

Abstract
c–Src is a membrane-associated tyrosine kinase that can be activated by many types of extracellular signals, and can regulate the function of a variety of cellular protein substrates. We demonstrate that epidermal growth factor (EGF) and β–adrenergic receptors activate c–Src by different mechanisms leading to the phosphorylation of distinct sets of c–Src substrates. In particular, we found that EGF receptors, but not β2–adrenergic receptors, activated c–Src by a Ral-GTPase-dependent mechanism. Also, c–Src activated by EGF treatment or expression of constitutively activated Ral–GTPase led to tyrosine phosphorylation of Stat3 and cortactin, but not Shc or subsequent Erk activation. In contrast, c–Src activated by isoproterenol led to tyrosine phosphorylation of Shc and subsequent Erk activation, but not tyrosine phosphorylation of cortactin or Stat3. These results identify a role for Ral–GTPases in the activation of c–Src by EGF receptors and the coupling of EGF to transcription through Stat3 and the actin cytoskeleton through cortactin. They also show that c–Src kinase activity can be used differently by individual extracellular stimuli, possibly contributing to their ability to generate unique cellular responses.
Keywords: β-adrenergic receptor/c-Src/EGF receptor/Ral/Stat3
Introduction
Many, if not most, signaling molecules can be modulated by a wide variety of extracellular signals and can interact directly with and influence the activity of a large number of cellular target proteins. Understanding how individual signaling inputs can generate unique cellular responses under these conditions is a major challenge in signal transduction research. The non-receptor tyrosine kinase, c–Src, represents a good paradigm for this problem (for a review, see Brown and Cooper,1996), since it is activated in response to diverse stimuli such as tyrosine kinase receptors, G–protein-linked receptors (Luttrell et al., 1997), integrins (Schlaepfer and Hunter, 1997) and estrogen receptors (Migliaccio et al., 1996). In addition, c–Src activity can promote the phosphorylation of a wide variety of proteins, such as cell surface receptors for epidermal growth factor (EGF) (Biscardi et al., 1999a) and glutamate, (Yu and Salter, 1999) adaptor proteins such as Shc, transcription factors such as Stat3, and components of the endocytic machinery such as clathrin (Wilde et al., 1999) and dynamin (Ahn et al., 1999). Many of these regulators and targets of c–Src can co-exist in the same cell, making it plausible that signaling specificity could arise if distinct subsets of c–Src targets were activated by different extracellular signals.
β2-adrenergic receptors activate c–Src by promoting its interaction with the adaptor protein arrestin (Luttrell et al., 1999). Activated c–Src then promotes the phosphorylation of either Shc or GAB1, which leads to the subsequent activation of the Ras/Raf/Erk signaling cascade. This cascade is thought to mediate the growth-promoting effects of stimulated G-protein-linked receptors observed in some cell types.
In contrast, very little is known about the mechanism of c–Src activation by EGF receptors, although more is known about the consequence of c–Src activation by the receptor. c–Src activity is critical for EGF-induced mitogenesis, and one of the important EGF-induced downstream targets of c–Src is the EGF receptor itself (Maa et al., 1995). The phosphorylation sites on the receptor have been identified, but all that is known about their function is that they do not promote the Ras/Raf/Erk signaling cascade (Biscardi et al., 1999a; Tice et al., 1999). c–Src is also thought to contribute to EGF effects on cell shape through its substrates that are components of the cytoskeleton, such as Fak, p130 CAS, cortactin, paxillin and p190 Rho-GTPase-activating protein (GAP) (for a review, see Belsches et al., 1997). c–Src also contributes to EGF effects on gene transcription through its ability to promote the tyrosine phosphorylation of Stat transcription factors (Bromberg et al., 1998; Turkson et al., 1998).
Until now, little evidence existed for the involvement of small GTP-binding proteins in c–Src activation. The possibility that Ral-GTPases might be involved could be inferred only from very indirect evidence. First, the two proteins have similar cellular distributions. The majority of both c–Src and Ral is present on the cytoplasmic face of intracellular vesicles, with a small fraction present in the plasma membrane at any time (Feig and Emkey, 1993; Weernink and Rijksen, 1995). Among intracellular vesicles, both proteins are enriched in endosomes and synaptic vesicles (Kaplan et al., 1992; Linstedt et al., 1992; Bielinski et al., 1993; Onofri et al., 1997; Foster-Barber and Bishop, 1998). Secondly, elevated Ral-specific guanine nucleotide exchange factor (Ral-GEF) activity (Goi et al., 1999) and elevated activity of the c–Src substrate, Stat3 (Ihara et al., 1997), generate the same phenotype in PC12 cells, inhibition of nerve growth factor (NGF)-induced neurite outgrowth.
Two highly similar (85% identity) GTPases, RalA and RalB, constitute a distinct family of Ras-related proteins (Bos, 1997), and as such they cycle between active GTP- and inactive GDP-bound forms. Ral proteins are activated by Ral-GEFs, many of which contain Ras-binding sites. A growing body of evidence supports the idea that activation of Ral-GEFs initiates a downstream signaling pathway distinct from that of Ras (Urano et al., 1996; Wolthuis et al., 1997, 1998b). However, Ras-independent activation pathways to Ral also exist (Hofer et al., 1998; Wolthuis et al., 1998a).
In general, elevated Ral-GEF activity is associated with enhanced cell proliferation (Urano et al., 1996; White et al., 1996; D'Adamo et al., 1997; Wolthuis et al., 1997; Goi et al., 1999). Ral-GEF activity is also involved in insulin regulation of the forkhead transcription factor (Kops et al., 1999). Ral GTPases interact with at least three distinct cellular proteins that could carry out these actions: phospholipase D (PLD) (Jiang et al., 1995; Luo et al., 1998), a GAP for CDC42 and Rac GTPases called RalBP1 (also called RLIP or RIP) (Cantor et al., 1995; Jullien-Flores et al., 1995; Park and Weinberg, 1995) and filamin–α (also called ABP–280), one of a family of actin-cross-linking proteins (Gorlin et al., 1990; Takafuta et al., 1998; Xie et al., 1998). Ral-GTP can generate filapodia-like structures possibly by recruiting filamin into the filapodial cytoskeleton (Ohta et al., 1999).
In this study, we demonstrate that Ral-GTPases play an important role in the activation of c–Src by EGF receptors, but not by β2–adrenergic receptors. We also show that signaling downstream from c–Src can differ, depending upon which extracellular signal is responsible for its activation in cells.
