reaction mixture provided a random mixture of homocoupling and cross-coupling adducts, significant residual trans-haloaryl Ni complex remained upon termination of the reaction, particularly in the case of electron-rich aryl halides. Treatment of this complex with a second dissimilar aryl halide produced exclusively the cross-coupled product.
In search of thermally stable active catalysts, Takagi investigated the use of Ni0 generated in situ from complexes of NiIICl2 and trialkylphosphates or bidendate 1,2-bis(diethylphosphino)ethane (dppe).81,82,-83 Investigations, with iodobenzene, bromobenzene, chlorobenzene, as well as a few substituted derivatives thereof, revealed that NiIICl2(PEt3)2 in HMPA as solvent to be the most effective trialkylphosphate-based pre-catalyst. In all cases additional ligand and KI electrolyte were present in the reaction mixture. The Ni0(PEt3)n catalyst prepared in situ from the Zn-mediated reduction of NiCl2(PEt3)2 also mediated the homocoupling of heteroaromatic halides such as 3-bromoform (80%), methyl 2-bromo-5-furancarboxylate (90%), and iodothiophene (83-87%) in very high yield. However, neither 2-bromothiophene nor iodopyridines could be coupled in appreciable yield. A similar mechanistic proposal was advanced, with the one difference that a Ni0(PR3)2 rather than a Ni0(PR3)3, active complex was proposed. Given the decreased lability of trialkylphosphates vs triarylphosphates from Ni, it is not apparent why fewer ligands per metal center would be expected.
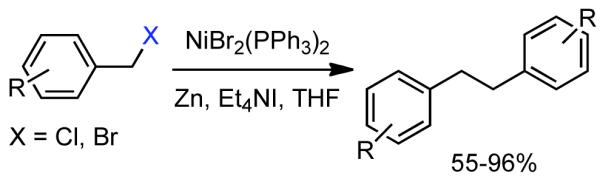
The final step-forward in Ni0-based homocoupling aryl halides was the employment of broadly soluble iodide sources, particularly Et4NI. In 1985, Iyoda reported that Ni0 generated in situ from NiIIBr2(PPh3)2 in the presence of Zn0 could mediate the rapid homocoupling of benzylchlorides and benzyl bromides at room temperature in less polar organic solvents such as THF or benzene if Et4NI was used as the iodide source (Scheme 10).84 A diversity of substituents was tolerated without any definitive reactivity trends, except to say that nitro groups were not tolerated. The relative insensitivity toward aromatic substitutions highlights the key difference between homocoupling at the aryl site vs the benzyl site, where electronic and steric effects are reduced. This technique was utilized in the synthesis of Ricardin B, a metabolite of liverwort Riccardia multifida that possesses a macrocyclic structure containing a bibenzyl linkage. In his synthesis, Iyoda, utilized the aforementioned catalytic conditions to achieve the intramolecular macrocyclization of benzylchloride.85
Scheme 10.
In conjunction with his disclosure of Et4NI as an iodide source in benzyl halide homocoupling, Iyoda also began to explore the homocoupling of aryl halides.86 NiIIBr2(PPh3)2 was determined to be the optimal catalyst for the homocoupling of bromobenzene in the presence of Zn0 and Et4NI in THF as solvent. Using these optimized conditions, bi-1,6-methano[10]annulenes were prepared from 7-bromobicyclo[4.4.1]undeca-1,3,5,7,9-pentaene and oligo- and poly(azulene)s were prepared from 1-bromoazulene and 1,3-dibromo azulene (Scheme 11). Iyoda also reported the synthesis of other interesting polycyclic aromatics starting from bromo-tropones via this strategy.87,88
Scheme 11.

Iyoda’s work on Et4NI promoted Ni0-catalyzed homocoupling culminated in a detailed investigation of the homocoupling of aryl and pyridyl halides.70 This methodology had several innate advantages that contributed to its widespread adoption. First, like the method developed by Colon it proceeded efficiently with less expensive and more readily available aryl chlorides. Additionally, it did not require significant excess ligand. But most importantly, Iyoda’s method was compatible in THF as solvent as opposed to less easily removed DMF or DMAc. In the course of the optimization the homocoupling technique for aryl bromides, Iyoda observed that only 10 mol% Et4NI was needed for the homocoupling of bromobenzene, methyl 4-bromobenzoate, or 4-bromoanisole, yet 100 mol% was needed to maximize the homocoupling yield of methyl 2-bromobenzoate. Interestingly, no Et4NI was needed when aryl iodides were employed, suggesting the sufficient iodide is generated in situ to form stabilized nickelate complexes or alternatively to accelerate electron transfer. Using this knowledge of appropriate Et4NI loading level, several para/meta and ortho-bis(substituted biphenyls) were prepared using 10 mol% Et4NI and 100 mol% Et4NI respectively. For para/meta-substituted aryl halides only 10 mol% Ni was required, while the ortho-substituted aryl halides required higher catalyst loading levels of 20-50 mol% (Scheme 12, top). Yields were only marginally better for aryl bromides than aryl chloride after similar reaction time under identical reaction conditions. Furthermore, an attempt to perform the homocoupling of para-chlorobromobenzene gave only polymer and yielded no isolable 4,4′-dichlorobiphenyl. Together these results indicate that under these homocoupling conditions the rates of chloro- and bromoarene homocoupling are comparable. Overall, the rate of homocoupling appeared to be I>>Br>Cl, which parallels the results of Semmelhack, but contradicts those of Colon. The contradiction of both Semmelhack and Iyoda’s rate observations with the observations of Colon, could simply stem from the fact that the former two ignore the role of autocatalysis and are measuring uncorrected rates. Aware that some N-heterocyclic arylbromides had been successfully homocoupled in the past using Ni0,62,89 Iyoda sought to provide a more general approach to the homocoupling of halo pyridine derivatives. Under similar conditions, the homocoupling of a variety of substituted bromo- and chloropyridines to produce bipyridines also proceeded smoothly (Scheme 12, bottom). Due to the propensity of bipyridines especially 2,2′-bipyridines to serve as ligand of Ni0/II, higher loading levels of catalyst were employed (30 mol%). Identical conditions were also able to furnish biquinolines from haloquinolines and biisoquinolines from haloisoquinolines (Scheme 12, bottom). At the time, biisoquinolines were a particularly challenging target using the state-of-the art Ullmann coupling techniques available at that time.
Scheme 12.

2.1.2. Homocoupling of Aryl Sulfonates to Produce Biaryls
The ability to homocouple aryl halides under mild reaction conditions with relatively inexpensive Ni-based catalytic systems is of great practical utility. Nevertheless, phenol derived arenes are typically less expensive and provide access to a variety of substitution patterns that may be difficult to access for aryl halides. Phenols can be readily converted to activated and nonactivated sulfonate leaving groups, such as triflates, tosylates, and mesylates by treatment with the corresponding sulfonyl halide or anhydride. The first efforts to expand Ni0-catalyzed homocoupling from aryl halides to activated aryl sulfonates were reported by Yamashita in 198690 and 1987.91 To homocouple aryl triflates, Yamashita employed the general conditions established by Colon,73,74 specifically NiIICl2 as the progenitor salt in the presence of PPh3 ligand, NaI additive, and excess Zn0 as reducing agent (Scheme 13). The rate of the reaction was increased up to two-fold through the use of ultrasonication in DMF at 60 °C. In general, a molar ratio of [Ar-OTf]o:[Zn0]o:[NiIICl2]o:[NaI]o:[PPh3]o = 1.0 : 1.5 : 0.08 : 0.6 : 0.6 was employed. Decreasing the excess of NaI and PPh3 relative to NiIICl2 progenitor salt, severely diminished the rate and yield of the reaction.
Scheme 13.

While triflates are the most reactive of conventional sulfonate leaving groups, they are expensive and the precursors are more difficult to handle. A method to homocouple the least expensive, most-atom efficient, yet nonactivated aryl mesylates is therefore preferred. The homocoupling of less reactive aryl sulfonates such as aryl tosylates and mesylates was suggested to proceed at a much lower rate,90 and later demonstrated for one example with low yield.91
Concurrent with his development of analogous methods to produce poly(p-phenylenes) via Ni-catalyzed polycondensation (See section 2.2.3.), Percec undertook a comprehensive investigation of the scope and reactivity of Ni0 catalyzed homocoupling of non-activated aryl sulfonates.92 Starting from methyl 4-hydroxybenzoate, six activated and nonactivated arylsulfonates were prepared: -OSO2CF3, -OSO2CH3, -OSO2Ph, -OSO2-p-PhCH3, -OSO2-p-PhF, and -OSO2-p-PhCl. All six substrates were subjected to homocoupling conditions using Ni0 generated in situ from NiIICl2(PPh3)2 in the presence of Zn0 and Et4NI in refluxing THF as solvent. Nearly quantitative GC yields were obtained for all substrates except for methyl 4-(((4-chlorophenyl)sulfonyl)oxy)benzoate, which suffered from diminished yield due to competitive cross-coupling with the aryl chloride on the sulfonate leaving group. The triflate, the phenylsulfonate, and the p-fluorophenylsulfonate exhibited similar reactivity achieving complete conversion in <5 h while the mesylate and the tosylate were somewhat slower achieving complete conversion in <10 h. Broad substrate scope was demonstrated for both aryl phenylsulfonates and more promisingly from aryl mesylates (Scheme 14). Electron withdrawing esters, ketones, and cyano groups were tolerated as well as electron donating, methyl, and methoxy groups. Polyaromatic mesylates such as methyl 6-((methylsulfonyl)oxy)-2-naphthoate were also effectively homocoupled. Lower yields were noted for p-fluorophenyl methanesulfonate, suggesting a competition between inductive and resonance effects. The order of reactivity was shown to be para > meta > ortho. Para- and meta-substituted aryl mesylates achieved high conversion in 10 h, while ortho-substitution required extended reaction times for maximum conversion. Neither pentafluorophenyl nor p-nitrophenyl methanesulfoante participated in the homocoupling pathway. Interestingly, under these reaction conditions, pentafluorophenyl methanesulfonate underwent complete demesylation to form the free-phenol. The fate of the p-nitrophenyl methanesulfonate was less certain, but competitive electron transfer reactions such as reduction of the aryl-nitro group or formation of nitro-Ni0 complexes93 are possible. The incompatibility of nitro groups with Ni0 mediated homocoupling had been well documented for aryl halides.
Scheme 14.

In 1986, Colon proposed a mechanism for the homocoupling of arylchlorides in polar aprotic solvents using Ni0-generated in situ in the presence of excess Zn0 (Scheme 8).74 While this mechanism was postulated for aryl chlorides, it can be envisioned for both aryl halide and aryl sulfonate leaving groups. Experimental observations for the homocoupling of aryl mesylates were more consistent with Colon’s mechanism than with other alternatives. As with the homocoupling of aryl chlorides reported by Colon, the homocoupling of aryl mesylates almost always resulted in minute quantities of monosubstituted biphenyl adducts resulting from an aryl exchange mechanism. In this subprocess the aryl-halide was cross-coupled with a PPh3 derived phenyl unit. Even if no aryl mesylate was provided, small levels of biphenyl were generated from NiIICl2(PPh3)2 alone. In some cases the reduction of the aryl mesylate was also observed. Reduction could occur either through nickel hydride formation or through radical hydrogenolysis.68,69 Percec, provided evidence that the prevalence of reduction is not determined only by the reductive potential of the mesylate, but also by the availability of abstractable proton sources such as adventitious moisture, thereby providing evidence for a radical pathway. For example, the homocoupling of 4-acetylphenyl-methansulfonate was plagued by 20% reductive side-product while sulfonates with lower reduction potential such as aryl p-fluorobenzenesulfonates under similar conditions did not exhibit such extensive levels of reduction. In the former case, the presence of an abstractable proton from the enolizable ketone could explain the higher levels of byproduct.
Optimization of reaction conditions indicated that the dipolar aprotic solvents typically employed for the homocoupling of aryl-halides were not as effective as less polar THF for the homocoupling of aryl-mesylates as they seemed to produce colloidal Ni-black and mediate a greater degree of reduction. This side-product could be diminished through the use of a greater excess of PPh3, but such efforts resulted in more sluggish reactions and lower overall yields. While, almost all reports of Ni0 catalyzed homocoupling have indicated that the presence of even adventitious moisture can retard the reaction or mediate reduction of halide, in the homocoupling of aryl mesylates catalyzed by Ni0 generated in situ from NiIICl2(PPh3)2 in the presence of Zn0 and 20% excess PPh3, high yield (90%) could be achieved using reagent grade THF without drying. The robustness of this homocoupling reaction was demonstrated by the fact that the addition of a further 5 mol% H2O relative to aryl mesylate reduced the yield by a meager 7%. If on the other hand, no excess PPh3 ligand was provided 5 mol% H2O will shut down the reaction completely. Of course, the rate of Ni0-catalyzed homocoupling is also affected by the catalyst loading level. High conversion required 10 mol% of Ni0, while 1-5 mol % loading level showed high residual aryl mesylate.
The optimization studies also provided some insight into the mechanism of Ni0-catalyzed homocoupling. Relatively high catalyst loading levels (1.5 equivalents of catalyst relative to the aryl) of Et4NI were required for efficient coupling, as yield increased with increasing loading level. It was apparent, that addition of Et4NI on its own was able to improve the stability of Ni0 complexes, while Et4NBr was shown to be far less effective. Perhaps as discussed by Colon, iodide stabilizes Ni0 through the formation of penta-coordinate nickelate intermediates. In the present case, even less PPh3 is used than with Colon’s systems and therefore, even higher levels of iodide may be required. The increased demand for Et4NI in the presence of lower ligand levels provides further support for the role of the halide as stabilizer for nickel to prevent decomposition, rather than as a reagent that promotes electron transfer.
In a later report, Percec used the Ni0-catalyzed homocoupling strategy to outline a general strategy for the synthesis of 2,2′-diaroyl-4,4′-dihydroxybiphenyls (Scheme 15).94 Here, 5-methoxy-2-[(methylsulfonyl)oxy]benzophenones were efficiently homo-coupled with using Ni0 generate in situ from NiIICl2(PPh3)2 in the presence of Zn0, Et4NI. Due to the retarding effects of the ortho-substituents, extra PPh3 (40%) was required to stabilize the Ni0 from premature decomposition. The resulting 2,2′-diaroyl-4,4′-dimethoxybiphenyls could be deprotected in the presence of BBr3 to reveal free phenols. The phenolic sites allow for further mesylation to prepare a bifunctional monomer for the synthesis of soluble poly(p-phenylenes) (See section 2.2.3). The yields for the Ni0-catalyzed homocoupling were lower than normal (45-65%) on account of the bulky ortho-aryl substituents. Nevertheless, the ability to generate coupling fragments efficiently from a phenolic precursor using inexpensive methanesulfonyl chloride imparts a distinct advantage to this route. A subsequent report, detailed the synthesis of a bismesylate monomer via the same route, starting from the phenol-derived precursor, 2-benzoyl-4-ethoxyphenyl methanesulfonate.95 Here, in the absence of additional PPh3 ligand, very low conversion was achieved using THF as solvent, and significant starting material was recovered. Previous work demonstrated that low conversion in the presence of ortho-substituents could be alleviated by the addition of excess ligand.94 Here, an alternative solution was provided which greatly improved the yield (56%) when the reaction was performed at higher temperature (100 °C) in dioxane. Higher yield was ultimately restricted by the competitive formation of demesylated, C-O bond cleavage, and phenyl transfer byproducts. Similar findings were noted, when 3-benzoyl-4-((methylsulfonyl)oxy)phenyl acetate was used as an alternative homocoupling electrophile.96
Scheme 15.

2.1.3. Ni-Catalyzed Homocoupling of Aryl Halides to Produce Poly(phenylenes) and other Polymers
The first poly(arylenes) prepared via homocoupling mediated by Ni0 were the poly(2,5-pyridinediyl)s reported by T. Yamamoto in 1988.97 In this early example, zero-valent nickel complexes previously explored by Semmelhack for the homocoupling of haloarenes such as Ni0(COD)2 in the presence of PPh3 ligand or Ni0(PPh3)4 were employed in stoichiometric or superstoichiometric quantities. The synthetic application of Ni0(COD)2 and Ni0(PPh3)4 were developed extensively by A. Yamamoto, and as a consequence their use in the polycondensation of aryl halides, heteroaryl halides, and related monomers is often dubbed “Yamamoto Coupling.” A variety of conjugated polymers have been prepared through this approach including poly(2,5-pyridinediyl)s,97,98 poly(2,2′-bipyridine-5,5′-diyl)s,98,99 poly(pyrazine)s,100 poly(p-phenylene)s,101,102 poly(thiophene)s,101,102,103,104 poly(furan)s,105 poly(2,7-[9H,10H]dihydrophenanthrenene)s,102 poly(9,10-anthracene)s, 102 poly(quinoline)s,106 poly(isoquinoline)s,106, high molecular weight poly(carbazole)s,107 copolymers,102 and many others (For selected examples see Scheme 16).108,109
Scheme 16.

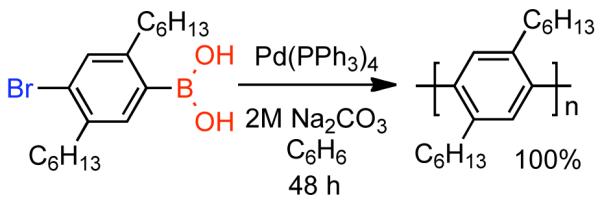
In the 1980s and 90s, the synthesis of poly(p-phenylene) (PPP) and related polymers represented a significant challenge.110,-115 Academic research groups were interested in PPP as an aromatic analogue of linear poly(ethylene) for fundamental studies into their conformation, crystallization, material properties and polymer physics, while industrial laboratories sought approaches to soluble PPPs with accessible synthesis, processing and device fabrication. Ultimately, interest in transition metal catalysis using designed aromatic monomers emerged as a possible solution. In 1989,116 Wegner, Feast and Schlüter reported the synthesis of soluble poly(para-2,5-di-n-hexylphenylene) via Pd0(PPh3)4 catalyzed Suzuki polycondensation117 (Scheme 17). Through this heterophase synthesis employing an AB para-bromophenylboronic acid monomer, soluble PPP with DP~28 was prepared.
Scheme 17.
In 1990, Colon and Kwiatkowski were the first to utilize NiIICl2 catalysis that they developed earlier for biaryl synthesis73,74 to the synthesis of poly(aryls) and poly(heteroaryls) (Scheme 18).118 This methodology can be distinguished from the method developed by T. Yamamoto, in that it is catalytic in nickel. Ni0 catalyst generated in situ from Zn0 reduction of NiIICl2 in the presence of PPh3 as ligand and DMAc as solvent provided high molecular weight poly(arylethersulfone) homopolymers, poly(arylethersulfone) copolymers, and poly(thiophene). Polymerization of 1-chloro-4-((4-(4-chlorophenoxy)phenyl)sulfonyl)benzene, the 1:1 adduct of p-chlorophenol and 4,4′-dichlorodiphenylsulfone, elucidated the regiochemistry of the polymerization. Sulfone activates the aryl chloride toward coupling while aryl ethers are deactivating. Therefore, the resulting polymer is composed largely of symmetrical biaryl subunits. Investigation into the robustness of the reaction revealed that complete removal of adventitious moisture is necessary to prevent Ni0/H2O mediated reduction119 of aryl halide reactants and intermediates. While the use of PPh3 ligand provides some tolerance toward O2, a significant excess of O2 will deactivate the catalyst. It was also found that Zn0 purity, particularly the complete exclusion of ZnIIO, is necessary for the reductant to mediate continuous catalyst renewal and for the polymerization to achieve high molecular weight polymers. Chain-end analysis revealed that without further modification, polymers produced via this method are almost exclusively aryl-Ni terminated. In most applications, the aryl-nickel end-groups are typically consumed via addition of chlorobenzene after complete consumption of the monomer. Related studies, demonstrated that the polymers could be end-capped with other functional aryl chlorides or propargyl chloride to produce chain-end functionalized polymers that can be cured at high temperature to form cross-linked coatings.120
Scheme 18.

Shortly after, Ueda employed a similar approach to prepare four high molecular weight poly(ether ketones) (polymers 1-4, Scheme 19).121 A similar catalytic system was employed, though bipyridine (bpy) was added as a coligand to suppress the Ni0/PPh3-catalyzed reduction of phenyl group transfer to aryl chlorides. For all monomers employed, quantitative conversion was achieved, though with unoptimized conditions molecular weights were low. Only polymer 4 (Scheme 19), was soluble throughout the course of the polymerization, and was therefore selected for optimization. A 1:1:2 ratio of bpy to NiIICl2 to PPh3 was typically employed, to form a Ni0(bpy)/(PPh3) complex in situ through reduction with superstoichiometric Zn0. Excess bpy was shown to limit MW by forming an overly stable Ni0(bpy)2 complex. Maximum molecular weight was achieved at 90 °C in DMAc as solvent, though DMF, NMP, HMPA, and DMSO as solvents also provided quantitative conversion. Under optimized conditions polymer 4 (Scheme 19) could be prepared with Mn = 28,000. Relatively high DP(n) ~ 236 was calculated for this polymer, based on chlorine analysis of the chain-ends. However, this DP(n) does not match well with the molecular weight, and as suggested by Colon and Kwiatkowski,118 most chains may be Ar-Ni terminated. If the Ar-Ni chain-ends are not accounted for the DP will be overestimated. Later, Ueda expanded this approach to the synthesis of poly(3-phenyl-2,5-thiophene) from the corresponding 2,5-dichlorothiophene.122 Ueda favored a mechanism involving slow metathesis of ArNiIL3. This approach was devoid of 2,4-coupling and branching side-reactions common to electrochemical polymerizations of thiophenes, with the main side-reaction being aryl-transfer from phosphine, which in the case of polycondensation, has the effect of terminating growing chains. This chain-termination via aryl transfer could be suppressed, just as it could in homocoupling to form biaryls, by the use of bpy as co-ligand and/or higher phosphine loading levels. 13C NMR studies confirmed an equal distribution of head-to-tail and head-to-head addition triads, indicating random regiochemistry in the polymerization.
Scheme 19.

In 1999, Percec utilized Ni0-catalysis to prepare side-chain liquid crystalline polymers (SCLCP)s based on a PPP backbone.123 SCLCPs were prepared from n-[(4-cyano-4′-biphenyl)oxy]alkyl-2,5-dichlorobenzoate using Ni0 prepared via the in situ reduction of NiIICl2(PPh3)2 with Zn0 (Scheme 20). PPPs with DP between 3 and 62 were obtained in good yield, with regioirregular orientation of the side-chains. For all alkyl chain lengths investigated (n = 2-12), NII LC phases where observed in between the glassy and isotropic domains, exhibiting clear even-odd effects in N-I phase transition temperature and enthalpy.
Scheme 20.

While the development of new methods for Ni0-catalyzed homocoupling of aryl halides has seemed to subside, new applications continue to surface. In 2004, Yagci reported the preparation of a dibromo macromonomer via the controlled ring-opening polymerization of poly(ε-caprolactone) (PCL) initiated by 2,5-dibromo-1,4-(dihydroxymethyl)benzene.124 The PCL functionalized dibromide was utilized in Pd0(PPh3)4 catalyzed polycondensation with 2,5-dihexylbenzene-1,4-boronic acid to form an alternating copolymer or NiIICl2(PPh3)2/Zn0/bpy catalyzed homopolymerization. Later, in an attempt to discern the effect of side-chain architecture on the structure morphology of PPPs, Dimerel prepared a diverse array of PPP oligomers with pendant grafted polymers via macromonomer approach.125 Para- and meta-dibromo arenes bearing poly(oxazoline) (POx) side-chains were prepared via the controlled ring-opening polymerization of methyl 2-oxazoline in acetonitrile using 1,4-dibromo-2-(bromomethyl)benzene or 1,3-dibromo-5-(bromomethyl)benzene as initiators. The POx bearing dibromides were used in tandem with the graft-PCL, PCL-b-PS, or PCL-b-POx dibromobenzenes of Yagci as co-monomers in NiIICl2/PPh3/bpy/Zn0 catalyzed polycondensation (Scheme 21). The various copolymers (DP ranging 17-25) exhibited phase separated morphologies that were visualized with AFM and supported WAXS studies.
Scheme 21.

2.1.4. Ni-Catalyzed Homocoupling of Aryl Sulfonates to Produce Poly(phenylenes) and other Polymers
In a preliminary report in 1991, Percec reported the Ni-catalyzed polymerization of bisaryltriflates to prepare poly(phenylene)s (Polymers 1, 2, 4, 5, Scheme 22).126 In a subsequent report, these approaches were detailed as convenient approaches to soluble poly(p-phenylenes).127 This approach greatly expanded the scope and feasibility of PPP synthesis as it could utilize hydroquinone derived monomers, as well as bishalides. Two methods for generating Ni0 in situ were investigated: (A) the NiIICl2/PPh3/bpy/Zn0 system reported by Colon and Kwiatkowski for poly(ether sulfones)118,121,122 and (B) the NiIICl2(PPh3)2/Zn/Et4NI/THF system originally developed by Iyoda for the homocoupling of aryl halides.70 Both methods were effective at mediating the polymerization of substituted 2,5-dibromo-, 2,5-dichloro-, and 2,5-bis(trifluoromethyl)sulfonylarenes to produce unbranched poly(p-phenylenes) (Polymers 1-3, Scheme 22.). It is important to note that in addition to using a precomplexed NiIICl2(PPh3)2 as the pre-catalyst, method (B) employs Et4NI as an additive. Et4NI as a source of iodide is thought to promote electron-transfer by bridging Ni and Zn species,70,54 but may also stabilize the active catalyst through formation of penta-coordinate nickelates. In polymerization experiments where excess Zn0 reductant is present, it was evident that the rate determining step was the reduction of ArNiII(PPh3)2X to ArNiI(PPh3)X, and therefore, potential electron-transfer accelerants such Et4NI greatly improve the overall rate and efficiency of the reaction.
Scheme 22.
The efficacy of the two methods was judged by their ability to produce high molecular weight polymers. It was apparent for method A in DMF that the molecular weight of the polymer increased according to leaving group -Cl>-Br>-OTf, while for method B in THF the order was reversed with triflates being the most effective. The observations of opposite reactivity trends in the two methods were not definitively explained. However, these trends suggest that in DMF the rate of oxidative addition to Ni0 is controlled by the electronegativity of the leaving group, while in THF the bond strength plays a more crucial role. The highest molecular weights ~6300 (DP ~ 47) was achieved using method B with methyl 2,5-bis[[(trifluoromethyl)sulfonyl]oxy]benzoate as monomer. The application of method B to the polycondensation of 2,5-bis[[(trifluoromethyl)sulfonyl]oxy]biphenyl and 2-tert-butyl-1,4-bis[[(trifluoromethyl)sulfonyl]oxy]benzene produced polymer of lower molecular weight, thereby suggesting that the steric effects of the ortho-substituent on the reactivity of the leaving group can be dramatic. In all cases the distribution of regioisomers and high degree of conformational isomerism present in the laterally substituted poly(p-phenylenes) provided solubility in most polar organic solvents (Scheme 23).
Scheme 23.

Following the disclosure by Percec that soluble poly(2-methoxycarbonylphenylene-1,4-dyl) could be prepared through the Ni0-catalyzed homocoupling of methyl 2,5-dichlorobenzoate,127 Kaeriyama devised an approach to convert the resulting polymer into poly(p-phenylene).128 Saponification of the methyl ester with NaOH provided poly(2-carboxyphenylene-1,4-diyl), which could be converted in nearly quantitative yield to poly(p-phenylene) by decarboxylation upon treatment CuIIO in refluxing quinoline.
Later, Percec reported the synthesis of other soluble PPPs containing alternating 4,4′-(1,1′-binaphthyl) and 4,4′-(3,3′-diphenyl)biphenyl moieties along the main chain (Scheme 24).129 The alternating sequence was achieved through the homopolymerization 4,4′-bis[5-trifluoromethanesulfonyloxy)-2-biphenylyl]-1,3′-binaphthyl, a bistriflate monomer containing an interior binaphthyl group and periphery biphenyl groups. Both of the aforementioned methods for the in situ generation of Ni0 catalyst were employed and provide similar conversions ~35% and degrees of polymerization DP = 8-9. Since each monomer repeat unit contains four main-chain phenyl units the resulting polymers are comparable to PPPs with DP = 32-36.
Scheme 24.

Using identical conditions to those developed for polymerization of bismesylates, low molecular weight oligomers of 4,4′′′-dichloro-l,1′:2′,1′′′-quaterphenyl and 4,4′′′-dichloro-l,1′:3 ′,1′′: 3′′,1′′′-quaterphenyl were prepared through Ni0-catalyzed homocoupling (Scheme 25, left and middle).130 The low molecular weight achieved for these meta- and ortho- kinked poly(p-phenylenes) were largely due to general insolubility. In the case of the meta-quaterphenyl, low molecular weight may also be partially attributable to the formation of cyclic trimers. More soluble polyaromatic PPPs were prepared via the Ni0-catalyzed polymerization of 2,5-bis(4-chloro-1-naphthyl)biphenyl (Scheme 25, right).131 In this case, higher molecular weight polymers Mn = 2700 due to the improved solubility of the polymer. Ultimately, efforts to make higher DP polymers from 2,5-bis(4-chloro-1-naphthyl)biphenyl seemed to be hampered by steric hindrance imposed by the naphthyl rings, which act as a pseudo-ortho substituent. Nevertheless, the white color of these polymers demonstrated an absence of secondary cyclization reactions which plague analogous cation-radical polymerizations and produce discoloring perylene and triphenylene units in the main-chain.132
Scheme 25.

In subsequent work, Percec expanded the Ni-catalyzed homocoupling procedure he developed for bismesylates92,94 to the polymerization to bismesylate monomers.133 In contrast to previous systems, the bismesylate monomers were readily prepared in high yield from the substituted hydroquinones by treatment with inexpensive methanesulfonyl chloride in the presence of pyridine as base. Initial selection of the most appropriate catalytic system was performed using methyl 2,5-bis[(methylsulfonyl)oxy]benzoate as the monomer (Scheme 26). The NiIICl2(PPh3)2/Zn/Et4NI/THF system reported previously bistriflates and bishalides126,127 achieved high yield and molecular weight, while the NiIICl2/PPh3/bpy/DMAc was less effective.118,121,122 Even higher yields and molecular weights could be achieved by switching to dioxane as solvent and increasing the reaction temperature. Other ligands besides PPh3 were explored such AsPh3, P(o-tolyl)3, and PCy3, but only oligomeric products were formed due to rapid decomposition of the catalyst. It was thought that improved molecular weight could be achieved by employing monomers with a larger branched substituent with greater configurational entropy, such as 2-ethylhexyl 2,5-bis[(methylsulfonyl)oxy]benzoate (Scheme 26). Unfortunately, the steric bulk of the larger substituent had a retarding effect on the polymerization, which could only be overcome by increasing the Zn0 loading level from 3.1 equivalents to 7.0 equivalents. Nevertheless, studies with the 2-ethylhexyl ester substituent revealed the universality of the catalyst system for dibromides, dichlorides, bis mesylates, bistriflates and bis(p-fluorobenzenesulfonates).
Scheme 26.

Homopolymers of methyl 2,5-bis[(methylsulfonyl)oxy]benzoate were limited to DP~30, due to premature precipitation during the polymerization caused by the diminished solubility of the higher molecular weight polymer. Copolymerization with 5-50% 2-ethylhexyl 2,5-bis[(methylsulfonyl)oxy]benzoate, isopropyl 2,5-bis[(methylsulfonyl)oxy]benzoate, or methyl 3,5-bis[(methylsulfonyl)oxy]benzoate increased solubility of the polymer allowing for higher DP (Scheme 26). The highest DP was achieved with a monomer feed ratio of 75% methyl 2,5-bis[(methylsulfonyl)oxy]benzoate and 25% 2-ethylhexyl 2,5-bis[(methylsulfonyl)oxy]benzoate. Similar, liquid crystalline poly(alkoxycarbonyl-m-phenylene)s and poly(alkoxycarbonyl-p-phenylene)s were prepared through similar Ni0-catalyzed polycondensation of alkoxy 2,5- and 3,5-dichlorobenzoates.134 The Ni0-catalyzed polycondensation was tolerant of a broad array of bismesylate monomers (Scheme 27). With the exception of poly(p-phenylene) and the poly(p-phenylene) bearing cyano or p-chlorobenzoyl substituents at the ortho-position, all of the resulting polymers were soluble. The highest molecular weight was achieved for the polymerization of 4′-fluoro-2,5-bis[(methylsulfonyl)oxy]benzophenone (DP~101). It is apparent that the homocoupling polymerization is influenced by the nature of the substituent ortho to the mesylate and that bulky groups such as tert-butyl are inhibitory. By reducing the rate of homocoupling, the bulky ligands amplify reduction and phenylation side reactions, leading to premature chain-termination. Side-reactions leading to triphenylphosphine incorporation into the main-chain were not detected.135,136-137
Scheme 27.

The solubility of PPPs is greatly enhanced by the configurational entropy that results from conformational and regioisomerism inherent to mono-functional bismesylate monomers. However, the ortho-substituent to the mesylates can reduce the rate of polymerization and ultimately limit conversion. Elimination of ortho-substituents to the mesylate, while maintaining some degree of configurational entropy was conceived as a potential method to enhance the reactivity while maintaining sufficient solubility to achieve high MW PPPs.138 Regioirregular PPPs derived from aryl bismesylates (1a-1f, Scheme 28) are generally soluble, whereas regioregular PPPs derived from 2,2′-disubstituted 4,4′-bis[(methylsulfonyl)oxy]biphenyls possessed less configurational entropy and were generally insoluble (2a-2f, Scheme 28). Copolymerization of the two classes of monomers provided a somewhat regioirregular structure with fewer ortho-substituents to the mesylates providing enhanced reactivity (Scheme 28). Through this compromise, the highest molecular weights PPPs (Mn = 34,790, DP= 176) were produced through the polymerization of –CO(p-FC6H4) substituted 1e and 2e.
Scheme 28.

The solubility, and resulting molecular weight of regioregular PPPs derived from 2,2′-disubstituted 4,4′-bis[(methylsulfonyl)oxy]biphenyls or 2,2′-disubstituted 4,4′-bis[(trifluoromethylsulfonyl)oxy]biphenyls monomers could be improved by selection of substituent groups, such as trifluoromethyl and trifluoromethoxy, that themselves possessed greater configurational entropy.139 While high molecular weight (Mn = 35,200, DP = 220) could be achieved for the homopolymerization of the aryl bistriflate, 2-(trifluoromethoxy)-1,4-bis[[trifluoromethyl)sulfonyl]oxy]benzene, similar 2,2′-disubstituted 4,4′-bis[(methylsulfonyl)oxy]biphenyls or 2,2′-disubstituted 4,4′-bis[(trifluoromethylsulfonyl)oxy]biphenyls were even more effective. The homopolymerization of 2,2′-bis(trifluoromethoxy)-4,4′-bis[[(trifluoromethyl)sulfonyl]oxy]biphenyl or the copolymerization of 2,2′-bis(trifluoromethoxy)-4,4′-bis[[(methylsulfonyl]oxy]biphenyl and 2,2′-bis(trifluoromethoxy)-4,4′-bis[[(methylsulfonyl]oxy]-biphenyl provided polymers with molecular weight Mn = 54,500 (DP = 340) and Mn = 55,200 (DP= 363), respectively. Expansion of the Ni0-catalyzed polymerization of bismesylates to the synthesis of other poly(arylenes)s derived from more symmetric monomers resulted in larger insoluble homopolymers (Scheme 29).140 Exceptions included the homopolymers derived from 2,2-bis(methylsulfonyloxyphenyl)propane, 2-(3-methylsulfonyloxyphenyl)-2-(4′methylsulfonyloxyphenyl)propane, and bulky phenolphthalein derivative (3,3-bis(4-methylsulfonyloxyphenyl)-1-[3H]-isobenzofuranone). These monomers provided relatively soluble polymers, but with low DPs of 7, 13, and 12, respectively. Alternatively the introduction of asymmetry through a 1-(ethyl)-ethyl linker group in 1-(4-methylsulfonyloxyphenyl)-2-(4-methylsulfonyloxy-4′-biphenylyl)butane) provided greatly enhanced solubility and polymer with significantly higher molecular weight. Copolymerization of these symmetric poly(arylenes) monomers with each other or with a branched arylbismesylate, 2-(ethylhexyl)-2,5-bis(methylsulfonyloxy)benzoate, also improved solubility and permitted access to higher molecular weight polymers.
Scheme 29.

Main-chain phosphorous-containing polymers are often used as thermally stable resins for fire-retardency applications. In 1997, McGrath prepared amorphous poly(arylene phosphine oxide)s (PAPO)s via the NiIICl2/PPh3/Zn0 mediated homocoupling of bis(4-chlorophenyl)phenylphosphine oxide (Scheme 30a).141 Interestingly, higher molecular weight polymer (Mn = 15,300) was produced when the bisdichloro was added to a mixture of preactivated Ni0-catalyst, whereas lower molecular weights (Mn = 9,280) were achieved when the monomer was mixed with NiIICl2, PPh3, Zn0 in DMAc and then elevated to the reaction temperature of 70 °C. The resulting PAPOs could be partially reduced to poly(arylene phosphine) via treatment with phenylsilane. In 2001, Sheares prepared poly(4′-fluoro-2,5-diphenyl sulfone) via the method described previously by Colon where Ni0 was produced in situ from the NiIICl2 in the presence of Zn0, PPh3 and bpy.142 A series of novel poly(p-phenylene)s were prepared via post-polymerization functionalization of aryl-fluoride via SNAr substitution. In a later work, Sheares used the same methods to prepare poly(4′-fluorophenyl-bis(4-phenyl)phosphine oxide) and related copolymers (Scheme 30b).143 Both dichloro and dimesylate monomers were compatible with Ni0-catalyzed homocoupling polymerization, but somewhat higher molecular weights were achieved for the dichloride monomer. The presence of the para-fluoro substituent on the arylphosphineoxide monomer or on the benzophenone comonomer allowed for the same facile post-polymerization functionalization via SNAr substitution with alkoxy, phenoxy, or amino nucleophiles. The substituted PAPOs and PAPO-co-PPPs were demonstrably more soluble in organic solvents.
Scheme 30.

