

Abstract
Background
Choroideremia is an X-linked recessive disorder characterized by vision loss with progressive atrophy of the retinal photoreceptors, retinal pigment epithelium (RPE), and choriocapillaris. Ectodermal dysplasia is a heterogeneous group of disorders characterized by a deficiency of two or more ectodermal derivatives. We report on the phenotypic and genetic characteristics of a 29-year-old woman with both choroideremia and ectodermal dysplasia.
Materials and Methods
Observational case report with physical and ophthalmic examination, fluorescein angiography (FA), visual field testing, electroretinography, and cytogenetic analysis. This study adhered to the tenets of the Declaration of Helsinki and The New York Eye and Ear Infirmary Institutional Review Board guidelines.
Results
Physical and ocular examination revealed hypotrichosis, hypohidrosis, full dentures, meibomian gland hypoplasia, and a decrease in corneal tear film. Visual acuity was hand motions in the right eye and 20/50 in the left eye. Fundus examination and fluorescein angiography were consistent with advanced choroideremia and revealed diffuse bilateral RPE and chorioretinal atrophy with sparing of the fovea. Visual field testing had less than 10-degree central islands in both eyes. Scotopic electroretinogram (ERG) was flat with a small flicker response. Cytogenetic analysis showed a complex translocation involving chromosomes X, 1, and 3: 46,X,t(X;1;3)(q13;q24;q21),inv(9)(p11q13). Selective inactivation of the normal X chromosome was present in blood and skin. Chromosomal analyses of the proband's family (mother and two brothers) were normal.
Conclusion
An X-autosome chromosomal translocation combined with non-random inactivation of the normal X-chromosome in a woman resulted in the phenotypic findings of choroideremia and ectodermal dysplasia.
Keywords: choroideremia, ectodermal dysplasia, CHM, EDA, chromosomal translocation
INTRODUCTION
Choroideremia (OMIM [Online Mendelian Inheritance in Man] 303100) is an X-linked recessive disease characterized by progressive atrophy of the retinal photoreceptors, RPE, and the choriocapillaris, leading to progressive vision loss.1 The abnormal gene is located on the X chromosome at Xq21. Most affected males develop nyctalopia in the first or second decade of life with peripheral visual field constriction and an abnormal ERG. Central vision is usually affected in the late stages of the disease. Female carriers are usually asymptomatic with a mottled mosaic fundus appearance due to random inactivation of one of the X chromosomes. This process, called lyonization, occurs in all placental mammals and is the reason choroideremia is rarely present in females.
Ectodermal dysplasia is a heterogeneous group of disorders characterized by absence or deficient function of at least two derivatives of the ectoderm including teeth, hair, nails, and/or sweat glands. Of the more than 150 different forms of ectodermal dysplasia, X chromosome mutations account for more than half of the cases (OMIM 305100); however, autosomal recessive forms have also been reported. Clinical manifestations of ectodermal dysplasia include hypohidrosis, hypotrichosis, and facial and dental malformations. Ocular manifestations of ectodermal dysplasia, which can result in loss of vision, typically involve the anterior segment of the eye. These include an absence or hypoplasia of the meibomian glands, corneal limbal stem cell and tear film deficiencies, lens opacities, and nasolacrimal duct abnormalities with an increased risk of corneal scarring, corneal ulcers, and recurrent dacryocystitis.
There are several syndromes of ectodermal dysplasia with associated ocular findings. These include Hay–Wells syndrome, or Ankyloblepharon/Ectodermal Dysplasia/Clefting (AEC) syndrome, Ectrodactyly/Ectodermal Dysplasia/Cleft syndrome (EEC),2 and Ectodermal Dysplasia/Ectrodactyly/Macular Dystrophy (EEM).3 We report on the phenotypic and genetic characteristics of a 29-year-old woman with both choroideremia and ectodermal dysplasia.
CASE PRESENTATION
A 29-year-old woman with ectodermal dysplasia presented with progressive nyctalopia and decreased vision for 10 years. External exam revealed hypotrichosis, hypohidrosis, lightly pigmented hair, fair skin, and full dentures (Figure 1). Best corrected visual acuity was hand motions with −4.25 – 1.25 × 150 in the right eye and 20/50 with −3.00 – 1.25 × 85 in the left eye. Slit lamp examination revealed sparse hypopigmented eyelashes, hypoplastic meibomian glands, blepharitis, and decreased tear film (Figure 2). Symmetric peripheral iris transillumination defects and minimal Y suture lens opacities were also present. Schirmer's I and II tests for aqueous tear production were 0.5 mm in both eyes. American Optical Hardy-Rand-Rittler Pseudoisochromic color vision testing (Southbridge, MA) demonstrated mild red-green and blue-yellow deficiencies in both eyes.
FIGURE 1.