Results
Ral activity modulates Stat tyrosine phosphorylation in PC12 cells
The first suggestion that Ral proteins influence tyrosine kinase signaling came from our recent finding that the effects of elevated Ral-GEF activity in PC12 cells mimic those of activated Stat3 (Ihara et al., 1997; Goi et al., 1999). Therefore, to test whether Ral signaling modulates tyrosine phosphorylation of Stat3, PC12 cells were transiently transfected with a vector encoding hemagglutinin (HA)-tagged Stat3 along with vectors encoding either an active Ral-GEF, Rgr, or a constitutively activated allele of RalA, RalA72L. HA-Stat3 was immunoprecipitated 2 days later and then immunoblotted with anti-phosphotyrosine-specific Stat3 antibodies. Both activators of the Ral signaling pathway increased tyrosine-phosphorylated Stat3 to levels that were comparable to those obtained upon treatment of cells with interleukin–6 (IL–6), a known activator of Stat3 in PC12 cells (Figure 1, top). Immunoblotting for total Stat3 showed that equal levels of the transcription factor were present in all samples (Figure 1, bottom).

Fig. 1. Ral signaling modulates tyrosine phosphorylation of Stat3 in PC12 cells. PC12 cells were transiently transfected with either control vector (C), vector expressing the Ral-specific exchange factor Rgr, a constitutively activated Ral allele, RalA72L, or a dominant-negative Ral allele, Ral28N, along with HA-tagged Stat3. After 48 h, the cells were treated with either buffer or IL–6 (10 ng/ml) for 10 min. The tagged Stat3 was then immunoprecipitated and blotted with anti-phosphotyrosine-specific Stat3 antibodies (p–Stat3). Total HA-Stat3 expression (Stat3) in all of the experiments is shown at the bottom. The data represent the results of at least three independent experiments.
Since these results argued that Ral has the capacity to promote tyrosine phosphorylation of Stat3, we next tested whether tyrosine phosphorylation of Stat3 by IL–6 in PC12 cells is dependent upon Ral-GEF activity. To suppress Ral-GEF's ability to activate Ral in cells, RalA28N, a dominant-negative (dn) form of Ral, which is analogous to Ras17N, was transiently transfected along with HA-Stat3. The cells were then stimulated with IL–6. Expression of dnRal blocked IL–6-induced tyrosine phosphorylation of HA-Stat3 (Figure 1, top). These results showed that Ral signaling participates in cytokine receptor-induced tyrosine phosphorylation of Stat3.
Ral activates c–Src in 293 epithelial cells and 3Y1 fibroblasts
A growing body of evidence supports the idea that tyrosine phosphorylation of Stat proteins can be mediated by the activation of the non-receptor tyrosine kinase c–Src (Yu et al., 1995; Cirri et al., 1997; Bromberg et al., 1998; Chaturvedi et al., 1998; Turkson et al., 1998). To test whether Ral regulates c–Src activity, HEK293 cells were used because they can be transfected at high enough efficiency (∼70%) to measure the effects of Ral on endogenous signaling proteins. The Ral-GEF, Rgr or RalA72L were transiently transfected into HEK293 cells and 48 h later c–Src was immunoprecipitated and assayed in vitro for tyrosine kinase activity on denatured enolase as substrate. Expression of Rgr or constitutively activated RalA enhanced c–Src activity by ∼2–fold, while EGF treatment increased its activity ∼3–fold (Figure 2A). Since only ∼70% of the cells were transfected with Rgr or Ral72L, while all of the cells were exposed to EGF, the three stimuli actually had comparable effects on c–Src activation.


Fig. 2. Ral activates c–Src kinase activity. (A) Top: 293 cells were transiently transfected with control vector (C), or vector encoding Rgr or RalA72L. After 24 h, the cells were serum starved (0.5%) for 12 h and then some cells were treated with EGF for 10 min. The cells were lysed and c–Src was immunoprecipitated. The immunoprecipitates were used in a kinase assay in vitro using denatured enolase as substrate. The labeled reaction was then run on SDS–gels, and exposed to a phosphoimager. The fold increase in enolase phosphorylation compared with control sample (C) is marked on top of the radioactive image. Bottom: data from at least three independent experiments were pooled and plotted with standard errors of the mean indicated. (B) 3Y1(EGFR) cells or 3Y1(EGFR) cells expressing RalA72L were serum starved for 12 h and then some cells were exposed to EGF for 10 min. c–Src was then immunoprecipitated and Src-like activity was assayed in an in vitro kinase assay as described above. Total c–Src levels in cell lysates are depicted at the bottom. The data are representative of two independent experiments. (C) Activated Ras (Ras61L) and Ras effector mutants that preferentially activate Ral-GEFs (Ras12V37G) or Raf (Ras12V35S) were transiently transfected into 293 cells and assayed for c–Src activity as described above. Total c–Src and Ras levels in cell lysates are shown at the bottom. The data are representative of two independent experiments.
3Y1 rat fibroblasts stably expressing the EGF receptor [3Y1(EGFR)] were also used to demonstrate an effect of Ral on c–Src activity. c–Src activity was measured in control cells and in cells stably expressing RalA72L. Serum-starved 3Y1(EGFR) cells expressing RalA72L displayed ∼1.6–fold higher c–Src activity than control cells. This increase was similar to that observed when control cells were stimulated with EGF (Figure 2B, top). Comparable levels of c–Src were detected in all samples (Figure 2B, bottom).
Since Ras is known to promote the GTP-bound state of Ral in cells, we also demonstrated that RasH61L stimulates c–Src activity to a degree similar to that of RalA72L in transiently transfected 293 cells (Figure 2C). RasH12V37G, an effector mutant of activated Ras that preferentially activates Ral-GEFs over other Ras effectors, also stimulated the kinase activity of c–Src in these cells. In contrast, RasH12V35S, which preferentially activates Raf kinase over other Ras effector proteins, did not activate c–Src in this system, demonstrating specificity between Raf and Ral-GEF signaling (Figure 2C). Comparable levels of Ras mutants were expressed in all transfected cells (Figure 2C, bottom)
Ral-GEF activity is required for EGF receptor, but not β2-adrenergic receptor, activation of c–Src
EGF activates c–Src, but the mechanism involved is not known. Therefore, we determined whether Ral activation by Ral-GEFs is required for EGF-induced c–Src activation by transfecting 293 cells transiently with dnRal or control vector. c–Src was then immunoprecipitated and its kinase activity measured in vitro as described above. In control cells, EGF led to an ∼2.7–fold increase in the activation of c–Src. However, in cells expressing dnRal, c–Src activation upon exposure to EGF was reduced by ∼75% (Figure 3A). This value is an underestimate of the true inhibitory effect of the protein since only ∼70% of cells were transfected with dnRal while all of the cells were stimulated with EGF. These findings were confirmed in 3Y1(EFGR) cells, where the stable expression of inhibitory RalA28N also suppressed EGF-induced activation of c–Src by ∼75% (Figure 3B). Importantly, EGF activation of Erk was not blocked in the same cells (Figure 3C), demonstrating clear specificity in the effects of Ral on EGF receptor signaling.