Zengin prepared similar main-chain nitrogen-containing polymer from bis-(4-trifluoromethane-sulfonyloxyphenyl)phenylamine monomer. Using Ni0 generated in situ from NiIICl2(PPh3)2 in the presence of Zn0 and Et4NI, poly(bis(4-phenyl)phenylamine) with Mn = 23,714 (Scheme 31, a) was achieved.144 The resulting polymer was doped with HCl to produce conducting/photo-luminescent polymers. Other conducting and PL polymers have been prepared via Ni0-catalyzed homocoupling of functional aryldihalides, such as poly(N,N’-phenyl-3,6-pyromellitic-dianhydride) (Scheme 31, b)145 and poly(9,10-dihydrophenanthrene-2,7-diyl) (Scheme 31, c).146 The former was prepared through Colon’s NiIICl2/PPh3/Zn0/DMF conditions, starting from a dibromo monomer, while the latter either through Semmelhack’s Ni0/(COD)2 method (Yamamoto Coupling) or through electrochemical generation of Ni0 in situ from NiIIBr2(bpy)3.
Scheme 31.

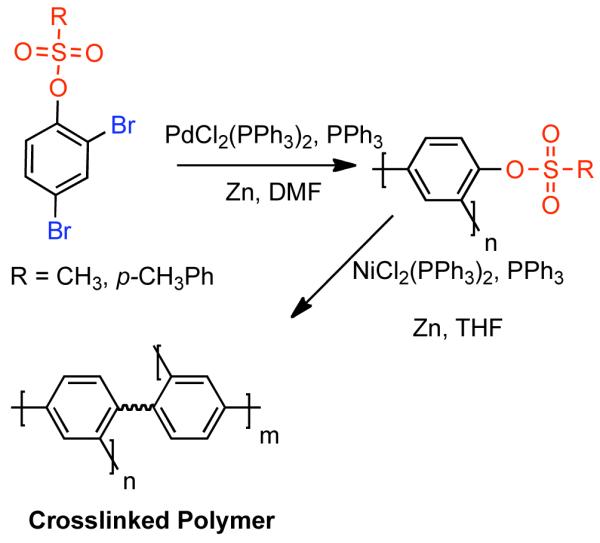
The homocoupling of mesylates has also been used in the post-polymerization functionalization of poly(arylenes). Bae prepared poly(2,4-phenyl sulfonates) via Pd0-catalyzed homocoupling of AB2-type dihaloaryl sulfonate monomers (Scheme 32).147 Subsequent treatment of the sulfonate-bearing poly(meta-phenylene) provided a crosslinked polymer network.
Scheme 32.
More recently, Sheares148 developed monomers were prepared that combined the structural elements of 2,2-bis(((trifluoromethanesulfonyl)oxy)phenyl) hexafluoropropane and p-chlorobenzophenone. In this fashion depending upon their periphery functionality, they could be polymerized using Ni0-catalyzed conditions of Percec, either through the bis(triflate), the p-chlorobenzophenone, or both to prepare isomeric hexafluoroisopropylidene-linked benzophenone-containing polymers (Scheme 33). The use of the tetrafunctional monomer provided tough-crosslinked films that could not previously be achieved with typical benzophenone-based polymers.
Scheme 33.

2.1.5 Homocoupling of Vinyl Halides and Sulfonates
Subsequent to his disclosure of aryl halide homocoupling,42 in 1972 Semmelhack reported that Ni0(COD)2 was also an effective catalyst for the homocoupling of vinyl bromides or 2-, and 3-haloacrylates to form symmetric 1,3-dienes (Scheme 34).149 Alkyl bromides and 2-bromostyrenes reacted rather sluggishly providing only fair yield, while electron-deficient 2-, and 3-haloacrylates provided nearly quantitative conversion and high yield.
Scheme 34.

The system developed by Kende for the generation of Ni0 in situ from the Zn0 mediated reduction of NiIICl2(PPh3)2 was a more versatile reagent used to mediate the homocoupling of aryl halides as well as the vinyl halides and allyl halides.47 β-bromostyrene and cinnamyl chloride were successfully homocoupled under these conditions to furnish trans-1,4-diphenylbutadiene (43% yield) and biscinnamyl (50% yield), respectively. Likewise, Takagi’s high-temperature Ni0(PEt3)n catalyst derived from the Zn0-mediated reduction of NiIICl2(PEt3)2 also effectively homocoupled vinyl halides such as 2-bromo-1,1-diphenylethene (97%), β-bromostyrene (85%), and 1-bromo-2-methylpropene (83%).81
During efforts directed toward the synthesis of natural product bibenzopyran-4-ol, Lin discovered a modified catalytic system capable of mediating the homocoupling of various vinyl iodides and bromides.150 The synthesis called for the homocoupling of 3-iodo-6-methoxybenzopyran-4-one (Scheme 35), using Cu-mediated Ullmann strategies or with Ni0 generated in situ from NiIICl2(PPh3)2 in the presence of Zn0. Using DMF as solvent with excess PPh3 ligand as additive, it was found that the use of K2CO3 as a base improved the yield and limited the degree of dehalogenation. Nevertheless, yields were still relatively low. It was surmised that protonation of neutral arylnickel intermediates was occurring and that K2CO3 deprotonated the resulting cationic byproduct providing re-entry to the catalytic cycle. To improve the yield further more powerful bases were examined. Ultimately, it was determined that NaH provided remarkable acceleration of the reaction and improved product selectivity. Both rate, yield, and selectivity could be further enhanced by switching the solvent from DMF to non-polar toluene. From the distribution of byproducts, a variant of Ullmann-type reactions was proposed and a bridged dinuclear organo-Ni intermediate was invoked, though the mechanism for its formation was not elucidated or supported (Scheme 35). It is surprising that NaH provided dramatic improvements to yield, rate, and selectivity, even if Zn0 was omitted. This may suggest that the role of NaH is at least in part similar to that of Zn0, a reductant, and that they reinforce each other. This is supported by Caubère’s work on NiCRA56-63 (See Section 2.2.1), where NaH mediated hydridic reduction of NiII species was shown to be a contributing reaction. Regardless of the mechanism of this transformation, a variety of α-iodo-α, β-unsaturated ketones, as well as 2-bromopyridine, α-bromostyrene, and β-bromostyrene were compatible with this catalytic system.
Scheme 35.

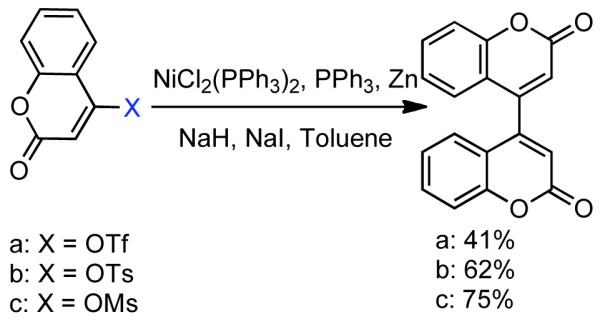
In a later report, Lin demonstrated the application of the NiIICl2(PPh3)2/PPh3/Zn0/NaH/toluene catalytic system for the homocoupling of 4-tosylcoumarins en route to biologically active biscoumarins.151 Kappe later explored a similar strategy for the synthesis of aza-analogues of biscoumarins.152,153 The compatibility of this catalytic system with tosylates, brings into question the involvement of halide-bridged intermediate (Scheme 35), though it is technically possible that the use of NaI as additive may facilitate the formation of this intermediate through displacement chemistry. Subsequently, Lin showed that the aforementioned catalytic system can efficiently homocouple 4-methanesulfonyl, 4-toluenesulfonyl, and 4-trifluoromethylsulfonyl coumarins (Scheme 36).154 The best yields and highest rates were achieved for the mesylates, while the triflates provided the lowest yields, and tosylates the most sluggish reactivity. In the case of the triflates, significant reduction byproduct was observed.
Scheme 36.
Given the convenience of generating 4-mesylcoumarin electrophiles, Lin also investigated the potential of using these intermediates directly in a modified cross-coupling reaction with aryl halides (Scheme 37).154 This cross-coupling is perhaps more appropriately described as an asymmetric homocoupling reaction, as it lacks the transmetallation step common to most Ni-catalyzed cross-coupling reactions. Lin determined that by adding the aryl halide, for instance 2-iodo-5-methoxybenzaldehyde, to a 4-mesylcoumarin such as 2-oxo-2H-chromen-4-yl methanesulfonate, and by eliminating iodide containing additives and NaH, the cross-coupled adduct could be enhanced while the two respective homo-coupling products minimized (Scheme 37). The methodology for cross-coupling was shown to be quite robust for a diversity of mesylcoumarins and arylbromides/iodides, providing an array of 4-aryl coumarins in 52-90% yield. Neither the substitution pattern of the 4-mesylcoumarin nor of the aryl halide had a pronounced effect on yield or product distribution, indicating unusual limited steric and electronic effects on reactivity. If however, instead of PPh3, dppe was used as a ligand, homocoupling of the aryl iodide dominated.
Scheme 37.

2.2. Cross-Coupling of Aryl and Vinyl Mesylates and Tosylates
In the previous chapter (Section 2.2), reactions involving Ni0 catalyzed homocoupling of sulfonates were surveyed. Typically, these reactions were used to produce symmetric biaryls or related polymers, though there were selected examples where cross-coupling was achieved through control of reagent feed and reaction conditions. Nevertheless, the synthesis of asymmetric molecules is typically more efficient when the cross-coupling reaction employs chemically dissimilar fragments. A number of Ni0 catalyzed techniques have emerged from the cross-coupling of aryl, vinyl, and alkyl sulfonates, including less reactive but often more desirable mesylates and tosylates.
2.2.1 Suzuki-Miyaura Cross-Coupling
Discovered in 1979,155 the Suzuki-Miyaura cross-coupling reaction has become a standard for the synthesis of asymmetric biaryls via Csp2-Csp2 cross-coupling, due in large part to mild reaction conditions and high functional groups tolerance as well as the stability, ease of handling, and low toxicity of the organoboron coupling partners. Suzuki-Miyaura cross-coupling was born as Pd – triarylphosphine chemistry for the cross-coupling of aryl halides and triflates.22,10,36,156,157 However, as the field matured, exploration of new ligands as well as less expensive but more reactive Ni-catalysts have led to the expansion of the scope into the realm of less active aryl tosylates and mesylates. While it will not be comprehensively covered in this review, it should be noted that the development of advanced Pd-catalysts have also recently allowed for Pd-catalyzed Suzuki-Miyaura coupling of aryl tosylates and mesylates with aryl boronic acids,158,159 boronate esters,159 and trifluoroborate salts.160,161,162,163
2.2.1.1. NiII phosphine catalysts to Ni0 with reducing agents
In a seminal 1995 report, Percec simultaneously reported the first Ni-catalyzed Suzuki-Miyaura cross-coupling and the first Ni or Pd catalyzed cross-coupling of arylboronic acids with aryl mesylates.164 At that time, PdIICl2(dppf) and Pd0(PPh3)4 were amongst the most effective catalysts for Suzuki-Miyaura cross-coupling of aryl halides and aryl boronic acids. Earlier work had indicated that Pd-catalyzed cross-coupling of aryl triflates with aryl boronic acids was possible165,166,167 often in the presence of LiCl additive.168 Nevertheless, preliminary studies on the Pd-catalyzed cross-coupling of aryl sulfonates with phenyl boronic acid, indicated that only electron-deficient aryl triflates could provide the asymmetric biaryls in high yield.164 Previously, Percec had employed Ni0 generated in situ from NiIICl2(PPh3)2 via Zn0 reduction as a catalyst for the homocoupling of aryl mesylates.92 The homocoupling of aryl sulfonates and Suzuki-Miyaura cross-coupling of aryl sulfonates were expected to share certain mechanistic elements. Of particular note, both pathways would involve the oxidative addition of aryl sulfonates to the zero-valent metal. As Ni0 is more nucleophilic than Pd0 it was expected that Ni0 may make a better catalyst for the cross-coupling of less reactive aryl sulfonates. Previously, NiIICl2(dppf) had been used as a catalyst in cross-coupling reactions with Grignard169 and organozinc reagents.170 This same catalyst was found to be effective for the cross-coupling of aryl boronic acids with aryl mesylates, tosylates, phenylsulfonates, p-fluorophenylsulfonates, and triflates.164 In making the transition to Ni-catalyzed Suzuki-Miyaura cross-coupling anhydrous conditions were needed, it is important to note that the use of aqueous Na2CO3 as base, a typical condition when a Pd catalyst is employed, was replaced by 3.0 equivalents K3PO4 in anhydrous organic solvents such as THF or dioxane165,166,167 in order to prevent water mediated deactivation of the Ni and associated reductive pathways. Lower levels of K3PO4 resulted in lower yields. It is important to note that all of the required conditions for homocoupling are still met, but that the change in catalyst ligand from PPh3 to dppf, the presence of the aryl boronic acid coupling partner and/or the action of K3PO4 as a base, completely suppressed this pathway. Using the catalytic system derived from NiIICl2(dppf), it was determined that no conversion was possible in the absence of Zn0 powder (Scheme 38). Either Zn0 was necessary for the reduction of NiIICl2(dppf) to the active Ni0 catalyst, is necessary for other reduction steps in the catalytic cycle, or both. Scouting experiments between aryl sulfonates and phenyl boronic acid using THF as solvent suggested better yields for electron-deficient aryl triflates such as methyl 4-[(trifluoromethylsulfonyl)oxy]benzoate (80%), and the lowest yield for electron-rich aryl mesylates such p-[(methylsulfonyl)oxy]toluene (33%). In the case of the electron-deficient triflate, ~20% reduction product was also observed. In the cross-coupling of phenylboronic acid and phenyl methanesulfonate, the use of dioxane as solvent allowed for higher reaction temperatures and consequently higher yields (Scheme 38). Attempts were made to optimize the cross-coupling of methyl 4-[(methylsulfonyl)oxy]benzoate (48%). Other catalysts such as NiIICl2(PPh3)2, NiIICl2(dppe), NiIICl2(dppp) were less effective than NiIICl2(dppf). Likewise, additives that aided Pd-catalyzed cross-coupling such as LiCl, or Ni-catalyzed homocoupling such as Et4NI or KBr, provided slightly reduced cross-coupling yield. However, switching solvents from THF to dioxane and running at elevated temperature, 95 °C, substantially improved the yield (67%). Similar conditions applied to the cross-coupling of phenyl mesylated with p-methoxyphenyl boronic acid provided were also quite promising (81% yield) (Scheme 39).
Scheme 38.
Scheme 39.
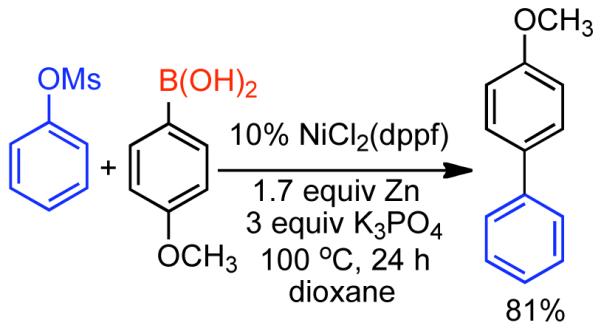
As with the in situ generation of Ni0 for the homocoupling of aryl halides and sulfonates, Zn0 is not the only effective reducing agent for the NiII pre-catalysts employed in Suzuki-Miyaura cross-coupling. Using the same NiIICl2(dppf) catalyst previously employed by Percec for the cross-coupling of aryl-mesylates, in 1996 Miyaura reported that n-BuLi could be employed for the in situ generation of the Ni0.171 In the first report on the NiIICl2(dppf)/n-BuLi system, its efficiency for the cross-coupling of aryl chlorides and aryl boronic acids was explored. In all cases 4.0 equivalents of n-BuLi were employed at 80 °C in dioxane. This represents a mild excess of reducing agent as 3.0 equivalents of n-BuLi were shown to generate the Ni0 catalyst with concomitant evolution of butane, butene and octane. Optimization of the reaction conditions for the cross-coupling of 3-chlorotoluene and phenyl boronic acid as demonstrated that indeed K3PO4 as base in dioxane at 80 °C provided the highest yield (89%). (Scheme 40). As in Percec’s study of Ni-catalyzed cross-coupling of aryl mesylates, other pre-catalysts such as NiIICl2(dppe), NiIICl2(dppb), NiIICl2(dppp), and NiIICl2(PPh3)2 were compatible but provided lower yields (18-77%), with often elevated levels of boronic acid homocoupling side-products (7-16%). The order of reactivity according to ligand was dppf > PPh3 > dppb > dppp > dppe. The reaction protocol using NiIICl2(dppf) pre-catalyst was efficient for both electron-rich and electron-deficient aryl chlorides and tolerated a diversity of substituent functional groups including nitriles, aldehydes, ketones, esters, amides and amines (Scheme 41).
Scheme 40.
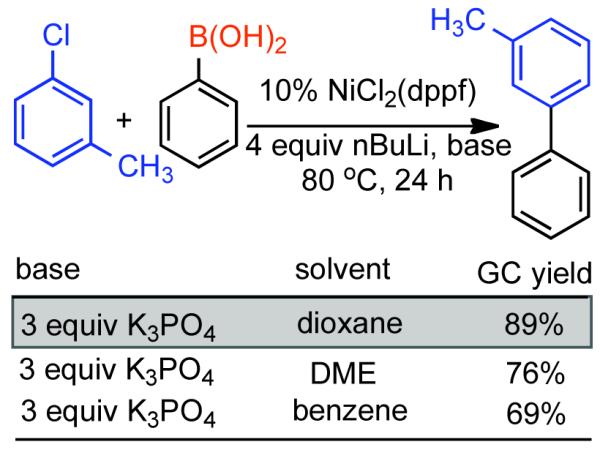
Scheme 41.
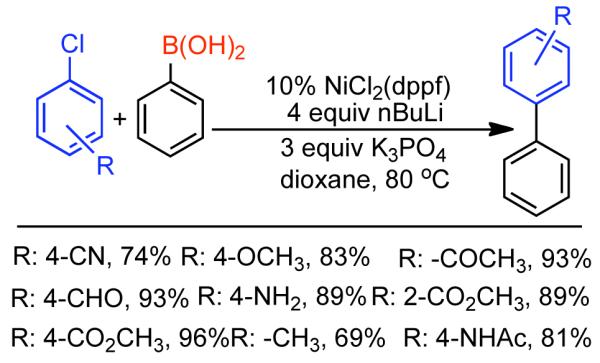
Although never exhaustively validated, the mechanism of Ni-catalyzed cross-coupling reaction was considered similar with the Pd-catalyzed cross-coupling proposed by Suzuki.172 The catalytic cycle in both cases involves three sequential steps: oxidative addition, transmetallation and reductive elimination. Zinc powder performs the reduction of the NiII pre-catalyst to the active Ni0 catalyst. Aryl mesylate undergoes oxidative addition to the Ni0 species to provide NiII σ-aryl complex. The oxidative addition product containing the weakly coordinated mesylate ion might undergo ligand exchange with base-provided the phosphate anion. This potentially more reactive oxo-nickel intermediate may further participate in a transmetallation reaction to provide a diaryl NiII species. In the final step, reductive elimination provides the cross-coupled product and regenerates the Ni0 catalyst (Scheme 42).
Scheme 42.

In a more comprehensive investigation of the n-BuLi/NiIICl2(dppf) mediated Suzuki-Miyaura cross-coupling,173 it was determined that while 10 mol% Ni-loading is sometimes required, the catalyst loading level could generally be reduced to 3 mol% without significant deterioration in yield, especially if additional phosphine ligand was employed to prevent catalyst decomposition. Miyaura found that additional co-ligand slowed the reaction somewhat but could in many cases provided higher yield after prolonged time. While the typical procedure was to use the same co-ligand as that bound to the pre-catalyst, Miyaura provided some of the first examples of mixed-ligand and demonstrated similar efficiency, including non-phosphine ligands such as AsPh3 and SbPh3. In addition to n-BuLi and by then well known Zn0 mediated generation of Ni0 in-situ, diisobutylaluminum hydride (DIBAH) was also found to be an efficient reducing agent. Mechanistic studies revealed that there was no effect of boronic acid substitution on reaction rate, nor was there much of a dependence on the nature of electron-donating substituents (ρ = 0.59). Nevertheless, for electron-withdrawing substituents, a strong accelerating effect was observed (ρ = 8.6). These results agreed with earlier studies of Casser174 into the oxidative addition of aryl chlorides to NiII, suggesting that oxidative addition is rate determining. These results were notably different from those for oxidative addition to Pd0, which had a marked dependence in the electron donating regime (ρ = 5.2) and was particularly sensitive to nitrogen-containing substituents.175 Comparative cross-coupling experiments of substituted aryl chloride with phenyl boronic acid demonstrated nearly uniformly higher yields for NiIICl2(dppf) than for Pd0(PPh3)4 especially for electron-donating or nitrogenous substituents, thereby providing strong support for the assertion that Ni0-catalysis is more robust for less reactive halide and pseudohalide leaving groups. The tolerance of Ni0-toward nitrogen and other heteroatoms lent itself to the successful cross-coupling of heteroaryl chlorides. 7-Chloroindole provided good cross-coupling yields with phenyl boronic acid using 3 mol% NiIICl2(dppf) (80% yield), while 2-chlorothiophene provided higher yield with 10 mol% NiIICl2(dppf) (88%) or with an extra equivalent of dppf (86%). Likewise, the cross-coupling of 2-methyl-6-(2-methylphenyl)benzoxazole (90% yield) and 2-(2-methylphenyl)-4,6-dimethoxy-1,3,5-triazine (69 % yield) with phenyl boronic acid were efficient when 3 mol% NiIICl2(dppf) and 3 mol% dppf co-ligand and 10 mol% NiIICl2(dppf) and 10 mol% dppf co-ligand were employed, respectively. On the other hand, 2-chloroquinoline (90% yield) was more effectively cross-coupled in the presence of 3 mol % NiCl2(PPh3)2 and 6 mol% of PPh3 co-ligand and 3-(2-methylphenyl)-4-methyl-7-methoxycoumarin (90% yield) seemed to benefit from a mixed ligand system composed of 3 mol% NiIICl2(dppf) and 6 mol% SbPh3.
In a subsequent study, Miyaura expanded the use of the n-BuLi reducing agent to the cross-coupling of aryl mesylates with aryl boronic acids.176, Initial screening of the reaction of tolyl boronic acid with p-methoxyphenyl mesylate, suggested that for less reactive aryl sulfonates refluxing toluene was a superior solvent to dioxane and that the 3 mol% NiIICl2(dppf) with an additional 3 mol% of dppf prevented catalyst decomposition and furnished higher yields (85%). Interestingly, it was found that while the addition of excess H2O to the reaction was deleterious,173 three equivalents of K3PO4 must be used in the form of the n-hydrate. Completely anhydrous K3PO4 provided significantly diminished yield (25%) (Scheme 43). Miyaura explored the scope of n-BuLi/NiIICl2(dppf) catalyzed cross-coupling. High yields were obtained for almost all meta and para substituents, while ortho substituents provided diminished yields. Aryl mesylates containing electron-withdrawing substituents exhibited significant rate enhancement, while electron-deficient aryl-boronic acids suffered from competitive protodeborylation. Therefore, in considering reaction design, if possible electron-withdrawing groups should be incorporated on the aryl sulfonate or aryl halide electrophile, though the reduced yield caused by protodeborylation could be somewhat alleviated if the solvent was changed to dimethoxy ethane. As with the n-BuLi/NiIICl2(dppf) catalyzed cross-coupling of aryl chlorides, some heteroaryls were also tolerated such as 4-methanesulfoxyindole (85% yield).
Scheme 43.
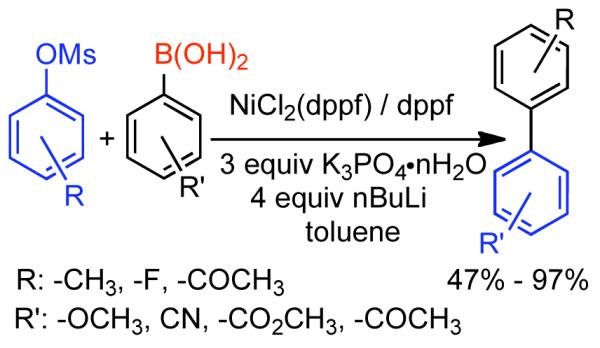
As mentioned before, Ni0-catalyzed homocoupling and Suzuki-Miyaura cross-coupling are sensitive to adventitious moisture and can lead to counter-productive reduction pathways. In addition to simply exhaustive reagent and solvent drying, it had been established that some degree of water tolerance could be achieved through the use of excess phosphine ligand. In 1999, Genêt also reported a water-soluble and water-stable mixed-ligand catalyst prepared through the in situ reduction of NiIICl2(dppe) in the presence of co-ligand sodium triphenylphosphinotrimetasulfonate (TPPTS) using Zn0 as reducing agent (Scheme 45a).177 This water-soluble catalyst mediated the coupling of electron-deficient and electron-rich aryl chlorides with phenyl boronic acids in good yield (70-81%) in mixtures of 1,4-dioxane, DMF, or NMP and water. Some evidence suggested that 1,4-dioxane was a more effective solvent for electron-deficient than for electron-rich aryl chlorides. Further evaluation of the cross-coupling electron-deficient 4-chloro-acetophenone in dioxane/water revealed efficient cross-coupling with a diversity of boronic acids, including 1-naphthylboronic acid, 1-thienyl boronic acid, cinnamyl boronic acid in generally good yield (72-99%). Only 3-aminophenyl-1-boronic acid (47%) and simultaneous ortho-substitution on the aryl boronic acid and aryl chloride (67%) provided lower yields.
Scheme 45.

2.2.1.2 NiII Phosphine Catalysts to Ni0 Without External Reducing Agents
NiII catalysts must be activated to their Ni0 active form via an external or internal reducing agent (See Chapter 2.3.2.). External reducing agents include Zn0 or NaH co-agents, while internal reducing agents are cross-coupling fragments capable of converting NiII to Ni0 prior to initiation of the catalytic cycle. The use of external reducing agents such as Zn0 powder and particularly pyrophoric reagents such as n-BuLi is often undesirable. The first external reducing agent-free and incidentally room temperature cross-coupling of aryl mesylates was reported by Kobayashi using lithium aryl178 or alkenyl organoborates179 as coupling partners. These lithium aryl and alkenyl boronates were prepared in situ from the corresponding boronates via treatment with alkyl lithium reagents. Kobayashi observed that lithium aryl and alkenyl borates themselves reduce the NiII precatalyst to active Ni0 in situ and allow for very efficient cross-coupling with aryl and alkenyl mesylates and tosylates. Using these lithium borate coupling fragments, both NiIICl2(dppf) or NiIICl2(PPh3)2 served as effective precatalysts. Kobayashi proposed that the relatively slow rate of cross-coupling of aryl-mesylates in Percec’s original studies were the result of the poor transmetallation with the boronic acids that could only be accelerated by switching from THF to a solvent with higher reflux temperature, namely dioxane. When lithium boronates were employed a cross-coupling partners, the high isolated yields could be achieved at room temperature, but only for aryl mesylates with electron-withdrawing groups as substituents (Scheme 44).
Scheme 44.

In 1997, Indolese made a critical observation that chloroarenes could be cross-coupled with aryl boronic without the use of an external reductant (Scheme 45c). Using the NiIICl2(dppf)/K3PO4 system developed by Percec for the Suzuki-Miyaura cross-coupling of aryl mesylates with aryl boronic acids, Indolese found that when applied to aryl chlorides in dioxane or anisole at 95 °C no external reducing agents was needed to activate the catalyst. 180 Interestingly, even lower catalyst loading was needed than previous studies, as low as 1 mol%. Enhancement of reaction yield was more effective through increasing the excess of aryl boronic acid than through higher catalyst loading. Preliminary screening studies on the cross-coupling of phenyl boronic acid and 4-chloroanisole indicated that NiIICl2(dppf), NiIIBr2(dppf), NiIICl2(dppb), and NiIICl2(PPh3)2 were similarly effective pre-catalysts. The use of more stable chelating ligands such as dppe and dppp provided diminished yields suggesting that some degree of phosphine dissociation is needed in the reaction. While it had been demonstrated previously that Ni σ-aryl complexes could mediate the “cross-coupling via homo-coupling” of aryl halides,74 they were not efficient catalysts for the cross-coupling of aryl chlorides with aryl boronic acids. Using NiIICl2(dppf) at typically 1 mol%, good to excellent yields were achieved for a variety of electron-deficient and electron-rich aryl chlorides. Homocoupling and aryl-transfer side products were typically at low and always under 10 %. As frequently observed in Ni0-catalyzed homo- and cross-coupling nitro groups were not tolerated. Later, Miyaura expanded on the use of NiIICl2(PPh3)2181 in reductant-free cross-coupling of aryl chlorides and introduced NiIICl2(PCy3)2 as a similarly competent catalyst. Interestingly, it was observed that while strong bases such as KOH and NaOH were compatible with the cross-coupling, but only if the catalyst was pre-reduced with diisobutylaluminium hydride (DIBAH). A significant retarding neighboring group effect for ortho-acyl groups or 2-halopyridines was also noted.
In 2001, Monteiro expanded the use of NiIICl2(PCy3)2 / 2 PCy3 as an efficient reducing-agent free catalytic system for the cross-coupling of aryl tosylates (Scheme 45d). 182 Like with aryl chlorides and NiIICl2(dppf),180 aryl boronic acids were apparently able to reduce the NiII pre-catalyst to the Ni0 active catalyst. A diversity of aryl electron-deficient (94 - 99% yield) and electron-rich (79 – 88 % yield) tosylates were transformed to the corresponding monofunctional biphenyl in very good yield via cross-coupling with phenyl boronic acid. Slightly lower yields (47-60%) were obtained for ortho-substituted aryl tosylates. Depending upon the boronic acid used, Hammett plots revealed a strong electronic effect on the aryl tosylate reactivity (ρ = 1.6-2.0). In contrast to the cross-coupling of aryl chlorides in the presence of reducing agent,173 an electronic effect of the aryl boronic acid on reaction rate was also observed (ρ = 0.76-0.85), suggesting that without a reducing agent the boronic acid is more intimately involved in the rate determining step of the reaction. Later, Monteiro determined that under similar conditions more reactive aryl iodides and bromides were also shown to participate in phosphine free cross-coupling with boronic acids using only 0.5 mol% NiCl2·6H2O and K3PO4 in dioxane at 100 °C (Scheme 45e). 183 Fair to good yields were achieved, but only after extended reactions times, which could exceed 100h. Previously, Leadbeater had found that phosphine-free but not ligand-free cross-coupling was possible with NiIICl2(NEt3)2 and NiIICl2(bpy) pre-catalysts in dioxane.184
In 2004, Percec reported the development of a universal catalytic system for the NiII-catalyzed Suzuki-Miyaura cross-coupling of aryl iodides, bromides, chlorides, mesylates and tosylates, with aryl boronic acids.185 In the past, solvent has been shown to have a strong effect on the outcome of Suzuki-Miyaura cross-coupling reactions. As substrate solubility can be an issue, it is definitely useful to have a general catalyst capable of mediating cross-coupling under diverse conditions. Five catalysts (NiIICl2(PPh3)2, NiIICl2(PCy3)2, NiIICl2(dppf), NiIICl2(dppe) and NiIICl2(dppb)) were tested in the cross-coupling phenyl boronic with p-methoxycarbonyl substituted aryl chlorides, mesylates, and tosylates in both toluene and dioxane as solvent. Using 5 mol% catalyst and 10 mol% monodentate or 5 mol % bidendate co-ligand, only NiIICl2(dppe)/dppe was highly active for all leaving groups and both solvents (Scheme 46). Using this ideal catalyst, higher yields were obtained with toluene as solvent and chloride as the leaving group. Lower catalyst and ligand loadings were possible, but did not always provided optimal yields. Screening of the reaction scope, revealed that NiIICl2(dppe)/dppe as well NiIICl2(PPh3)2/2PPh3 and NiIICl2(PCy3)2/2PCy3 were very effective for mediating the cross-coupling of electron-deficient mesylates such as those bearing cyano-, ester-, acetate- or aromatic functionalities in both dioxane and toluene. However, NiIICl2(dppe)/dppe was relatively ineffective for the cross-coupling of electron-rich aryl mesylates. Quite surprisingly, it was found that the use of the mixed-ligand system NiIICl2(dppe)/PPh3 provided consistently high cross-coupling yields for electron-deficient and electron-rich aryl mesylates, chlorides, bromides, and iodides with phenylboronic acid. While a few earlier examples of mixed-ligand Ni complexes were mentioned previously, never before was their reactivity found to be so uniformly superior as demonstrated in this publication.
Scheme 46.
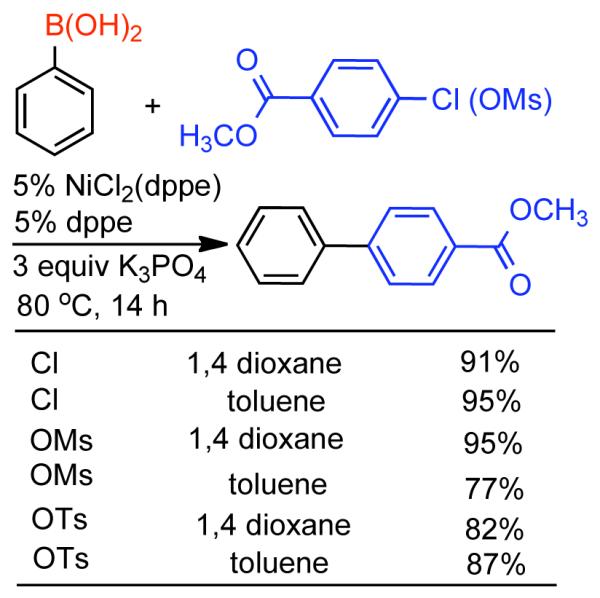
More recently, a catalytic system based on NiII-(σ-aryl) complex was reported to be very efficient in the Suzuki cross-coupling of aryl tosylates with arylboronic acids without the need of a reducing agent (Scheme 47).186 Very high yields for both electron-deficient and electron-rich tosylates were achieved with broad tolerance toward substituent functionality, save for aldehydes and nitro-bearing tosylates for which no product formation was observed. The success of NiII-(σ-aryl complex) in the cross-coupling of aryl tosylates and aryl boronic acids is an interesting contrast to earlier failures for aryl-chloride cross-coupling.180
Scheme 47.

2.2.1.3 N-Heterocyclic Carbene based Ni(II) Catalyst for Suzuki-Miyaura Cross-coupling
N-Heterocyclic carbene (NHC) based Ni catalysts have recently received significant interest for their use in different cross-coupling reactions due to their air-stability and ease of manipulation.187,188,189 (Scheme 48).
Scheme 48.

NHC based NiII catalysts were demonstrated to be very effective for the Suzuki-Miyaura protocol. Chen tested the catalytic activity of complexes I-III from Scheme 48 and found I and II to provide the highest yields in the cross-coupling of aryl chlorides with boronic acids (78%-98% yield).187 Nickel pincer complexes based on NHC ligands, such as complex VI (Scheme 49) showed increased reactivity towards cross-coupling of both aryl/alkenyl tosylates and mesylates as well as aryl bromides and chlorides with boronic acids.188,189 Fair to good isolated yields were obtained with improved reactivity in the electron-deficient aryl tosylates. Electron-rich and sterically hindered tosylates provided lower yields.
Scheme 49.
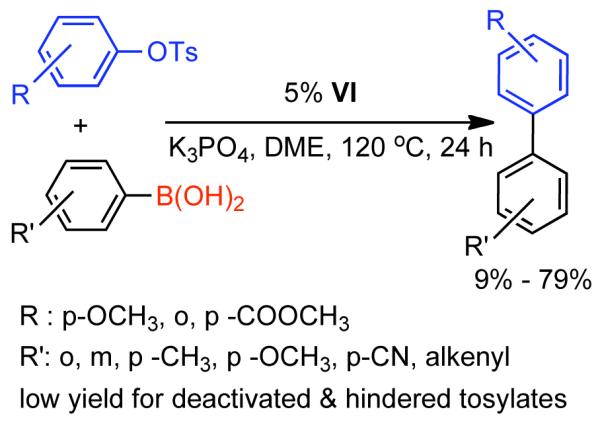
2.2.1.4 Ni0 Catalysts for Suzuki-Miyaura Cross-Coupling
In addition to in situ reduction by an external reducing agent or by the boronic acid, active Ni0-catalysts can also be supplied directly to the reaction. The earliest Ni0 catalysts employed directly for Suzuki-Miyaura coupling of aryl halides were the heterogeneous nickel on charcoal (Ni0/C) first reported by Lipshutz in 2000.190,191 The active catalyst was obtained by the reduction of a NiII(NO3)2 salt impregnated on charcoal with added triphenylphosphine as ligand and 4 equiv of n-BuLi as reducing agent. The catalyst was easily obtained in situ prior to the Suzuki coupling at room temperature by stirring the salt, ligand and reducing agent in dioxane. Aryl and heteroaryl chlorides were effectively coupled in dioxane at reflux with aryl boronic acids under 5-10 % Ni0/C, 4 equivalents of PPh3 ligand, K3PO4 as base and LiBr as an additive. The reaction conditions were suitable for aldehyde, ketone, nitrile and quinoline substrates. The yield of coupling products was also sensitive to the amount and the nature of the phosphine ligand. Bidentate ligands (BINAP, dppf, dppe) inhibited the coupling while monodentate phosphine ligand PPh3 was more effective. The impregnation of the phosphine ligand on the Ni0/C catalyst was performed by exposing the heterogeneous catalyst to 4 equiv of PPh3 in THF at room temperature in the absence of the aryl chloride substrate and boronic acid. Filtration of the solution provided the active catalyst containing only half of the initial quantity of PPh3 ligand. Therefore the Ni0/C/PPh3 contains only two phosphine ligands and is assumed to release one of them in the oxidative addition step. The active catalyst obtained using this procedure is less expensive and more “green” than many of its homogeneous analogues due to easy handling and recycling. Inductively coupled plasma atomic emission spectroscopy (ICP-AES) detected only trace levels of leaching from the charcoal matrix to the solution. This observation also suggests that the Ni0-mediated catalysis is occurring at the surface.
Later work, also explored homogenous Ni0 catalysts. Ni0(COD)2, the original catalyst used by Semmelhack in Ni0-catalyzed homocoupling of aryl halides is sufficiently stable to be isolated in inert atmosphere, however it is particularly sensitive to oxygen and requires delicate handling. Nevertheless, Ni0(COD)2 possesses somewhat unique reactivity in Suzuki-Miyaura cross-coupling reactions as it is supplied in the active oxidative state and can directly undergo the first step of oxidative addition. Not surprisingly, several groups have reported the use of Ni0(COD)2 in Suzuki-Miyaura cross-coupling as a very efficient catalyst for less reactive aryl chlorides, tosylates.192,193,194 and mesylates.195 High reactivity at room temperature was observed, however phosphine co-ligands (PCy3,192,195 PPh3,193 or ferrocenylmethylphosphine194), bathophenanthroline (BP)196 or imidazolium-carbene197 were required to stabilize the Ni0 intermediates during the catalytic cycle. Alternatively, colloidal Ni0 obtained from reduction of NiII(OAc)2 with NaBH4 required tetrabutylammonium bromide (TBAB)198 as stabilizing surfactant. The phosphine-free TBAB-stabilized Ni0 was recoverable, but only effective at mediating the cross-coupling of aryl iodides and bromides.
The first Suzuki-Miyaura cross-coupling reaction of aryl tosylates employing Ni0(COD)2 was reported by Hu.192 As Ni0(COD)2 required no activation, cross-coupling proceeded at room temperature. Different phosphine ligands were tested for activity as co-ligand with the best results provided by PCy3. The investigation of the effect of solvent and base resulted in K3PO4 and THF as the optimum bases and solvent. The protocol was compatible with aryl tosylates bearing either activating or deactivating groups as substituents. (Scheme 51a). PPh3, a less expensive and more stable ligand than PCy3 was demonstrated to be efficient for the cross-coupling of aryl chlorides with electron-donating or electron-withdrawing substituents (Scheme 51b). Sterically hindered substrates such as 1-chloro-2,6-dimethylbenzene were inactive under these conditions. Ferrocenylmethylphosphine or its polymer-bound analogues also stabilize Ni0(COD)2 derived intermediates in the cross-coupling of aryl tosylates.194 While more easily recovered and reused than the homogenous catalyst, the polymer-bound ligand provided lower yields.
Scheme 51.