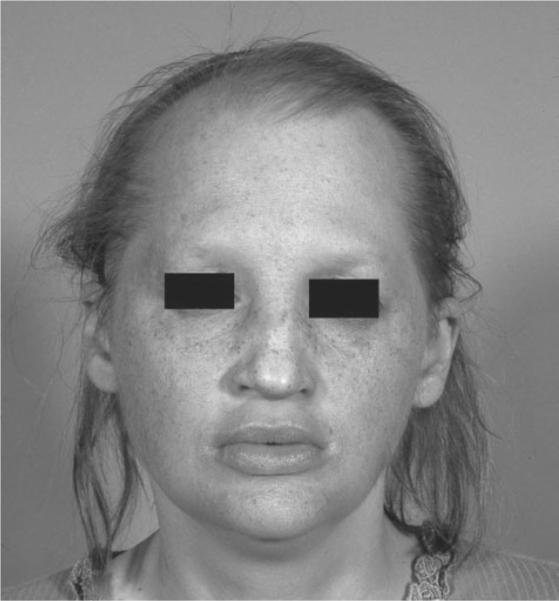
External examination revealed alopecia with lightly pigmented hair, fair skin, and full dentures.
FIGURE 2.
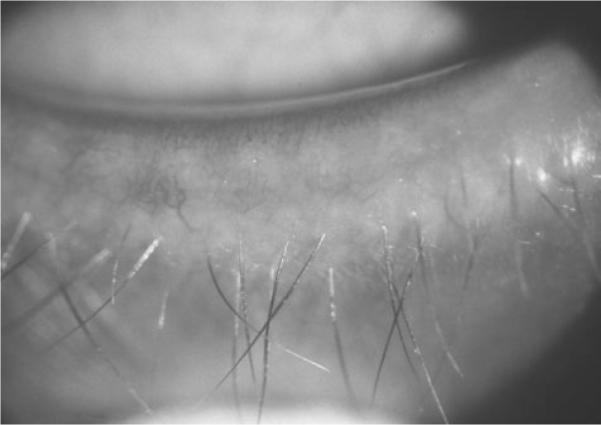
External examination revealed a sparse number of hypopigmented eyelashes, a decreased number of meibomian glands, and evidence of blepharitis.
Fundus examination revealed diffuse RPE and choroidal atrophy with sparing of a small island in the macula of both eyes (Figure 3). There were multiple focal areas of punctate RPE hyperplasia in the periphery. The optic nerve had slight waxy pallor and the retinal blood vessels were attenuated. Consistent with the findings of Noble et al., fluorescein angiography (FA) confirmed the diffuse choriodal atrophy and defined the area of spared RPE and choroid centrally (Figure 4).4 Goldmann visual fields demonstrated less than 10-degree central islands in both eyes. Electro-physiologic testing revealed a flat scotopic ERG with small flicker response.
FIGURE 3.
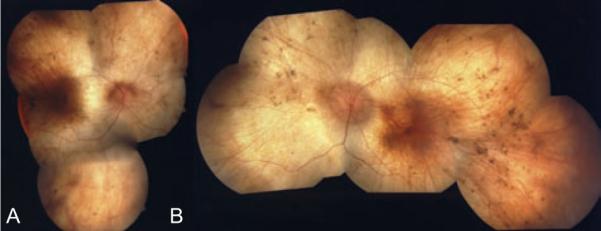
Fundus examination revealed diffuse RPE and choroidal atrophy with sparing of a small island in the macula of both eyes. Figure A is the right eye and Figure B is the left eye.
FIGURE 4.
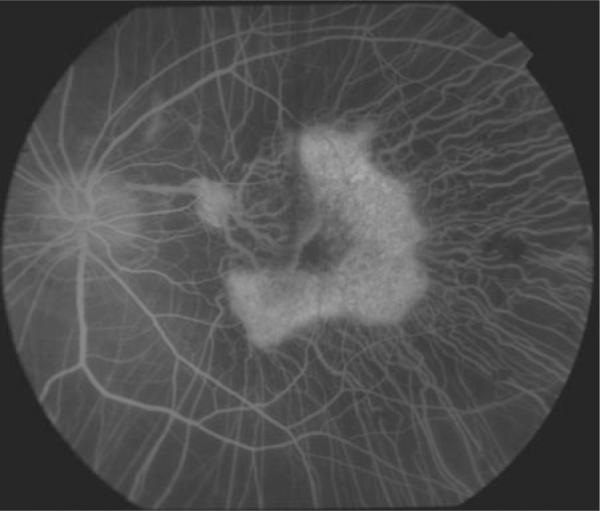
Fluorescein angiography revealed diffuse choriodal atrophy and defines the area of spared RPE and choroid centrally.
GENETIC AND LABORATORY TESTING
Cytogenetic analysis revealed an abnormal karyotype with the presence of complex translocations and a pericentric inversion (Figure 5): 46,X,t(X;1;3) (q13;q24;q21),inv(9)(p11q13). There was inactivation of the normal X-chromosome in blood and skin. Genetic analysis of the proband's family including two brothers and her mother revealed normal chromosomes. The patient had a normal plasma ornithine level.
FIGURE 5.