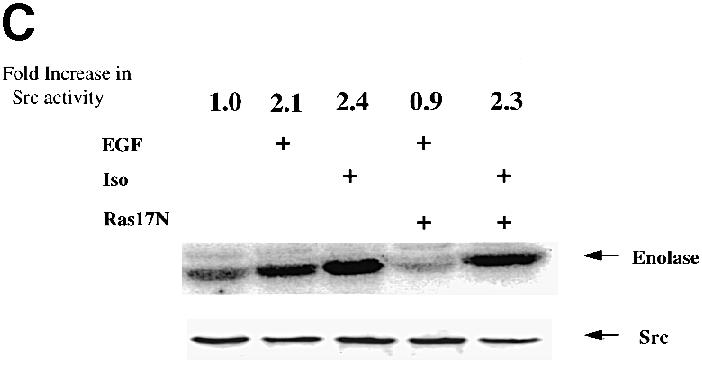
Fig. 3. Inhibition of Ral-GEF or Ras-GEF activities suppresses EGF- but not isoproterenol-induced activation of c–Src. (A) 293 cells were transiently transfected with either control vector or vector encoding dominant-negative RalA28N. After 48 h, cells were serum starved and some cells were treated with buffer, EGF or isoproterenol for10 min. c–Src was immunoprecipitated and assayed in vitro as described in Figure 2. The fold increase in c–Src activity is indicated on top of the radioactive image. Total c–Src levels in cell lysates are shown at the bottom. The data are representative of at least three independent experiments. (B) 3Y1(EGFR) cells or 3Y1(EGFR) cells expressing RalA28N were serum starved and then some cells were treated with EGF for 10 min. c–Src was then immunoprecipitated and assayed in vitro as described in Figure 2. Total c–Src levels in cells lysates are shown below. Below that are immunoblots of these lysates using phospho-specific ERK antibodies. (C) The experiments were performed as in (A) except that Ras17N was substituted for Ral28N. The data are representative of at least three independent experiments.
Ligands that activate G-protein-linked receptors also activate c–Src. In particular, β2–adrenergic receptors have recently been shown to activate c–Src by promoting its binding to the adaptor protein arrestin. To determine whether β2–adrenergic receptor activation of c–Src is also dependent upon Ral activity, 293 cells were transfected as above except that the cells were stimulated with the β2–adrenergic receptor agonist, isoproterenol. In contrast to EGF stimulation, isoproterenol stimulation of c–Src activity (2.9–fold) was not affected by the expression of dnRal (Figure 3A).
Since EGF activates Ral through the activation of Ras (Bos, 1998), the dependence of EGF-induced c–Src activation on Ras was determined by the expression of dnRas17N in 293 cells as described above. EGF activation of c–Src was suppressed by expression of dnRas17N, consistent with EGF activating c–Src through a Ras/Ral-mediated signaling cascade (Figure 3C). In contrast, expression of dnRas17N had no effect on β2-adrenergic receptor activation of c–Src. The experiments with both dnRal and dnRas demonstrate that EGF receptors and β2–adrenergic receptors activate c–Src by different mechanisms.
Ral and β2–adrenergic receptors lead to the phosphorylation of different subsets of potential c–Src targets
c–Src is capable of activating a wide variety of signaling molecules and signaling pathways through its tyrosine kinase activity. These include Stat transcription factors (Garcia and Jove, 1998), components of the actin cytoskeleton such as cortactin (Belsches et al., 1997), and the Shc adaptor protein that can lead to the activation of the Ras/Raf/Erk signaling cascade (Luttrell et al., 1996; Cary and Guan, 1999). To determine whether the mechanism of c–Src activation can influence the specificity of downstream signaling from c–Src, Ral and β–adrenergic receptors were compared for their ability to promote the phosphorylation of known targets of c–Src.
Using PC12 cells transfected with HA-Stat3, we already demonstrated that Ral has the potential to influence this c–Src-responsive transcription factor (see Figure 1). To confirm that Ral activity controls the tyrosine phosphorylation of endogenous Stat3, complex formation between c–Src and tyrosine-phosphorylated Stat3 was assessed. RalA72L was transfected transiently into 293 cells, c–Src was immunoprecipitated from serum-starved cells and then the sample was immunoblotted with anti-Stat3 antibodies. RalA72L promoted the association of c–Src with Stat3 to a level comparable to that seen after EGF treatment (Figure 4A). Antibodies specific for tyrosine-phosphorylated Stat3 were used to confirm that RalA72L expression induced the tyrosine phosphorylation of endogenous Stat3 (Figure 4A). Similar conclusions were obtained when serum-starved 3Y1(EGFR) cells stably expressing RalA72L were compared with control 3Y1(EGFR) cells (Figure 4B).

Fig. 4. Ral activity regulates the c–Src-induced tyrosine phosphorylation of Stat3. (A) 293 cells were transiently transfected with vector control, or vector expressing Ral28N or Ral72L. After 24 h, cells were serum starved for 12 h and some cells were exposed to EGF for 10 min. c–Src was immunoprecipitated and the precipitates were immunoblotted with either anti-Stat3 antibodies (upper lanes) or anti-phosphotyrosine-specific Stat3 antibodies (lower lanes). The data are representative of two independent experiments. (B) 3Y1(EGFR) cells or 3Y1(EGFR) cells expressing either RalA72L or RalA28N were serum starved for 12 h and then some cells were exposed to EGF for 10 min. c–Src was then immunoprecipitated from cell lysates and immunoblotted with anti-phosphotyrosine-specific Stat3 antibodies.
Moreover, expression of dnRal either transiently in 293 cells (Figure 4A) or stably in 3Y1(EGFR) cells (Figure 4B) suppressed EGF-induced association of c–Src with tyro– sine-phosphorylated Stat3. Together, these experiments clearly show that Ral activity plays a major role in coupling EGF receptors to c–Src-mediated tyrosine phosphorylation of the Stat3 transcription factor.
Src is also known to phosphorylate regulators of the actin cytoskeleton, such as the actin-binding protein cortactin. Transient expression of activated RalA72L into 293 cells increased the tyrosine phosphorylation of cortactin, and transient expression of dnRal in these cells suppressed the tyrosine phosphorylation of cortactin induced by EGF (Figure 5A). Thus, Ral also couples EGF receptors to Src effects on the cytoskeletal protein, cortactin.