As will be described in a later subchapter, Percec recently reported the neopentylglycolborylation of aryl bromides and iodides and subsequent cross-coupling with aryl iodides, bromides, mesylates and tosylates under complementary Ni/Pd or Ni/Ni protocols.195 6% of Ni(COD)2 with 18% PCy3 ligand was found to be very efficient for the cross-coupling of aryl neopentylglycolboronate esters with both electron-rich and electron-deficient aryl mesylates and tosylates (Scheme 53). These studies demonstrated for the first time that Ni0 could mediate the Suzuki-Miyaura cross-coupling of aryl-boronate esters with aryl halide and aryl sulfonates.
Scheme 53.

Recently, Molander reported the first Ni-catalyzed cross-coupling of aryl and heteroaryltrifluoroborates with aryl mesylates (Scheme 54).199 Model studies with electrophiles derived from 2-naphthol utilizing 10 mol% Ni(COD)2 and 20 mol% PCy3HBF4 in 1:1 t-BuOH/H2O provided an order of reactivity of OMs>OTs~OSO2NMe2>OPiv>OBoc>OCONE2>OBz>>OMe = 0. Given these reactivity trends, cost-effective aryl mesylates were chosen as the primary electrophillic substrate.
Scheme 54.

2.2.2. Negishi Cross-Coupling
Boronic acids and esters are certainly not the only transmetallating reagents that can be employed in Ni0-catalyzed cross-coupling reactions. In the late 1970s, following the breakthrough discovery of Kumada200 that Ni0 can catalyze the cross-coupling of aryl halides with Grignard reagents (See section 2.3.3), Negishi discovered that organozinc, organoaluminum and organozirconium reagents could be used in the Pd or Ni catalyzed cross-coupling with aryl, alkenyl, alkyl halides. 20,21,15,55,201,202 While multiple organometallic transmetallating reagents were found to be compatible, the “Negishi Reaction” typically employs organozinc55 reagents, including aryl- or heteroaryl-, benzyl, alkynyl, or alkylzincs.
Pd-catalyzed Negishi cross-coupling has been intensively developed. Nevertheless, the use of Ni-catalysts as cost effective alternatives has recently emerged and have found extensive use particularly in the field of alkyl-alkyl couplings.203,204,205,206,207,208 This subchapter will cover only aspects of the Ni-catalyzed Negishi cross-coupling of aryl- or heteroarylzinc reagents with aryl/vinyl mesylates with some of examples of aryl/vinyl halide electrophiles for historical context.
2.2.2.1. Aryl (Heteroaryl)-Aryl Coupling
The driving force for the aryl-aryl Negishi cross-coupling was the use of biaryls compounds for a broad range of applications ranging from liquid crystal displays to biologically active natural products used in commercially available pharmaceuticals.209,201
While sensitive to moisture and air, organozinc reagents have a high rate of transmetallation, which was thought to be the rate-determining step in their cross-coupling. Arylzinc substrates can be obtained either by direct zinc insertion through transmetallation of the corresponding Grignard or aryllithium or through an I/Zn exchange reaction.210,211 In 1977 Negishi reported the first synthesis of biaryls via the Ni or Pd-catalyzed cross-coupling of arylzinc reagents and aryl bromides and iodides. Ni0 (PPh3)4 catalyst (5% mol) was prepared in situ from NiII(acac)2, PPh3 and (i-Bu)2AlH in a ratio 1:4:1. Various electrophilic functional groups such as nitrile and ester were tolerated giving cross-coupling products in excellent yields (85%-95%) even at room temperature (Scheme 55). The cross-coupling reaction was also compatible with less reactive aryl halides containing electron-donating groups such as methyl or methoxy.
Scheme 55.
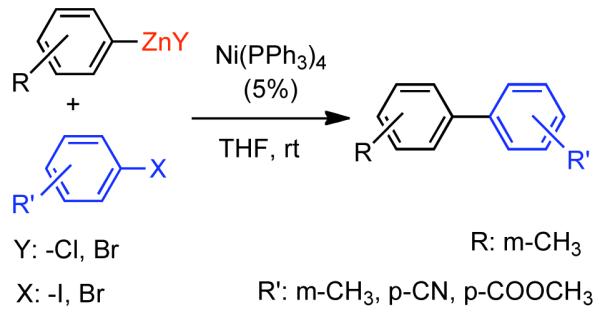
The Ni0 catalysts reported previously, Ni0(PPh3)4 and Ni0(COD)2, are not stable in air. Leadbeater reported the use of biscyclopentadienyl nickel (NiIICp2 or nickelocene) as a more robust precatalyst.212 Tertiary monophosphine and monophosphite ligands (PPh3, PCl3 and P(OiPr)3) react easily with NiIICp2 providing the Ni0 catalyst in situ. Aryl chlorides, although typically less reactive in Negishi cross-coupling are cost and atom effective as compared to the corresponding iodo- and bromo-derivatives. Miller213 reported efficient Negishi cross-coupling of ortho- and para-substituted aryl and heteroaryl chlorides with organozinc reagents using NiII(acac)2 and dppf or PPh3 as coligand. The reaction proceeded at room temperature with 2-5% catalyst loading in very good yields. Grignard-sensitive functionalities, such as nitrile, ketone or ester groups were not disturbed by the arylzinc reagents (Scheme 56).
Scheme 56.
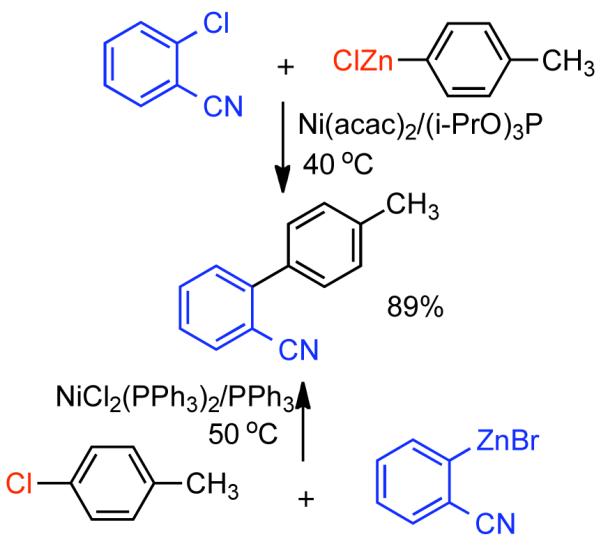
Ortho-, meta- and para-substituted aryl triflates were successfully coupled with arylzinc reagents in the presence of similar catalyst-ligand combinations 5% NiIICl2(PPh3)2, 5% NiII(acac)2/10% i-PrMgCl or PPh3.214,215 Knochel developed a catalytic system based on NiIICl2 using a combination of diethyl phosphite (EtO)2P(O)H and DMAP as ligands and an optimum 8:1 mixture of THF and N-ethylpyrrolidine (NEP) as solvent.216,217 The catalyst loading was reduced to levels as low as 0.0025 mol% and 0.01 mol% (EtO)2P(O)H or DMAP ligands. Different ligands and solvent mixtures were screened to minimize homocoupling of aryl halide and improve yields with less reactive electron-rich arylzinc halides. Without the co-ligand, the reaction was found to be very slow and only traces of products were obtained, while solvents such as THF, NMP, DMF, DMSO < DMPU produced mostly homocoupling byproduct, due to the low solubility of the organozinc reagent in the pure solvents. The order of reagent mixing was also found to play an important role in the reaction rate. If the Grignard reagent was added to the solution of ZnBr2 premixed with NEP to generate the organozinc reagent, the reaction was considerably faster than if the Grignard solution was added first to the ZnBr2. In the later case increased precipitation was observed diminishing the overall yield. In general heteroaryl zinc bromides were more reactive than the corresponding heteroaryl zinc chloride. Traces of biaryls were observed during the preparation arylzinc reagents from the corresponding aryl Grignard by treatment with ZnBr2. Apparently, adventitious ppm levels of nickel in commercial ZnBr2 is able to mediate a small level of homocoupling.
Knochel’s Ni-catalyzed Negishi cross-coupling methodology is very efficient and works well with aryl or alkenyl bromides, chlorides, triflates, and nonaflates as electrophiles and is compatible with esters, ketones and heteroatoms (Scheme 57). Nitriles were less effective as substrates due in part to coordination with the catalyst. Although successful for triflates and nonaflates due to higher reactivity, these conditions could not be applied to aryl tosylates, mesylates and phosphates. Additionally, the cross-coupling was sensitive to steric effects.217
Scheme 57.

Unprotected amides218 as well as amines, alcohols and phenols219 were found compatible with Negishi cross-coupling conditions when 1% Pd(OAc)2 and 2% S-Phos were used as catalyst and ligand respectively (Scheme 58). Arylzinc iodides were efficiently cross-coupled with bromo anilines, alcohols or phenols but not with corresponding protic aryl chlorides. In the case of the chloroaniline, quantitative deprotonation of the aniline prior to coupling and only 17 % cross-coupling product was observed. Ortho- or meta-substitution with diverse functionality including ester, cyano or trifluoromethyl groups on either the bromoaniline or organozinc reagent was not detrimental to yield. The reaction was also found to be suitable for alkyl zinc or benzyl zinc reagents. This protocol exhibited clear limitations. Primary or secondary amines induced deactivation of the catalyst due to the donor capacity of nitrogenous substituents. More significant, no reactivity of the organozinc reagents with aryl chlorides was observed and the reaction was also sensitive to steric hindrance. Knochel also observed that this Ni-catalyzed approach was effective for the cross-coupling of organozinc reagents with functionalized bromo anilines. Optimal yields were obtained using 2 mol% NiII(acac)2/3 mol% PPh3 or 2,2′-bipyridine. As in the Pd-catalyzed Negishi procedure, slow addition of the zinc reagent over 90 min via syringe pump was required for satisfactory yields of biphenyl products.
Scheme 58.

The scope of the Negishi coupling was extended later to heterobiaryls synthesis from cross-coupling of furyl,220,221,222,223,224,225 imidazolyl,226 indolyl227,228,229 and pyridyl230,231 zinc halides with aryl bromide and iodide using Pd catalysts (Scheme 59).
Scheme 59.
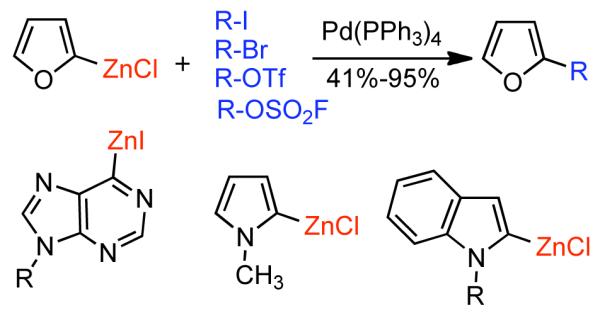
NiIICl2(dppp)232 and NiIIBr2(bpy)233 catalysts were successfully used for the cross-coupling of aryl pyridines and thienyl zinc halides in fair to good isolated yields providing an alternative low cost method for the synthesis of aryl-heteroaryl or heteroaryl-heteroaryl products. N-Heterocyclic carbenes (NHCs) PdII 234,235,236 or NiII 187,237,238 complexes have been recently employed in the Negishi aryl-aryl cross-coupling (Scheme 60). NHCs as an alternative to phosphine ligands gained recognition due to their high shelf stability and ease of handling.239,240 The PdII or NiII precatalysts are facile to prepare and can be used to furnish in biphenyl compounds in good yield using very low catalyst loadings. The first Negishi cross-coupling of aryl halides, triflates, tosylates and mesylates with alkyl or aryl zinc reagents using the Pd-NHC methodology based on PEPPSI-IPr system was reported by Organ.241 Alkyl mesylates and tosylates reacted in very high yields. However, aryl tosylates and mesylates were not active under PEPPSi-IPr protocol. A one-pot protocol was also applied by Knochel,235 in which the organozinc reagents were generated in situ from aryl or heteroaryl bromides and iodides in the presence of LiCl and Zn0 dust. In this way, the handling of air and water sensitive organozinc reagents was avoided. The catalyst loading was low (0.5 mol % of PEPPSI) while the reaction time varied from 0.5 to 24 h depending on the substrate. The first use of (NHC)-NiII catalysts in the Negishi coupling was reported by Chen237. Two catalytic systems containing mononuclear and binuclear NiII complexes with NHCs were obtained and their reactivity was compared. Heteroarene (NHC)-binuclear NiII complexes (I) showed higher reactivity as compared to the mononuclear complexes, perhaps a result of strong bimetallic cooperativity. The catalyst loading was higher as compared to analogous PdII but still relatively low (0.1-4.0 mol%). NHC-NiII catalysts showed good activity for unactivated para-, meta- or ortho- mono-or di-substituted aryl chloride or vinyl chloride electrophiles.
Scheme 60.
2.2.2.1. Aryl (Heteroaryl)-Alkyl (Alkenyl) Coupling
Extensive studies to expand the scope of the Negishi protocol to aryl-alkyl and alkyl-alkyl cross-coupling were performed by Knochel, Fu and Cardenas. Inspired by his earlier results in 1995,242 where primary unsaturated alkyl halides were found extremely active in Negishi cross-coupling due to presumably intermediate complex between NiII and olefin that facilitate transmetallation and reductive elimination, Knochel in 1998 described the cross-coupling of arylzinc reagents with alkyl iodides in the presence of 10% NiII(acac)2 catalyst and trifluoromethyl styrene as promoter (Scheme 61).243
Scheme 61.
Other additives such as electron-deficient arenes, styrenes and ketones were found to assist reductive elimination by binding to the Ni catalyst.203 Ammonium salts such as tetrabutylammonium iodide accelerate the Negishi cross-coupling for otherwise inactive benzyl zinc substrates and provided good yields for cross-coupling of benzylic zinc bromide with several examples of primary aryl and alkenyl triflates (Scheme 62).204
Scheme 62.
Diethyl phosphite and DMAP as co-ligands in THF-NEP as solvent improved significantly the efficiency of Negishi cross-coupling between alkenyl electrophiles such as halides or triflates with arylzinc reagents (Scheme 63). Under these conditions, catalyst loading could be reduced to as low as 1 mol%.217 Protocols using slightly higher catalyst loading (e.g. 2.5 mol% NiII(acac)2 and 5.0 mol% bis-(2-diphenylphosphinophenyl)ether (DPE-Phos) ligand and THF-NEP as solvent was successful for cross-coupling of aminoalkylzinc halide with aryl or heteroaryl triflates to produce aminoalkyl (hetero)arenes in 78 – 92% yields.244
Scheme 63.

Knochel used solid-phase resins (Rink-MBHA) to improve the Negishi Ni-catalyzed cross-coupling efficiency of resin bound ortho-substituted aryl halides and nonaflates with functionalized alkylzinc iodides. High purities and good yields (70 - 92 %) were obtained using a similar Negishi protocol with NiII(acac)2 as catalyst, Bu4NI as additive and THF-NMP as solvent. The desired cross-coupling product could ultimately be cleaved from the resin using TFA: CH2Cl2 (1:1) mixture.245
Aryl-alkyl and aryl-alkenyl cross-coupling products can be either generated from arylzinc reagents with the corresponding alkyl halides or by cross-coupling of an alkyl or alkenyl zinc halide with the aryl halide.246 Huo reported an efficient method for the direct insertion of Zn0 metal as dust, granule or shot for synthesis of alkyl zinc bromides and chlorides by activation with 1-5 mol% iodine.247 Previous methods required highly reactive Rieke-Zn for oxidative insertion248,249 or milder methods in which the Zn0 dust was activated with 1,2-dibromoethane and Me3SiCl and then reacted with alkyl bromides, using catalytic amount of alkali iodide.250 The alkyl zinc reagent were cross-coupled at 80 °C using 2 mol% NiIICl2(PPh3)2 in DMA or NMP as solvents. Bromides salts such as LiBr and Bu4NBr were very effective for mediating Zn0 insertion and efficient cross-coupling of alkyl chlorides with 4-chorobenzonitrile (Scheme 64).
Scheme 64.
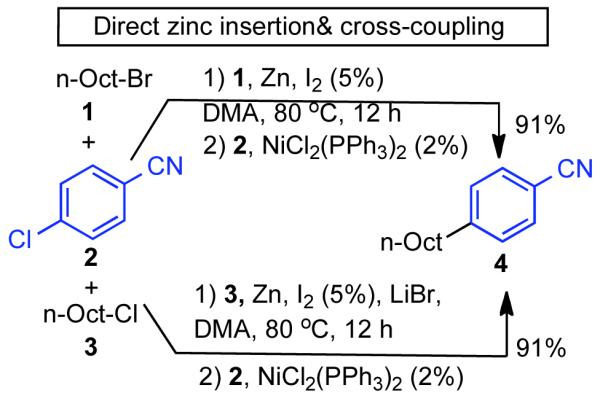
Zn0 insertion occurred in other polar aprotic solvents such as DMF, DMSO and DMPU, but not in THF, dioxane, diethyl ether, DME or acetonitrile. The author suggested a dual role for the iodine. Due to matched redox potentials an initial reaction between iodine and Zn0 provides a clean, reactive surface of the metal, while the I− anion may also induce a halogen exchange with alkyl bromide to provide a more reactive substrate alkyl iodide. Cardenas investigated the Ni-catalyzed cross-coupling including experimental and DFT studies on the mechanism.246 Substituted aryl iodides were successfully cross-coupled at room temperature with organozinc reagents in the presence of 3% NiIICl2(bpy)4 catalyst and 3% bpy as ligand. Diverse substituent functionalities worked well such as esters, nitriles, amines or acids. As typical for Ni-catalyzed homo- and cross-coupling nitro derivatives did not participate. Chlorides and tosylates were not reactive under these conditions providing for selective cross-coupling in chloroiodoarenes. Based on previous reports in the literature and DFT calculations the authors suggest a possible radical pathway. Oxidative addition of the haloarene on Ni0 complex should provide a NiII complex in the initiation process followed by a propagation step involving a NiI-NiIII catalytic cycle. Two paths are proposed: one involving the transmetallation on NiI before oxidative addition of the aryl halide or oxidative addition of the active catalyst to the aryl halide resulting in NiIII complex followed by transmetallation. The two catalytic cycles were investigated through DFT calculations of the activation energies during the presumed catalytic cycle. [NiI(bpy)I] and iodine was used as a model to minimize the number of possible isomeric species. Lower activation energies were obtained for the first catalytic path suggesting that the actual mechanism involves transmetallation of the NiI intermediate with alkyl zinc halide followed by oxidative addition towards diorgano-NiIII intermediate (Scheme 65).
Scheme 65.
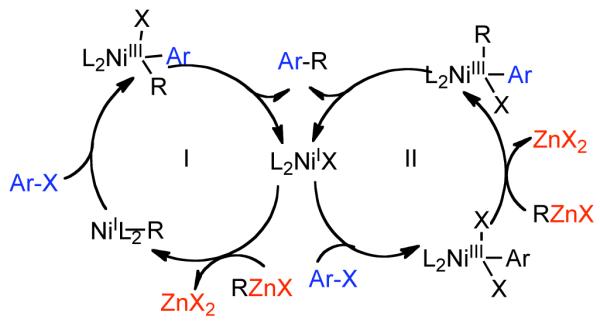
Styrene derivatives are important monomers and building blocks for fine chemical and their synthesis by vinylation of aryl halides using Pd vs Ni-cross-coupling was recently reviewed by Denmark.251 Two general strategies are employed for the synthesis of styrene derivatives the first one by cross-coupling of vinyl electrophiles with aryl zinc reagents or by cross-coupling of aryl electrophiles with vinyl zinc reagents. Yamakawa reported the first Ni-catalyzed cross-coupling of aryl halides with vinyl zinc bromide to produce styrene derivatives using NiII(acac)2 precatalyst or Ni0(COD)2 catalyst and Xantphos as a ligand. Vinylation of various aryl bromides and chlorides with vinyl zinc bromide was achieved in good yield for activated aryl halides but was slow for electron donating aryl bromides (Scheme 66).
Scheme 66.
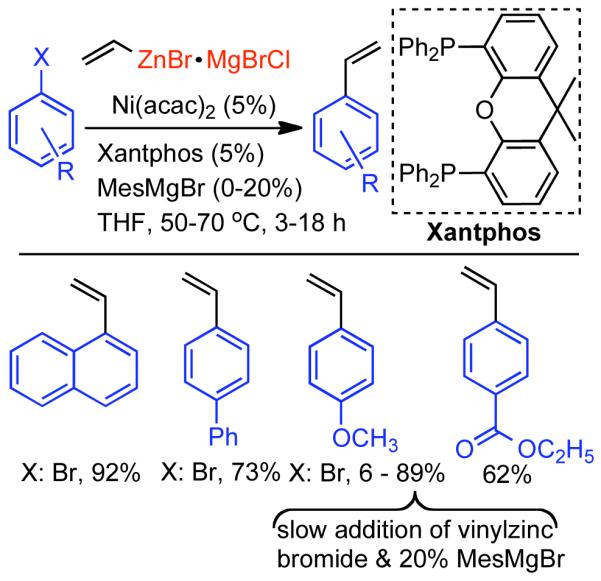
Catalytic carbonylative cross-coupling reactions are important for the synthesis of enones. Ni-catalyzed carbonylative reaction between enol triflates and aryl, alkenyl or alkyl organozinc reagents were recently reported by Chen using cost-effective NiIICl2 precatalyst, 4,4′-dimethoxy-2,2′-bipyridine as ligand in the presence of CO (1 atm) (Scheme 67).252 Good yields were obtained using various cyclic enol triflates in conjunction with diverse diaryl or dialkyl zinc reagents.
Scheme 67.
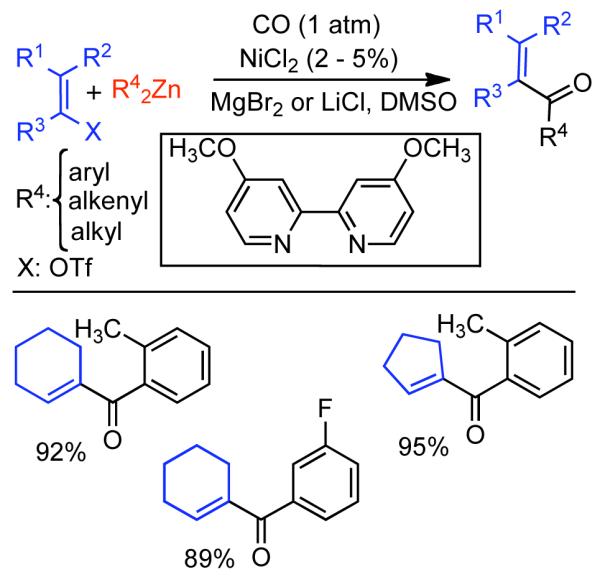
Effort was invested on establishing catalyst control over the stereochemistry of cross-coupling. Stereoselective cross-coupling reactions include both enantioselective catalysis and diastereoselective synthesis. In the first case, enantiomerically pure catalyst is used to control the stereochemistry of an active center, while in diastereoselective cross-coupling a stereocenter in the substrate influences the formation of another stereocenter within the molecule. Gagne used Fu methodology for the Ni-catalyzed Negishi coupling and applied it to carbohydrates in the first Ni-catalyzed cross-coupling of aryl and alkyl zinc reagents with secondary sp3-glycosyl halides. 253 The (iPr-PyBox) pincer ligand reported by Fu was not suitable for glycosyl electrophiles due to competitive β-elimination, while the unsubstituted PyBox ligand was found to be more effective and afforded the cross-coupling product in moderate yields. Mannosyl halide was cross-coupled in excellent yields giving high α-selectivity as compared to only slightly higher β-selectivity in the cross –coupling of glucosyl halides. Screening of catalyst and ligand combinations demonstrated that Ni0(COD)2/tBu-Terpy/DMF was a better catalyst for achieving β-gluco selectivity, providing good control over the stereochemistry and tolerance to different functionalities such as halides, acetals, ethers and esters (Scheme 68). It is evident that the stereochemical outcome of the cross-coupling can be controlled by the appropriate choice of NHC ligand. Heteroaryl zinc halide substrates, such as those derived from furans and 3-thiophenes, but not pyridines, were also effective in stereocontrolled cross-coupling. The substrates themselves played an important role in the control of stereochemistry where C2 axial substituted mannosides induced high α-stereoselectivity and glucose and galactose having a less defined anomer preference resulted in reduced α:β selectivity.
Scheme 68.

Two mechanisms have been proposed for the Ni-catalyzed Negishi cross-coupling of aryl halides, both involving the generation of carbon-centered radical prior to the formation of the Ni-C bond.254,255,256 In the first mechanism the carbon-centered radical was generated via an inner sphere halogen abstraction, while in the second it could be formed by outer sphere single electron transfer (OSET). Both mechanisms suggests step-wise oxidative addition through one-electron transfer rather than a two-electron process (Scheme 69). However, the generation of the glucosyl or mannosyl radicals should provide α-selective in both cases due to strong anomeric effects within the substrate. Therefore, the dependence of stereoselectivity on the catalyst suggests a more complex mechanism.
Scheme 69.

2.2.3. Kumada Cross-Coupling
2.2.3.1. Aryl (Heteroaryl)-Aryl Coupling
The first report on cross-coupling of aryl or olefinic halides with aryl Grignard reagents catalyzed by NiII(acac)2 and NiIICl2(dppp) or NiIICl2(dppe) was described simultaneously by Kumada200 and Corriu, respectively.257 This “Kumada-Corriu” or “Kumada” reaction quickly became a valuable tool for C-C bond formation and was extended to many other substrates using Ni,200,258,259 Pd,260 Fe and Cu based catalysts.19 Aryl iodides, bromides and notably chlorides were demonstrated to efficiently cross-couple with Grignard reagents in very good yields in the presence of Ni catalysts based on nickel halide phosphines or nickel-on-charcoal (Ni/C) where direct reduction of the NiII/C to Ni0/C was obtained via Grignard reagent.261,262,191 Aryl triflates were also proven to be efficient in the asymmetric synthesis of chiral biaryls using Pd-based chiral catalyst (PdIICl2[(s)-Phephos])263 (Scheme 70). Synthesis of the chiral biaryls was also accomplished in up to 95% ee using Ni-catalyst based on (S)-1-[(R)-2-(diphenyl-phosphino)ferrocenyl]ethyl methyl ether ((S)-(R)-PPFOMe) and aryl bromides as substrates (Scheme 70).264
Scheme 70.
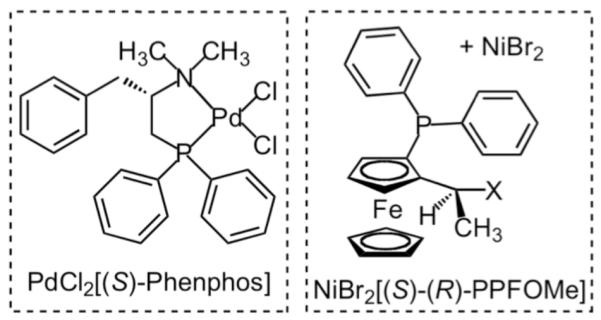
Although aryl halide and triflate electrophiles were known to be effective in Kumada cross-coupling, aryl mesylates were generally regarded as inactive. In a 1995 report, Percec demonstrated the efficient oxidative addition of aryl mesylates to Ni0 and successful transmetallation with Grignard reagent and subsequent reductive elimination to produce aryl-aryl coupling products.265 The active catalyst was generated in situ from NiIICl2(dppf) and 1 equivalents of Zn0 powder as reducing agent to produce the aryl-aryl cross-coupling products in moderate yields (31%-83%) (Scheme 71).
Scheme 71.

In the search for more efficient, cost effective and environmental friendly conditions for Kumada cross-coupling reactions, two parameters were investigated: catalyst/ligand combinations for efficient coupling at minimal catalyst loading and mild reaction conditions and new electrophilic substrates as coupling partners to the Grignard reagents that provide direct transformations and access to environmentally friendly substrates. While most NiII catalysts utilized in Kumada cross-coupling are nickel complexes with monodentate (PPh3, PCy3) or bidentate (dppp, dppf, dppe) phosphine ligands, immobilized NiII catalyst were less explored, but nevertheless would provide the advantage of easy removal and handling. Several examples of heterogeneous catalysts active for Kumada cross-coupling such as polymer supported nickel,266 nickel-on-charcoal (Ni/C),261,262 nickel/nanoporous materials,267 N-heterocyclic carbene-based NiII complex268 or silica immobilized NiII catalyst (Scheme 72) were reported.269
Scheme 72.
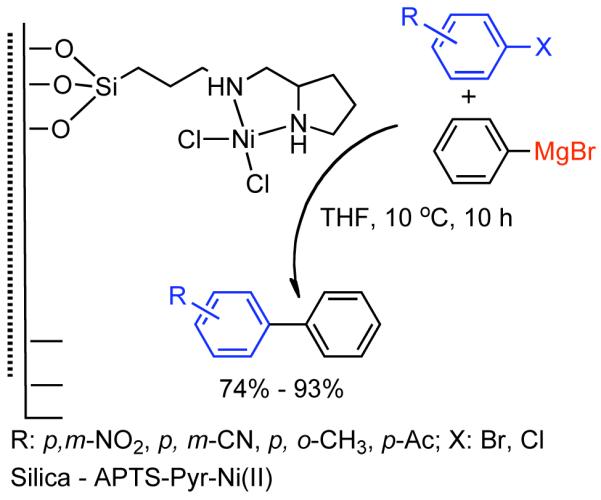
NHC ligands have strong σ-donating properties stabilizing various oxidation states of Ni resulted in the catalytic cycle. While dissociation of the ligand is prevalent in Ni-phosphine complexes, NHC-based Ni-complexes are more stable. Ni-NHCs were investigated as active catalysts for Kumada cross-coupling (Scheme 73). The active catalyst was either obtained in-situ270,271,272 or was synthesized beforehand and used subsequently in the cross-coupling.273,274,268,237,275,276 Aryl triflates and halides including fluorides were successfully coupled with aryl Grignard in medium to excellent yields. Activation of the relatively intractable C-F bond was reported by Herrmann using a Ni-NHC catalyst (bis[1,3-di(2′,6′-diisopropylphenyl)imidazolin-2-ylidene]nickel0) obtained in situ from Ni0(COD)2 and the sterically hindered NHC.277 Pd0/II- and Ni0/II-NHC catalysts based on other diaminocarbenes were reported by Fürstner and their catalytic performance was demonstrated in Suzuki, Heck, Kumada and amination reactions indicating their universality.278
Scheme 73.

The high activity of polynuclear Ni-NHC complexes was explained as a result of Ni-Ni bimetallic cooperation in catalysis. Nakamura reported other examples of systems where bimetallic cooperation between Ni and Mg was responsible for high catalytic activity.279,280 The design of the system was presented as a push-pull mechanism between a d10 metal and a Lewis acidic main group metal that synergistically promotes carbon-halogen bond cleavage. The bimetallic cooperation is orchestrated through the design of a bidentate ligands L1-L2 that can simultaneously coordinate to a group 10 metal (M1) (Ni0 or Pd0) and a Lewis acidic metal M2 (Scheme 74).
Scheme 74.
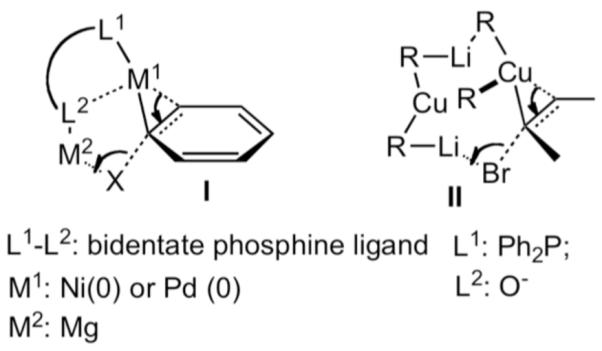
Hydroxyphosphine ligands (PO) were found to significantly accelerate and facilitate cross-coupling of Grignard reagents with less active substrates such as aryl fluorides, chlorides, triflates, polyfluorides, polychlorides and even carbamates and phosphates.279 The cooperative role of the ligand in the cross-coupling reactions was investigated by Nakamura for both Ni-catalyzed Kumada reactions279,280 and asymmetric Cu-catalyzed C-C bond formation.281,282,283 Even very low ligand and catalyst loading (0.05-1.00 mol%) were able to considerably accelerate the reaction and promote rapid (in 1-3h) cross-coupling in very good yields (91-98%) (Scheme 75). Aryl sulfide substrates dramatically reduced the reaction rate suggesting that they interact with the catalyst as ligands to form a species that does not directly participate in the catalytic cycle.279
Scheme 75.

Nakamura studied the mechanism of Kumada cross-coupling and design of bimetallic catalytic systems.279 In order to understand the origin of the high efficiency of the Ni/PO system, he measured the 12C/13C kinetic isotope effects (KIEs) in the first irreversible step of the catalytic cycle of o-tolyl halide (X = F, Cl, Br, I) with phenylmagnesium bromide. Two extremes in the KIEs distribution were expected, one is an asymmetric distribution when complexation with the π bond occurs, while high KIE values at the ipso carbon are expected when C-X bond cleavage (oxidative addition) is prevalent. The data were compared with the KIEs obtained for the conventional nickel- or palladium-bisphosphine catalysis. The results obtained showed an unequal distribution of the KIEs on the six carbon atoms from the aromatic ring. This outcome contradicts the mechanism of single-electron transfer in the oxidative addition proposed by Kochi for the stoichiometric reaction of an electron-rich Ni0-phosphine complex with an aryl iodide or bromide. The KIEs values were also different depending on the nature of the aryl halide. Aryl iodides had smaller KIEs values compared to aryl bromides and chlorides. In addition, the KIE values of the Ni/PO catalytic system were much different from those obtained for the conventional Ni-bisphosphine catalysis but similar to the KIEs of Pd-bisphosphine catalysis and the organocopper catalysis. Supplementary DFT calculations favored the bimetallic synergy and the hypothetical transition state I from Scheme 74 where a Ni/Mg species showed a strong preference for simultaneous interaction with the aryl iodide. DFT calculations supported the efficacy of the Ni/PO system as compared to the Ni-bisphosphines. A mechanism (Scheme 76) consistent with both KIE data and DFT calculations was proposed.284,279
Scheme 76.

Recently, Matsubara reported a novel T-staped NiI-NHC, NiCl(IPr)2 (IPr = 1,3-bis(2,6-diisopropylphenyl)imidazolin-2-ylidene), isolated from the reaction of Ni0-(NHC)2 with phenyl chloride and its use in efficient cross-coupling with Grignard reagents (Scheme 77).285 Unlike many other Ni-NHCs, the catalyst is not air-stable, but could be isolated by recrystallization from mixture of hexane : THF at −30 °C under inert atmosphere. Depending on the solvent, two forms were generated. The dimeric complex 3 was obtained after dissolution of compound 2 in C6D6, although excess of the NHC ligand did not provided structure 3, which demonstrated an equilibrium between the dimeric structure and the ligand. Single crystals of the active catalyst were analyzed by X-ray diffraction and revealed a 3-coordinate planar T-shape structure.
Scheme 77.