Fig. 5. Ral activity regulates the tyrosine phosphorylation of cortactin, but not that of Shc or the activation of Erk. (A) 293 cells were transiently transfected with either control vector, RalA72L or RalA28N and then serum starved for 12 h. Some cells were then exposed to EGF for 10 min. Cell lysates were immunoprecipitated with α–cortactin antibodies and then immunoblotted with α–phospho– tyrosine-specific antibodies. The data are representative of two independent experiments. Total cortactin levels in cell lysates are displayed below. (B) 293 cells were treated as in (A) except that cell lysates were immunoprecipitated with α–Shc antibodies and then immunoblotted with anti-phosphotyrosine antibodies. (C) Cells were treated as in (A) except that cell lystates were immunoblotted with α–phospho-specific Erk antibodies.
c–Src can also activate the Erk kinase by promoting the tyrosine phosphorylation of the Shc adaptor protein (Luttrell et al., 1996; Schlaepfer and Hunter, 1997). However, expression of Ral72L in transiently transfected 293 cells did not enhance the tyrosine phosphorylation of Shc (Figure 5B). It also had no effect on Erk activity, as assessed by reactivity of an activation-specific phospho-Erk antibody (Figure 5B). In addition, Figure 3C already showed that the expression of dnRal had no detectable effect on EGF activation of Erk in 3Y1(EGFR) cells. The same result is shown here in 293 cells (Figure 5C). Thus, although Ral can activate c–Src in 293 and 3Y1(EGFR) cells, this fraction of c–Src is not capable of activating the Shc/Ras/Erk signaling cascade.
Unlike Ral-activated c–Src, β–adrenergic receptor- activated c–Src has previously been shown to promote Shc tyrosine phosphorylation and subsequent Erk activation. However, isoproterenol addition to 293 cells did not promote the association of c–Src with tyrosine-phosphorylated Stat3 or the tyrosine phosphorylation of cortactin (Figure 6), even though under these conditions it did activate c–Src (see Figure 3A and C) and Erk (Figure 6). Therefore, EGF/Ral and β–adrenergic receptor promoted the phosphorylation of distinct subsets of potential c–Src substrates (see Figure 7).

Fig. 6. Isoproterenol stimulation leads to Erk activation but not to cortactin or Stat3 tyrosine phosphorylation. 293 cells were serum starved for 24 h and then exposed to either EGF or isoproterenol for 10 min. Top: cortactin was immunoprecipitated and then the immunoprecipitates were blotted with anti-phosphotyrosine-specific antibodies. Middle: c–Src was also immunoprecipitated from the lysates and then blotted with anti-Stat3 antibodies. Bottom: cell lysates were run directly on SDS–gels and then blotted with phospho-specific Erk antibodies. The results are representative of at least two independent experiments.

Fig. 7. EGF and β–adrenergic receptors activate c–Src by different mechanisms that lead to the tyrosine phosphorylation of different sets of c–Src substrates. EGF receptors, but not β–adrenergic receptors (βAR), activate c–Src by a Ras- and Ral-dependent signaling pathway. Importantly, Ral-induced c–Src activation leads to Stat3 and cortactin tyrosine phosphorylation, but not Shc phosphorylation and Erk activation. In contrast, βAR activation of c–Src leads to Shc but not cortactin or Stat3 tyrosine phosphorylation. Ral can also be activated by Ras-independent pathways, such as through calcium, which would allow c–Src to activate downstream targets without activating Ras and its multiple effectors. Overall, these findings suggest how different receptors may generate specific cellular effects through a common kinase.
Discussion
The results presented here demonstrate that individual cell surface receptors can influence distinct subsets of the many potential downstream targets of c–Src. For example, c–Src activation by an EGF receptor/Ral-GTPase signaling pathway promotes Stat3 and cortactin tyrosine phosphorylation in the cells we studied. However, c–Src activated in this way does not lead to Shc tyrosine phosphorylation and subsequent activation of the Ras/Raf/Erk signaling pathway. In contrast, c–Src activated by β2–adrenergic receptors participates in the induction of Shc phosphorylation and Erk activation, but not in the tyrosine phosphorylation of Stat3 and cortactin in these cells. We also showed that EGF and β2–adrenergic receptors activate c–Src by different mechanisms. The former, but not the latter, functions through a Ras- and Ral-dependent signaling pathway. These findings may begin to explain how signaling molecules such as c–Src, which affect diverse target proteins, can generate unique cellular effects in response to individual stimuli. It will be interesting to determine if the phenomena observed here can be generalized. For example, do c–Src proteins activated by other stimuli, such as platelet-derived growth factor (Walker, 1993; Weernink and Rijksen, 1995), integrins (Schlaepfer and Hunter, 1997), calcium (Rusanescu et al., 1995), thrombin and estradiol (Migliaccio et al., 1996) also phosphorylate distinct sets of target proteins?
The discovery that Ral regulates c–Src was unexpected. Src has been thought of as an upstream regulator of the Ras/Raf/Erk signaling cascade for quite some time (Smith et al., 1986), yet Ral is best known as a downstream target of Ras. A major conclusion of this study is that although c–Src can sometimes be an upstream regulator of Ras, such as after β2-adrenergic receptor activation, it does not play such a role after EGF- and Ral-induced stimulation, at least in the cells studied here (see Figure 7). In fact, this would be redundant since EGF has a more direct route to Erk through direct phosphorylation of Shc. Thus, failure of Ral-regulated Src to activate Erk in response to EGF stimulation may not be of particular importance in the context of EGF signaling. However, Ral can be regulated by signals such as calcium that function by Ras-independent pathways (Hofer et al., 1998; Wolthuis et al., 1998a; M'Rabet et al., 1999) (see Figure 7). In such instances, the selective activation of c–Src substrates upon Ral activation could contribute to signaling flexibility by allowing c–Src to be activated without Ras and subsequent Erk activation.
Ral-GTPases have the potential to influence at least three downstream signaling molecules, RalBP1 (or RLIP and RIP), filamin (or ABP280) and PLD1. Which of these proteins is involved in c–Src activation, and how specificity in c–Src substrate phosphorylation is achieved remain to be determined. For β2–adrenergic receptors, activation of c–Src occurs as a consequence of binding to arrestin via the SH3 domain of c–Src. Why c–Src activated in this way leads to the tyrosine phosphorylation of Shc but not of Stat3 or cortactin also remains to be determined. One possibility being explored is that the EGF receptor/Ral and β2–adrenergic receptor/arrestin signaling pathways activate different pools of c–Src, whose access to c–Src substrates differ.
One of the c–Src-responsive signaling molecules phosphorylated upon β–adrenergic stimulation is Shc. Since phosphorylated Shc leads to Ras activation, one would expect β2–adrenergic receptors to promote the tyrosine phosphorylation of cortactin and Stat3 through Ras- mediated Ral activation (see Figure 7). The fact that this does not occur is probably due to the fact that Ral does not activate c–Src upon β2–adrenergic receptor stimulation. This can be inferred from our finding that expression of dnRal did not significantly suppress isoproterenol-induced c–Src activation. Like c–Src, Ral may also have the capacity to activate only a subset of potential downstream targets under distinct circumstances. Alternatively, Ral activation by ligands may be necessary for c–Src activation, but not sufficient. The c–Src activation we observed upon expression of a constitutively activated allele of Ral or Ral-GEF may reflect an extreme situation that obviates the need for a complementary signal that is normally supplied by EGF receptor but not β2-adrenergic receptor activation.
Experiments in three different cell types, PC12 pheochromocytoma cells, 293 epithelial cells and 3Y1 fibroblasts, were used to confirm that Ral can regulate c–Src. In 293 and 3Y1 cells, c–Src activity was measured directly, while in PC12 cells the tyrosine phosphorylation of transfected Stat3 was suggestive of c–Src activity. In each case, elevation of Ral-GTP levels activated c–Src, while suppression of Ral-GTP levels by expression of dnRal inhibited EGF- or IL–6-induced c–Src activation. The effect of dnRal on EGF receptor function was quite specific since EGF activation of Erk was not affected in the same cell lysates where c–Src activation was blocked. Previous studies focusing on Ral- and Ral-GEF-induced cell proliferation (Urano et al., 1996; White et al., 1996; Wolthuis et al., 1997), fos or E2F promoter activation (Okazaki et al., 1997; Wolthuis et al., 1997; Gille, 1999) and phosphorylation of the forkhead transcription factor (Kops et al., 1999) showed clear effects by Ral-GEFs but little or no effects by an activated allele of Ral. Thus, these studies left open the possibility that these effects were due to a function of Ral-GEFs other than Ral activation. This is not the case for c–Src activation detected here, since a constitutively activated allele of RalA generated effects comparable to growth factor stimulation or Ral-GEF expression.
Although the major part of this study involved the investigation of the role of Ral in c–Src activation by the EGF receptor, we also showed that in PC12 cells, dnRal blocked IL–6-induced tyrosine phosphorylation of Stat3. A large literature supports the idea that the Janus tyrosine kinases (JAKs) mediate Stat phosphorylation by interleukins (Ihle, 1995), suggesting that Ral proteins can also regulate this family of tyrosine kinases. However, c–Src can activate JAKs (Campbell et al., 1997), raising the possibility that in PC12 cells Ral regulates JAKs through c–Src in response to IL–6 stimulation. However, JAKs may not be involved at all since there is evidence for a primary role for Src in Stat activation during IL–3 and EGF stimulation of cells (Chaturvedi et al., 1998; Olayioye et al., 1999). Further analysis of Ral function in interleukin signaling may add insight into the relationship between Src, JAK and Stat proteins.
Unlike their Ras counterparts, constitutively activated Ral alleles have not been considered as potential oncogenes because expression of the mutant protein by itself has not been found to enhance cell proliferation. However, Ral72L expression has been shown to enhance modestly focus formation induced by expression of a weak Raf allele in NIH-3T3 cells (Urano et al., 1996). Moreover, we have recently observed that constitutively activated Ral expression in 3Y1 cells overexpressing the EGF receptor enhanced colony formation in semi-solid media (Lu et al., 2000). In the present study, we showed that these cells displayed elevated c–Src activity. This alteration may contribute to the cells' enhanced growth properties, since a series of investigations have documented the synergistic effects of c–Src and EGF receptor activities on the transformed phenotype of tissue culture cells (Roche et al., 1995; Belsches et al., 1997).
A variety of human tumor cell types, including carcinoma of the colon and mammary gland (Ottenhoff-Kalff et al., 1992; Muthuswamy and Muller, 1994; Muthuswamy et al., 1994; Watson and Miller, 1995; Garcia et al., 1997; Biscardi et al., 1999b), display elevated c–Src and Stat3 activity. Our finding that Ral can activate c–Src suggests that the presence of a constitutively activated mutant allele of Ral or amplification of a Ral-GEF could contribute to the deregulation of c–Src and Stat3 found in tumors. In addition, many breast cancers display elevated levels of EGF receptors or the related HER–2 receptors. Our finding that expression of constitutively activated Ral enhances soft agar growth of cells overexpressing EGF receptors (Lu et al., 2000) suggests that its presence could enhance the oncogenic potential of neoplastic cells containing amplified EGF or ERB2 genes (Garcia et al., 1997; Biscardi et al., 1998).
Materials and methods
Reagents
Antibodies against Stat3 were from Santa Cruz Biotech. Phospho-specific antibodies to Stat3 and Erk were from New England Biolabs. Cortactin antibodies were from UBI, while RalA antibodies were from Transduction Laboratories. c–Src antibody, GD11, was a gift from T.Parsons (University of Virginia). EGF and IL–6 were from Upstate Biotech, and isoproterenol was from Sigma.
Cell culture
293 cells were grown in Dulbecco's modified Eagle's medium (DMEM) plus 10% fetal calf serum (Hyclone). 3Y1(EGFR) cells were grown in DMEM plus 10% calf serum (Hyclone), while PC12 cells were grown in DMEM plus 10% iron-enriched calf serum plus 5% horse serum.
Transfections
PC12 cells (70% confluent 60 mm dish) were transfected with Pfx–1 lipid (Invitrogen). DNA (11 μg) + Pfx–1 (33 μl) + serum-free DMEM was added to cells for 4 h. The medium was replaced with complete culture medium and incubated for 36 h. The medium was then replaced with serum-free DMEM for 12 h before stimulation with ligands. Transfection efficiency was ∼15%. 293 cells (70% confluent 60 mm dish) were transfected using the calcium phosphate method. DNA (4 μg) + 438 μl of H2O + 62 μl of 2 M CaCl2 + 50 μl of 2× HBS (50 mM HEPES, 10 mM KCl, 12 mM dextrose, 280 mM NaCl, 1.5 mM Na2HPO4) was added to cells in complete medium for 12 h. The medium was then changed and cells were incubated for 48 h. The medium was then replaced with serum-free DMEM for 12 h before exposing the cells to ligands.