Significant attention has been devoted to the development of new electrophilic coupling partners for the Kumada reaction that would provide access to environmentally friendly substrates and would reduce the number of steps required for the transformation. While aryl halides and triflates were intensively used as electrophiles as well direct C-H activation, phenol substrates and their derivatives such as carbamates, phosphates and in particular phenolates are desirable since they can be easy synthesized from abundant and cost-effective phenols. Previous use of phenols as precursors to electrophiles required their transformation into active functionalities such as triflate, mesylates, tosylates, aryl carboxylates and carbamates. Very recently Shi reported the use of lithium, potassium, sodium or magnesium phenolates as coupling partners in Kumada reaction to generate biaryls in high yields (See Chapter 8).286 Here, the Lewis acidic metal cations were proposed to generate tightly coordinated polymetallic intermediates. Aryl cyanides have also been successfully used as electrophiles in the Kumada cross-coupling behaving as pseudo-halides in the C-C bond formation. Miller reported the cross-coupling of benzonitriles with Grignard reagents using NiIICl2(PMe3)2 as catalyst in refluxing THF.287,288,289 The competing attack on the cyano functionality was suppressed using t-BuOLi as an additive, however, the reported yields were lower than the cross-coupling of classic aryl halides electrophiles. Nevertheless, this procedure could be applied to electron-rich and deficient benzonitriles as well as to heteroaromatic nitriles. Shi has also explored an intriguing variant of the Kumada cross-coupling employing diaryl sulfates as electrophiles.290 In this approach, phenols were readily converted to the corresponding disulfates through treatment with N,N’-sulfuryldiimidazole. It was found that NiIICl2(PCy3)2 in the presence of excess PCy3 ligand was the most effective catalytic system providing efficient cross-coupling within 2 h at ambient temperature (Scheme 78). Both, aryl units on the disulfate are cross-coupled indicating that magnesium monosulfate salt identified as the byproduct from the first cross-coupling can undergo subsequent oxidative addition to Ni. This approach may be advantageous in some circumstances, as unlike analogous reactions with monosulfates, the Kumada cross-coupling of aryl disulfates is 100% carbon-efficient producing non-toxic MgSO4 and MgBr2 as the major byproducts. However, it will be necessary to consider the stoichiometric quantities of N,N’-sulfuryldiimidazole in considering carbon and cost efficiency.
Scheme 78.

2.2.3.2. Aryl (Heteroaryl)-Alkyl Coupling
Later developments involved the extension of the Kumada protocol to cross-coupling of saturated alkyl Grignard reagents with alkyl or aryl chlorides and tosylates to generate aryl-alkyl or alkyl-alkyl coupled products. Kambe reported the first aryl-alkyl coupling and observed a remarkable effect of 1,3-butadiene in stabilizing the catalyst in the cross-coupling reaction of alkyl bromides, chlorides and tosylates with alkyl or aryl Grignard reagents (Scheme 79).291,292 The optimized catalyst/additive used was 1 mol% NiIICl2 and 10% 1,3-butadiene, while nickel complexes based on phosphine ligands such as NiIICl2(dppp) or NiIICl2(PPh3)2) provided the cross-coupling product in very low yields. Aryl and secondary alkyl Grignard reagents cross-coupled with the corresponding alkyl bromide and tosylates in moderate to good yield. Interestingly, aryl substituted bromide remained intact when the substrate contained both aryl and alkyl bromide functionally, thereby demonstrating selective cross-coupling with Grignard reagent at the alkyl site (Scheme 79).
Scheme 79.
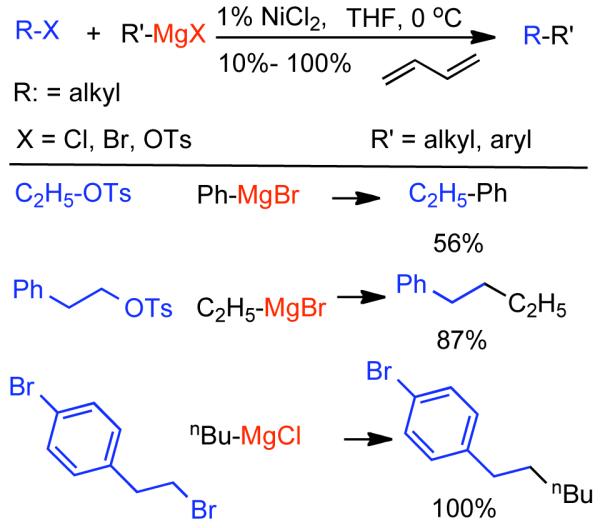
A mechanism was proposed involving the reduction of the NiIICl2 to Ni0 in the presence of the Grignard reagents followed by complexation with 1,3-butadiene to generate a bis-π-allyl nickel complex that further reacts with the Grignard reagent to give complex 3 followed by reductive elimination to generate the cross-coupling product (Scheme 80).
Scheme 80.

Ni complexes containing a pincer amidobis(amine) ligand were found to have a broader scope than the Pd-complexes due to their high reactivity for secondary alkyl halides whereas Pd catalysis was limited to only primary alkyl halides substrates. Hu reported an efficient Ni-pincer catalyst with high reactivity towards functionalized alkyl bromides and significantly extended the scope of the Kumada coupling for non-activated alkyl halides.293 (Scheme 81)
Scheme 81.

2.2.4. Chain-Growth Condensation Polymerization via Kumada Coupling
In 1992, McCullough294 and Rieke295 independently reported the NiIICl2(dppp) and NiIICl2(dppe) catalyzed condensation polymerization of (5-bromo-4-hexylthiophen-2-yl)magnesium bromide or 5-bromo-2-bromozincio-3-hexylthiophene to produce regioregular poly(3-hexylthiophene) (PHT). These and related methodologies have been extensively employed in the synthesis of thiophene, phenylene, fluorene, and related polymer based electronic materials.4 A survey of all such applications is beyond the scope of this review as such work has been devoted to aryl and heteroaryl halides. Nevertheless, there is one recent development that is important to consider in future exploration of Ni-catalyzed cross-coupling polymerization of aryl-phenol derived electrophiles. In 2004, both Yokoyama296,297,298,299 and McCullough300 reported that under conditions of low NiIICl2/L-catalyst loading these Kumada-type and Negishi-type polymerization reactions could be converted from a step-growth condensation polymerization to a chain-growth polymerization. It is thought that chain-growth occurs under these scenarios by intramolecular transfer of the catalyst from the site of rate-determining reductive elimination301 to the new chain-end (Scheme 82). This chain-growth condensation polymerization has been applied to the synthesis of poly(thiophene)s, poly(thiophene) copolymers, and poly(phenylene)s,302 and poly(fluorene)s303 with sterically controlled regioregularity,304 predictable molecular weight evolution and lower polydispersity than could be achieved via analogous step-growth polymerization.
Scheme 82.

2.2.5 Other Transmetallating Reagents
2.2.5.1. Organosilanes
Alkynylsilanes have been used with success for Sonogashira cross-coupling reaction. Generally this reaction proceeds in the presence of homogenous Pd catalysis. However, because of high contamination of the product and difficulty in purification, efforts were made to use a cheaper, but still highly reactive catalyst such as Ni. Wang and coworkers reported in 2004 an ultrafine particle Ni0 catalyzed reaction of a terminal alkynylsilane with aryl iodides, vinyl iodides and aryl bromides in heterogeneous catalytic conditions. They successfully cross-coupled 1-phenyl-2-(trimethysilyl)acetylene with p-iodotoluene by using 100 nm Ni0 as catalyst in the presence CuI and PPh3 in isopropanol solution at 80 °C using of KOH as base (Scheme 83), neat, or under microwave irradiation in the presence of alumina (Scheme 84).305,306
Scheme 83.
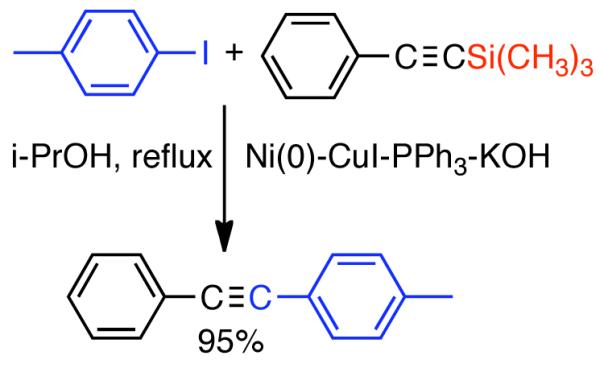
Scheme 84.
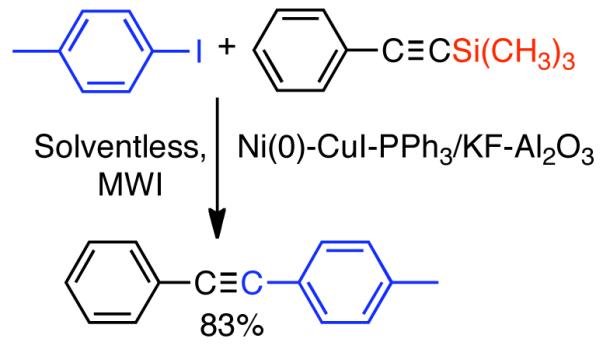
This nanoparticulate Ni0 proved to be an inexpensive alternative to the Pd–catalyzed reactions, and resulted in good yields of the desired cross-coupling product. In the search for cheaper metal catalysts, Heyon group developed bimetallic core/shell nanoparticles (NPs) with a Ni rich core/Pd rich shell structure307. They showed the catalyst was active for the cross-coupling reaction of aryl bromide with ethynyltrimethylsilane (Scheme 85). The NP catalysts can be reused and recycled at lest five times without losing the catalytic activity.
Scheme 85.
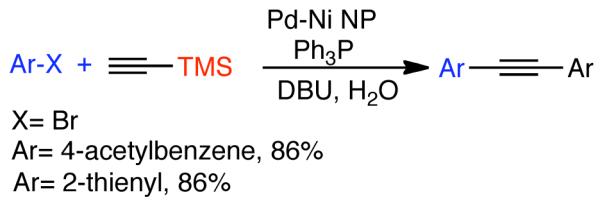
To date there have been no examples of Ni-catalyzed Hiyama reactions involving aryl or vinyl sulfonates. However, recent examples of Pd-catalyzed Hiyama cross-coupling of aryl and vinyl mesylates308 and tosylates161,309 have been reported.
2.2.5.2 Organostannanes
The cross-coupling reaction of organostannanes with organic electrophiles is known as the Stille reaction. This method uses frequently palladium catalysts and provides good result only for halides and triflates.25 Searching for alternative to palladium catalyst and to extend this reaction for other organic electrophiles such as aryl mesylates, Percec et al.265 tested the applicability of (tri-n-butylphenyl)stannane for Ni0 catalyzed reactions of p-(methoxycarbonyl)phenyl mesylate (Scheme 86). The reaction proceed in the presence of NiIICl2(PPh3)3 or NiIICl2(dppf) in THF and in both cases were obtained lower yields (23-24%) of the cross-coupled product and large amount of homo-coupled side product (Scheme 86). The explanation of the side reaction was the poor reactivity of organostannane towards the ArNiIIL2X species resulted from the oxidative addition of aryl mesylate to Ni0. Lee group studied also the nickel-catalyzed cross-coupling and carbonylative cross-coupling of organostannanes using hypervalent iodonium salts310 and they found that iodonium salt has a good reactivity in the presence of NiII(acac)2 (10mol%) and NMP as solvent (Scheme 87).
Scheme 86.
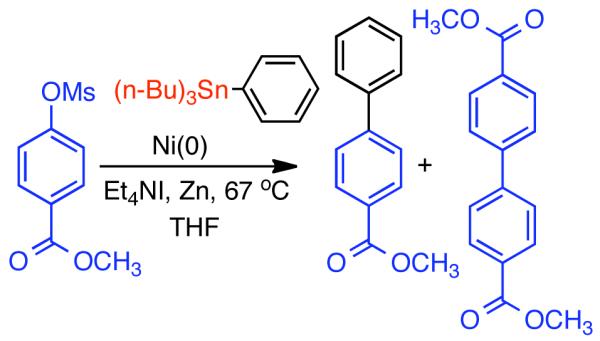
Scheme 87.
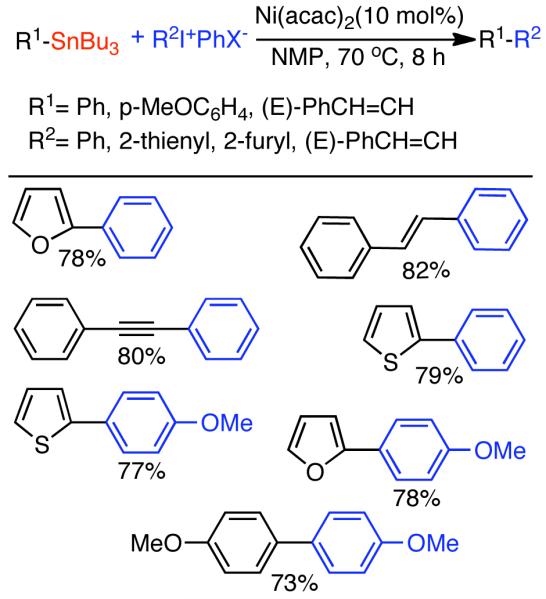
2.2.5.3 Organoindium Reagents
Recently Wu explored the use of organoindium reagents in the cross-coupling of aryl sulfonates (Scheme 88).311 5 mol % NiIICl2(PCy3)3 in the presence of excess of PCy320 mol%) was found to be an optimal catalyst from the cross-coupling of aryl mesylates, tosylates, and phenyl sulfonates with triarylindium reagents. Decreases in ligand or catalyst loading resulted in significantly diminished yield. It was proposed that a mechanism akin to standard Suzuki-Miyaura coupling applied, wherein triarylindium transmetallates the oxidative addition adduct. As the catalyst is supplied as the NiII-complex it is apparent that the triarylindium reagents are capable of mediating the reduction to Ni0.
Scheme 88.

2.3 Functionalization of Sulfonates
The previous chapters covered the homocoupling (Section 2.2.) and cross-coupling (Section 2.3.3) of aryl and vinyl sulfonates utilizing Ni-catalysts. However, transformation of sulfonates is not limited to the construction of C-C bonds. In many cases, C-O derived sulfonates can be catalytically functionalized312 with various heteroatom bearing groups or alternatively reduced to C-H. Functionalization of sulfonates can provide periphery moieties required in the final product or alternatively install a handle for subsequent coupling reaction, ultimately allowing for homocoupling or cross-coupling derived exclusively from phenolic or enolic starting materials and employing only low-cost Ni-catalysts.
2.3.1. C-B Bond Formation
The utility for boronic acids, esters and trifluoroborate313,314,315 salts as synthetic intermediates,22 catalysts316, sensors317, building blocks for functionalized materials318, and in medicine,319 has lead to considerable development in the art of C-B bond formation. Ultimately, a general, robust and cost-effective, methodology is sought. The increased usage of aryl and vinyl boronic acids, esters and trifluoroborate salts in the Suzuki-Miyaura cross-coupling reaction, has contributed significantly to the current fascination with C-B chemistry.
The chemistry of C-B bond formation dates back to the late 19th century when diaryl mercury compounds and boron trichloride were used to generate boronic acids.319 Due to the limited scope of the chemistry, as well as safety and environmental concerns, this method was never applied to large-scale synthesis. Later, in the early 20th century an alternative methodology involving the trapping of arylmetal intermediates derived from aryl halides with electrophilic borates at low temperatures was reported (Scheme 89). 320,321 Amongst the most common aryl metal intermediates are Grignard and organolithium reagents. Due to the attractive costs of the reagents, this method has been frequently applied to large-scale synthesis in both academic and industrial settings. However, the substrate scope is limited to arylhalides, typically aryl bromides and iodides. Moreover, this method does not tolerate certain electrophilic or protic functional groups such as carboxylic esters and hydroxyl groups. Sometimes the functional group intolerance can be mitigated by the use of the recent “in-situ quench method”. For example, n-BuLi was added to a solution of 3-bromopyridine and triisopropyl borate. In this case, the rapid quenching of the aryllithium intermediate prevented decomposition and providing improved yield. Nevertheless, for some substrates such as aryl carboxylic esters, the yield remained inadequate.322
Scheme 89.
Besides the trapping of arylmetal intermediates with borate, direct activation and borylation of C-H bond is quite attractive (Scheme 90). The first example of the direct C-H borylation was elaborated by Hartwig in 1995 using stoichiometric levels of (CO)5MnBcat (cat = catechol), (CO)5ReBCat or Cp(CO)2FeBcat.323 Early catalytic examples of direct C-H borylation were provided by Hartwig and Smith in 1999, using Cp*Re(CO)3 catalyst under a mercury light in the presence of B2pin2 as boron source,324 or Cp*Ir(PMe3)(H)(BPin),325 respectively. Further refinement of this protocol using a thermally activated Cp*Rh(η4-C6Me6) catalyst326 and more convenient and economical HBpin as the boron source327,328,329,330 considerably improved the regioselectivity of the reaction.331,332,333,334 The direct C-H borylation is not regiospecific and while typically providing meta-borylated products can be coaxed to producing ortho-borylated products under directing conditions.
Scheme 90.
Another, and decidedly regiospecific, approach to the synthesis of aryl boronates is transition metal catalyzed coupling of aryl halides, sulfonates and sulfates with diboronyl reagents (Scheme 91). In 1995, Miyaura used a Pd catalyst, to generate arylboronic esters via the coupling of aryl bromides, iodides and triflates with diboronyl esters such as B2pin2.335 The corresponding boronic acids were obtained after acid workup. The standard catalyst used in this procedure was PdIICl2(dppf) in conjunction with a base. This protocol exhibits advantages as compared to arylmetal approaches derived from the correspondingly more mild conditions that were more tolerant toward carbonyl-containing substrates. However, the high cost of Pd-catalysts, diboronyl reagents, and aryl bromides, iodides and triflates can be an obstacle to its implementation. Not surprisingly, efforts have been made broaden the scope of the reaction so as to employ less expensive and more available boron sources, less expensive substrates such as aryl chlorides, mesylates and tosylates, as well as the use of non-precious metal based Ni-catalysts. Given the availability, structural diversity, and relatively low cost of phenol-derived substrates their use as precursors for organoboron compounds is appealing 336 This chapter will focus on the C-B bond formation through aryl sulfonates and sulfates catalyzed by Ni.
Scheme 91.
In 2000, Tour reported the first example of Ni-catalyzed borylation, in the context of generating polyborylated flame-retardant materials.337 Inspired by Miyaura’s work on Pd0 catalysts, they applied a similar ligand to a more reactive Ni0 catalyst. Surveying an array of bidentate phosphine ligands (including dppm, dppe, dppp, and dppb), Tour found that NiIICl2(dppp) was the most effective catalyst for the diborylation and triborylation of the two investigated substrates 1,4-dibromobenzene and 1,3,5-tribromobenzene. The use of NEt3 limited side-reactions and facilitated the C-B bond formation.338 (Scheme 92) A later attempt to use 2,6-bis (methylphospholyl)pyridine ligand and its cationic PdII and NiII complexes for synthesis of arylboronic esters did increase the turnover number of PdII catalyst. However, no activity of NiII complex was found.339
Scheme 92.
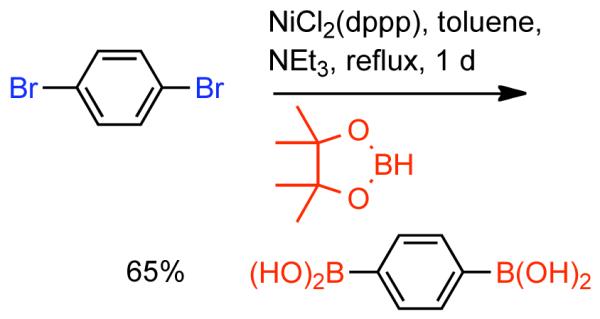
Previously, Miyaura has demonstrated the use of activated triflates for the synthesis of aryl boronates.335 However, less expensive340 albeit less reactive sulfonates such as mesylates and tosylates were not compatible with the original Pd-catalyzed protocols due to sluggish oxidative addition. In the early 1990s, Percec discovered that Ni0 catalysts formed in situ from NiII precatalysts via reduction with Zn0, could react with aryl mesylates and tosylates and undergo cross-coupling and homocoupling reactions in good yields.92,94,164,265 The high reactivity of Ni catalyst was explained by the higher electron density of Ni0 compared to Pd0. Inspired by the activation of less reactive mesylates and tosylates via Ni, Percec’s group continued their investigation to generate aryl boronate esters using Ni catalysts and aryl halides and sulfonates as substrates. Early work was carried out using aryl halides as the substrate.341 Initial studies were performed using HBpin prepared in situ from pinacol and BH3·DMS.342 However, it was discovered that significantly less expensive neopentylglycol when treated with BH3·DMS could produce in situ a novel boron source, neopentylglycolborane (Scheme 93).343 The preparation of arylneopentyglycolboronate esters was found to have advantages as compared to the corresponding arylpinacolboronate esters, including improved crystallinity for easy purification as well as more facile conversion to the corresponding boronic acid. In fact, mild conditions employing transesterification with diethanolamine were elaborated to convert the arylneopentylglycolboronate esters to their corresponding boronic acids even in the presence of labile methyl carboxylates (Scheme 94).344,345
Scheme 93.
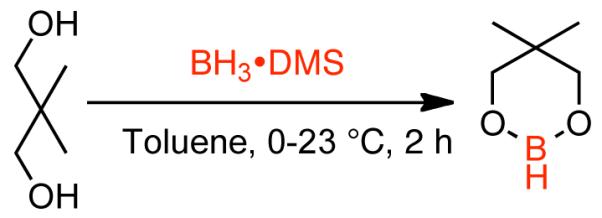
Scheme 94.

The first Ni-catalyzed neopentylglycolborylations were found to be most effective using NiIICl2(dppp) in the presence of dry Et3N as base. In this initial report, it was found that electron-rich and electron-deficient aryl bromides and iodides, including fused-aromatic substrates and sensitive functionality such as methyl esters could be efficiently converted to their corresponding aryl neopentylglycolboronates. The reaction conditions were sensitive to the solvent and were most successful in toluene or anisole, whereas related Pd0 catalysts for borylation,346 were efficient in dioxane. Excess ligand diminished the amount of hydrodehalogenated byproduct, presumably through the stabilization of the in situ formed Ni0 complex. (Scheme 95)
Scheme 95.

The resulting arylneopentylglycolboronate esters were found to be compatible with sequential Ni-catalyzed cross-coupling with aryl chlorides, bromides and iodides. Electron-deficient aryl neopentylglycolboronate esters cross-coupled effectively in the presence of mild K3PO4·nH2O base, whereas electron-rich boronate esters required stronger NaOH as base. The use of NaOH was incompatible with certain functional groups, thereby limiting somewhat the range of biaryls that could be produced.
Therefore, further development of the Ni-catalyzed neopentylglycolborylation focused on process improvements to overcome some of the original limitations. Firstly, it was observed that the formation of neopentylglycolborane and its subsequent activation of the NiII-precatalyst were thermally separable, allowing for the preparation of the neopentylglycolborane directly in the reaction media. Additionally, it was found that the incompatibility of electron-rich aryl neopentylglycolboronates with cross-coupling under mild conditions could be remedied by the use of Ni0(COD)2/PCy3 or complimentary Pd catalysis. Together, these approaches could be combined to produce a one pot-synthesis of biaryls by adding the Pd-catalyst and a second aryl halide, mesylate or tosylate substrate after completion of the initial Ni-catalyzed borylation reaction. In the course of pursuing improvements to the original Ni-catalyzed borylation, the compatibility with aryl sulfonate substrates was also explored. Percec had previously demonstrated that aryl mesylates could, under appropriate Ni-catalyzed conditions, be harnessed for homocoupling and cross-coupling reactions. Notably, Ni-catalyzed approaches to the Suzuki cross-coupling of aryl mesylates with aryl boronic acids was developed. Therefore, the development of a Ni-catalyzed borylation of aryl mesylates, would provide a convenient route to diversely functionalized biaryls that did not require the use of expensive aryl halide intermediates. It was found that anisole mesylate could be borylated, but only in very low yield 8% (Scheme 96).195 While this was the first example of Ni-catalyzed borylation of aryl sulfonates, the yield needed significant improvement.
Scheme 96.
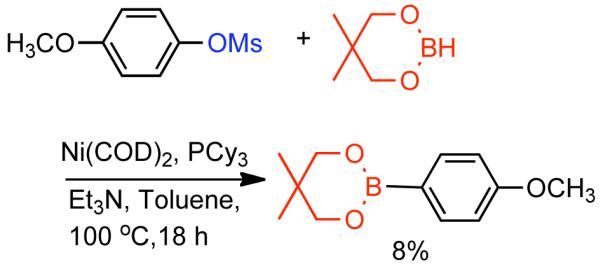
Subsequently, Percec found that the use of mixed ligand catalysts, notably NiIICl2(dppp) in the presence of dppf co-ligand, could improve the activity of the catalyst allowing for the neopentylglycolborylation of aryl chlorides.347 In addition to the electron-withdrawing and electron-donating functionality demonstrated previously for the neopentylglycolborylation of aryl bromides341 and aryl iodides,341 other functionality such as cyano, ketone, methylcyano, phenylsulfone, and bisimide were effectively tolerated. Bisborylation of dichlorides347 or borylation of protic substrates provided incomplete or no conversion to product, respectively. (Scheme 97)
Scheme 97.

In addition to the NiIICl2(dppp)/dppf mixed-ligand catalyst system, a recent study of a vast array of mixed-ligand Ni-catalysts provided additional privileged catalysts for the neopentylgycolborylation of relatively intractable ortho-substituted aryl halides.348 Substrates with electron-withdrawing, including esters and fluoro groups, and electron-donating functional groups were efficiently borylated. Likewise, pseudo-ortho substituted 2-bromo, and 2-iodothiophene could be borylated in high yield, with catalyst loading level as low as 3 mol% NiIICl2(dppf). Nevertheless, the isolated yield was low for some substrates. In some cases, low yields were due to difficulties in separation or isolation of the viscous oil. In many cases this could be alleviated by converting the crude product to the corresponding crystalline potassium aryltrifluoroborate. For other substrates, most notably for methyl 2-halobenzoates, significant levels of protodeborylated and hydrodehalogenated products were produced, causing a lower yield. Mechanistic studies revealed that protodeborylation of arylneopentyglycolborylates requires moisture or air in the presence of a NiII complex, and that such protodeborylation only occurs for bulky, electron-deficient ortho-substiutents such as methyl ester groups. Arylneopentylglycolboronates bearing electron-donating or para-substitutents do not undergo protodeborylation under similar conditions. Therefore, it is likely that the formation of methyl benzoate during the neopentylglycolborylation of methyl 2-halobenzoate is the result of NiII–catalyzed protodeborylation. While not found to be a major problem in most substrates investigated, it was also determined that homocoupling of the aryl neopentylglycolboronate was possible in the presence of NiII-catalyst and air, while homocoupling of the aryl halide can occur in the absence of neopentyglycolborone. Therefore, if arylneopentylglycolborylation of a new substrate is unsatisfactory, efforts to exclude adventitious moisture or air should be made, and fresh-preparation of the neopentylglycolborane may provide marked improvements.
Percec further demonstrated the power of the mixed-ligand NiIICl2(dppp)/dppf catalyst system in the first general borylation of aryl mesylates and tosylates.349 High yields were obtained for unsubstituted aryl mesylates. For ortho and para substituted aryl mesylates, the yield was lower (1-33% and 24-65%, respectively) (Scheme 98). However, the reactivity differences between electron-withdrawing and electron-donating substituents was comparatively insignificant.
Scheme 98.

Recalling his earlier studies in Ni-catalyzed homo- and cross-coupling, Percec found that in situ reduction of the NiII precatalyst to its active form with Zn0 powder considerably accelerated the reaction and greatly improved the yield. A diversity of electron-rich and electron-deficient mesylates were found to be completely borylated in 1-2 h with high isolated yields (75-97%).349 Notably, aldehyde, ketone, and N-heterocyclic (pyridines) functionality were not tolerated by these conditions resulting in notable byproduct formation. However, sterically hindered ortho-substituted mesylates could be converted to the corresponding boronate esteres in good yield (Scheme 99). Other co-ligands were also studied but provided lower yield. The catalyst loading level could be reduced from 10 mol% to as low as 2 mol% NiIICl2(dppp) and 4 mol% dppf and still generate good yields but only after longer reaction times (~7 h).349
Scheme 99.

Ni0, particularly the powerful mixed-ligand catalysts,347,348,349 were far more reactive than Pd0-systems toward unreactive substrates such as aryl chlorides and aryl mesylates and tosylates.349 To date, no successsful Pd-catalyzed borylation of aryl mesylates or tosylates has been reported. However, recently bulky electron donating ligands, such as Buchwald ligands, were able to affect efficient Pd-catalyzed Suzuki-Miyaura cross-coupling of aryl sulfonates159,350,161, while bis(di-tert-butyl(4-dimethylaminophenyl)-phosphine) dichloropalladium(II) has been developed as an effective catalyst for the cross-coupling of aryl, heteroaryl, and vinyl boronic acids with σ-cyanohydrin triflates.351
2.3.2. Homocoupling of Boronic Acids
In Ni-catalyzed Suzuki-Miyaura cross-coupling of aryl halides and pseudohalides with aryl boronic acids, NiII-precatalysts are converted to the active Ni0 catalyst via reduction by an external agent such as Zn0 (Section 2.2.1.1.) or via excess boronic acid (Section 2.2.1.2.) (Scheme 100). If the boronic acid mediates the reduction of the precatalyst, two equivalents of boronic acid per equivalent of NiII-complex transmetallate to form an ArNiIIAr complex. Activation to the Ni0 precatalyst is achieved by reductive elimination to produce homocoupled biaryls. Using stoichiometric NiII, quantitative homocoupling is trivial. However, it is also possible to perform the homocoupling catalytically. In 2006, Tao reported the homocoupling of various aryl boronic acids in the presence of the most simple NiII-catalyst, NiIICl2·6H2O, in the presence of K2CO3 as base in pyridine.352 Catalyst (Scheme 100) turnover is achieved by either oxidative addition of Ni0 to the aryl-boronic acid or more likely through the oxidation of Ni0 to NiII. The latter pathway is supporting by the compatibility of the reaction with aerobic conditions. Homocoupling of boronic acids can also be achieved using similar PdII or other transition metal catalysts such as Au,353 Cu,354 Pd,355,356,357,358,359,360,361,362,363,364 and Rh.365,366 However, well-established methods for Ni-catalyzed homocoupling of aryl halides and sulfonates are available. Moreover, as most hard-metallation or transition-metal catalyzed methods for installing boronic acid or ester functionality utilize aryl halide or sulfonate starting materials, it is typically more efficient to use the former halide/pseudohalide homocoupling as opposed to boronic acid homocoupling.
Scheme 100.

2.3.3. C-N Bond Formation
Aryl nitrogen derivatives are very common derivatives and can be found in wide range of materials such as natural products367, functional materials368,369, conjugated polymers370,371, dyes372, and pharmaceuticals373. Although used as building blocks in many applications their synthesis has been a great challenge to organic chemists. Traditional methods such as reductive amination required severe reaction conditions that were not compatible with sensitive functional groups. This deficiency could be resolved only by inefficient protection and deprotection of such functionalities. Another approach to aryl C-N bond formation is the Ullmann reaction,374 but this too entailed harsh conditions in addition to stoichiometric amounts of copper. This functionalization procedure has many drawbacks, such as highly variable yield, frequent side reactions such as diarylation, and the protocol is limited to a relatively narrow range of substrates. Therefore, there has been considerable effort to develop mild transition metal catalysts for aryl C-N bond formation in particular for transition metal catalyzed amination based on Pd37,375,376, Ni and Cu. In this subchapter we will cover those approaches employing Ni-catalysts.
The first example of nickel catalyzed amination dates back to 1975 when Cramer reported the Ni-catalyzed displacement of aryl halides.377 The reaction was carried out using phenyl chloride with methylamine in the presence of NiIICl2 and required high temperatures and an excess of methylamine as the solvent. However, the yields obtained were quite modest and further effort was needed to develop more efficient procedures. In 1995, Christau and Desmurs studied Ni-catalyzed amination of aryl bromides.378 Modern modification of this reaction was inspired by corresponding Pd-catalyzed amination technology. The first practical Ni-catalyzed amination was reported by Buchwald in 1996 while investigating Ni-catalyzed coupling of aryl chlorides with anilines.379 Ni0(COD)2 was used as the catalyst in the presence of dppf as co-ligand and sodium t-butoxide as base and in the temperature range 70-100°C (Scheme 101). The mild reaction conditions provided tolerance toward often incompatible functional groups such as ethers, nitriles and acetyls. Side reactions including the reduction of aryl chlorides were observed while aldehyde substrates provided amides as final products. NiIICl2(1,10-phenanthroline) was also tested in order to avoid air sensitive Ni0(COD)2 catalyst, but the yields were very modest and not reproducible.
Scheme 101.
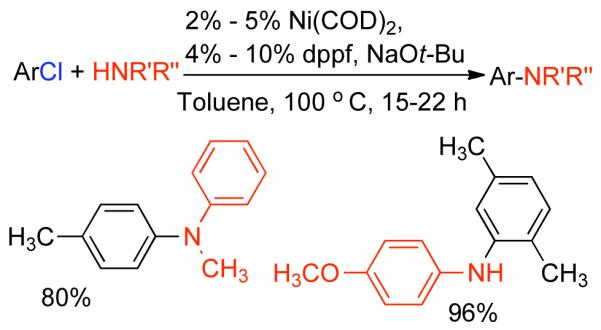
2.3.3.1. Ni/NHC Catalysts
In 1998, Fort used air stable Ni-CRA catalysts, previously developed by Caubere, to replace the air sensitive Ni(COD)2 complex. The Ni-NHC catalyst was prepared in situ by reduction of NiII(OAc)2 with NaH and in the presence of t-AmOH and bpy. The freshly prepared Ni0 catalyst was useful for the amination of aryl chlorides with cyclic secondary amines providing the cross-coupling products in moderate yields.380 Reduction and homocoupling byproducts were typically observed, but their levels could be reduced using higher levels of Ni catalyst. Further optimization of this system reduced the catalyst loading to 20% and broadened the amination scope to acyclic secondary amines.381 However the reaction was sensitive to the nature of the substrates and the electronic effects of the substituents. Electron-deficient aryl halides were favored while electron rich substituents required higher catalyst loading and longer reaction time (Scheme 102). The reaction protocol was successfully applied in moderate yields for the diamination of dichloro aryl derivatives, and 1,3-dichlorobenzene with heteroamines 2,6-and 3,5-diaminopyridines. The diamination required an increase in the amount of heteroamine and higher temperatures to reflux.382 Triamination was achieved on 1,3,5-trichlorobenzene with cyclic secondary amines in 60-75% yield. 383 However, since stoichiometric amount of NaH was used, no compatibility with any sensitive functional groups was reported.
Scheme 102.
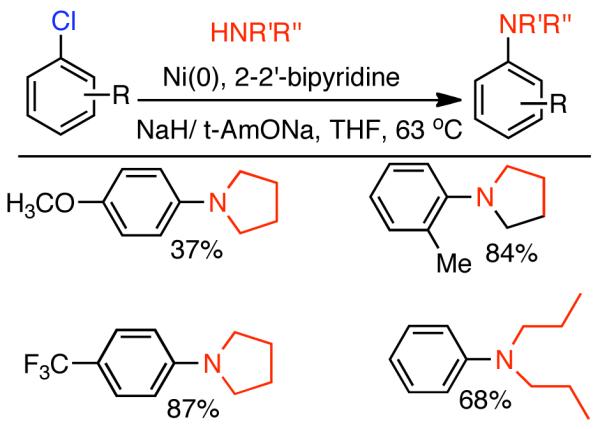
In 2001, Fort provided the first examples of Ni-NHC catalyzed amination reactions.384 NHC’s are better σ donor compared to phosphine ligands and thus facilitate oxidative addition. The most effective Ni-NHC were 1,3-bis(2,6-diisopropylphenyl) imidazol-2-ylidene (SIPr) and IPr (Scheme 103) in conjunction with NiII(acac)2 as the metal source. t-BuONa was used as a base and played the role of both deprotonating the imidazolium as well as activating sodium hydride to reduce NiII to Ni0.
Scheme 103.
By using NHC ligands, Fort was able to reduce the catalyst loading to 2 mol% and shorten the reaction time without lowering the yield. Even more important, it was found that the loading level of NaH used in reaction could be reduced.385 NaH provides a strongly basic environment with low tolerance for sensitive functional groups, therefore reducing the amount of NaH showed great potential of enlarging the substrate scope. By reducing the amount of NaH, secondary cyclic and acyclic amines and anilines were successfully cross-coupled and effective diamination of diarylhalides was achieved with good yield. (Scheme 104)
Scheme 104.

Using this methodology, intramolecular amination of aryl chlorides was performed for the synthesis N-heterocyclic compounds.386 The method was applied to the synthesis of five-, six- and seven-membered rings. (Scheme 105)
Scheme 105.
Moreover, benzoxazines, benzoxapines, benzo[e]indolizidine, and 5,6-dihydroindolo[2,1-a]isoquinoline were synthesized in high yields using this straightforward protocol (Scheme 106). In 2005, arylation of aromatic diamines was also achieved using IPr instead of SIPr and dioxane as solvent (Scheme 107).387 To date, no activation of aryl sulfonates or sulfates has been reported by Ni-NHC catalyst systems.
Scheme 106.
Scheme 107.
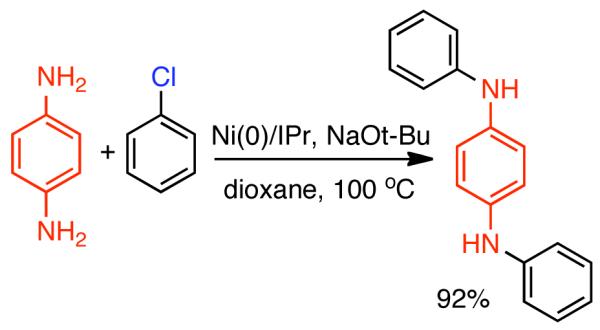
2.3.3.2. Ni/C Catalysts
In addition to Ni-NHC catalysts, another robust and easily handled Ni catalyst is Ni/C. 4 years after Buchwald’s disclosure of Ni-catalyzed amination reaction, Lipshutz applied the catalyst system to their charcoal supported Ni0 system.388 Ni0/C was prepared by reducing NiII with n-BuLi in presence of a ligand. A survey of Ni0/C catalysts with different phosphine ligands, revealed dppf to be the most effective ligand for the amination of p-chlorotoluene with phenylamine. The reaction proceeds to 93% conversion after 18 h in dioxane or toluene. Lithium t-butoxide was found to be the only effective base (Scheme 108) (Scheme 109). Further screening of substrates demonstrated successful amination of both electron-deficient and electron-rich aryl chlorides with secondary and primary heteroaryl or aryl amines including anilines. As was found in Pd0 catalyzed amination,375 electron deficient aryl chlorides were more reactive while electron-rich aryl halides required longer time and higher catalyst loading.
Scheme 108.

Scheme 109.
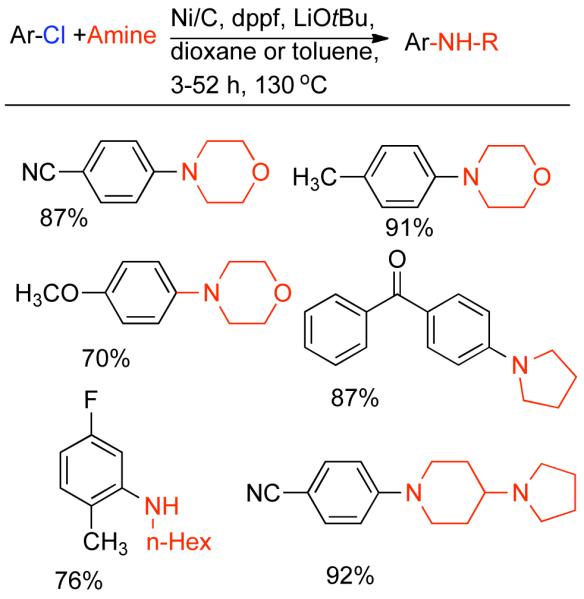
Further work aimed to develop methods of reducing NiII in the absence of n-BuLi.389 Lithium t-butoxide in combination with the amine was found to reduce NiII directly to Ni0. Without adding external reducing reagent, the reaction proceeded relatively slow, and a 15 min to 1 h induction period was observed. Although this system showed slow activation, comparable conversions to those from the n-BuLi system were ultimately achieved. A study of the mechanism of amination was performed to determine the order of reaction. A zero-order rate dependence was obtained for the oxidative addition of aryl chlorides suggesting this step was not the rate-determined step. The catalyst was suggested to be homogenous instead of heterogeneous390 while charcoal served as a reservoir of Ni0. Additionally this catalytic system was effective even for larger scales of over 15 mmol,389 while microwave irradiation could be employed to shorten the reaction time391 (Scheme 110). Using microwave heating, Lipshutz observed good yields for the amination of aryl halides including heteroaromatic halides. Highly hindered 2-chloroanisole showed slow reactivity, however increasing the catalyst loading and using higher ligand concentration resolved this problem.
Scheme 110.
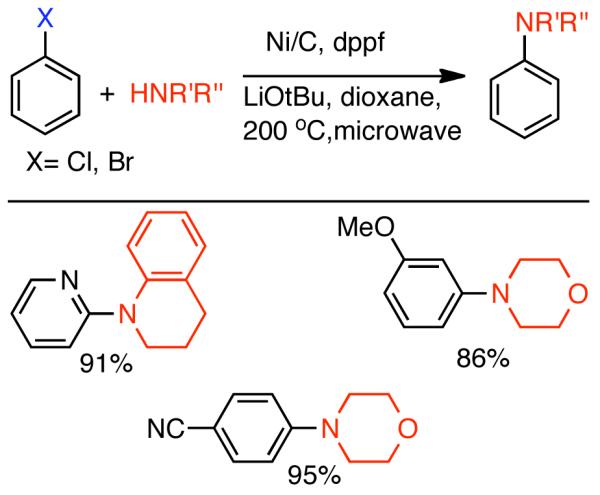
Microwave irradiation was applied to the synthesis of trisubstituted aromatic compounds, which were precursors to the preparation of oxazolidinone antibacterial linezolid (Scheme 111).392 Bimetallic supported heterogeneous catalysts are very attractive in the search for effective catalysts for complex reactions. Cu-Ni/C was tested in amination procedure although the results did not show great improvement to either yield or substrate scope.393 Compared to traditional homogenous catalysts, supported Ni catalysts are attractive because they can be recycled. However, since arenes bearing common functional groups such as ketones and esters are not effectively aminated, the use of such catalysts is limited. Additionally the reaction was only successful for activated aryl halides although the use of aryl tosylates and mesylates was possible as mentioned in a recent review by Lipshutz.394
Scheme 111.

2.3.3.3. Ni-(σ-aryl) Complexes as Catalysts
In the two preceding methods, it was critical to generate Ni0 species in situ, for example by the action of reducing agents such as Zn0,164 NaH, t-BuLi or other organometallic compounds. Inspired by the Kumada-Corriu reaction, Yang proposed an amination route using Grignard reagents both as a base and as a reducing reagent.395 Using this protocol, they reported the first Ni-catalyzed arylation of diarylamines with unactivated aryl bromides and iodides in moderate to good yields. The amine was first treated with the Grignard reagent in THF. The THF was removed and replaced with toluene before NiIICl2(PPh3)2, PPh3 and aryl halide were added (Scheme 112).
Scheme 112.
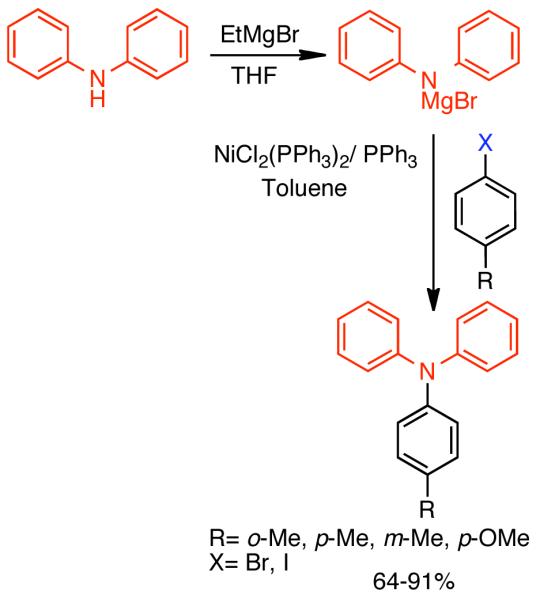
The main drawback of the Grignard-first protocol was its low tolerance to electrophilic or protic functional groups. For such functionality inefficient protection/deprotection strategies are required. An improvement to this reaction was demonstrated by the use of Ni-(σ-aryl) complexes. Ni-(σ-aryl) complexes are an intermediate of oxidative addition cycle. By introducing this complex as catalyst, they were able to generate Ni0 without an external reducing agent.396,397 For the co-ligand of the Ni-(σ-aryl) complexes, phosphine ligands such as PPh3 or nitrogen-based ligand 1,10-phenanthroline and bpy were explored. However, none of the phosphine ligands or nitrogen-based ligands provided acceptable results. When NHC was used as a co-ligand in THF or dioxane as solvent high reactivity was obtained with catalyst loading as low as 3 mol% in the presence of 3 mol% ligand (Scheme 113).
Scheme 113.
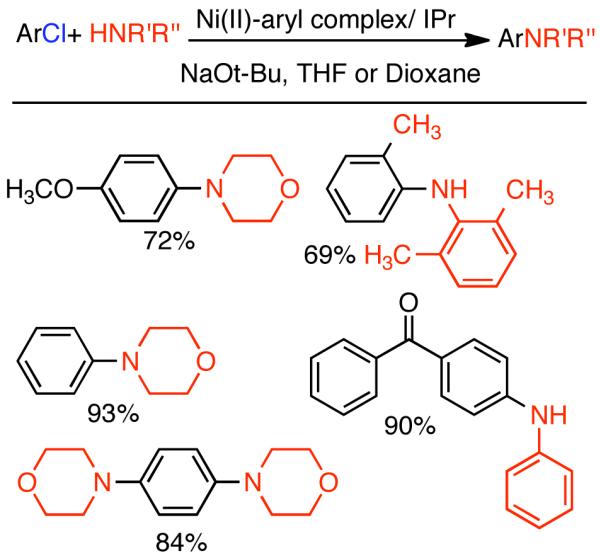
In 2008, Yang successfully harnessed aryl tosylates using IPr carbene ligand and obtained the aminated products in isolated yield as high as 96% for specific substrates providing the first example of Ni-catalyzed amination of tosylates.398 NaOt-Bu was an effective base, while weaker bases did not affect transmetallation. As reported by Fort, amination of electron-rich substrates is difficult to achieve. While electron-withdrawing groups activated amination of aryl halides, electron-withdrawing substituents on aryl tosylates provided side products such as decomposition into phenols. Amination using aniline was also successful although the phenol formation was also detected. The Ni-catalyzed amination of tosylates was found to be sensitive to steric effects and thus lower yields were obtained for the amination of 2-naphthyl tosylate and 1-naphthyl tosylates (Scheme 114). The study of the reactivity of tosylates indicated trade-offs in the amination procedure and required a compromise between reactivity and selectivity. Although a fast transmetallation process requires stronger bases, their presence can affect the sulfonate bond and attack it at the S-O site to produce undesired side reactions.
Scheme 114.
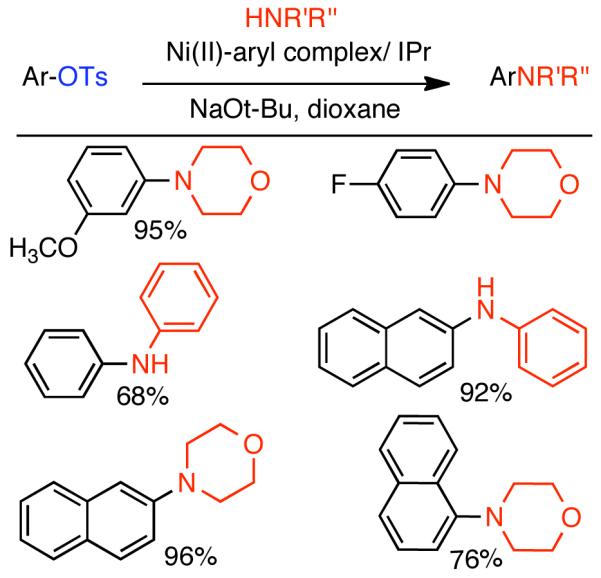
2.3.3.4. Other Transformations
In 2000, Bolm reported the Pd or Ni catalyzed cross-coupling of arylsfulonates with N-aryl sulfoximines.399 PdII(OAc)2/BINAP proved to be an effective catalyst for the sulfoximination of aryl nonaflates and triflates, whereas Ni0(COD)2/BINAP was necessary for the analogous transformation with less reactive aryl tosylates (Scheme 115).
Scheme 115.
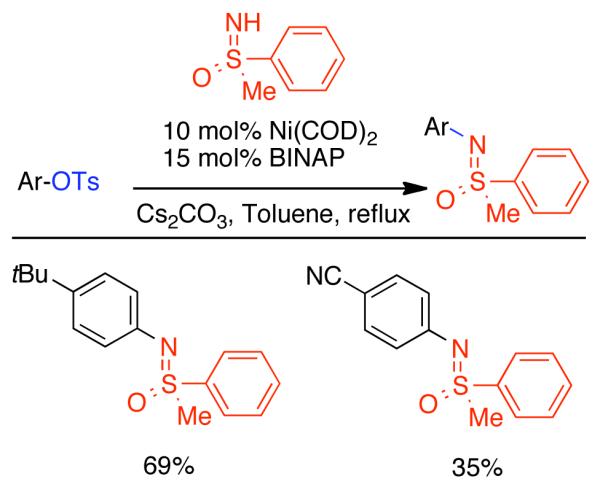
2.3.4. Reduction and Deoxygenation
The selective reduction of aryl C-O bonds can be a challenge, but nevertheless is an important transformation with wide applications in total synthesis.400,401 Traditional procedures require harsh conditions such as high temperature and pressure. An alternative is to first convert the phenols into the corresponding tosylates, mesylates or triflates and then reduce the sulfonates in the presence of transition metal catalysts. As has been observed in various homo- and cross-coupling reactions of aryl sulfonates, reduction of sulfonates was detected as a side reaction. Not surprisingly then, Pd, Ni and Cu based catalysts are frequently used in reduction reactions. This subchapter will be focused on Ni catalyzed reduction and deoxygenation of aryl and vinyl sulfonates.
In 1999 Sasaki reported one of the first procedures for the Ni0-catalyzed reduction of aryl triflates.402 In this approach Ni0 was generated in situ using Zn0 as a reducing agent in the presence of dppp or PPh3 as the most effective phosphine ligands. It is important to note that the source of H in the reduction to arenes is provided by the solvent (Scheme 116).
Scheme 116.
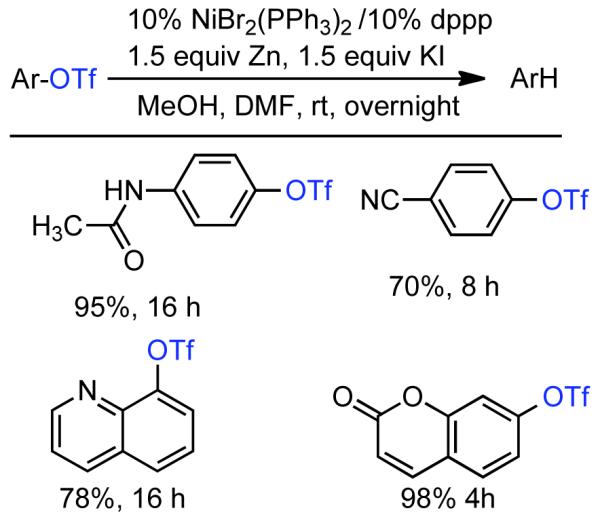
Further optimization of the reaction conditions provided access aryl mesylates as available substrates.403 While dppp in methanol was effective for reduction of tosylates, dppb showed better reactivity for mesylates in methanol/DMF (30%) mixture. The reduction was not affected by the nature of substituents on the aromatic unit nor any related steric effects providing very good yields for all substrates (Scheme 117).
Scheme 117.
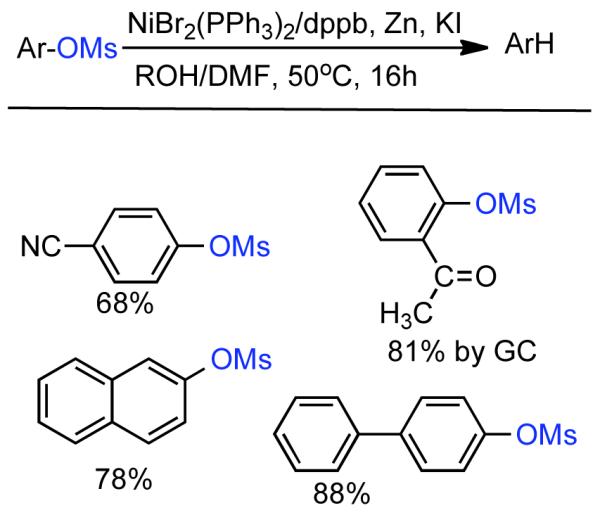
Yus demonstrated that vinyl triflates were also successfully reduced into alkenes using Ni0 formed in situ using excess of lithium, and catalytic amount of DTBB as reducing reagent (Scheme 118).404 Deuterium-labeled derivatives showed the source of H comes from H2O in NiCl2 · H2O (Scheme 118). When excess of Ni salts were added, further reduction of alkenes was observed (Scheme 118). For chiral molecules and cis-, trans-olefins, the symmetry was retained after reduction. Aryl triflates failed to give a good yield due to the competitive cleavage of S-O bonds to produce phenol as side product.405 Yus reported the expansion of the scope of the reduction to alkyl but not aryl mesylates.406
Scheme 118.

In 2006, Kogan published the first room temperature reduction of aryl tosylates with borane hydrides catalyzed by Ni.407 A variety of tosylates were reduced using NaBH4 in THF in the presence of NiIICl2(PPh3)2 catalyst at room temperature. In the case of carbonyl and nitrile containing electron-deficient substrates, dimethylamine borane was used as a reducing agent in order to avoid reduction of the sensitive functional groups. Kogan found PCy3 performed better than other phosphine ligands while dppb showed a particularly low conversion for aryl tosylates. This Ni-catalyzed reduction performed admirably with lower catalyst loading levels than previously reported (Scheme 119).
Scheme 119.

Lipshutz tested his heterogeneous Ni/C catalyst in the reduction of mesylates and tosylates and obtained good results.408 The Ni/C catalyst was prepared using Me2NH·BH3 as reducing agent and PPh3 as a ligand. Microwave irradiation decreased the reaction time considerably to 45 min and in some cases provided quantitative reduction (Scheme 120). This mild reaction showed very good tolerance to other sensitive functional groups such as esters, amides, and ketones. Moreover, since borane was synthesized prior to substrate addition, the reaction media was not particularly basic and could accommodate reduction of peptidic adducts providing quantitative conversion before decomposition would occur394 (Scheme 121). Despite the mild reaction some substrates are still susceptible for side reactions.
Scheme 120.

Scheme 121.
2.3.5. Cyanation
Insertion of a cyanide group is an important reaction in organic synthesis, as the resulting cyano-group can impart the desired functionality and properties to the compound or itself be utilized as a handle in diverse transformations. However, no efficient way of transforming aryl C-O bonds into aryl C-CN bond existed until the early 20th century due to the strong bond of sp2 C with O. Even for aryl halide substrates, the cyanation reaction can only proceed in the presence of transition metals such as Pd409 and Ni. Cassar410,411 and Sakakibara412,413 performed early work on Ni catalyzed cyanation of aryl halides. However, as mentioned in previous chapters, cyanation of a phenol group is important because it provides another route of synthesis of aryl cyanide when the corresponding aryl halide is not available. This subchapter is focusing on cyanation of aryl and vinyl sulfonates.
In 1989, in the course of pursuing biotransformations in organic synthesis, Widdowson tried to apply a Pd-catalyzed cyanation protocol and obtained unsatisfactory results. He applied the more reactive Ni catalysts to promote the cyanation process, which led to the first discovery of Ni0 catalyzed cyanation.414 The procedure required KCN as a cyanide source and utilized Ni0(PPh3)4 formed in situ from the reduction of NiIIBr2(PPh3)2 with Zn powder. A variety of phenols were transformed into the corresponding aryl nitriles in two steps. Even high-hindered substrates such as the triflate derived from 2,4,6-trimethylphenol was successful converted into the nitrile in moderate yields although for aryl halides, side reactions such as homocoupling were observed. Attempts to perform dicyanation were also not successful (Scheme 122), perhaps as result of problems with the solubility of the triflates. In general less solubility substrates produced the coupling products in much lower yields.
Scheme 122.
Soon after, Sakakibara reported related work on Ni-catalyzed cyanation of aryl triflates.415 The cyanide source was also same KCN with NiIIBr2(PPh3)2 as a precatalyst that is reduced in situ to Ni0 by Zn0 powder. This protocol provided quantitative transformation of the phenol to benzonitrile. Side reactions such as the cleavage of the S-O bond were not detected. As in the case of Ni-catalyzed amination and reduction, electron rich-substrates did not participate as effectively as electron-deficient substrates, providing lower yield. The sluggish reactivity of electron-rich substrates could be mitigated by increasing the reaction temperature to 80°C and changing the catalyst ligand to dppf. However the reaction was not sensitive to steric hindrance and was efficient for chlorides when dppf was used to suppress the homocoupling side reactions (Scheme 123).
Scheme 123.
In the early work of Sakakibara,412,413 it was observed that the order of addition of the cyanide and the aryl halide substrates as well as the solubility of cyanide is playing an important role in the reaction yield. They claimed that when higher solubility NaCN salt was used as a nitrile source a bromide atom of NiIIBr2(PPh3)2 was replaced by cyano group and that the formed product NiII(CN)2(PPh3)3 was further reduced by Zn0 powder. By employing controlled reagent addition, or by using the less soluble cyanide salts the cyanation of aryl halides and heteroaromatic halides was accomplished.416 In 1995, Percec successfully addressed this problem.265 It was found that in the presence of a cyanide source, the in situ generated Ni0 catalytic system he had developed for the homo- and cross-coupling of aryl mesylates, could also mediate the cyanation of aryl mesylates. Et4NI was used to facilitate electron transfer from Zn0 to Ni0, while less soluble KCN was used to avoid the poisoning of Ni0 by cyanide. An excess of PPh3 co-ligand was added to limit homocoupling and phosphine transfer reactions. Interestingly, changing to dipolar aprotic solvents such as CH3CN and DMF completely suppressed the homocoupling. No excess of I ion was needed in the presence of excess ligand. Alternatively, increasing the reaction temperature was an effective way of reducing the formation of homocoupling byproducts, resulting in increased yield. Ultimately, optimized reaction conditions were established employing NiIICl2(PPh3)2 (0.1 equiv), PPh3 (0.2 equiv), Zn0 (1.0 equiv), KCN (1.5 equiv), with DMF as solvent at 80 °C (Scheme 124).
Scheme 124.
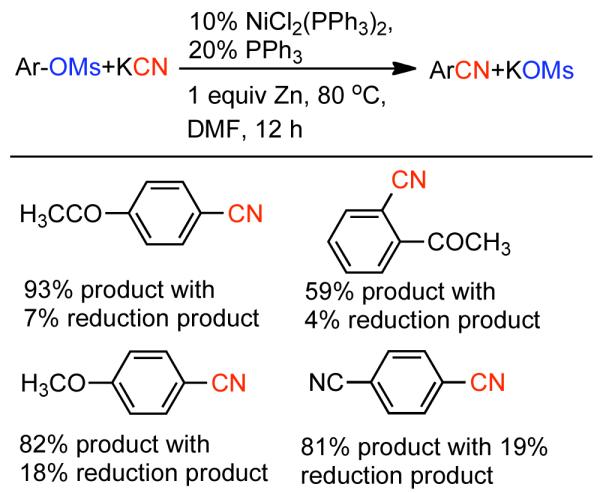
Further advances in Ni-catalyzed cyanation included the use of microwave irradiation, which considerably reduced the reaction time from hours to minutes.417,418,419 No current literature on Ni0 catalyzed cyanation of aryl tosylates was found. Recent work in Pd-catalyzed cyanation has focused on the use of less toxic cyanide sources such as potassium hexacyanoferrate (II),420 which provides safer delivery of cyanide as well as slow controlled liberation of cyanide ions. The development of Ni-catalyzed cyanations utilizing safe cyanide sources remains an unaddressed challenge.
2.3.6. Thioetherification
In 1995, Percec demonstrated that aryl mesylates could be effectively thioetherified in the presence of a Ni-catalyst (Scheme 125).265 Phenyl mesylate was successfully cross-coupled with sodium benzenethiolate in the presence of 10 mol% NiIICl2(dppf) and 20 mol% dppf co-ligand with stoichiometric Zn0 as reductant to provide diphenyl sulfide in 94% yield. The excess dppf co-ligand was required to otherwise rapid catalyst decomposition. Relatively good yields could also be obtained using NiIICl2(dppf) as the pre-catalyst in the presence of 20 mol% PPh3 co-ligand, but poor yields were obtained if the active catalysts was generated in situ from NiIICl2(PPh3)2 due to premature catalyst decomposition. Attempts to generate dissymmetric diarylsulfides using electron-deficient aryl mesylate coupling partners bearing cyano, ester, or ketone functionality in conjunction with sodium benzenethiolate were met with limited success (20-32% yield of asymmetric biaryl). In each case, up to 36% diphenylsulfide was generated as byproduct. Interestingly, only 0.7 % yield diphenylsulfide could be generated if the mesylate was not present, suggesting that oxidative addition of the aryl mesylate is required for byproduct formation. A plausible mechanism was proposed, wherein the desired product Ar-S-Ph can oxidatively add to Ni0-complex to generate ArS-NiII-Ph(dppf). Displacement of Ar-S with sodium benzenethiolate, could provide PhS-NiII-Ph(dppf), which upon reductive elimination would generate diphenyl sulfide.
Scheme 125.
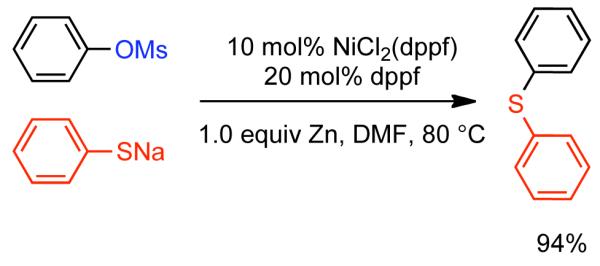
3.Nickel-Catalyzed Cross-Coupling Of Aryl Sulfamates
It is evident that the C-OSO2R functional group, where R is an alkyl or aryl substituent provides an excellent handle for carbon-carbon bond formation. Recent work has expanded the versatility of the - -OSO2R leaving group to include aryl sulfamates.
3.1 Kumada Coupling of Aryl Sulfamates
Snieckus has shown that the O-sulfamate moiety is an effective group for directed ortho metallation group (DoM) and a cross-coupling partner for Kumada-Corriu reactions. In an early report,421 Snieckus demonstrated that aryl O-carbamates can participate as a coupling partner with Grignard reagents, using NiII(acac)2 as catalyst and Et2O as solvent, at room temperature in good yields. Based upon these promising results with Ni-catalysts, Snieckus explored various catalysts in conjunction with O-sulfamates in Kumada cross-coupling. Through catalyst screening it was found that in particular, Ni-NHC complex, NiClCpIMes, was an extraordinarily efficient catalyst for the cross-coupling of aryl O-sulfamates with p-tolylmagnesium bromide.422 The power of this catalyst was proposed to be due to the synergy of the strongly Lewis basicity of the NHC ligand, which allows for effective oxidative addition, and the relatively high steric bulk that provides accelerating reductive elimination. This procedure was applied to the construction of simple biaryls, N-protected anilines and azabiaryls in fair to good yield (Scheme 126).
Scheme 126.

Later Du Bois prepared benzene-fused cyclic sulfamates from ortho-substituted phenols and showed that they are efficient partners in Ni-catalyzed cross-coupling reactions with aryl and alkyl-Grignard reagents.423 In screening studies, 5 mol% NiIICl2(dppp) was found to be a superior catalyst for the cross-coupling with MeMgBr (Scheme 127). Higher yields were obtained in benzene and Et2O, but not in THF. N-alkyl substituents on the benzoxathiazine such as n-Pr, i-Bu or Bn did not affect the reaction performance, whereas allyl N-protecting groups decreased the yield considerably.
Scheme 127.
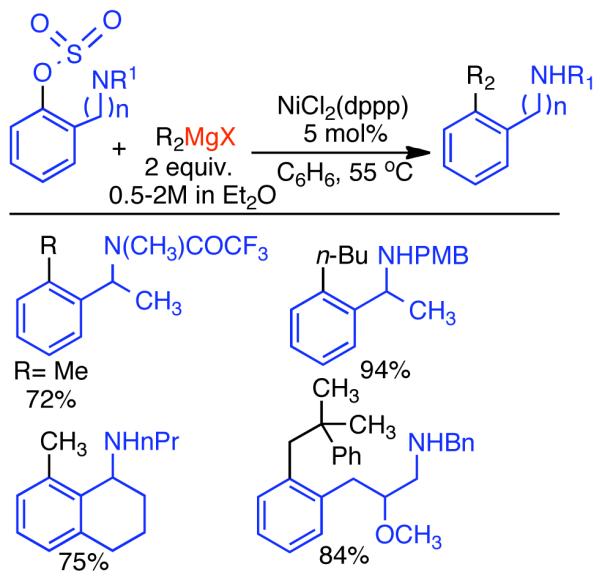
3.2 Suzuki-Miyaura Coupling of Aryl Sulfamates
Garg recently reported the first successful Suzuki-Miyaura cross-coupling of aryl sulfamates with arylboronic acids. NiIICl2(PCy3)2 was employed as an air and water insensitive catalyst, using K3PO4 as base, in toluene at 130 °C.424 Naphthyl sulfamates, nonfused aromatic substrates, as well as the heteroaromatic and vinyl sulfamates, bearing electron-withdrawing or electron-donating substituents, were converted to biaryls products in good yields (Scheme 128). The steric hindrance of ortho-substituted sulfamates could be overcome by increasing the amount of catalyst. This Ni-catalyzed cross-coupling of sulfamates was applied to the synthesis of anti-inflammatory drug flurbiprofen by allowing insertion of multiple functional groups onto the aromatic ring (Scheme 129).
Scheme 128.

Scheme 129.
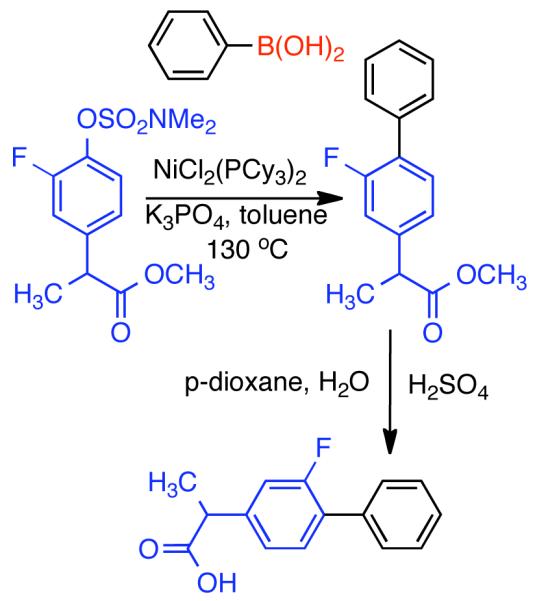
4. Nickel-Catalyzed Cross-Couplings of Aryl Ethers
4.1. Kumada Couplings
Aryl and vinyl ethers, although typically considered “inert”, have been employed in a range of Ni-catalyzed cross-coupling reactions. Wenkert reported the earliest example of ether cross-couplings in 1979.425 In this seminal contribution, vinyl methyl ethers were shown to undergo Kumada coupling with phenyl and methyl Grignard reagents in good yield (Scheme 130). For example, a vinyl ether could be smoothly converted to an arylated or methylated product using NiIICl2(PPh3)2 as catalyst. The corresponding reactions involving aryl methyl ethers were less successful, although it was found that naphthyl-derived substrates cross-coupled efficiently with phenylmagnesium bromide to deliver arylated products. Of note, the corresponding reaction using methylmagnesium bromide did not proceed under the identical reaction conditions. In later work, Wenkert further explored the Kumada coupling of enol and aryl ethers using NiIICl2(PPh3)2 as catalyst, paying particular attention to the stereochemistry of associated with the ring-opening of dihydropyrans and dihydrofurans.426
Scheme 130.
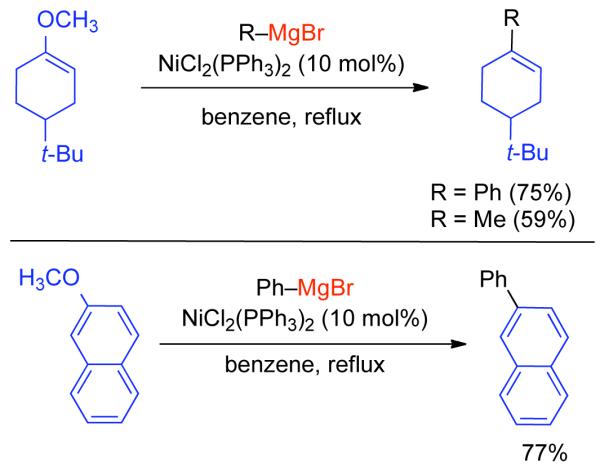
In 2004, Dankwardt significantly expanded the scope of aryl ether cross-couplings by switching ligands to either PCy3 or PhPCy2 and shifting to ethereal solvents.427 Using this modified protocol, a range of simple aryl ethers, including a TMS ether, were converted to biaryl products in good to excellent yield (Scheme 131). The reaction was tolerant of free hydroxyl groups, in addition to nitrogen-containing heterocycles. Coupling of imidazole- and indole-derived substrates with phenyl magnesium bromide gave arylated products. The latter case proceeded efficiently even at ambient temperature. Ultimately, Dankwardt’s efforts enable the arylation of a range of aryl ethers, although the corresponding coupling with alkyl Grignard reagents was not possible.
Scheme 131.

Shi’s laboratory has recently discovered conditions that allow for the cross-coupling of aryl ethers with methyl magnesium bromide, thus enabling the formation of sp3-sp2 C–C bonds.428 With PCy3 as ligand and toluene as solvent, a variety of ethers derived from 2-naphthol were converted to methylated products in good to excellent yields (Scheme 132). Non-fused aromatic ethers cross-coupled less efficiently compared to naphthyl-based substrates, and required higher temperatures (110 °C vs. 80 °C). Indeed, a competition experiment provided the mono-coupled product in 90% yield.
Scheme 132.

Shi has also reported the Kumada coupling of benzylic ethers.429 This unique transformation allows for the Ni-catalyzed formation of sp3-sp3 C–C bonds. Methyl, t-butyl, phenyl, and trimethylsilyl ethers could be converted to 2-ethylnaphthalene at ambient temperature by employing the dppf ligand (Scheme 133). As shown in Scheme 134, Ni-catalyzed methylation of benzylic ethers and aryl ethers can be carried out sequentially by modulating the choice of ligand and temperature.
Scheme 133.
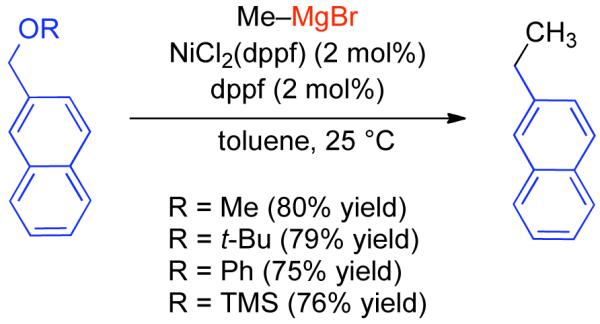
Scheme 134.

Non-standard aryl and vinyl ethers have also found use as electrophiles in Ni-caralyzed Kumada coupling. In 1980, Kumada, described the cross-coupling of silyl enol ethers with Grignard reagents using NiII(acac)2, NiIICl2(PPh3)2, and NiIICl2(dppf)430. Later, Johnstone observed that NiIICl2/L (L = PPh3, dppe, or dppe) or Ni0(PPh3)4 were effective catalysts for Kumada cross-coupling of aryl 5-1-phenyltetrazolyl ethers with alkyl and aryl Grignard reagents and proposed a mechanism involving coordination of tetrazolyl nitrogen to the incoming organomagnesium bromide.431
4.2. Suzuki-Miyaura Couplings
In 2008, Chatani demonstrated the first Suzuki coupling of aryl ethers.432 By employing a Ni(COD)2/PCy3 catalyst system, methyl ethers were cross-coupled with aryl boronic esters to give biaryl products. The transformation proceeded most efficiently with fused aromatic substrates, and a variety of boronic esters could be utilized. For example, 2-methoxynaphthalene was transformed to biaryls with high yields (Scheme 135). Electron-deficient phenol derivatives could also be employed, as demonstrated by the conversion of an ester-containing substrate to a biaryl product. The scope of the reaction was later expanded to facilitate the Suzuki coupling of vinyl methyl ethers.433
Scheme 135.

4.3. C–N Bond Formation
Chatani has discovered conditions that enable the amination of methyl ethers using nickel catalysis. The amination of 2-methoxynaphthalene proceeds readily using Ni(COD)2 and the N-heterocyclic carbene ligand IPr in toluene at 120 °C for a range of secondary amines (Scheme 136).434 Cyclic amines of varying sizes were tolerated, as were acyclic amines and substrates possessing additional heteroatoms. Phenolic methyl ethers could also be employed in the amination reaction, albeit with lower yields (ca. 40%).
Scheme 136.

4.4. C–O Reduction
In a recent report, Martin435 found that aryl methyl ethers could be reduced to the corresponding arene by using catalytic (5-10 mol%) Ni(COD)2/PCy3 in the presence of stoichiometric tetramethyldisiloxane (TMDSO) as a hydride source (Scheme 137). This technique can be broadly applied to substituted arenes, naphathalenes, biaryls, or heterobiaryls and provides a useful approach toward removing methoxy groups from a substrate after their use as an ortho-directing groups has been fulfilled.
Scheme 137.

5. Nickel-Catalyzed Cross-Couplings of Aryl and Vinyl Phosphates
5.1. Kumada Couplings
One of the earliest reports of the nickel-catalyzed cross-coupling of phenolic derivatives was published in 1981.436 In this seminal study, Kumada demonstrated that vinyl phosphates could be coupled with trimethylsilylmethyl magnesium halides in the presence of a Ni(acac)2. For example, a vinyl phosphate readily prepared from cyclohexanone, was converted to the corresponding allylsilane in 81% yield (Scheme 138). The methodology was also tolerant of acyclic vinyl phosphate substrates. Since Kumada’s report, Claesson437 and Bäckvall438 have expanded the scope of this methodology to include the cross-coupling of dienyl phosphates with aryl and alkyl Grignard reagents. Nicolaou has also demonstrated the nickel-catalyzed Kumada coupling of a lactam-derived ketene aminal phosphate with trimethylsilylmethyl magnesium chloride.439
Scheme 138.
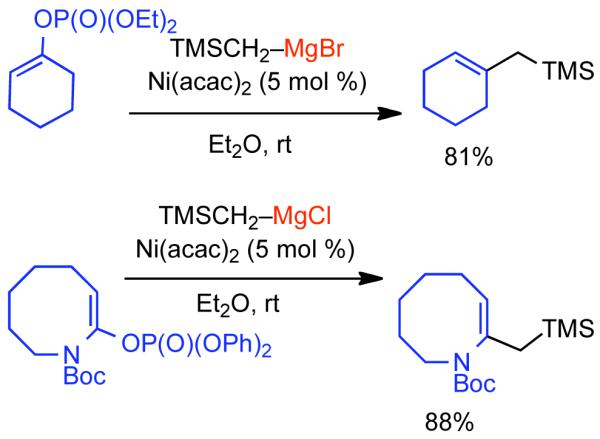
Kumada’s conditions for vinyl phosphate coupling also proved amenable to the corresponding reactions of aryl phosphates.440 As shown in Scheme 139, a naphthyl phosphate underwent Ni-catalyzed Kumada coupling to deliver arylated products. Both aryl and alkyl Grignard reagents could be utilized in this methodology. Furthermore, in addition to naphthyl-derived substrates, non-fused aromatics cross-coupled in synthetically useful yields.
Scheme 139.
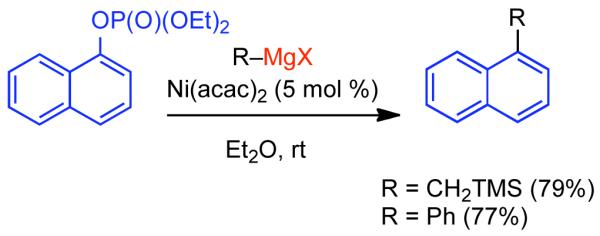
Nakamura has recently reported the Kumada coupling of aryl phosphates using a versatile hydroxyphosphine ligand (Scheme 140).279 Even electron-rich substrates and ortho-substituted derivatives could be employed using this methodology. On the basis of experimental and computational studies, the authors attribute the high catalytic activity to an in situ-formed bimetallic species derived from the nickel precatalyst, the hydroxyphosphine ligand, and the Grignard reagent.
Scheme 140.