Immunoprecipitations
Cells were washed three times in ice-cold phosphate-buffered saline and lysed in NP–40 lysis buffer [50 mM Tris pH 7.5, 150 mM NaCl, 1 mM EDTA, 0.5% NP–40, 1 mM sodium orthovanadate, 1 mM phenylmethylsulfonyl fluoride (PMSF), 10 μg/ml aprotinin, 10 μg/ml leupeptin and 15% glycerol] or RIPA buffer (10 mM Tris pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% NP–40, 0.1% SDS, 0.1% sodium deoxycholate, 1 mM sodium orthovanadate, 1 mM PMSF, 10 μg/ml aprotinin and 10 μg/ml leupeptin). The lysis buffer for Src and Stat co-immunoprecipitation consisted of 50 mM Tris pH 7.5, 137 mM NaCl, 0.1% Triton X–100, 2 mM EGTA, 1 mM sodium orthovanadate, 1 mM PMSF, 10 μg/ml aprotinin and 10 μg/ml leupeptin. The lysates (1mg of protein) were incubated on ice for 15 min and then centrifuged at 10 000 g for 10 min. The resulting supernatants were immunoprecipitated with primary antibody–protein A–Sepharose complexes at 4°C for 90 min. Immunoprecipitates were washed four times with NP–40 lysis buffer or RIPA buffer before being loaded onto SDS–gels or applied to Src kinase assays.
Src kinase assays
Cells were lysed in RIPA buffer (10 mM Tris–HCl pH 7.4 plus 150 mM NaCl, 1 mM EDTA, 1% NP–40, 0.1% SDS, 0.1% sodium deoxycholate, 1 mM orthovanadate, 1 mM PMSF, 10 μg/ml aprotinin and 10 μg/ml leupeptin) or NP–40 lysis buffer, and pp60srcwas immunoprecipitated with anti-Src monoclonal antidody or anti-filamin antibody. The immune complexes were washed three times with lysis buffer and twice with kinase buffer (40 mM HEPES pH 7.4, 10 mM MgCl2, 3 mM MnCl2). Immunoprecipitates were then incubated with 10 μCi of [γ–32P]ATP (600 Ci/mm) in 30 μl of kinase with 0.5 μM ATP plus 1 mM dithiothreitol and 2.5 μg of acid-treated rabbit muscle enolase (Sigma) for 5 min on ice.
The reactions were stopped by the addition of SDS-containing sample buffer. The samples were analyzed by SDS–PAGE with 10% gel. The gel was dried and 32P–containing proteins were visualized by autoradiography and quantitated on a phosphoimager.
DNA constructs
PMT3-Ral72, PMT3-Ral28N and PMT3-Ras 61L were described previously. PCAGGS-neo HA-Stat3 was obtained from K.Nakajima, Osaka University Medical School.
Acknowledgments
Acknowledgements
We thank Dr Tom Parsons for his generous gift of c–Src antibodies and K.Nakajima for HA-Stat3. This work was supported by a PHS Grant from the NIGMS to L.A.F.
References
- Ahn S., Maudsley, S., Luttrell, L.M., Lefkowitz, R.J. and Daaka, Y. (1999) Src-mediated tyrosine phosphorylation of dynamin is required for β2-adrenergic receptor internalization and mitogen-activated protein kinase signaling. J. Biol. Chem., 274, 1185–1188. [DOI] [PubMed] [Google Scholar]
- Belsches A.P., Haskell,M.D. and Parsons,S.J. (1997) Role of c–Src tyrosine kinase in EGF-induced mitogenesis. Front. Biosci., 2, D501–D518. [DOI] [PubMed] [Google Scholar]
- Bielinski D.F., Pyun, N.Y., Linko-Stentz, K., Macara, I. and Fine, R.E. (1993) Ral and Rab3a are major GTP-binding proteins of axonal rapid transport and synaptic vesicles and do not redistribute following depolarization stimulated synaptosomal exocytosis. Biochim. Biophys. Acta, 1151, 246–256. [DOI] [PubMed] [Google Scholar]
- Biscardi J.S., Belsches, A.P. and Parsons, S.J. (1998) Characterization of human epidermal growth factor receptor and c–Src interactions in human breast tumor cells. Mol. Carcinogen., 21, 261–272. [DOI] [PubMed] [Google Scholar]
- Biscardi J.S., Maa, M.C., Tice, D.A., Cox, M.E., Leu, T.H. and Parsons, S.J. (1999a) c–Src-mediated phosphorylation of the epidermal growth factor receptor on Tyr845 and Tyr1101 is associated with modulation of receptor function. J. Biol. Chem., 274, 8335–8343. [DOI] [PubMed] [Google Scholar]
- Biscardi J.S., Tice, D.A. and Parsons, S.J. (1999b) c–Src, receptor tyrosine kinases, and human cancer. Adv. Cancer Res., 76, 61–119. [DOI] [PubMed] [Google Scholar]
- Bos J.L. (1997) Ras-like GTPases. Biochim. Biophys. Acta, 1333, M19–M31. [DOI] [PubMed] [Google Scholar]
- Bos J.L. (1998) All in the family? New insights and questions regarding interconnectivity of Ras, Rap1 and Ral. EMBO J., 17, 6776–6782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromberg J.F., Horvath, C.M., Besser, D., Lathem, W.W., and Darnell, J.E.,Jr (1998) Stat3 activation is required for cellular transformation by v-src. Mol. Cell. Biol., 18, 2553–2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown M.T. and Cooper, J.A. (1996) Regulation, substrates and functions of src. Biochim. Biophys. Acta, 1287, 121–149. [DOI] [PubMed] [Google Scholar]
- Campbell G.S., Yu, C.L., Jove, R., and Carter-Su, C. (1997) Constitutive activation of JAK1 in Src-transformed cells. J. Biol. Chem., 272, 2591–2594. [DOI] [PubMed] [Google Scholar]
- Cantor S., Urano, T. and Feig, L.A. (1995) Identification and characterization of RalBP1, a potential downstream target of Ral GTPases. Mol. Cell. Biol., 15, 4578–4584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cary L.A. and Guan,J.L. (1999) Focal adhesion kinase in integrin-mediated signaling. Front. Biosci., 4, D102–D113. [DOI] [PubMed] [Google Scholar]
- Chaturvedi P., Reddy, M.V. and Reddy, E.P. (1998) Src kinases and not JAKs activate STATs during IL-3 induced myeloid cell proliferation. Oncogene, 16, 1749–1758. [DOI] [PubMed] [Google Scholar]
- Cirri P., Chiarugi, P., Marra, F., Raugei, G., Camici, G., Manao, G. and Ramponi, G. (1997) c–Src activates both STAT1 and STAT3 in PDGF-stimulated NIH3T3 cells. Biochem. Biophys. Res. Commun., 239, 493–497. [DOI] [PubMed] [Google Scholar]
- D'Adamo D.R., Novick, S., Kahn, J.M., Leonardi, P. and Pellicer, A. (1997) rsc: a novel oncogene with structural and functional homology with the gene family of exchange factors for Ral. Oncogene, 14, 1295–1305. [DOI] [PubMed] [Google Scholar]
- Feig L.A. and Emkey,L.A. (1993) Ral gene products and their regulation. In Lacal,J.C. and McCormick,F. (eds), The ras Superfamily of GTPases. CRC Press, London, UK, pp. 247–258. [Google Scholar]
- Foster-Barber A. and Bishop, J.M. (1998) Src interacts with dynamin and synapsin in neuronal cells. Proc. Natl Acad. Sci. USA, 95, 4673–4677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia R. and Jove, R. (1998) Activation of STAT transcription factors in oncogenic tyrosine kinase signaling. J. Biomed. Sci., 5, 79–85. [DOI] [PubMed] [Google Scholar]
- Garcia R., Yu, C.L., Hudnall, A., Catlett, R., Nelson, K.L., Smithgall, T., Fujita, D.J., Ethier, S.P. and Jove, R. (1997) Constitutive activation of Stat3 in fibroblasts transformed by diverse oncoproteins and in breast carcinoma cells. Cell Growth Differ., 8, 1267–1276. [PubMed] [Google Scholar]
- Gille H. and Downward, J. (1999) Multiple Ras effector pathways contribute to G(1) cell cycle progression. J. Biol. Chem., 274, 22033–22040. [DOI] [PubMed] [Google Scholar]
- Goi T., Rusanescu, G., Urano, T. and Feig, L.A. (1999) Ral-specific guanine nucleotide exchange factor activity opposes other Ras effectors in PC12 cells by inhibiting neurite outgrowth. Mol. Cell. Biol., 19, 1731–1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorlin J.B., Yamin, R., Egan, S., Stewart, M., Stossel, T.P., Kwiatkowski, D.J. and Hartwig, J.H. (1990) Human endothelial actin-binding protein (ABP-280, nonmuscle filamin): a molecular leaf spring. J. Cell Biol., 111, 1089–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofer F., Berdeaux, R. and Martin, G.S. (1998) Ras-independent activation of Ral by a Ca2+-dependent pathway. Curr. Biol., 14, 839–842. [DOI] [PubMed] [Google Scholar]
- Ihara S., Nakajima, K., Fukada, T., Hibi, M., Nagata, S., Hirano, T. and Fukui, Y. (1997) Dual control of neurite outgrowth by STAT3 and MAP kinase in PC12 cells stimulated with interleukin-6. EMBO J., 16, 5345–5352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihle J.N. (1995) Cytokine receptor signalling. Nature, 377, 591–594. [DOI] [PubMed] [Google Scholar]
- Jiang H., Luo, J.-Q., Urano, T., Lu, Z., Foster, D.A. and Feig, L.A. (1995) Involvement of Ral GTPase in v-Src-induced phospholipase D activation. Nature, 378, 409–412. [DOI] [PubMed] [Google Scholar]
- Jullien-Flores V., Dorseuil, O., Romero, F., Letourneur, F., Saragosti, S., Berger, R., Tavitian, A., Gacon, G. and Camonis, J.H. (1995) Bridging Ral GTPase to Rho pathways. J. Biol. Chem., 270, 22473–22477. [DOI] [PubMed] [Google Scholar]
- Kaplan K.B., Swedlow, J.R., Varmus, H.E. and Morgan, D.O. (1992) Association of p60c-src with endosomal membranes in mammalian fibroblasts. J. Cell Biol., 118, 321–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kops G.J., de Ruiter, N.D., De Vries-Smits, A.M., Powell, D.R., Bos, J.L. and Burgering, B.M. (1999) Direct control of the forkhead transcription factor AFX by protein kinase B. Nature, 398, 630–634. [DOI] [PubMed] [Google Scholar]
- Linstedt A.D., Vetter, M.L., Bishop, J.M. and Kelly, R.B. (1992) Specific association of the proto-oncogene product pp60c-src with an intracellular organelle, the PC12 synaptic vesicle. J. Cell Biol., 117, 1077–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z., et al. (2000)Phospholipase D and RalA cooperate with the epidermal growth factor receptor to transform 3Y1 rat fibroblasts. Mol. Cell. Biol., 20, 462–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J.Q., et al. (1998)Functional association between arf and RalA in active phospholipase D complex. Proc. Natl Acad. Sci. USA, 95, 3632–3637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luttrell L.M., Hawes, B.E., van Biesen, T., Luttrell, D.K., Lansing, T.J. and Lefkowitz, R.J. (1996) Role of c–Src tyrosine kinase in G protein-coupled receptor- and Gβγ subunit-mediated activation of mitogen-activated protein kinases. J. Biol. Chem., 271, 19443–19450. [DOI] [PubMed] [Google Scholar]
- Luttrell L.M., Della Rocca, G.J., van Biesen, T., Luttrell, D.K. and Lefkowitz, R.J. (1997) Gβγ subunits mediate Src-dependent phosphorylation of the epidermal growth factor receptor. A scaffold for G protein-coupled receptor-mediated Ras activation. J. Biol. Chem., 272, 4637–4644. [DOI] [PubMed] [Google Scholar]
- Luttrell L.M., et al. (1999)β-arrestin-dependent formation of β2 adrenergic receptor–Src protein kinase complexes. Science, 283, 655–661. [DOI] [PubMed] [Google Scholar]
- M'Rabet L., Coffer, P.J., Wolthuis, R.M., Zwartkruis, F., Koenderman, L. and Bos, J.L. (1999) Differential fMet-Leu-Phe- and platelet-activating factor-induced signaling toward Ral activation in primary human neutrophils. J. Biol. Chem., 274, 21847–21852. [DOI] [PubMed] [Google Scholar]
- Maa M.C., Leu, T.H., McCarley, D.J., Schatzman, R.C. and Parsons, S.J. (1995) Potentiation of epidermal growth factor receptor-mediated oncogenesis by c–Src: implications for the etiology of multiple human cancers. Proc. Natl Acad. Sci. USA, 92, 6981–6985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migliaccio A., Di Domenico, M., Castoria, G., de Falco, A., Bontempo, P., Nola, E. and Auricchio, F. (1996) Tyrosine kinase/p21ras/MAP-kinase pathway activation by estradiol–receptor complex in MCF-7 cells. EMBO J., 15, 1292–1300. [PMC free article] [PubMed] [Google Scholar]
- Muthuswamy S.K. and Muller, W.J. (1994) Activation of the Src family of tyrosine kinases in mammary tumorigenesis. Adv. Cancer Res., 64, 111–123. [DOI] [PubMed] [Google Scholar]