The Kumada coupling of aryl and vinyl phosphates has been employed in drug discovery (Scheme 141). Tsukuba Research Laboratories carried out the nickel-catalyzed cross-coupling of an aryl phosphate with ethylmagnesium chloride to deliver the alkylated product in 81% yield.441 In turn, the alkylated product served as a precursor to a series of compounds, which were found to inhibit the generation of the interleukin-1 (IL-1) cytokine. Subsequently, scientists at Abbott Laboratories reported the conversion of a vinylphosphate to an allylsilane using nickel catalysis.442 The allylsilane was elaborated to the depicted homoallylic alcohol, en route to a class of hydroxyethylene dipeptide isosteres.
Scheme 141.

Nickel-catalyzed Kumada couplings of phosphate derivatives have also been utilized in natural product total synthesis (Scheme 142 and Scheme 143). In their synthesis of the diterpene fuscol, Yamada and co-workers reported the coupling of a vinyl phosphate substrate with methyl magnesium iodide using Ni(acac)2 as catalyst.443 The transformation proceeded readily at 0 °C, thus providing an efficient means to install the C13 methyl group of the natural product. In 2003, Lu et al. demonstrated the use of an aryl phosphate ester in natural product synthesis (Scheme 140).444 The aryl phosphate was converted to the corresponding alkylated product under nickel-catalyzed Kumada conditions. This intermediate could be used to access both cryptotanshinone and tanshinone IIA, natural products that were being examined for their ability to inhibit cdc25 protein phosphatases.445
Scheme 142.

Scheme 143.

5.2. Suzuki-Miyaura Couplings
In contrast to the nickel-catalyzed Kumada coupling of phosphate esters, reports of the analogous Suzuki coupling are relatively scarce. The first nickel-catalyzed Suzuki coupling of a vinyl phosphate was described by Yang in 1999.446 It was shown that cyclohexenylphosphate underwent smooth cross-coupling with a variety of arylboronic acids to deliver arylated products in good yields (Scheme 144). Substitution on the arylboronic acid was tolerated at the para, ortho, and meta positions and electron-rich and electron-poor species could be utilized. In all cases, the requisite Ni0 precatalyst was generated by mixing NiIICl2(dppf) with n-butyllithium prior to the cross-coupling reaction.
Scheme 144.

Skrydstrup later expanded the scope of the vinyl phosphate Suzuki coupling.447,448 The nickel-catalyzed coupling of vinyl phosphates with aryl boronic acids proceeds most efficiently with the PCy3 ligand to deliver a variety of 1,1-disubstitued alkenes (Scheme 145). The scope of the transformation was found to be broad with respect to the vinyl phosphate substituents, as a number of functionalized aromatics could be employed, in addition to heterocycles and alkyl substituents. Considerable variation in the aryl boronic acid fragment was also tolerated.
Scheme 145.

The Suzuki coupling of aryl phosphate esters has not been reported. However, Han has recently achieved the cross-coupling of a related electrophilic species, namely, the aryl phosphoramide group.449 In this study, aryl “BOP” coupling partners were readily derived from phenols and bis(2-oxo-3-oxazolidinyl)phosphinic chloride (Scheme 143). Suzuki coupling of various substrates afforded biaryl products in high yield. Of note, naphthyl derivatives and heterocyclic substrates derived from 3-, and 4-hydroxypyridine could also be cross-coupled efficiently using this methodology.
5.3. Negishi Couplings
The nickel-catalyzed Negishi coupling of vinyl phosphates has been reported as a tool to synthesize a number of substituted coumarins.450 Upon examining several Pd and Ni catalysts, Yang and co-workers found that 1% NiIICl2(dppe) enabled the desired cross-coupling, which proceeded at ambient temperatures (Scheme 147). A range of organozinc reagents could be utilized, including aryl-, heteroaryl-, vinyl- and alkylzinc species, to deliver cross-coupled products in synthetically useful yields.
Scheme 147.

6. Nickel-Catalyzed Cross-Couplings of Aryl and Vinyl Esters
6.1. Suzuki-Miyaura Couplings
In 2008, the Garg and Shi laboratories simultaneously reported the Suzuki coupling of esters derived from phenols and naphthols.336,451,452 The two methodologies operate under similar conditions, although Garg’s method utilizes aryl boronic acid coupling partners, whereas Shi’s technology employs aryl boroxines. In Garg’s studies, it was found that aryl pivalates were optimal substrates for the coupling with boronic acids (Scheme 148). Naphthyl, phenyl, and heterocyclic pivalates could be utilized in this methodology. The scope with respect to the aryl boronic acid component was also shown to be fairly broad. It should be noted that the precatalyst employed, NiIICl2(PCy3)2,453,454 is commercially available, air-stable, and bench-top friendly.455 Moreover, a tandem acylation/cross-coupling variation of this transformation was developed, which allowed for the one-pot conversion of 1-naphthol to a biaryl product.336
Scheme 148.

As mentioned above, Shi’s protocol for the Suzuki coupling of phenolic esters utilizes aryl boroxines instead of aryl boronic acids.451 Under optimal conditions, with 0.88 equivalents of water, acetate, pivalate, and benzoate ester derivatives could be cross-coupled with aryl boroxines (Scheme 150). Non-fused aromatic substrates were also tolerated, provided that pivalate esters were employed as the substrates (Scheme 147). A computational mechanistic study on the nickel-catalyzed cross-coupling of aryl esters is available, which suggests that transmetallation is likely the rate-determining step in these processes.456 Recently, Molander has reported that in addition to boronic acids and boroximes, aryl and heteroaryl potassium trifluoroborate salts were effective partners for Ni(COD)2/PCy3HBF4 catalyzed cross-coupling of aryl pivolates (See section 2.2.1.4.).199 Nevertheless, it was observed that in all cases explored, ary mesylates were more reactive, providing higher yield in equivalent time.
Scheme 150.

Garg and Shi have independently demonstrated the utility of aryl ester cross-couplings through sequential cross-coupling sequences. In Shi’s example,451 Suzuki coupling of a readily available aryl pivalate furnished the biaryl product in 75% yield (Scheme 151). Subsequent Baeyer–Villiger oxidation, hydrolysis, and acylation provided a new aryl pivalate substrate in 62% yield over 3 steps. Finally, Suzuki coupling gave rise to a triaryl product. Garg’s example showcases the directing ability of aryl pivalates, in addition to the low reactivity of these substrates toward Pd0 (Scheme 152).336 A naphthyl pivalate underwent regioselective para bromination. Exposure of the resulting bromopivalate to Pd-catalyzed Suzuki coupling conditions with an indolyl boronic ester led to selective coupling of the aryl bromide. Of note, the robust pivalate group remained intact, despite the harsh basic conditions employed (i.e., aqueous K3PO4, 90 °C). Next, the aryl pivalate underwent Suzuki coupling under nickel-catalyzed conditions to afford a triaryl product in 88% yield.
Scheme 151.

Scheme 152.

The scope of the nickel-catalyzed ester coupling extends beyond aryl systems, as demonstrated by the cross-coupling of vinyl acetates and pivalates. For example, the pivalate derived from tetralone was cross-coupled in 79% yield (Scheme 150). Although most examples of vinyl acetate and pivalate coupling involve styrenyl systems, an estrone-derived vinyl acetate has also been coupled under Shi’s Suzuki conditions.457 Interestingly, in this latter example, the aryl acetate moiety in the substrate underwent hydrolysis rather than cross-coupling.
6.2. Negishi Couplings
The scope of pivalate cross-coupling has been extended to include the Negishi coupling of 2-naphthol derivatives and electron-deficient aryl pivalates.458 For example, 2-naphthylpivalate could be cross-coupled with a range of aryl zinc reagents to furnish 2-aryl naphthalenes (Scheme 154). The corresponding arylation of styrenyl vinyl pivalates was also demonstrated.
Scheme 154.

6.3. C–N Bond Formation
In addition to serving as tools for carbon–carbon bond formation, aryl pivalates have been utilized to access carbon–nitrogen bonds using methodology disclosed by Chatani.459 Although catalyst systems comprised of Ni0/PCy3 were not effective, the use of Ni(COD)2 and the N-heterocyclic carbene ligand IPr facilitated the desired transformation (Scheme 155). The scope of the methodology was broad with respect to both the pivalate and amine coupling partners.
Scheme 155.

7. Nickel-Catalyzed Cross-Couplings of Aryl and Vinyl Carbamates and Carbonates
7.1. Kumada Couplings
Although typically considered inert substrates, aryl and vinyl carbamates participate in cross-coupling reactions. The earliest examples of carbamate coupling were described by Kocienski and Dixon in 1989.460 The authors found that a vinyl carbamate substrate could be coupled with alkyl Grignard reagents in the presence of NiII precatalysts (Scheme 156). Methylation and butylation delivered the corresponding olefins with retention of alkene stereochemistry. In the latter case, the NiIICl2(dppe) complex was found to suppress undesired reduction of the substrate that had been observed when using NiII(acac)2. Betzer and coworkers have reported similar results using vinyl, aryl, and alkyl Grignard reagents, where the identity of the NiII precatalyst was found to influence product distribution (i.e., desired cross-coupling versus reduction or homocoupling).461
Scheme 156.

Snieckus has demonstrated that aryl carbamates undergo Kumada coupling with aryl and alkyl Grignard reagents under conditions similar to those utilized for vinyl carbamate coupling.421 The transformation is tolerant of fused aromatic carbamate substrates, in addition to non-fused aromatic derivatives, and heterocycles (Scheme 154). Of note, the authors found that ortho-substituted substrates could be utilized, which is advantageous considering that ortho-substituted aryl carbamates are easily accessible through directed-metallation chemistry31 and, more recently, via Pd-mediated C–H functionalization.34 Snieckus’ methodology has proven quite robust and scalable, as demonstrated in a high-yielding synthesis of 2,7-dimethylnaphthalene beginning from 2,7-dihydroxynaphthalene, which was carried out on 200 mmol scale.462 Recently,279 Nakamura has utilized a hydroxyphosphine ligand to achieve aryl carbamate Kumada couplings.
7.2. Suzuki-Miyaura Couplings
In 2009, the Garg and Snieckus laboratories simultaneously reported the nickel-catalyzed Suzuki coupling of aryl carbamates.424,463 Garg’s method allows for the cross-coupling of aryl carbamates with aryl boronic acids to deliver biaryls (Scheme 158). Fused aromatic substrates provided the highest yields, whereas non-fused aromatics typically afforded products in ca. 50% yield. The optimal reaction conditions also facilitated the Suzuki coupling of naphthyl carbonates.
Scheme 158.

Snieckus’ variant of the aryl carbamate Suzuki coupling possesses a wide substrate scope.463 By employing a mixture of triaryl boroxine and aryl boronic acid (10:1) ratio at 150 °C, naphthyl substrates, non-fused aromatics, and heterocyclic substrates could be used in the methodology (Scheme 159). Most strikingly, several ortho-substituted aromatics could be cross-coupled in synthetically useful yields. This tactic, prefaced by the introduction of ortho substituents using directed metallation, provided access to several polysubstituted aromatic and heteroaromatic compounds. More recently, Shi has disclosed an alternative protocol for the Suzuki coupling of aryl carbamates using triaryl boroxines and 1 equiv of water.464 This modified protocol functioned at lower temperatures (i.e., 110 °C), provided biaryls in high yields ranging from 62–95%, and proved amenable to the corresponding cross-coupling of vinyl carbamate substrates.
Scheme 159.

7.3. C–N Bond Formation
A single example of carbamate amination has been reported in the literature. In Chatani’s study of aryl pivalate amination (see Section 6.3),459 it was found that an aryl carbamate derived from phenol coupled efficiently with morpholine (Scheme 157). The transformation proceeded at 80 °C and furnished the aminated product in 99% yield after 3 h.
Scheme 157.

8. Nickel-Catalyzed Cross-Coupling of Phenols
8.1. Kumada Couplings of 2-Naphthols
Shi and coworkers recently described the unprecedented cross-coupling of 2-naphthol derivatives.286 In this transformation, 2-naphthol derivatives were treated sequentially with methyl magnesium bromide and an aryl Grignard reagent in the presence of NiIIF2 and PCy3 at 120 °C to deliver arylated products (Scheme 161). Although the reaction only proceeds with 2-naphthol derivatives, the scope with respect to substituents on the naphthol is quite broad, as is the range with respect to the aryl Grignard reagents. Cross-coupling products were obtained in 67- 92 % yield depending on the nature of the substrate. Several phenoxide substrates were tested and it was found that bulky substituents decreased the rate of the reaction resulting in lower yields.
Scheme 161.

The cross-coupling is thought to proceed by initial formation of a magnesium naphtholate complex, which is susceptible to Ni0-promoted oxidative addition. Single crystals grown from 2-NaphtylOMgBr demonstrate a dimeric structure with both oxygen atoms coordinating two magnesium ions into a 4-member ring structure. However, a catalytic cycle involving NiII/NiIV could not be excluded. Shi’s methodology is atom economical and provides the first example of phenolate cross-coupling. The catalyst for this reaction was obtained in situ from the complexation NiF2 and PCy3.The halide from the Grignard reagent was found to play an important role in the cross-coupling, with best results generated by Br−. A mechanism of the phenoxide coupling was proposed to involve oxidative addition, transmetalation with the formation of a 6-member ring transition state followed by reductive elimination to generate the biaryl product (Scheme 162).
Scheme 162.
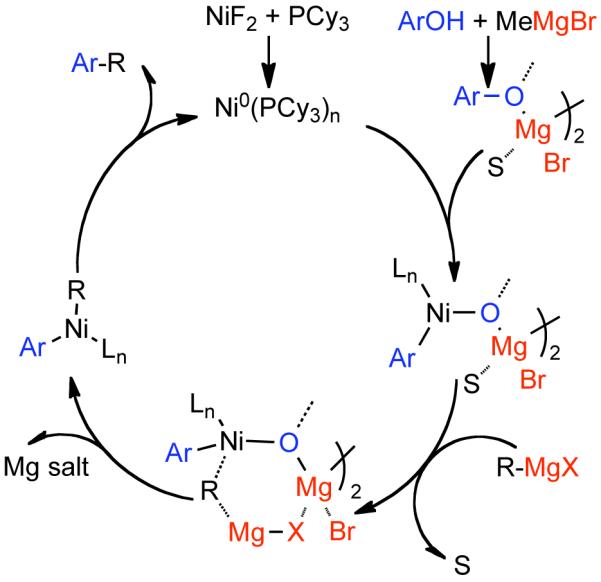
9. Ni-Catalyzed Activation of Other Inert Bonds
9.1. Aryl Fluorides
All previous subchapters in this review addressed Ni-catalyzed homocoupling, cross-coupling or functionalization of C-O bonds. Similar reactions of C-X were discussed for historical context and in some cases comparison of mechanism or utility were also provided. The primary advantage of the C-O bond is its ubiquity and the diverse and selective and cost-effective chemistry for installing or manipulating its functionality. Nevertheless, the primary characteristic of the C-O bond in comparison to most C-X bonds, is its significantly higher bond strength. This of course cannot be said for the highly polarized C-F bond, which have in fact significantly higher bond dissociation energies than C-O, and their activation for cross-coupling is far from trivial. Due to the generally inert-nature of the C-F bond, they could provide for a truly orthogonal functional group for the synthesis of complex organic frameworks.465
Some progress has been realized toward the cross-coupling of C-F electrophiles particularly through the use of Ni-NHC catalysts. Early success in Ni-catalyzed cross-coupling of C-F electrophiles was demonstrated by Herrmann in 2001,277 following an earlier report indicating low levels of C-F activation of 1-chloro-2-fluorobenzene under similar conditions.270 Using 5 mol% of a Ni-NHC catalyst bis[1,3-di(2′,6′-diisopropylphenyl)imidazolin-2-ylidene]nickel0 or Ni-NHCs generated in situ from 5 mol% NiII(acac)2 and 5 mol% of the NHC ligand bis[1,3-di(2′,6′-diisopropylphenyl)imidazolin-2-ylidene], efficient Kumada cross-coupling of aryl-fluorides functionalized with electron-donating or electron-rich substituents in the para or ortho positions with phenyl, t-butylphenyl, or mesityl Grignard reagents could be achieved (Scheme 163). Limited steric influence is demonstrated by the tolerance of ortho-substituents on the aryl-fluoride and the compatibility with bulky mesityl Grignard. It was shown that for the in situ generated Ni-catalyst, a better yield were generated using 1:1 mixtures of the NiII(acac)2 and the NHC ligand than corresponding 1:2 mixtures indicating that the Ni0-mono NHC complex is the active form of the catalyst. Hammett analysis indicates strong negative polarization of the aromatic ring in the transition state, consistent with an oxidative addition mechanism. Lower yields for some substrates was due in large part to the homocoupling of the Grignard reagent, which is not entirely unexpected for such unreactive electrophiles. A similar example of Kumada cross-coupling of 4-fluorotoluene using an Ni-NHC complex in the presence of a phosphorous tetradecyl(trihexyl)phosphonium decanoate ionic liquid has been provided by Walsby and Clyburne.466 Using conditions optimized for the phenylether electrophiles, the Kumada cross-coupling of 1-fluoronaphthalene and fluorobenzene was also shown to proceed using 5 mol% NiIICl2(PCy3)2, in 93 % yield and 100% GC conversion, respectively.428 Hindered N-substituted secondary phosphine oxides (Scheme 164a),467 hindered phosphine oxides (Scheme 164b)468 as well as triarylphosphites (Scheme 164c)469 and pincer complexes (Scheme 164d)273 were also effective ligands for the Ni-catalyzed Kumada cross-coupling of aryl fluorides.
Scheme 163.

Scheme 164.
Mongin found that more conventional catalysts such as NiIICl2(dppe) or NiII(acac)2 in the presence of dppf or dppp could mediate the room-temperature Kumada cross-coupling of fluoroazines and fluorodiazines with various aryl Grignard reagents in good yield (Scheme 165).470 Surprisingly, these simple conditions were also able to mediate the cross-coupling of 4-fluorotoluene with aryl Grignards in fair yield. In addition to the polar oxidative addition pathway proposed by Herrmann, the authors suggested that in the case of fluoroazines, an addition-elimination pathway involving nucleophilic aromatic substitution to form a nickelate complex, followed by subsequent fluoride elimination. Interestingly using as low as 0.05 mol% NiIICl2(dppp) Kumada cross-coupling of fluorobenzene, difluorobenzenes, and trifluorobenzenes with tolyl magnesium bromide could be affected in fair to good yield.471 Cross-couplings with polyfluorinated arenes provided a distribution of mono-, di- and often tri-coupled products.
Scheme 165.

More recently, Shi reported very effective Kumada cross-coupling of 1-fluoronaphthalene and fluorobenzene with methyl magnesium bromide using 5 mol% of NiIICl2(PCy3)2 (Scheme 166).428 The same catalyst was also effective for the Kumada cross-coupling of methyl magnesium bromide with activated methoxy-substituted arenes (See Chapter 4.1). In another report, Shi discovered that in similar Negishi cross-coupling reactions of aryl pivaloates utilizing the same catalyst, the C-F bond remains inert if incorporated on the arylzinc reagent, but could provide double arylation if found on the electrophile.458
Scheme 166.

There are even a few examples of Ni-catalyzed coupling of Grignard reagents that favor alkyl or aryl fluorides. Mechanistically divergent from Kumada coupling of organofluorides, Kambe has described an unusual transformation wherein vinyl magnesium chlorides are alkylatively dimerized (Scheme 167).472 Since oxidative addition is not productive, this alkylative dimerizaiton favors oxidatively inert alkyl fluorides. In a later study, Kanabe found that NiIICl2(dpppbz) (dpppz = 1,2-bis[di(4-isopropylphenyl)phosphine]benzene) was a very effective catalyst for the Kumada cross-coupling of ortho-dihalophenols with alkyl Grignard reagents (Scheme 168).473 It was observed that in this ortho-directed cross-coupling, 2-fluorophenols were more reactive than their corresponding 2-chloro or 2-bromophenols.
Scheme 167.
Scheme 168.

Treatment of fluoropyridines with Ni0(COD)2 in the presence of PEt3 provides an isolable ArNiIIF(PEt3)2, which can be used as a catalyst for the cross-coupling of polyfluoropyridines474 and polyfluoropyrimidines475 with Bu3SnCH=CH2 (Scheme 169). Cross-coupling occurs exclusively at the position ortho to the nitrogen via an addition/elimination mechanism.
Scheme 169.
Following Herrmann’s earlier disclosure, Robins found that a similar catalytic system, 10 mol % Ni0(COD)2 + 10 mol % of the NHC, IPr HCl could mediate the Suzuki cross-coupling of 4-fluoropurine nucleosides with aryl boronic acids in fair to good yield (Scheme 170).197 As 4-fluoropurine nucleosides are readily prepared, this strategy has application toward the rapid synthesis of diverse novel nucleosides. Attempts to perform similar transformations using a Pd-catalyst were unsuccessful due to an unavoidable insertion of oxygen into the aryl-aryl C-C bond to form a biaryl ether.
Scheme 170.

Eventually, attention turned to perfluorinated arenes. Radius found that bimetallic Ni-NHC complex476 [Ni2(iPr2Im)4(COD)] could undergo oxidative addition to the para C-F bond in perfluorotoluene to form an isolable NiII adduct.477 This process could be harnessed in the subsequent Suzuki cross-coupling of perfluorotoluene or perfluorobiphenyl with phenyl, 4-methoxyphenyl, 4-tolyl-, and 4-biphenyl boronic acid in 44-83% yield (Scheme 171). Only 2 mol% of the bimetallic catalyst (or 4 mol% Ni) was necessary and in all cases C-F activation occurred para to the substituent. In a subsequent study, Radius explored the nature of the selectivity of the Ni-NHC complexes for C-F vs C-H bond activation.478 DFT studies indicated that a plausible mechanism that explains the high reactivity and chemoselectivity might involve precoordination of perfluoroarene by the bimetallic catalyst prior to oxidative addition.
Scheme 171.

Ultimately, Ni-NHC complexes in the absence of a coupling partner can be used to mediate mild defluorination of lightly fluorinated arenes. Using 1:1 catalytic complexes of the Ni-NHC and sodium alkoxide Et2CHONa the effective reduction of fluoroarenes such as 1/2-fluoronapthalenes, 3/4-fluorotoluenes, 2/3/4-fluoroanisoles, 3-fluoro-N-methylaniline, or 2/3-fluoropyridines could be mediated with variable yield. The presence of a β-hydride in the sodium alkoxide was found to be critical for the reaction. It was proposed that the sodium alkoxide attacks the oxidative addition product of Ni and the aryl fluoride. Subsequent β-hydride elimination provides the hydrodefluorinated product. Amongst the surveyed Ni-catalysts, the Ni-NHC complex formed from Ni(0) clusters and IMes·HCl was the most effective catalyst, though IPr·HCl was also quite competent.
9.2. Aryl Cyanides
While perfluorinated arenes are readily preparable and there are notable advances in mild selective fluorination to prepare lightly fluorinated arenes, other relatively “inert” functional groups such as cyanide are more readily installed. In 2007, Radius observed that the bimetallic Ni-NHC, [Ni2(iPr2Im)4(COD)], which had previously been shown to mediate C-F bond cleavage and facilitate the cross-coupling of aryl fluorides, could also mediate aryl and alkyl C-CN bond cleavage.479 Later, in an effort to develop a cross-coupling reaction between the carboxylate group of cyano-substituted aryl pivalates and phenyl boroxines, Shi found that NiIICl2(PCy3)2 mediated low but nevertheless slightly higher levels of cyano than pivalate activation.480 Further optimization of the catalyst system showed promising catalyst activity using 10 mol% NiIICl2(PCy3)2, excess PCy3 ligand (20 mol%), using potassium tert-butoxide as base in the presence of CuF2 as an additive. A screen of various boron transmetallating reagents in the cross-coupling of 2-naphthylnitrile revealed fair yields with boroxines, boronate esters, and boronic acids (43-68%), while poor yields (<5%) were obtained using potassium trifluoroborates. The highest yields were obtained using neopentylglycolboronate esters, and a subsequent study of reaction scope revealed that electron-deficient and electron rich aryl, naphthyl and heteroaryl cyanides and neopentylglycolboronates in fair to good yield (31-82%). The worst yields were obtained for ortho-substituted arylneopentylglycolboronates. Despite variable yield, the ability of Ni to activated relatively “inert” bonds under orthogonal conditions, allowed for the sequential synthesis of asymmetric polyarylated benzene (Scheme 172). Starting from 3-chloro-5-methoxybenzonitrile, Pd-catalyzed Suzuki-Miyaura coupling of the chloride with 4-butylphenylboronic acid provided a biphenyl substituted with both cyano and methoxy groups. NiIICl2(PCy3)2 in the presence of excess PCy3, potassium tert-butoxide, and CuF2 provided selective cross-coupling of the more reactive cyano group with 4-methylphenylneopentylglycolboronate ester. The remaining aryl methyl ether successfully underwent Ni-catalyzed Kumada cross-coupling of phenylmagnesium bromide, providing the triarylated benzene in 36 % overall yield.
Scheme 172.

10. Conclusions and Outlook
Synthetic organic chemistry is most certainly in its maturity. While there will continue to be landmark discoveries of routes to particularly complicated medicinal natural products and their analogues, or to the preparation of relatively intractable or high-energy designed molecules, few would question the collective ability to ultimately prepare in some fashion any viable481 structure. More questionable is our ability to do so in a practical and economical fashion. Cross-coupling and homocoupling chemistry will continue to be indispensable tools for the construction of small molecules and polymers.
As detailed in this review, Ni is emerging as an extraordinarily versatile catalyst for homocoupling cross-coupling, functionalization and defunctionalization reactions. Like the later group 10 metals, Ni is compatible with cross-coupling of halocarbon electrophiles such as C-Br and C-I, but is more reactive toward C-Cl and even “inert” C-F bonds. Additionally, Ni is particularly effective for reactions involving C-O derived electrophiles such as relatively reactive triflates and nonaflates, but also toward the less reactive mesylates and tosylates, as well as sulfamates, phosphates, phosphoramides, certain esters, carbonates and carbamates, and even particularly activated ethers and phenols. In cross-coupling reactions, a vast assortment of coupling partners have been explored running the gamut of transmetallating reagents derived from B, Mg, In, Si, Sn, and Zn. Ni-catalyzed functionalization with amides, boronates and diborons, cyanates, and thioethers is now well known, as is defunctionalization by reduction or deoxygenations. Ni reactions involving these less reactive electrophiles are typically achieved using simpler and less-expensive catalytic systems than those required to mediate the same chemistry with later transition metals. Together with inherently lower cost of Ni metal and in particular C-O derived electrophiles, the use of Ni-catalysts for such transformations are often more attractive to large scale processes. Moreover, the capacity of Ni to undergo oxidative addition to less reactive phenol- and enol-derived electrophiles, allows entry into an abundant library of structurally diverse building blocks.
More and more, Ni and other early transition metal catalysts are being suitable and often more effective alternatives to precious metal catalysts for key C-C bond formation reactions and critical functional group transformations. Through the continued exploration of Ni-catalysis it is expected that more readily available and often bioderived C-O based electrophiles can be used in the place of more expensive, less diverse, and often toxic halide equivalents. While sulfonates, particularly inexpensive mesylates, were amongst the first such C-O electrophiles to be developed and find academic and industrial utility in Ni-catalyzed chemistry, recent advances have allowed for more ready use of esters, ethers, and even phenols. Further development of such methodologies may ultimately allow for general and highly practical cross-coupling and functionalization reactions using unmodified feedstock chemicals, where both electrophile and transmetallating reagents are derived from phenolic or enolic precursors using only Ni-catalysis. Additionally, the ability of Ni to activate almost any electrophile under similar or orthogonal482 conditions should allow for efficient sequential, iterative, or even one-pot strategies for the synthesis of complex molecular structures. While Ni has been somewhat overlooked in the favor of more popularly studied and therefore well understood Pd, Ni is now back in the limelight and hopefully its full potential will be unlocked in the years to come.
Scheme 50.
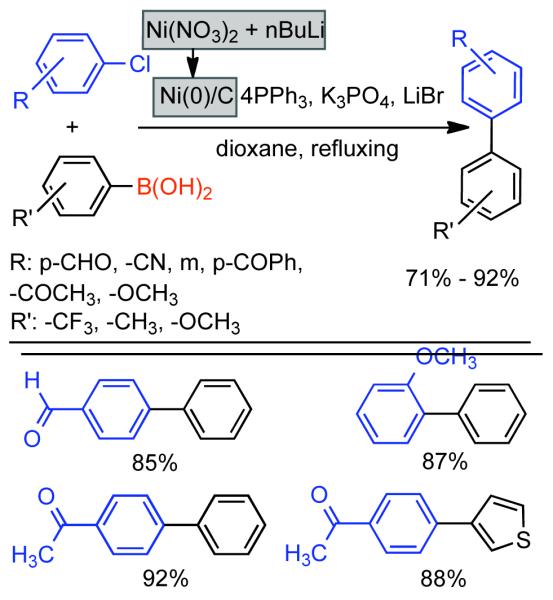
Scheme 52.
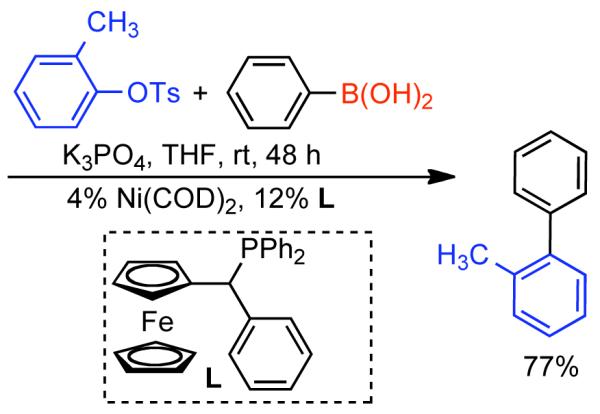
Scheme 146.

Scheme 149.
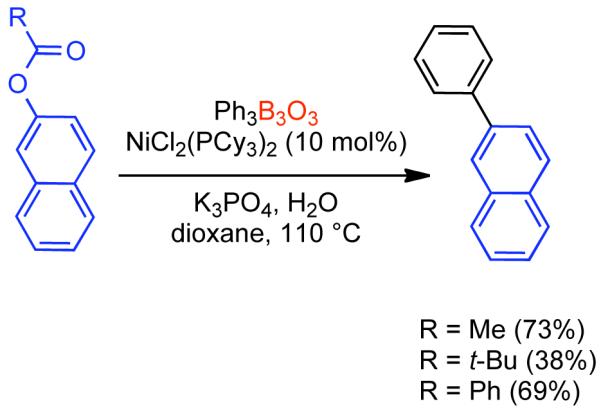
Scheme 153.

Scheme 160.
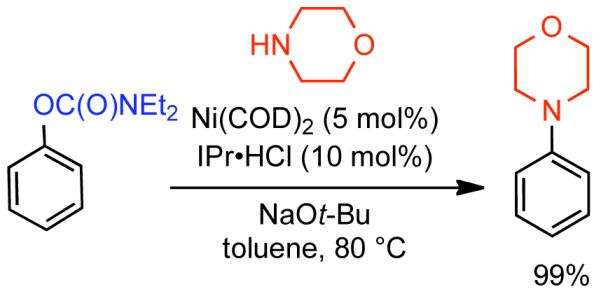
11. Acknowledgements
Financial support by the National Science Foundation (DMR-0548559, DMR-0520020 and DMR-0935165) and the P. Roy Vagelos Chair at Penn are gratefully acknowledged (V.P), in addition to the National Institutes of Health GM-079922 (N.K.G). B.M.R gratefully acknowledges funding from an NSF Graduate Research Fellowship, an ACS Division of Organic Chemistry Graduate Fellowship (Roche), and a University of Pennsylvania Dissertation Fellowship. K.Q. thanks the Foote family for a graduate fellowship.
12. Abbreviations
- acac
acetylacetonato
- bpy
2,2′-bipyridine
- COD
1,4-cyclooctadiene
- CRA
Complex reducing agent
- DFT
Density Functional Theory
- DIBAH
diisobutylaluminium hydride
- DMPU
1,3-Dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone
- DMS
dimethylsulfide
- DoM
directed ortho metallation
- DP
degree of polymerization
- dppb
1,2-bis(diethylphosphino)butane
- dppe
1,2-bis(diethylphosphino)ethane
- dppm
1,2-bis(diethylphosphino)methane
- dppp
1,2-bis(diethylphosphino)propane
- dppf
1,1′-bis(diethylphosphino)ferrocene
- HMPA
hexamethylphosphoramide
- HMPT
hexamethylphosphorus triamide
- ICP-AES
inductively coupled plasma atomic emission spectroscopy
- IPr
1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene
- IMes
1,3-dimesitylimidazol-2-ylidene
- KIE
kinetic isotope effect
- NEP
N-ethylpyrrolidine
- Ni/C
Nickel on charcoal
- NiCRA
Ni-complex reducing agent
- NHC
N-heterocyclic carbene
- NP
nanoparticles
- OSET
outer-sphere electron transfer
- -OSO2CF3
triflate
- -OSO2CH3
mesylate
- -OSO2CF2CF2CF2CF3
nonaflate
- -OSO2-p-PhCH3
tosylate
- PAPO
poly(arylenephosphine oxide)
- PCL
poly(ε-caprolactone)
- PHT
poly(3-hexylthiophene)
- PL
photoluminescent
- POx
poly(oxazoline)
- PPP
poly(p-phenylene)
- SCLCP
side-chain liquid crystalline polymers
- SIPr
1,3-bis(2,6-diisopropylphenyl)-4,5-dihydroimidazol-2-ylidene
- SNAR
substitution nucleophilic aromatic
- TBAB
tetrabutylammonium bromide
- TPPTS
sodium triphenylphosphinotrimetasulfonate
Biography
Author Profiles

Brad M. Rosen was born in Philadelphia, Pennsylvania. He received his A.B. and A.M. in Chemistry from Harvard University in 2005, where he worked on the synthesis of cyclic enediyne antibiotics under the direction of Professor Andrew G. Myers. In 2009, he received his Ph.D. from the University of Pennsylvania. Under the direction of Professor Virgil Percec, his doctoral studies focused on the development of Ni-catalyzed cross-coupling and borylation, synthesis and retrostructural analysis of self-assembling dendrons, and the elaboration of single-electron transfer living radical polymerization. He is the recipient of a Rohm and Haas Graduate Research Fellowship, an NSF Graduate Research Fellowship (2005–2008), an ACS Division of Organic Chemistry Graduate Fellowship (2008–2009, Sponsored by Roche, Inc.), a University of Pennsylvania Dissertation Fellowship (2009), and the 2010 Miller Award for the best Doctoral Thesis from the Department of Chemistry at the University of Pennsylvania. Since December 2009, he is a Principal Investigator at DuPont Central Research and Development, Experimental Station, Wilmington, Delaware.

Kyle Quasdorf was born and raised in Alden, Iowa. He graduated from Iowa State University in 2007 with a B.S. in Chemistry. While at Iowa State, he worked in the laboratory of Dr. Walter S. Trahanovsky studying reactions in supercritical fluids. In 2007, he began graduate studies at UCLA and works under the direction of Professor Neil Garg as a Foote Graduate Fellow. Kyle’s graduate research involves the discovery of Ni-catalyzed cross coupling reactions of phenolic derivatives and the total synthesis of heterocyclic natural products.

Daniela A. Wilson (neé Apreutesei) was born in Comanesti, county of Bacau, Romania. She received her B.S. degree (Hon) in chemistry from “A. I. Cuza” University of Iasi, Romania, in 2001 and an M.S. degree (Hon) in environmental chemistry from the same university in 2003. In 2001 she started the Ph.D. program in parallel at “Gh. Asachi” Technical University of Iasi, Department of Organic Chemistry, Romania, under the supervision of Professor Dan Scutaru. During that time she joined in 2003 the group of Professor Shin’ichi Nakatsuji, University of Hyogo, Himeji, Japan, as an exchange Ph.D. student and the group of Dr. Georg Mehl, University of Hull, Hull, U.K., as a Marie Curie Fellow in 2004–2005. In 2006 she defended her Ph.D. thesis with summa cum laudae distinction. Since 2007, she is a postdoctoral researcher in the group of Professor Virgil Percec at the University of Pennsylvania. Her research interests include synthetic organic chemistry, organometallic chemistry, and supramolecular chemistry.

Na Zhang was born in Jiangxi, China. She received her B.S. in Chemical Physics from University of Science and Technology of China in 2009. In the same year, she started her Ph.D program in department of chemistry, University of Pennsylvania, under the supervision of Professor Virgil Percec. Her research interests include nickel catalyzed borylation and cross-coupling reactions.

Ana-Maria Resmerita was born in Barlad, country of Vaslui, Romania. She received a B.S. in macromolecular chemistry from “Gh.Asachi” Technical University of Iasi, Romania in 2005 and in 2006 M.S. in biopolymer materials from the same university. In parallel she started her PhD degree at the same university in the Department of Natural and Synthetic Polymers under the supervision of Professor Nicolae Hurduc. She defended her PhD thesis in 2008. Since 2009 she is a postdoctoral researcher in the group of Professor Virgil Percec at the University of Pennsylvania. Her research interests include organic synthesis, polymer synthesis, and supramolecular chemistry.

Neil K. Garg was born in Fishkill, NY, USA in 1978. He received his BS in Chemistry from New York University in 20, where he performed undergraduate research with Professor Marc Walters. During his undergraduate years, he spent several months in Strasbourg, France while conducting research with Professor Mir Wais Hosseini at Université Louis Pasteur. Garg attended the California Institute of Technology for graduate studies and worked under the direction of Professor Brian Stoltz. In 2005, he received his Ph. D. and then spent two years in Professor Larry Overman’s research laboratory at the University of California, Irvine as an NIH postdoctoral scholar. Professor Garg started his independent career at UCLA in 2007, where his laboratory is focused in the areas of transition metal catalysis and natural product synthesis.

Virgil Percec was born and educated in Romania (Ph.D., 1976). He defected from his native country in 1981 and after short postdoctoral appointments at the University of Freiberg, Germany, and the University of Akron, U.S.A., he joined the Department of Macromolecular Science at Case Western Reserve University in Cleveland (1982) as an Assistant Professor. He was promoted to Associate Professor in 1984, to Professor in 1986, and to Leonard Case Jr. Chair in 1993. In 1999 he moved to the University of Pennsylvania as P. Roy Vagelos Professor of Chemistry. Percec’s research interests lies at the interface between organic, bioorganic, supramolecular, polymer chemistry, and liquid crystals, where he contributed over 640 refereed publications, 50 patents, and over 10 endowed and invited lectures. His list of awards includes Honorary Foreign Member to the Romanian Academy (1993), Humboldt Award (1997), NSF Research Award for Creativity in Research (1990, 1995, 20), PTN Polymer Award from The Netherlands (2002), the ACS Award in Polymer Chemistry (2004), the Staudinger–Durrer Medal from ETH (2005), the International Award of the Society of Polymer Science from Japan (2007), and the H. F. Mark Medal from the Austrian Research Institute for Chemistry and Technology (2008). He is a Fellow of IUPAC (2001), PMSE Division of ACS (2003), AAAS (2004), RSC (2008) and a Honorary member of the Israel Society of Chemistry (2009). He is the editor of the Journal of Polymer Science, Part A: Polymer Chemistry (since 1996) and of the book series Liquid Crystals and serves on the Editorial Boards of 20 international journals.
13. References
- (1).Corey EJ, Cheng X-M. The Logic of Chemical Synthesis. Wiley-Interscience; New York: 1995. [Google Scholar]
- (2).Nicolaou KC, Sorenson EJ. Classics in Total Synthesis. Wiley-VCH; New York: 1996. [Google Scholar]
- (3).Wilson RJ, Danishefsky SJ. J. Org. Chem. 2006;71:8329. doi: 10.1021/jo0610053. [DOI] [PubMed] [Google Scholar]
- (4).Grimsdale AC, Chan KL, Martin RE, Jokisz PG, Holmes AB. Chem. Rev. 2009;109:897. doi: 10.1021/cr000013v. [DOI] [PubMed] [Google Scholar]
- (5).Lehn JM. Proc. Natl. Acad. Sci. U. S. A. 2002;99:4763. doi: 10.1073/pnas.072065599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Rosen BM, Wilson CJ, Wilson DA, Peterca M, Imam MR, Percec V. Chem. Rev. 2009;109:6275. doi: 10.1021/cr900157q. [DOI] [PubMed] [Google Scholar]
- (7).de Meijere A, Diederich F. Metal Catalyzed Cross-Coupling Reactions Volumes 1 & 2. Wiley-VCH; New York: 2004. [Google Scholar]
- (8).Beller M, Bolm C. Transition Metals for Organic Synthesis: Building Blocks and Fine Chemicals, 2 Volume Set. Wiley-VCH; New York: 2004. [Google Scholar]
- (9).Hegedus L, Soderberg B. Transition Metals in the Synthesis of Complex Organic Molecules. 3rd Ed. University Science Books; 2009. [Google Scholar]
- (10).Hassan J, Sevignon M, Gozzi C, Schulz E, Lemaire M. Chem. Rev. 2002;102:1359. doi: 10.1021/cr000664r. [DOI] [PubMed] [Google Scholar]
- (11).Denmark SE, Regens CS. Acc. Chem. Res. 2008;41:1486. doi: 10.1021/ar800037p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Sherry BD, Fürstner A. Acc. Chem. Res. 2008;41:1500. doi: 10.1021/ar800039x. [DOI] [PubMed] [Google Scholar]
- (13).Würtz S, Glorius F. Acc. Chem. Res. 2008;41:1523. doi: 10.1021/ar8000876. [DOI] [PubMed] [Google Scholar]
- (14).Hartwig J. Acc. Chem. Res. 2008;41:1534. doi: 10.1021/ar800098p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Fu GC. Acc. Chem. Res. 2008;41:1555. doi: 10.1021/ar800148f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Heck RF. Acc. Chem. Res. 1979;12:146. [Google Scholar]
- (17).Heck RF. Palladium Reagents. Academic Press; London: 1990. [Google Scholar]
- (18).Hiyama T, Shirakawa E. Top. Curr. Chem. 2002;219:61. [Google Scholar]
- (19).Terao J, Kambe N. Acc. Chem. Res. 2008;41:1545. doi: 10.1021/ar800138a. [DOI] [PubMed] [Google Scholar]
- (20).Negishi EI. Bull. Chem. Soc. Jpn. 2007;80:233. [Google Scholar]
- (21).Negishi EI. Bull. Chem. Soc. Jpn. 2007;80:1598. [Google Scholar]
- (22).Miyaura N, Suzuki A. Chem. Rev. 1995;95:2457. [Google Scholar]
- (23).Miyaura N. Top. Curr. Chem. 2002;219:11. [Google Scholar]
- (24).Sonogashira K. J. Organomet. Chem. 2002;653:46. [Google Scholar]
- (25).Stille JK. Angew. Chem. Int. Ed. 1986;25:508. [Google Scholar]
- (26).Fugami K, Kosugi M. Top. Curr. Chem. 2002;219:87. [Google Scholar]
- (27).Tsuji J. Palladium Reagents and Catalysts: New Perspectives for the 21st Century. 2nd Ed. Wiley; New York: 2004. [Google Scholar]
- (28).Rappoport Z, editor. The Chemistry of Phenols. John Wiley & Sons Ltd; Chichester: 2003. [Google Scholar]
- (29).Scifinder Scholar, version 2007a. Chemical Abstract Services; Columbus, OH: 2009. [Google Scholar]
- (30).Smith MB, March J. March’s Advanced Organic Chemistry. 6th ed. John Wiley & Sons, Inc.; New Jersey: 2007. p. 670. [Google Scholar]
- (31).Snieckus V. Chem. Rev. 1990;90:879. [Google Scholar]
- (32).Xiao B, Fu Y, Xu J, Gong T-J, Dai J-J, Yi J, Liu L. J. Am. Chem. Soc. 2010;132:468. doi: 10.1021/ja909818n. [DOI] [PubMed] [Google Scholar]
- (33).Bedford RB, Webster RL, Mitchell CJ. Org. Biomol. Chem. 2009;7:4853. doi: 10.1039/b916724m. [DOI] [PubMed] [Google Scholar]
- (34).Zhao X, Yeung CS, Dong VM. J. Am. Chem. Soc. 2010;132:5837. doi: 10.1021/ja100783c. [DOI] [PubMed] [Google Scholar]
- (35).Prices as of July 23, 2010.
- (36).Martin R, Buchwald SL. Acc. Chem. Res. 2008;41:1461. doi: 10.1021/ar800036s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Yang BH, Buchwald SL. J. Organomet. Chem. 1999;576:125. [Google Scholar]
- (38).Wilke G. Angew. Chem. 1963;75:10. [Google Scholar]
- (39).Yamamoto A, Morifuji K, Ikeda S, Saito T, Uchida Y, Misono A. J. Am. Chem. Soc. 1965;87:4652. [Google Scholar]
- (40).Yamamoto T, Yamamoto A, Ikeda S. J. Am. Chem. Soc. 1971;93:3350. [Google Scholar]
- (41).Saito T, Uchida Y, Misono A, Yamamoto A, Morifuji K, Ikeda S. J. Am. Chem. Soc. 1966;88:5198. [Google Scholar]
- (42).Semmelhack MF, Helquist PM, Jones LD. J. Am. Chem. Soc. 1971;93:5908. [Google Scholar]
- (43).Semmelhack MF, Ryono LS. J. Am. Chem. Soc. 1975;97:3874. [Google Scholar]
- (44).Semmelhack MF, Helquist P, Jones LD, Keller L, Mendelson L, Ryono LS, Smith JG, Stauffer RD. J. Am. Chem. Soc. 1981;103:6460. [Google Scholar]
- (45).Tolman CA, Seidel DH, Gerlach DH. J. Am. Chem. Soc. 1972;94:2669. [Google Scholar]
- (46).Tolman CA, Seidel DH, Gosser LW. J. Am. Chem. Soc. 1974;96:53. [Google Scholar]
- (47).Kende AS, Liebeskind LS, Braitsch DM. Tetrahedron Lett. 1975:3375. [Google Scholar]
- (48).Klein A, Budnikova YH, Sinyashin OG. J. Organomet. Chem. 2007;692:3156. [Google Scholar]
- (49).Jennings PW, Pillsbury DG, Hall JL, Brice VT. J. Org. Chem. 1976;41:719. [Google Scholar]
- (50).Mori M, Hashimato Y, Ban Y. Tetrahedron Lett. 1980;21:631. [Google Scholar]
- (51).Mori M, Ban Y. Tetrahedron Lett. 1976;17:1803. [Google Scholar]
- (52).Mori M, Ban Y. Tetrahedron Lett. 1976;17:1807. [Google Scholar]
- (53).Troupel M, Rollin Y, Sibille S, Perchion J, Fauvarque J-F. J. Organomet. Chem. 1980;202:435. [Google Scholar]
- (54).Zembayashi M, Tamao K, Yoshida J, Kumada M. Tetrahedron Lett. 1977:4089. [Google Scholar]
- (55).Negishi E, King AO, Okukado N. J. Org. Chem. 1977;42:1821. [Google Scholar]
- (56).Caubere P. Angew. Chem. Int. Ed., Engl. 1983;22:599. [Google Scholar]
- (57).Guillaumet G, Mordenti L, Caubère P. J. Organomet. Chem. 1975;102:43. [Google Scholar]
- (58).Guillaumet G, Mordenti L, Caubère P. J. Orgaomet. Chem. 1975;102:353. [Google Scholar]
- (59).Loubinoux R, Vanderesse R, Caubère P. Tetrahedron Lett. 1977:3951. [Google Scholar]
- (60).Vanderesse R, Brunet JJ, Caubère P. J. Org. Chem. 1981;46:1270. [Google Scholar]
- (61).Vanderesse R, Brunet JJ, Caubère P. J. Organomet. Chem. 1984;264:263. [Google Scholar]
- (62).Vanderesse R, Lourak M, Forti Y, Caubère P. Tetrahedron Lett. 1986;27:5483. [Google Scholar]
- (63).Brunet JJ, Besozzi D, Courtois A, Caubère P. J. Am. Chem. Soc. 1982;104:7130. [Google Scholar]
- (64).Rieke RD, Wolf WJ, Kujundzic N, Kavaliunas AV. J. Am. Chem. Soc. 1977;99:4159. [Google Scholar]
- (65).Matsumoto H, Inaba S-I, Rieke RD. J. Org. Chem. 1983;48:840. [Google Scholar]
- (66).Rieke RD. Acc. Chem. Res. 1977;10:301. [Google Scholar]
- (67).Kavaliunas AV, Rieke RD. J. Am. Chem. Soc. 1980;102:5944. [Google Scholar]
- (68).Tsou TT, Kochi JK. J. Am. Chem. Soc. 1979;101:6319. [Google Scholar]
- (69).Tsou TT, Kochi JK. J. Am. Chem. Soc. 1979;101:7547. [Google Scholar]
- (70).Iyoda M, Otsuka H, Sato K, Nisato N, Oda M. Bull. Chem. Soc. Jpn. 1990;63:80. [Google Scholar]
- (71).Yamamoto T, Wakabayashi S, Osakada K. J. Organomet. Chem. 1992;428:223. [Google Scholar]
- (72).Chao CS, Cheng CH, Chang CT. J. Org. Chem. 1983;48:4904. [Google Scholar]
- (73).Colon I, Maresca LM, Kwiatkowski GT. U. S. Patent 4 263 466. 1981
- (74).Colon I, Kelsey DR. J. Org. Chem. 1986;51:2627. [Google Scholar]
- (75).Nakamura A, Otsuka S. Tetrahedron Lett. 1974:463. [Google Scholar]
- (76).Fahey DR, Mahan JE. J. Am. Chem. Soc. 1976;98:4499. [Google Scholar]
- (77).Lewin M, Aizenshtat Z, Blum J. J. Organomet. Chem. 1980;184:255. [Google Scholar]
- (78).Kikukawa K, Takagi M, Matsuda T. Bull. Chem. Soc. Jpn. 1979;52:1493. [Google Scholar]
- (79).Schiavon G, Bontempelli G, Corrain B. J. Chem Soc., Dalton Trans. 1981:1074. [Google Scholar]
- (80).Armatore C, Jutland A. Organometallics. 1988;7:2203. [Google Scholar]
- (81).Takagi K, Hayama N, Inokawa S. Chem. Lett. 1979:917. [Google Scholar]
- (82).Takagi K, Hayama N, Inokawa S. Bull. Chem. Soc. Jpn. 1980;53:3691. [Google Scholar]
- (83).Takagi K, Hayama N, Sasaki K. Bull. Chem. Soc. Jpn. 1984;57:1887. [Google Scholar]
- (84).Iyoda M, Sakaitani M, Otsuka H, Oda M. Chem. Lett. 1985;127 [Google Scholar]
- (85).Iyoda M, Sakaitani N, Otsuka H, Oda M. Tetrahedron Lett. 1985;26:4677. [Google Scholar]
- (86).Iyoda M, Sato K, Oda M. Tetrahedron Lett. 1985;26:3829. [Google Scholar]
- (87).Iyoda M, Sato K, Oda M. J. Chem. Soc., Chem. Commun. 1985:1547. [Google Scholar]
- (88).Iyoda M, Sato K, Oda M. Tetrahedron Lett. 1987;28:625. [Google Scholar]
- (89).Tiecco M, Testafferi L, Tingoli M, Chianelli D, Montanucci M. Synthesis. 1984:736. [Google Scholar]
- (90).Yamashita J, Inoue Y, Kondo T, Hashimoto H. Chem. Lett. 1986:407. [Google Scholar]
- (91).Inoue Y, Yamashita J, Kondo T, Hashimoto H. Nippon Kagaku Kaishi. 1987:197. [Google Scholar]
- (92).Percec V, Bae J-Y, Zhao M, Hill DH. J. Org. Chem. 1995;60:176. [Google Scholar]
- (93).Berman RS, Kochi JK. Inorg. Chem. 1980;19:248. [Google Scholar]
- (94).Percec V, Bae J-Y, Zhao M, Hill DH. J. Org. Chem. 1995;60:1066. [Google Scholar]
- (95).Bae J-Y, Percec V. J. Ind. Eng. Chem. 1998;4:64. [Google Scholar]
- (96).Bae J-Y. J. Ind. Eng. Chem. 1998;4:319. [Google Scholar]
- (97).Yamamoto T, Ito T, Kubota K. Chem. Lett. 1988:153. [Google Scholar]
- (98).Yamamoto T, Maruyama T, Zhou Z-H, Ito T, Fukuda T, Yoneda Y, Begum F, Ikeda T, Sasaki S, Takezoe H, Fukuda A, Kubota K. J. Am. Chem. Soc. 1994;116:4832. [Google Scholar]
- (99).Choi B-K, Yamamoto T. Electrochem. Commun. 2003:566. [Google Scholar]
- (100).Yamamoto T, Fujiwara Y, Fukumoto H, Nakamura Y, Koshihara S-Y, Ishikawa T. Polymer. 2003;44:4487. [Google Scholar]
- (101).Yamamoto T, Morito A, Maruyama T, Zhou Z-H, Kanbara T, Sanechika K. Polym. J. 1990;22:187. [Google Scholar]
- (102).Yamamoto T, Morita A, Miyazaki Y, Maruyama T, Wakyama H, Zhou Z-H, Nakamura Y, Kanbara T, Saksaki S, Kubota K. Macromolecules. 1992;25:1214. [Google Scholar]
- (103).Tan L, Curtis MD, Francis AH. Macromolecules. 2002;35:4628. [Google Scholar]
- (104).Nanos JI, Kampf JW, Curtis MD, Gonzalez L, Martin DC. Chem. Mater. 1995;7:2232. [Google Scholar]
- (105).Politis JK, Nemes JC, Curtis MD. J. Am. Chem. Soc. 2001;123:2537. doi: 10.1021/ja003588i. [DOI] [PubMed] [Google Scholar]
- (106).Kanbara T, Saito N, Yamamoto T, Kubota K. Macromolecules. 1991;24:5883. [Google Scholar]
- (107).Zhang Z-B, Fujiki M, Tang H-Z, Montonaga M, Torimitsu K. Macromolecules. 2002;35:1988. [Google Scholar]
- (108).Yamamoto T, Lee B-L. Macromolecules. 2002;35:2993. [Google Scholar]
- (109).Yamamoto T. J. Organometallic Chem. 2002;653:195. [Google Scholar]
- (110).Percec V, Pugh C, Cramer E, Okita S, Weiss R. Makromol. Chem., Macromol. Symp. 1992;54/55:113. [Google Scholar]
- (111).Gin DL, Conticello VP. Trends Polym. Sci. 1996;4:217. [Google Scholar]
- (112).Feast WJ, Tsibouklis J, Pouwer KL, Groenendaal L, Meijer EW. Polymer. 1996;37:5017. [Google Scholar]
- (113).Percec V, Hill D. In: Step Growth Polymers for High-Performance Materials, New Synthetic Methods. Labadie JW, Hedrick JL, editors. 1996. p. 2. ACS Symposium Series 624. [Google Scholar]
- (114).Berresheim AJ, Muller M, Müllen K. Chem. Rev. 1999;99:1747. doi: 10.1021/cr970073+. [DOI] [PubMed] [Google Scholar]
- (115).Kraft A, Grimsdale AC, Holmes AB. Angew. Chem. Int. Ed. 1998;37:402. doi: 10.1002/(SICI)1521-3773(19980302)37:4<402::AID-ANIE402>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- (116).Rehahn M, Schlüter AD, Wegner G, Feast WJ. Polymer. 1989:1054. [Google Scholar]
- (117).Schlüter AD. J. Polym. Sci. Part A: Polym. Chem. 2001;39:1533. [Google Scholar]
- (118).Colon I, Kwiatkowski GT. J. Polym. Sci. Part A: Polym. Chem. 1990;28:367. [Google Scholar]
- (119).Colon I. J. Org. Chem. 1982;47:2622. [Google Scholar]
- (120).Colon I, Merriam CN. U. S. Patent 4,4,00,566. 1984
- (121).Ueda M, Ichikawa F. Macromolecules. 1990;23:926. [Google Scholar]
- (122).Ueda M, Miyaji Y, Ito K, Oba Y, Sone T. Macromolecules. 1991;24:2694. [Google Scholar]
- (123).Percec V, Asandei AD, Hill DH, Crawford D. Macromolecules. 1999;32:2597. [Google Scholar]
- (124).Yurteri S, Cianga I, Degirmenci M, Yagci Y. Polym. Int. 2004;53:1219. [Google Scholar]
- (125).Demirel AL, Yuteri S, Cianga I, Yagci Y. J. Polym. Sci. Part A: Polym. Chem. 2007;45:2091. [Google Scholar]
- (126).Percec V, Pugh C, Cramer E, Weiss R. Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.) 1991;32:329. [Google Scholar]
- (127).Percec V, Okita S, Weiss R. Macromolecules. 1992;25:1816. [Google Scholar]
- (128).Chaturvedi V, Tanaka S, Kaeriyama K. J. Chem. Soc., Chem. Commun. 1992:1658. [Google Scholar]
- (129).Percec V, Okita S, Bae J. Polym. Bull. 1992;29:271. [Google Scholar]
- (130).Percec V, Okita S. J. Polym. Sci. Part A: Polym. Chem. 1993;31:877. [Google Scholar]
- (131).Percec V, Okita S. J. Polym. Sci. Part A: Polym. Chem. 1993;31:1087. [Google Scholar]
- (132).Percec V, Okita S. J. Polym. Sci. Part A: Polym. Chem. 1993;30:1037. [Google Scholar]
- (133).Percec V, Bae J-Y, Zhao M, Hill DH. Macromolecules. 1995;28:6726. [Google Scholar]
- (134).Kaeriyama K, Kouyama S, Sekita M, Nakayama T, Tsukahara Y. Macromol. Rapid Commun. 1999;20:50. [Google Scholar]
- (135).Wallow TI, Seery TAP, Goodson FE, II., Novak BM. Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.) 1994;35:710. [Google Scholar]
- (136).Wallow TI, Novak BM. J. Org. Chem. 1994;59:5034. [Google Scholar]
- (137).Novak BM, Wallow TI, Goodson F, Loos K. Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.) 1995;36(1):693. [Google Scholar]
- (138).Percec V, Zhao M, Bae J-Y, Hill DH. Macromolecule. 1996;29:3727. [Google Scholar]
- (139).Grob MC, Feiring AE, Auman BC, Percec V, Zhao M, Hill DH. Macromolecules. 1996;29:7284. [Google Scholar]
- (140).Percec V, Zhao M, Bae J-Y, Asandei AD, Hill DH. Polym. Bull. 1997;38:515. [Google Scholar]
- (141).Ghassemi H, McGrath JE. Polymer. 1997;38:3139. [Google Scholar]
- (142).Bloom PD, Sheares VV. Macromolecules. 2001;34:1627. [Google Scholar]
- (143).Rusch-Salazar LA, Sheares VV. J. Polym. Sci. Part A: Polym. Chem. 2003;41:2277. [Google Scholar]
- (144).Zengin H, Zengin G, Topping CM, Smith DW. J. Polym. Sci. Part A: Polym. Chem. 2007;45:1860. [Google Scholar]
- (145).Rhee TH, Choi T, Chung EY, Suh DH. Macromol. Chem. Phys. 2001;202:906. [Google Scholar]
- (146).Saito N, Kanbara T, Sato T, Yamamoto T. Polym. Bull. 1993;30:285. [Google Scholar]
- (147).Lee T-S, Kim J, Bae J-Y. Polymer. 2004;45:5065. [Google Scholar]
- (148).Andjelkovic DD, Sheares VV. Macromolecules. 2007;40:7148. [Google Scholar]
- (149).Semmelhack MF, Helquist PM, Gorzynski JD. J. Am. Chem. Soc. 1972;94:9234. [Google Scholar]
- (150).Lin G-Q, Hong R. J. Org. Chem. 2001;66:2877. doi: 10.1021/jo001563w. [DOI] [PubMed] [Google Scholar]
- (151).Lei J-G, Lin G-Q. Chin. J. Chem. 2002;20:1263. [Google Scholar]
- (152).Hashim J, Glasnov TN, Kremsner JM, Kappe CO. J. Org. Chem. 2006;71:1707. doi: 10.1021/jo052283p. [DOI] [PubMed] [Google Scholar]
- (153).Hashim J, Kappe CO. Adv. Synth. Catal. 2007;349:2353. [Google Scholar]
- (154).Lei J-G, Xu M-H, Lin G-Q. Synlett. 2004:2364. [Google Scholar]
- (155).Miyaura N, Yamada K, Suzuki A. Tetrahedron Lett. 1979:3437. [Google Scholar]
- (156).Littke AF, Fu GC. Angew. Chem. Int. Ed. 2002;41:4176. doi: 10.1002/1521-3773(20021115)41:22<4176::AID-ANIE4176>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- (157).Herrmann WA. Angew. Chem. Int. Ed. 2002;41:1290. doi: 10.1002/1521-3773(20020415)41:8<1290::aid-anie1290>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- (158).Nguyen HN, Huang X, Buchwald SL. J. Am. Chem. Soc. 2003;125:11818. doi: 10.1021/ja036947t. [DOI] [PubMed] [Google Scholar]
- (159).Bhayana B, Fors BP, Buchwald SL. Org. Lett. 2009;11:3954. doi: 10.1021/ol9015892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (160).Zhang L, Meng T, Wu J. J. Org. Chem. 2007;72:9346. doi: 10.1021/jo7019064. [DOI] [PubMed] [Google Scholar]
- (161).So CM, Lau CP, Chan ASC, Kwong FY. J. Org. Chem. 2008;73:7731. doi: 10.1021/jo8014819. [DOI] [PubMed] [Google Scholar]
- (162).So CM, Lau CP, Kwong FY. Angew. Chem. Int. Ed. 2008;47:8059. doi: 10.1002/anie.200803193. [DOI] [PubMed] [Google Scholar]
- (163).Chow WK, So CM, Lau CP, Kwong FY. J. Org. Chem. 2010;75:5109. doi: 10.1021/jo100846t. [DOI] [PubMed] [Google Scholar]
- (164).Percec V, Bae JY, Hill DH. J. Org. Chem. 1995;60:1060. [Google Scholar]
- (165).Oh-e T, Miyaura N, Suzuki A. Synlett. 1990:221. [Google Scholar]
- (166).Watanabe T, Miyaura N, Suzuki A. Synlett. 1992:207. [Google Scholar]
- (167).Oh-e T, Miyaura N, Suzuki A. J. Org. Chem. 1993;58:2201. [Google Scholar]
- (168).Huth A, Beetz I, Schumann I. Tetrahedron. 1989;45:6679. [Google Scholar]
- (169).Yuan K, Scott WJ. Tetrahedron Lett. 1991;32:189. [Google Scholar]
- (170).Park K, Yuan K, Scott WJ. J. Org. Chem. 1993;58:4866. [Google Scholar]
- (171).Saito S, Sakai M, Miyaura N. Tetrahedron Lett. 1996;37:2993. [Google Scholar]
- (172).Miyaura N, Yamada K, Suginome H, Suzuki A. J. Am. Chem. Soc. 1985;107:972. [Google Scholar]
- (173).Saito S, Ohtani S, Miyaura N. J. Org. Chem. 1997;62:8024. doi: 10.1021/jo9707848. [DOI] [PubMed] [Google Scholar]
- (174).Foá M, Casser L. J. Chem. Soc., Dalton Trans. 1975:2572. [Google Scholar]
- (175).Portnoy M, Milstein D. Organometallics. 1993;12:1665. [Google Scholar]
- (176).Ueda M, Saitoh A, Oh-Tani S, Miyaura N. Tetrahedron. 1998;54:13079. [Google Scholar]
- (177).Galland JC, Savignac M, Genet JP. Tetrahedron Lett. 1999;40:2323. [Google Scholar]
- (178).Kobayashi Y, Mizojiri R. Tetrahedron Lett. 1996;37:8531. [Google Scholar]
- (179).Kobayashi Y, William AD, Mizojiri R. J. Organomet. Chem. 2002;653:91. [Google Scholar]
- (180).Indolese AF. Tetrahedron Lett. 1997;38:3513. [Google Scholar]
- (181).Inada K, Miyaura N. Tetrahedron. 2000;56:8657. [Google Scholar]
- (182).Zim D, Monteiro AL. Org. Lett. 2001;3:3049. doi: 10.1021/ol016526l. [DOI] [PubMed] [Google Scholar]
- (183).Zim D, Lando VR, Dupont J, Monteiro AL. Tetrahedron Lett. 2002;43:4009. [Google Scholar]
- (184).Leadbeater ME, Resouly SM. Tetrahedron. 1999:11889. [Google Scholar]
- (185).Percec V, Golding GM, Smidrkal J, Weichold O. J. Org. Chem. 2004;69:3447. doi: 10.1021/jo049940i. [DOI] [PubMed] [Google Scholar]
- (186).Fan X-H, Yang L-M. Eur. J. Org. Chem. 2010:2457. [Google Scholar]
- (187).Zhou YB, Xi ZX, Chen WZ, Wang DQ. Organometallics. 2008;27:5911. [Google Scholar]
- (188).Kuroda JI, Inamoto K, Hiroya K, Doi T. Eur. J. Org. Chem. 2009:2251. [Google Scholar]
- (189).Inamoto K, Kuroda J, Kwon E, Hiroya K, Doi T. J. Organomet. Chem. 2009;694:389. [Google Scholar]
- (190).Lipshutz BH, Sclafani JA, Blomgren PA. Tetrahedron. 2000:2139. [Google Scholar]
- (191).Lipshutz BH, Chrisman W, Tasler S, Spliethoff B, Tesche B. J. Org. Chem. 2003;68:1177. doi: 10.1021/jo020296m. [DOI] [PubMed] [Google Scholar]
- (192).Tang Z-Y, Hu Q-S. J. Am. Chem. Soc. 2004;126:3058. doi: 10.1021/ja038752r. [DOI] [PubMed] [Google Scholar]
- (193).Tang Z-Y, Hu Q-S. J. Org. Chem. 2006;71:2167. doi: 10.1021/jo052369i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (194).Tang Z-Y, Spinella S, Hu Q-S. Tetrahedron Lett. 2006:2427. [Google Scholar]
- (195).Wilson DA, Wilson CJ, Rosen BM, Percec V. Org. Lett. 2008;10:4879. doi: 10.1021/ol801972f. [DOI] [PubMed] [Google Scholar]
- (196).Zhou J, Fu GC. J. Am. Chem. Soc. 2004;126:1340. doi: 10.1021/ja039889k. [DOI] [PubMed] [Google Scholar]
- (197).Liu J, Robins MJ. Org. Lett. 2005;7:1149. doi: 10.1021/ol050063s. [DOI] [PubMed] [Google Scholar]
- (198).You E, Li P, Wang L. Synthesis. 2006;9:1465. [Google Scholar]
- (199).Molander GA, Beaumard F. Org. Lett. 2010;12:4022. doi: 10.1021/ol101592r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (200).Tamao K, Sumitani K, Kumada M. J. Am. Chem. Soc. 1972;94:4374. [Google Scholar]
- (201).Phapale VB, Cardenas DJ. Chem. Soc. Rev. 2009;38:1598. doi: 10.1039/b805648j. [DOI] [PubMed] [Google Scholar]
- (202).Netherton MR, Fu GC. Adv. Synth. Catal. 2004;346:1525. [Google Scholar]
- (203).Giovannini R, Studemann T, Dussin G, Knochel P. Angew. Chem. Int. Ed. 1998;37:2387. doi: 10.1002/(SICI)1521-3773(19980918)37:17<2387::AID-ANIE2387>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- (204).Piber M, Jensen AE, Rottlander M, Knochel P. Org. Lett. 1999;1:1323. [Google Scholar]
- (205).Zhou JR, Fu GC. J. Am. Chem. Soc. 2003;125:14726. doi: 10.1021/ja0389366. [DOI] [PubMed] [Google Scholar]
- (206).Fisher C, Fu GC. J. Am. Chem. Soc. 2005;127:4594. doi: 10.1021/ja0506509. [DOI] [PubMed] [Google Scholar]
- (207).Son S, Fu GC. J. Am. Chem. Soc. 2008;130:2756. doi: 10.1021/ja800103z. [DOI] [PubMed] [Google Scholar]
- (208).For a recent review on Negishi alkyl-alkyl couplings, see: Fu GC. Acc. Chem. Res. 2008;41:1555. doi: 10.1021/ar800148f.
- (209).Stanforth SP. Tetrahedron. 1998;54:263. [Google Scholar]
- (210).Diederich F, Stang P, editors. Metal-Catalyzed Cross-Coupling Reactions. Wiley-VCH; Weinheim: 1998. [Google Scholar]
- (211).Knochel P, Perea JJA, Jones P. Tetrahedron. 1998;54:8275. [Google Scholar]
- (212).Leadbeater NE. J. Org. Chem. 2001;66:7539. doi: 10.1021/jo0158377. [DOI] [PubMed] [Google Scholar]
- (213).Miller JA, Farrell RP. Tetrahedron Lett. 1998;39:6441. [Google Scholar]
- (214).Quesnelle CA, Familoni OB, Snieckus V. Synlett. 1994:349. [Google Scholar]
- (215).Koch K, Chambers RJ, Biggers MS. Synlett. 1994:347. [Google Scholar]
- (216).Gavryushin A, Kofink C, Manolikakes G, Knochel P. Org. Lett. 2005;7:4871. doi: 10.1021/ol051615+. [DOI] [PubMed] [Google Scholar]
- (217).Gavryushin A, Kofink C, Manolikakes G, Knochel P. Tetrahedron. 2006;62:7521. [Google Scholar]
- (218).Manolikakes G, Dong ZB, Mayr H, Li JS, Knochel P. Chem. Eur. J. 2009;15:1324. doi: 10.1002/chem.200802349. [DOI] [PubMed] [Google Scholar]
- (219).Manolikakes G, Hernandez CM, Schade MA, Metzger A, Knochel P. J. Org. Chem. 2008;73:8422. doi: 10.1021/jo8015852. [DOI] [PubMed] [Google Scholar]
- (220).Negishi E, Luo FT, Frisbee R, Matsushita H. Heterocycles. 1982;18:117. [Google Scholar]
- (221).Pelter A, Rowlands M, Clements G. Synthesis. 1987:51. [Google Scholar]
- (222).Campbell JB, Firor JW, Davenport TW. Synth. Comm. 1989;19:2265. [Google Scholar]
- (223).Arcadi A, Burini A, Cacchi S, Delmastro M, Marinelli F, Pietroni B. Synlett. 1990:47. [Google Scholar]
- (224).Hall JH, Chien JY, Kauffman JM, Litak PT, Adams JK, Henry RA, Hollins RAJ. Heterocyclic Chem. 1992;29:1245. [Google Scholar]
- (225).Roth GP, Fuller CE. J. Org. Chem. 1991;56:3493. [Google Scholar]
- (226).Stevenson TM, Prasad ASB, Citineni JR, Knochel P. Tetrahedron Lett. 1996;37:8375. [Google Scholar]
- (227).Sakamoto T, Kondo Y, Takazawa N, Yamanaka H. Tetrahedron Lett. 1993;34:5955. [Google Scholar]
- (228).Sakamoto T, Kondo Y, Takazawa N, Yamanaka H. Heterocycles. 1993;36:941. [Google Scholar]
- (229).Sakamoto T, Kondo Y, Takazawa N, Yamanaka H. J. Chem. Soc., Perkin Trans 1. 1996:1927. [Google Scholar]
- (230).Amat M, Hadida S, Bosch J. Tetrahedron Lett. 1993;34:5005. [Google Scholar]
- (231).Amat M, Hadida S, Sathyanarayana S, Bosch J. Tetrahedron Lett. 1996;37:3071. [Google Scholar]
- (232).Folli U, Iarossi D, Montorsi M, Mucci A, Schenetti L. J. Chem. Soc. Perkin Trans. 1. 1995:537. [Google Scholar]
- (233).Gosmini C, Lasry S, Nedelec JY, Perichon J. Tetrahedron. 1998;54:1289. [Google Scholar]
- (234).Hadei N, Kantchev EAB, O’Brien CJ, Organ MG. Org. Lett. 2005;7:3805. doi: 10.1021/ol0514909. [DOI] [PubMed] [Google Scholar]
- (235).Hadei N, Kantchev EAB, O’Brien CJ, Organ MG. J. Org. Chem. 2005;70:8503. doi: 10.1021/jo051304c. [DOI] [PubMed] [Google Scholar]
- (236).Sase S, Jaric M, Metzger A, Malakhov V, Knochel P. J. Org. Chem. 2008;73:7380. doi: 10.1021/jo801063c. [DOI] [PubMed] [Google Scholar]
- (237).Zhang X, Liu B, Liu A, Xie W, Chen W. Organometallics. 2009;28:1336. [Google Scholar]
- (238).Xi ZX, Zhou YB, Chen WZ. J. Org. Chem. 2008;73:8497. doi: 10.1021/jo8018686. [DOI] [PubMed] [Google Scholar]
- (239).Marion N, Nolan SP. Acc. Chem. Res. 2008;41:1440. doi: 10.1021/ar800020y. [DOI] [PubMed] [Google Scholar]
- (240).Diez-Gonzáles S, Marion N, Nolan SP. Chem. Rev. 2009;109:3612. doi: 10.1021/cr900074m. [DOI] [PubMed] [Google Scholar]
- (241).Organ MG, Avola S, Dubovyk I, Hadei N, Kantchev EAB, O’Brien CJ, Valente C. Chem. Eur. J. 2006;12:4749. doi: 10.1002/chem.200600206. [DOI] [PubMed] [Google Scholar]
- (242).Devasagayaraj A, Studemann T, Knochel P. Angew. Chem. Int. Ed. 1995;34:2723. [Google Scholar]
- (243).Giovannini R, Knochel P. J. Am. Chem. Soc. 1998;120:11186. [Google Scholar]
- (244).Melzig L, Gavryushin A, Knochel P. Org. Lett. 2007;9:5529. doi: 10.1021/ol702499h. [DOI] [PubMed] [Google Scholar]