- Muthuswamy S.K., Siegel, P.M., Dankort, D.L., Webster, M.A. and Muller, W.J. (1994) Mammary tumors expressing the neu proto-oncogene possess elevated c–Src tyrosine kinase activity. Mol. Cell. Biol., 14, 735–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta Y., Suzuki, N., Nakamura, S., Hartwig, J.H. and Stossel, T.P. (1999) The small GTPase RalA targets filamin to induce filopodia. Proc. Natl Acad. Sci. USA, 96, 2122–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okazaki M., Kishida, S., Hinoi, T., Hasegawa, T., Tamada, M., Kataoka, T. and Kikuchi, A. (1997) Synergistic activation of c-fos promoter activity by Raf and Ral-GDP dissociation stimulator. Oncogene, 14, 515–521. [DOI] [PubMed] [Google Scholar]
- Olayioye M.A., Beuvink, I., Horsch, K., Daly, J.M. and Hynes, N.E. (1999) ErbB receptor-induced activation of stat transcription factors is mediated by Src tyrosine kinases. J. Biol. Chem., 274, 17209–17218. [DOI] [PubMed] [Google Scholar]
- Onofri F., Giovedi, S., Vaccaro, P., Czernik, A.J., Valtorta, F., De Camilli, P., Greengard, P. and Benfenati, F. (1997) Synapsin I interacts with c–Src and stimulates its tyrosine kinase activity. Proc. Natl Acad. Sci. USA, 94, 12168–12173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottenhoff-Kalff A.E., Rijksen, G., van Beurden, E.A., Hennipman, A., Michels, A.A. and Staal, G.E. (1992) Characterization of protein tyrosine kinases from human breast cancer: involvement of the c-src oncogene product. Cancer Res., 52, 4773–4778. [PubMed] [Google Scholar]
- Park S.H. and Weinberg, R.A. (1995) A putative effector of Ral has homology to Rho/Rac GTPase activating proteins. Oncogene, 11, 2349–2355. [PubMed] [Google Scholar]
- Roche S., Koegl, M., Barone, M.V., Roussel, M.F. and Courtneidge, S.A. (1995) DNA synthesis induced by some but not all growth factors requires Src family protein tyrosine kinases. Mol. Cell. Biol., 15, 1102–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusanescu G., Qi, H., Thomas, S.M., Brugge, J.S. and Halegoua, S. (1995) Calcium influx induces neurite growth through a Src–Ras signaling cassette. Neuron, 15, 1415–1425. [DOI] [PubMed] [Google Scholar]
- Schlaepfer D.D. and Hunter, T. (1997) Focal adhesion kinase overexpression enhances ras-dependent integrin signaling to ERK2/mitogen-activated protein kinase through interactions with and activation of c–Src. J. Biol. Chem., 272, 13189–13195. [DOI] [PubMed] [Google Scholar]
- Smith M.R., DeGudicibus, S.J. and Stacey, D.W. (1986) Requirement for c-ras proteins during viral oncogene transformation. Nature, 320, 540–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takafuta T., Wu, G., Murphy, G.F. and Shapiro, S.S. (1998) Human β-filamin is a new protein that interacts with the cytoplasmic tail of glycoprotein Ibα. J. Biol. Chem., 273, 17531–17538. [DOI] [PubMed] [Google Scholar]
- Tice D.A., Biscardi, J.S., Nickles, A.L. and Parsons, S.J. (1999) Mechanism of biological synergy between cellular Src and epidermal growth factor receptor. Proc. Natl Acad. Sci. USA, 96, 1415–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turkson J., Bowman, T., Garcia, R., Caldenhoven, E., De Groot, R.P. and Jove, R. (1998) Stat3 activation by Src induces specific gene regulation and is required for cell transformation. Mol. Cell. Biol., 18, 2545–2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urano T., Emkey, R. and Feig, L.A. (1996) Ral-GTPases mediate a distinct downstream signaling pathway from Ras that facilitates cellular transformation. EMBO J., 16, 810–816. [PMC free article] [PubMed] [Google Scholar]
- Walker F., deBlaquiere, J. and Burgess, A.W. (1993) Translocation of pp60c-src from the plasma membrane to the cytosol after stimulation by platelet-derived growth factor. J. Biol. Chem., 268, 19552–19558. [PubMed] [Google Scholar]
- Watson C.J. and Miller, W.R. (1995) Elevated levels of members of the STAT family of transcription factors in breast carcinoma nuclear extracts. Br. J. Cancer, 71, 840–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weernink P.A. and Rijksen, G. (1995) Activation and translocation of c–Src to the cytoskeleton by both platelet-derived growth factor and epidermal growth factor. J. Biol. Chem., 270, 2264–2267. [DOI] [PubMed] [Google Scholar]
- White M.A., Vale, T., Camonis, J.H., Schaefer, E. and Wigler, M.H. (1996) A role for the Ral guanine nucleotide dissociation stimulator in mediating Ras-induced transformation. J. Biol. Chem., 271, 16439–16442. [DOI] [PubMed] [Google Scholar]
- Wilde A., Beattie, E.C., Lem, L., Riethof, D.A., Liu, S.H., Mobley, W.C., Soriano, P. and Brodsky, F.M. (1999) EGF receptor signaling stimulates SRC kinase phosphorylation of clathrin, influencing clathrin redistribution and EGF uptake. Cell, 96, 677–687. [DOI] [PubMed] [Google Scholar]
- Wolthuis R.M., de Ruiter, N.D., Cool, R.H. and Bos, J.L. (1997) Stimulation of gene induction and cell growth by the Ras effector Rlf. EMBO J., 16, 6748–6761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolthuis R.M., Franke, B., van Triest, M., Bauer, B., Cool, R.H., Camonis, J.H., Akkerman, J.W. and Bos, J.L. (1998a) Activation of the small GTPase Ral in platelets. Mol. Cell. Biol., 18, 2486–2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolthuis R.M., Zwartkruis, F., Moen, T.C. and Bos, J.L. (1998b) Ras-dependent activation of the small GTPase Ral. Curr. Biol., 8, 471–474. [DOI] [PubMed] [Google Scholar]
- Xie Z., Xu, W., Davie, E.W. and Chung, D.W. (1998) Molecular cloning of human ABPL, an actin-binding protein homologue. Biochem. Biophys. Res. Commun., 251, 914–919. [DOI] [PubMed] [Google Scholar]
- Yu C.L., Meyer, D.J., Campbell, G.S., Larner, A.C., Carter-Su, C., Schwartz, J. and Jove, R. (1995) Enhanced DNA-binding activity of a Stat3-related protein in cells transformed by the Src oncoprotein. Science, 269, 81–83. [DOI] [PubMed] [Google Scholar]
- Yu X.M. and Salter, M.W. (1999) Src, a molecular switch governing gain control of synaptic transmission mediated by N-methyl-d-aspartate receptors. Proc. Natl Acad. Sci. USA, 96, 7697–7704. [DOI] [PMC free article] [PubMed] [Google Scholar]