- (245).Jensen AE, Dohle W, Knochel P. Tetrahedron. 2000;56:4197. [Google Scholar]
- (246).Phapale VB, Guisan-Ceinos M, Bunuel E, Cardenas DJ. Chem. Eur. J. 2009;15:12681. doi: 10.1002/chem.200901913. [DOI] [PubMed] [Google Scholar]
- (247).Huo S. Org. Lett. 2003;5:423. doi: 10.1021/ol0272693. [DOI] [PubMed] [Google Scholar]
- (248).Zhu L, Wehmeyer RM, Rieke RD. J. Org. Chem. 1991;56:1445. [Google Scholar]
- (249).Guijarro A, Rosenberg DM, Rieke RD. J. Am. Chem. Soc. 1999;121:4155. [Google Scholar]
- (250).Jubert C, Knochel P. J. Org. Chem. 1992;57:5425. [Google Scholar]
- (251).Denmark SE, Butler CR. Chem. Commun. 2009:20. doi: 10.1039/b809676g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (252).Wang Q, Chen C. Tetrahedron Lett. 2008;49:2916. [Google Scholar]
- (253).Gong H, Gagne MR. J. Am. Chem. Soc. 2008;130:12177. doi: 10.1021/ja8041564. [DOI] [PubMed] [Google Scholar]
- (254).Jones GD, Martin JL, McFarland C, Allen OR, Hall RE, Haley AD, Brandon RJ, Kanavolova T, Desrochers PJ, Pulay P, Vicic DA. J. Am. Chem. Soc. 2006;128:13175. doi: 10.1021/ja063334i. [DOI] [PubMed] [Google Scholar]
- (255).Phapale VB, Buñuel E, García-Iglesias M, Cárdenas DJ. Angew. Chem. Int. Ed. 2007;46:8790. doi: 10.1002/anie.200702528. [DOI] [PubMed] [Google Scholar]
- (256).Lin X, Phillips DL. J. Org. Chem. 2008;73:3680. doi: 10.1021/jo702497p. [DOI] [PubMed] [Google Scholar]
- (257).Corriu KJP, Masse JP. J. Chem. Soc., Chem. Commun. 1972:144. [Google Scholar]
- (258).Tamao K, Sumitani K, Kiso Y, Zembayashi M, Fujioka A, Kodama S, Nakajima I, Minato A, Kumada M. Bull. Chem. Soc. Jpn. 1976;49:1958. [Google Scholar]
- (259).Kumada M. Pure Appl. Chem. 1980;52:669. [Google Scholar]
- (260).Hayashi T, Konishi M, Kumada M. Tetrahedron Lett. 1979:1871. [Google Scholar]
- (261).Lipshutz BH, Tomioka T, Blomgren PA, Sclafani JA. Inorg. Chim. Acta. 1999:164. [Google Scholar]
- (262).Tasler S, Lipshutz BH. J. Org. Chem. 2003;68:1190. doi: 10.1021/jo020297e. [DOI] [PubMed] [Google Scholar]
- (263).Hayashi T, Niizuma S, Kamikawa T, Suzuki N, Uozumi Y. J. Am. Chem. Soc. 1995;117:9101. [Google Scholar]
- (264).Hayashi T, Hayashizaki K, Kiyoi T, Ito Y. J. Am. Chem. Soc. 1988;110:8153. [Google Scholar]
- (265).Percec V, Bae JY, Hill DH. J. Org. Chem. 1995;60:6895. [Google Scholar]
- (266).Styring P, Grindon C, Fisher CM. Catal. Lett. 2001;77:219. [Google Scholar]
- (267).Park SY, Kang M, Yie JE, Kim JM, Lee I-M. Tetrahedron Lett. 2005;46:2849. [Google Scholar]
- (268).Xi H, Liu B, Chen W. J. Org. Chem. 2008;73:3954. doi: 10.1021/jo800197u. [DOI] [PubMed] [Google Scholar]
- (269).Chen XF, Wang L, Liu J. Synthesis-Stuttgart. 2009:2408. [Google Scholar]
- (270).Bohm VPW, Weskamp T, Gstottmayr CWK, Herrmann WA. Angew. Chem. Int. Ed. 2000;39:1602. doi: 10.1002/(sici)1521-3773(20000502)39:9<1602::aid-anie1602>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- (271).Wolf J, Labande A, Daran JC, Poli R. J. Org. Chem. 2006;691:433. [Google Scholar]
- (272).Wolf J, Labande A, Natella M, Daran JC, Poli R. J. Mol. Catal. A.: Chem. 2006;259:205. [Google Scholar]
- (273).Inamoto K, Kuroda J, Sakamoto T, Hiroya K. Synthesis. 2007:2853. [Google Scholar]
- (274).Matsubara K, Ueno K, Shibata Y. Organometallics. 2006;25:3422. [Google Scholar]
- (275).Berding J, Lutz M, Spek AL, Bouwman E. Organometallics. 2009;28:1845. [Google Scholar]
- (276).Berding J, van Dijkman TF, Lutz M, Spek AL, Bouwman E. Dalton Trans. 2009:6948. doi: 10.1039/b905036a. [DOI] [PubMed] [Google Scholar]
- (277).Bohm VPW, Gstottmayr CWK, Weskamp T, Herrmann WA. Angew. Chem. Int. Ed. 2001;40:3387. doi: 10.1002/1521-3773(20010917)40:18<3387::aid-anie3387>3.3.co;2-y. [DOI] [PubMed] [Google Scholar]
- (278).Kremzow D, Seidel G, Lehmann CW, Furstner A. Chem. Eur. J. 2005;11:1833. doi: 10.1002/chem.200400928. [DOI] [PubMed] [Google Scholar]
- (279).Yoshikai N, Matsuda H, Nakamura E. J. Am. Chem. Soc. 2009;131:9590. doi: 10.1021/ja903091g. [DOI] [PubMed] [Google Scholar]
- (280).Yoshikai N, Mashima H, Nakamura E. J. Am. Chem. Soc. 2005;127:17978. doi: 10.1021/ja056327n. [DOI] [PubMed] [Google Scholar]
- (281).Nakamura E, Mori S. Angew. Chem. Int. Ed. 2000;39:3750. doi: 10.1002/1521-3773(20001103)39:21<3750::aid-anie3750>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- (282).Yoshikai N, Nakamura E. J. Am. Chem. Soc. 2004;126:12265. doi: 10.1021/ja046616w. [DOI] [PubMed] [Google Scholar]
- (283).Yoshikai N, Iida R, Nakamura E. Adv. Synth. Catal. 2008;350:1063. [Google Scholar]
- (284).Yoshikai N, Matsuda H, Nakamura E. J. Am. Chem. Soc. 2008;130:15258. doi: 10.1021/ja807000a. [DOI] [PubMed] [Google Scholar]
- (285).Miyazaki S, Koga Y, Matsumoto T, Matsubara K. Chem. Commun. 2010;46:1932. doi: 10.1039/b924716e. [DOI] [PubMed] [Google Scholar]
- (286).Yu D-G, Li B-J, Zheng S-F, Guan B-T, Wang B-Q, Shi Z-J. Angew. Chem. Int. Ed. 2010;49:4566. doi: 10.1002/anie.200907359. [DOI] [PubMed] [Google Scholar]
- (287).Miller JA. Tetrahedron Lett. 2001;42:6991. [Google Scholar]
- (288).Miller JA, Dankwardt JW. Tetrahedron Lett. 2003;44:1907. [Google Scholar]
- (289).Penney JM, Miller JA, Dankwardt JW. Tetrahedron Lett. 2004;45:4989. [Google Scholar]
- (290).Guan B-T, Lu X-Y, Zheng Y, Yu D-G, Wu T, Li K-L, Li B-J, Shi Z-J. Org. Lett. 2010;12:396. doi: 10.1021/ol9028308. [DOI] [PubMed] [Google Scholar]
- (291).Terao J, Watanabe H, Ikumi A, Kuniyasu H, Kambe N. J. Am. Chem. Soc. 2002;124:4222. doi: 10.1021/ja025828v. [DOI] [PubMed] [Google Scholar]
- (292).Singh SP, Terao J, Kambe N. Tetrahedron Lett. 2009;50:5644. [Google Scholar]
- (293).Vechorkin O, Hu X. Angew. Chem. Int. Ed. 2009;48:2937. doi: 10.1002/anie.200806138. [DOI] [PubMed] [Google Scholar]
- (294).McCullough RD, Lowe RD. J. Chem. Soc. Chem. Commun. 1992:70. [Google Scholar]
- (295).Chen TA, Rieke RD. J. Am. Chem. Soc. 1992;114:10087. [Google Scholar]
- (296).Yokoyama A, Miyakoshi R, Yokozawa T. Macromolecules. 2004;37:1169. [Google Scholar]
- (297).Yokozawa T, Yokoyama A. Chem. Rec. 2005;5:47. doi: 10.1002/tcr.20032. [DOI] [PubMed] [Google Scholar]
- (298).Miyakoshi R, Yokoyama A, Yokozawa T. J. Polym. Sci. Part A: Polym. Chem. 2007;46:753. [Google Scholar]
- (299).Yokozawa T, Yokoyama A. Chem. Rev. 2009;109:5595. doi: 10.1021/cr900041c. [DOI] [PubMed] [Google Scholar]
- (300).Sheina EE, Liu J, Iovu MC, Laird DW, McCullough RD. Macromolecules. 2004;37:3526. [Google Scholar]
- (301).Lanni EL, McNeil AJ. J. Am. Chem. Soc. 2009;131:16575. doi: 10.1021/ja904197q. [DOI] [PubMed] [Google Scholar]
- (302).Miyakoshi R, Shimono K, Yokoyama A, Yokozawa T. J. Am. Chem. Soc. 2006;128:16012. doi: 10.1021/ja067107s. [DOI] [PubMed] [Google Scholar]
- (303).Huang L, Wu S, Qu Y, Geng Y, Wang F. Macromolecules. 2008;41:8944. [Google Scholar]
- (304).Boyd SD, Jen AK-Y, Luscombe CK. Macromolecules. 2009;42:9387. [Google Scholar]
- (305).Wang L, Li P, Zhang Y. Chem. Commun. 2004:514. doi: 10.1039/b314246a. [DOI] [PubMed] [Google Scholar]
- (306).Wang M, Li P, Wang L. Synth. Commun. 2004;34:2803. [Google Scholar]
- (307).Uk Son S, Jang Y, Park J, Na BH, Park HM, Yun HJ, Lee J, Hyeon T. J. Am. Chem. Soc. 2004;126:5026. doi: 10.1021/ja039757r. [DOI] [PubMed] [Google Scholar]
- (308).So CM, Lee HW, Lau CP, Kwong FY. Org. Lett. 2009;11:317. doi: 10.1021/ol802493z. [DOI] [PubMed] [Google Scholar]
- (309).Zhang L, Qing J, Yang P, Wu J. Org. Lett. 2008;10:4971. doi: 10.1021/ol802049t. [DOI] [PubMed] [Google Scholar]
- (310).Kang SK, Ryu H-C, Lee S-W. J. Chem. Soc., Perkin Trans. 1999;1:2661. [Google Scholar]
- (311).Zhang L, Luo Y, Wu J. Synlett. 2010:1845. [Google Scholar]
- (312).Togni A, Grützmacher H, editors. Catalytic Heterofunctionalization. Wiley-VCH; New York: 2001. [Google Scholar]
- (313).Molander GA, Canturk B. Angew. Chem. Int. Ed. 2009;48:9240. doi: 10.1002/anie.200904306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (314).Darses S, Genet J-P. Chem. Rev. 2009;108:288. doi: 10.1021/cr0509758. [DOI] [PubMed] [Google Scholar]
- (315).Doucet H. Eur. J. Org. Chem. 2008;12:2013. [Google Scholar]
- (316).Ishihara K, Yamamoto H. Eur. J. Org. Chem. 1999:527. [Google Scholar]
- (317).James TD, Shinkai S. Host-Guest Chemistry. Vol. 218. Springer-Verlag Berlin; Berlin: 2002. [Google Scholar]
- (318).Niu WJ, O’Sullivan C, Rambo BM, Smith MD, Lavigne JJ. Chem. Commun. 2005:4342. doi: 10.1039/b504634c. [DOI] [PubMed] [Google Scholar]
- (319).Hall DG. Boronic acids: preparation and applications in organic synthesis and medicine. Wiley-VCH Verlag GmbH; Weinheim: 2005. [Google Scholar]
- (320).William S, Johnson JR. J. Am. Chem. Soc. 1931;53:711. [Google Scholar]
- (321).Bean FR, Johnson JR. J. Am. Chem. Soc. 1932;54:4415. [Google Scholar]
- (322).Li WJ, Nelson DP, Jensen MS, Hoerrner RS, Cai DW, Larsen RD, Reider PJ. J. Org. Chem. 2002;67:5394. doi: 10.1021/jo025792p. [DOI] [PubMed] [Google Scholar]
- (323).Waltz KM, He X, Muhoro C, Hartwig JF. J. Am. Chem. Soc. 1995;117:11357. [Google Scholar]
- (324).Chen HY, Hartwig JF. Angew. Chem. Int. Ed. 1999;38:3391. doi: 10.1002/(sici)1521-3773(19991115)38:22<3391::aid-anie3391>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- (325).Iverson CN, Smith MR. J. Am. Chem. Soc. 1999;121:7696. [Google Scholar]
- (326).Chen HY, Schlecht S, Semple TC, Hartwig JF. Science. 2000;287:1995. doi: 10.1126/science.287.5460.1995. [DOI] [PubMed] [Google Scholar]
- (327).Shimada S, Batsanov AS, Howard JAK, Marder TB. Angew. Chem. Int. Ed. 2001;40:2168. doi: 10.1002/1521-3773(20010601)40:11<2168::AID-ANIE2168>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- (328).Cho JY, Tse MK, Holmes D, Maleczka RE, Smith MR. Science. 2002;295:305. doi: 10.1126/science.1067074. [DOI] [PubMed] [Google Scholar]
- (329).Ishiyama T, Nobuta Y, Hartwig JF, Miyaura N. Chem. Commun. 2003:2924. doi: 10.1039/b311103b. [DOI] [PubMed] [Google Scholar]
- (330).Tzschucke CC, Murphy JM, Hartwig JF. Org. Lett. 2007;9:761. doi: 10.1021/ol062902w. [DOI] [PubMed] [Google Scholar]
- (331).Boebel TA, Hartwig JF. J. Am. Chem. Soc. 2008;130:7534. doi: 10.1021/ja8015878. [DOI] [PubMed] [Google Scholar]
- (332).Robbins DW, Boebel TA, Hartwig JF. J. Am. Chem. Soc. 2010;132:4068. doi: 10.1021/ja1006405. [DOI] [PubMed] [Google Scholar]
- (333).Kawamorita S, Ohmiya H, Hara K, Fukuoka A, Sawamura M. J. Am. Chem. Soc. 2009;131:5058. doi: 10.1021/ja9008419. [DOI] [PubMed] [Google Scholar]
- (334).Ishiyama T, Isou H, Kikuchi T, Miyaura N. Chem. Commun. 2010;46:159. doi: 10.1039/b910298a. [DOI] [PubMed] [Google Scholar]
- (335).Ishiyama T, Murata M, Miyaura N. J. Org. Chem. 1995;60:7508. [Google Scholar]
- (336).Quasdorf KW, Tian X, Garg NK. J. Am. Chem. Soc. 2008;130:14422. doi: 10.1021/ja806244b. [DOI] [PubMed] [Google Scholar]
- (337).Morgan AB, Jurs JL, Tour JM. J. Appl. Polym. Sci. 2000;76:1257. [Google Scholar]
- (338).Murata M, Oyama T, Watanabe S, Masuda Y. J. Org. Chem. 2000;65:164. doi: 10.1021/jo991337q. [DOI] [PubMed] [Google Scholar]
- (339).Melaimi M, Thoumazet C, Ricard L, Le Floch P. J. Organomet. Chem. 2004;689:2988. [Google Scholar]
- (340).Phenyl trifluoromethanesulfonate is $89.5/5 mL, while phenyl tosylate is $49.70/25 g.
- (341).Rosen BM, Huang C, Percec V. Org. Lett. 2008;10:2597. doi: 10.1021/ol800832n. [DOI] [PubMed] [Google Scholar]
- (342).Tucker CE, Davidson J, Knochel P. J. Org. Chem. 1992;57:3482. [Google Scholar]
- (343).Pinacol: $0.50/g; neopentylglycol $0.02/g (Aldrich).
- (344).Rosen BM, Wilson DA, Wilson CJ, Peterca M, Won BC, Huang CH, Lipski LR, Zeng XB, Ungar G, Heiney PA, Percec V. J. Am. Chem. Soc. 2009;131:17500. doi: 10.1021/ja907882n. [DOI] [PubMed] [Google Scholar]
- (345).Rosen BM, Peterca M, Huang C, Zeng X, Ungar G, Percec V. Angew. Chem. Int. Ed. 2010;49:7002. doi: 10.1002/anie.201002514. [DOI] [PubMed] [Google Scholar]
- (346).Ishiyama T, Miyaura N. J. Organomet. Chem. 2000;611:392. [Google Scholar]
- (347).Moldoveanu C, Wilson DA, Wilson CJ, Corcoran P, Rosen BM, Percec V. Org. Lett. 2009;11:4974. doi: 10.1021/ol902155e. [DOI] [PubMed] [Google Scholar]
- (348).Moldoveanu C, Wilson DA, Wilson CJ, Leowanawat P, Resmerita A-M, Liu C, Rosen BM, Percec V. J. Org. Chem. 2010;75:5438. doi: 10.1021/jo101023t. [DOI] [PubMed] [Google Scholar]
- (349).Wilson DA, Wilson CJ, Moldoveanu C, Resmerita AM, Corcoran P, Hoang LM, Rosen BM, Percec V. J. Am. Chem. Soc. 2010;132:1800. doi: 10.1021/ja910808x. [DOI] [PubMed] [Google Scholar]
- (350).Zhang LA, Meng TH, Wu J. J. Org. Chem. 2007;72:9346. doi: 10.1021/jo7019064. [DOI] [PubMed] [Google Scholar]
- (351).He A, Falck JR. J. Am. Chem. Soc. 2010;132:2524. doi: 10.1021/ja910582n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (352).Liu G, Du QL, Xie JJ, Zhang KL, Tao XC. Chin. J. Catal. 2006;27:1051. [Google Scholar]
- (353).Zhang X, Zhao HT, Wang JH. J. Nanosci. Nanotech. 2010;10:5153. doi: 10.1166/jnn.2010.2409. [DOI] [PubMed] [Google Scholar]
- (354).Kirai N, Yamamoto Y. Eur. J. Org. Chem. 2009;12:1864. [Google Scholar]
- (355).Mitsudo K, Shiraga T, Tanaka H. Tetrahedron Lett. 2008;49:6593. [Google Scholar]
- (356).Yamamoto Y. Synlett. 2007:1913. [Google Scholar]
- (357).Prastaro A, Ceci P, Chiancone E, Boffi A, Fabrizi G, Cacchi S. Tetrahedron Lett. 2010;51:2550. [Google Scholar]
- (358).Wu N, Li XN, Xu X, Wang YM, Xu YY, Chen X. Lett. Org. Chem. 2010;7:11. [Google Scholar]
- (359).Mitsudo K, Shiraga T, Kagen D, Shi DQ, Becker JY, Tanaka H. Tetrahedron. 2009;65:8384. [Google Scholar]
- (360).Jin Z, Guo SX, Gu XP, Qiu LL, Song HB, Fang JX. Adv. Synth. Cat. 2009;351:1575. [Google Scholar]
- (361).Amatore C, Cammoun C, Jutland A. Eur. J. Org. Chem. 2008:4567. [Google Scholar]
- (362).Yadav JS, Gayathri KU, Ather H, Rehman HU, Prasad AR. J. Mol. Cat. A. – Chem. 2007;271:25. [Google Scholar]
- (363).Punna S, Diaz DD, Finn MG. Synlett. 2004:2351. [Google Scholar]
- (364).Koza DJ, Carita E. Synthesis. 2002:2183. [Google Scholar]
- (365).Yue Y, Yamamoto H, Yamame M. Synlett. 2009:2831. [Google Scholar]
- (366).Vogler T, Studer A. Adv. Synth. Cat. 2008;350:1963. [Google Scholar]
- (367).Negwer M. In: Organic-chemical drugs and their synonyms: (an international survey) Federal Republic of Germany; Akademie Verlag, editor. VCH Publishers; New York, NY: 1994. 7th rev. and enl. ed. Berlin. [Google Scholar]
- (368).Law KY. Chem. Rev. 1993;93:449. [Google Scholar]
- (369).Wang HL, MacDiarmid AG, Wang YZ, Gebler DD, Epstein AJ. Synth. Met. 1996;78:33. [Google Scholar]
- (370).Wang YZ, Gebler DD, Fu DK, Swager TM, MacDiarmid AG, Epstein AJ. Synth. Met. 1997;85:1179. [Google Scholar]
- (371).Gebler DD, Wang YZ, Fu DK, Swager M, Epstein AJ. J. Chem. Phys. 1998;108:7842. [Google Scholar]
- (372).Ono N. The nitro group in organic synthesis. Wiley-VCH; New York: 2001. [Google Scholar]
- (373).Kienle M, Dubbaka SR, Brade K, Knochel P. Eur. J. Org. Chem. 2007:4166. [Google Scholar]
- (374).Lindley J. Tetrahedron. 1984;40:1433. [Google Scholar]
- (375).Hartwig JF. Angew. Chem. Int. Ed. 1998;37:2047. [Google Scholar]
- (376).Fors BP, Watson DA, Biscoe MR, Buchwald SL. J. Am. Chem. Soc. 2008;130:13552. doi: 10.1021/ja8055358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (377).Cramer R, Coulson DR. J. Org. Chem. 1975;40:2267. [Google Scholar]
- (378).Christau HJ, Desmurs JR. Ind. Chem. Libr. 1995;7:240. [Google Scholar]
- (379).Wolfe JP, Buchwald SL. J. Am. Chem. Soc. 1997;119:6054. [Google Scholar]
- (380).Brenner E, Fort Y. Tetrahedron Lett. 1998;39:5359. [Google Scholar]
- (381).Brenner E, Schneider R, Fort Y. Tetrahedron. 1999;55:12829. [Google Scholar]
- (382).Desmarets C, Schneider R, Fort Y. Tetrahedron Lett. 2000;41:2875. [Google Scholar]
- (383).Desmarets C, Schneider R, Fort Y. Tetrahedron. 2001;57:7657. [Google Scholar]
- (384).Gradel B, Brenner E, Schneider RL, Fort Y. Tetrahedron Lett. 2001;42:5689. [Google Scholar]
- (385).Desmarets C, Schneider R, Fort Y. J. Org. Chem. 2002;67:3029. doi: 10.1021/jo016352l. [DOI] [PubMed] [Google Scholar]
- (386).Omar-Amrani R, Thomas A, Brenner E, Schneider R, Fort Y. Org. Lett. 2003;5:2311. doi: 10.1021/ol034659w. [DOI] [PubMed] [Google Scholar]
- (387).Kuhl S, Fort Y, Schneider R. J. Organomet. Chem. 2005;690:6169. [Google Scholar]
- (388).Lipshutz BH, Ueda H. Angew. Chem. Int. Ed. 2000;39:4492. [PubMed] [Google Scholar]
- (389).Tasler S, Lipshutz BH. J. Org. Chem. 2003;68:1190. doi: 10.1021/jo020297e. [DOI] [PubMed] [Google Scholar]
- (390).Lipshutz BH, Tasler S, Chrisman W, Spliethoff B, Tesche B. J. Org. Chem. 2003;68:1177. doi: 10.1021/jo020296m. [DOI] [PubMed] [Google Scholar]
- (391).Lipshutz BH, Frieman BA, Lee CT, Lower A, Nihan DM, Taft BR. Chem. Asian J. 2006;1:417. doi: 10.1002/asia.200600031. [DOI] [PubMed] [Google Scholar]
- (392).Mallesham B, Rajesh BM, Reddy PR, Srinivas D, Trehan S. Org. Lett. 2003;5:963. doi: 10.1021/ol026902h. [DOI] [PubMed] [Google Scholar]
- (393).Lipshutz BH, Nihan DM, Vinogradova E, Taft BR, Bogkovic ZV. Org. Lett. 2008;10:4279. doi: 10.1021/ol801676u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (394).Butler T, Swift E, Lipshutz BH. Org. Biomol. Chem. 2008;6:19. doi: 10.1039/b714600k. [DOI] [PubMed] [Google Scholar]
- (395).Chen C, Yang LM. Org. Lett. 2005;7:2209. doi: 10.1021/ol050608i. [DOI] [PubMed] [Google Scholar]
- (396).Chen C, Yang LM. Tetrahedron Lett. 2007;48:2427. [Google Scholar]
- (397).Chen C, Yang LM. J. Org. Chem. 2007;72:6324. doi: 10.1021/jo0709448. [DOI] [PubMed] [Google Scholar]
- (398).Gao CY, Yang LM. J. Org. Chem. 2008;73:1624. doi: 10.1021/jo7022558. [DOI] [PubMed] [Google Scholar]
- (399).Bolm C, Hildebrand JP, Rudolph J. Synthesis. 2000:911. [Google Scholar]
- (400).Sissouma D, Collet SC, Guingant AY. Synlett. 2004:2612. [Google Scholar]
- (401).Evans DA, Wood MR, Trotter BW, Richardson TI, Barrow JC, Katz JL. Angew. Chem. Int. Ed. 1998;37:2700. doi: 10.1002/(SICI)1521-3773(19981016)37:19<2700::AID-ANIE2700>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- (402).Takagi K, Sasaki K, Sakakibara Y. Bull. Chem. Soc. Jpn. 1991;64:1118. [Google Scholar]
- (403).Sasaki K, Kubo T, Sakai M, Kuroda Y. Chem. Lett. 1997:617. [Google Scholar]
- (404).Radivoy G, Alonso F, Yus M. Tetrahedron. 1999;55:14479. [Google Scholar]
- (405).Alonso F, Radivoy G, Yus M. Russ. Chem. Bull. 2003;52:2563. [Google Scholar]
- (406).Alonso F, Yus M. Chem. Soc. Rev. 2004;33:284. doi: 10.1039/b315131j. [DOI] [PubMed] [Google Scholar]
- (407).Kogan V. Tetrahedron Lett. 2006;47:7515. [Google Scholar]
- (408).Lipshutz BH, Frieman BA, Butler T, Kogan V. Angew. Chem. Int. Ed. 2006;45:800. doi: 10.1002/anie.200502887. [DOI] [PubMed] [Google Scholar]
- (409).Sundermeier M, Zapf A, Beller M. Eur. J. Inorg. Chem. 2003:3513. [Google Scholar]
- (410).Cassar L, Ferrara S, Foa M. Advances in Chemistry Series. 1974:252. [Google Scholar]
- (411).Cassar L, Ferrara S, Foa M. Abstr. Pap. J. Am. Chem. Soc. 1973:24. [Google Scholar]
- (412).Sakakibara Y, Yadani N, Ibuki I, Sakai M, Uchino N. Chem. Lett. 1982:1565. [Google Scholar]
- (413).Sakakibara Y, Okuda F, Shimobayashi A, Kirino K, Sakai M, Uchino N, Takagi K. Bull. Chem. Soc. Jpn. 1988;61:1985. [Google Scholar]
- (414).Chambers MRI, Widdowson DA. J. Chem. Soc., Perkin Trans. 1. 1989:1365. [Google Scholar]
- (415).Takagi K, Sakakibara Y. Chem. Lett. 1989:1957. [Google Scholar]
- (416).Sakakibara Y, Ido Y, Sasaki K, Sakai M, Uchino N. Bull. Chem. Soc. Jpn. 1993;66:2776. [Google Scholar]
- (417).Alterman M, Hallberg A. J. Org. Chem. 2000;65:7984. doi: 10.1021/jo0009954. [DOI] [PubMed] [Google Scholar]
- (418).Arvela RK, Leadbeater NE. J. Org. Chem. 2003;68:9122. doi: 10.1021/jo0350561. [DOI] [PubMed] [Google Scholar]
- (419).Zhang A, Neumeyer JL. Org. Lett. 2003;5:201. doi: 10.1021/ol027256p. [DOI] [PubMed] [Google Scholar]
- (420).Schareina T, Zapf A, Beller M. J. Organomet. Chem. 2004;689:4576. [Google Scholar]
- (421).Sengupta S, Leite M, Raslan DS, Quesnelle C, Snieckus V. J. Org. Chem. 1992;15:4066. [Google Scholar]
- (422).Macklin TK, Snieckus V. Org. Lett. 2005;7:2519. doi: 10.1021/ol050393c. [DOI] [PubMed] [Google Scholar]
- (423).Wehn PM, Du Bois J. Org. Lett. 2005;7:4685. doi: 10.1021/ol051896l. [DOI] [PubMed] [Google Scholar]
- (424).Quasdorf KW, Reiner M, Petrova KV, Garg NK. J. Am. Chem. Soc. 2009;131:17748. doi: 10.1021/ja906477r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (425).Wenkert E, Michelotti EL, Swindell CS. J. Am. Chem. Soc. 1979;101:2246. [Google Scholar]
- (426).Wenkert E, Michelotti EL, Swingdell CS, Tingoli M. J. Org. Chem. 1984;49:4894. [Google Scholar]
- (427).Dankwardt JW. Angew. Chem. Int. Ed. 2004;43:2428. doi: 10.1002/anie.200453765. [DOI] [PubMed] [Google Scholar]
- (428).Guan B-T, Xiang S-K, Wu T, Sun Z-P, Wang B-Q, Zhao K-Q, Shi Z-J. Chem. Commun. 2008:1437. doi: 10.1039/b718998b. [DOI] [PubMed] [Google Scholar]
- (429).Guan B-T, Xiang S-K, Wang B-Q, Sun Z-P, Wang Y, Zhao K-Q, Shi Z-J. J. Am. Chem. Soc. 2008;130:3268. doi: 10.1021/ja710944j. [DOI] [PubMed] [Google Scholar]
- (430).Hayashi T, Katsuro Y, Kumada M. Tetrahedron Lett. 1980;21:3915. [Google Scholar]
- (431).Johnstone RAW, McClean WN. Tetrahedron Lett. 1988;29:5553. [Google Scholar]
- (432).Tobisu M, Shimasaki T, Chatani N. Angew. Chem. Int. Ed. 2008;47:4866. doi: 10.1002/anie.200801447. [DOI] [PubMed] [Google Scholar]
- (433).Shimasaki T, Konno Y, Tobisu M, Chatani N. Org. Lett. 2009;11:4890. doi: 10.1021/ol901978e. [DOI] [PubMed] [Google Scholar]
- (434).Tobisu M, Shimasaki T, Chatani N. Chem. Lett. 2009;38:710. [Google Scholar]
- (435).Álvarez-Bercedo P, Martin R. J. Am. Chem. Soc. 2010 doi: 10.1021/ja106943q. DOI: 10.1021/ja106943q. [DOI] [PubMed] [Google Scholar]
- (436).Hayashi T, Fujiwa T, Okamoto Y, Katsuro Y, Kumada M. Synthesis. 1981:1001. [Google Scholar]
- (437).Sahlberg C, Quader A, Claesson A. Tetrahedron Lett. 1983;24:5137. [Google Scholar]
- (438).Sofia A, Karlström E, Itami K, Bäckvall J-E. J. Org. Chem. 1999;64:1745. doi: 10.1021/jo982060h. [DOI] [PubMed] [Google Scholar]
- (439).Nicolaou KC, Shi G-Q, Namoto K, Bernal F. Chem. Commun. 1998:1757. [Google Scholar]
- (440).Hayashi T, Katsuro Y, Okamoto Y, Kumada M. Tetrahedron Lett. 1981;22:4449. [Google Scholar]
- (441).Tanaka M, Chiba K-I, Okita M, Kaneko T, Tagami K, Hibi S, Okamoto Y, Shirota H, Goto M, Obaishi H, Sakurai H, Machida Y, Yamatsu I. J. Med. Chem. 1992;35:4665. doi: 10.1021/jm00103a003. [DOI] [PubMed] [Google Scholar]
- (442).Baker WR, Pratt JK. Tetrahedron. 1993;49:8739. [Google Scholar]
- (443).Iwashima M, Nagaoka H, Kobayashi K, Yamada Y. Tetrahedron Lett. 1992;33:81. [Google Scholar]
- (444).Jiang Y-Y, Li Q, Lu W, Cai J-C. Tetrahedron Lett. 2003;44:2073. [Google Scholar]
- (445).Huang WG, Jiang Y-Y, Li Q, Li JY, Lu W, Cai J-C. Tetrahedron. 2005;61:1863. [Google Scholar]
- (446).Nan Y, Yang Z. Tetrahedron Lett. 1999;40:3321. [Google Scholar]
- (447).Hansen AL, Ebran J-P, Gøgsig TM, Skrydstrup T. Chem. Commun. 2006:4137. doi: 10.1039/b609064h. [DOI] [PubMed] [Google Scholar]
- (448).Hansen AL, Ebran J-P, Gøgsig TM, Skrydstrup T. J. Org. Chem. 2007;72:6464. doi: 10.1021/jo070912k. [DOI] [PubMed] [Google Scholar]
- (449).Zhao Y-L, Li Y, Li Y, Gao L-X, Han F-S. Chem. Eur. J. 2010;16:4991. doi: 10.1002/chem.201000420. [DOI] [PubMed] [Google Scholar]
- (450).Wu J, Yang Z. J. Org. Chem. 2001;66:7875. doi: 10.1021/jo010452+. [DOI] [PubMed] [Google Scholar]
- (451).Guan B-T, Wang Y, Li B-J, Yu D-G, Shi Z-J. J. Am. Chem. Soc. 2008;130:14468. doi: 10.1021/ja8056503. [DOI] [PubMed] [Google Scholar]
- (452).Gooβen LJ, Gooβen K, Stanciu C. Angew. Chem. Int. Ed. 2009;48:3569. doi: 10.1002/anie.200900329. [DOI] [PubMed] [Google Scholar]
- (453).Stone PJ, Dori Z. Inorg. Chim. Acta. 1971;5:434. [Google Scholar]
- (454).Barnett KW. J. Chem. Educ. 1974;51:422. [Google Scholar]
- (455).Quasdorf KW, Garg NK. Encyclopedia of Reagents for Organic Synthesis. 2010 in press. [Google Scholar]
- (456).Li Z, Zhang S-L, Fu Y, Guo Q-X, Liu L. J. Am. Chem. Soc. 2009;131:8815. doi: 10.1021/ja810157e. [DOI] [PubMed] [Google Scholar]
- (457).Sun C-L, Wang Y, Zhou X, Wu Z-H, Li B-J, Guan B-T, Shi Z-J. Chem. Eur. J. 2010;16:5844. doi: 10.1002/chem.200902785. [DOI] [PubMed] [Google Scholar]
- (458).Li B-J, Li Y-Z, Lu X-Y, Liu J, Guan B-T, Shi Z-J. Angew. Chem. Int. Ed. 2008;47:10124. doi: 10.1002/anie.200803814. [DOI] [PubMed] [Google Scholar]
- (459).Shimasaki T, Tobisu M, Chatani N. Angew. Chem. Int. Ed. 2010;49:2929. doi: 10.1002/anie.200907287. [DOI] [PubMed] [Google Scholar]
- (460).Kocienski P, Dixon NJ. Synlett. 1989:52. [Google Scholar]
- (461).Porée F-H, Clavel A, Betzer J-F, Pancrazi A, Ardisson J. Tetrahedron Lett. 2003;44:7553. [Google Scholar]
- (462).Dallaire C, Kolber I, Gingras M. Org. Synth. 2002;78:42. [Google Scholar]
- (463).Antoft-Finch A, Blackburn T, Snieckus V. J. Am. Chem. Soc. 2009;131:17750. doi: 10.1021/ja907700e. [DOI] [PubMed] [Google Scholar]
- (464).Xi L, Li B-J, Wu Z-H, Lu X-Y, Guan B-T, Wang B-Q, Zhao K-Q, Shi Z-J. Org. Lett. 2010;12:884. doi: 10.1021/ol9029534. [DOI] [PubMed] [Google Scholar]
- (465).Amii H, Uneyama K. Chem. Rev. 2009;109:2119. doi: 10.1021/cr800388c. [DOI] [PubMed] [Google Scholar]
- (466).Ramnial T, Taylor SA, Bender ML, Gorodetsky B, Lee PTK, Dickie DA, McCollum BM, Pye CC, Walsby CJ, Clyburne JAC. J. Org. Chem. 2008;73:801. doi: 10.1021/jo701289d. [DOI] [PubMed] [Google Scholar]
- (467).Ackerman L, Born R, Spatz JH, Althammer A, Gshrei CJ. Pure Appl. Chem. 2006;78:209. [Google Scholar]
- (468).Ackerman L, Born R, Spatz JH, Meyer D. Angew. Chem. Int. Ed. 2005;44:7216. doi: 10.1002/anie.200501860. [DOI] [PubMed] [Google Scholar]
- (469).Dankwart JW. J. Organomet. Chem. 2005;690:932. [Google Scholar]
- (470).Mongin F, Mojovic L, Guillamet B, Trécourt F, Quéguiner G. J. Org. Chem. 2002;67:8991. doi: 10.1021/jo026136s. [DOI] [PubMed] [Google Scholar]
- (471).Saeki T, Takashima Y, Tamao K. Synlett. 2005:1771. [Google Scholar]
- (472).Terao J, Watabe H, Kambe N. J. Am. Chem. Soc. 2005;127:3656. doi: 10.1021/ja042565r. [DOI] [PubMed] [Google Scholar]
- (473).Wang J-R, Manabe K. Org. Lett. 2009;11:741. doi: 10.1021/ol802824b. [DOI] [PubMed] [Google Scholar]
- (474).Braun T, Perutz RN, Sladek MI. Chem. Commun. 2001:2254. [PubMed] [Google Scholar]
- (475).Steffen A, Sladek MI, Braun T, Neumann B, Stammler H-G. Organometallics. 2005;24:4057. [Google Scholar]
- (476).Schaub T, Radius U. Chem. Eur. J. 2005;11:5024. doi: 10.1002/chem.200500231. [DOI] [PubMed] [Google Scholar]
- (477).Schaub T, Backes M, Radius U. J. Am. Chem. Soc. 2006;128:15964. doi: 10.1021/ja064068b. [DOI] [PubMed] [Google Scholar]
- (478).Schaub T, Fischer P, Steffen A, Braun T, Radius U, Mix A. J. Am. Chem. Soc. 2008;130:9304. doi: 10.1021/ja074640e. [DOI] [PubMed] [Google Scholar]
- (479).Schaub T, Doring C, Radius U. Dalton Trans. 2007:1993. doi: 10.1039/b702959d. [DOI] [PubMed] [Google Scholar]
- (480).Yu D-G, Yu M, Guan B-T, Li B-J, Zheng Y, Wu Z-H, Shi Z-J. Org. Lett. 2009;11:3374. doi: 10.1021/ol901217m. [DOI] [PubMed] [Google Scholar]
- (481).Hoffmann R, von Ragué Schleyer P, Schaeffer HF. Angew. Chem. Int. Ed. 2008;47:7164. doi: 10.1002/anie.200801206. [DOI] [PubMed] [Google Scholar]
- (482).Tobisu M, Chatani N. Angew. Chem. Int. Ed. 2009;48:3565. doi: 10.1002/anie.200900465. [DOI] [PubMed] [Google Scholar]