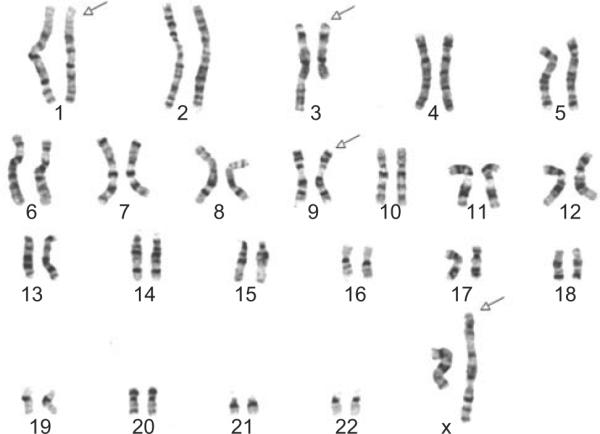
Karyotype from peripheral blood lymphocytes with arrows pointing to affected chromosomes X, 1, 3, and 9. Complex translocation of chromosomes X, 1, 3, and a pericentric inversion of chromosome 9: 46,X,t(X;1;3)(q13;q24;q21),inv(9)(p11q13).
DISCUSSION
Ectodermal dysplasia is a heterogeneous group of conditions characterized by absent or deficient function of at least two derivatives of the ectoderm. Our patient had the most common type of ectodermal dysplasia: hypohidrotic ectodermal dysplasia. Her reduced ability to sweat (hypohidrosis) and distinctive facial features (prominent forehead, thick lips, flat nasal bridge) are characteristic of this condition.5 This form is estimated to affect at least 1 in 17,000 people worldwide.6 About 95% are X-linked and involve mutations in the Ectodysplasin (EDA) gene which maps to Xq12–q13.1. Autosomal dominant and autosomal recessive forms also exist that involve the EDAR (2q11–q13) and EDARADD genes (1q42.2–q43), respectively.5
Associated retinal findings are not common with any of the over 150 types of ectodermal dyplasia. One rare autosomal recessive form, EEM syndrome (Ectodermal dyplasia, Ectrodactyly, Macular dystrophy) has an associated macular dystrophy. Hayakawa et al.3 described a 41-year-old woman with EEM syndrome who had profuse pigmentation of the posterior pole and macula with associated choriocapillaris atrophy. Similarly, Moloney et al.7 reported two siblings with ectodermal dysplasia and a macular choroidal dystrophy described as total choroidal atrophy. Wald et al.2 described one case of EEC (Ectodermal dysplasia, Ectrodactyly, and Clefting of the lip and palate) with bilateral retinal detachments that resembled stage 5 retinopathy of prematurity. Our patient's retinal findings consistent with choroideremia have not been previously reported.
The choroideremia gene (CHM) encodes a protein called Rab Escort Protein (REP-1) that is required for prenylation of rab geranylgeranyl transferase, an enzyme involved in intracellular transport.8 REP-1 deficiency leads to degeneration of photoreceptors, RPE, and choriocapillaris resulting in loss of vision. Molecular genetic studies have shown a wide variety of point, nonsense, frame-shift, and splice-site mutations involving the REP-1 gene located at Xq21.
Since choroideremia is an X-linked recessive (XR) disease, women are rarely affected due to lyonization, or random X-chromosome inactivation. Female carriers of the CHM gene may have a patchwork mosaic of RPE atrophy, but are usually free of visual symptoms. However, there are several genetic conditions in which a female may phenotypically present with an choroidermia. She may inherit two mutant CHM genes from an affected father and a carrier mother resulting in homozygosity or compound heterozygosity for mutant CHM alleles. Or, she may have Turner Syndrome or a Turner's Syndrome variant with a missing (45×) or partially deleted X-chromosome with an inherited CHM gene. These two scenerios were not present in our patient since she had no family history of choroideremia and did not have Turner Syndrome. Our patient had an X-autosome translocation with skewed X-inactiviation as the cause. In this case, since there were two breaks in the X-chromosome in this complex rearangement, the breaks interrupted and therefore inactivated both the CHM and EDA genes.
In our case, initial random X-activation at the blastocyst stage was likely followed by selection for cells with full autosomal complement. This is because X-activation of the translated X-autosome chromosome can extend to inactivate the attached autosome, a potentially lethal condition for the affected cell. A cell with monosomy for extensive autosomal loci would not survive. Sex chromosome monosomy is better tolerated since most X-chromosome genes are transcribed from a single X-chromosome in an individual cell. Thus, it is usual to see non-random inactivation of the normal X-chromosome to preserve genetic balance in the situation of an X-autosome translocation.
The diagnosis of choroideremia is typically based on clinical findings in affected males and carrier females in the family. The classic symptoms of impaired dark adaptation, signs of diffuse chorioretinal degeneration, peripheral field loss, a pattern of rod-cone degeneration followed by a flat ERG, and X-linked family history is considered diagnostic.9 Deoxyribonucleic acid (DNA) and immunoblot analysis are available for diagnostic confirmation, carrier testing, and/or preimplantation or prenatal diagnosis.10 DNA analysis can be done by targeted mutation analysis, sequence analysis, or duplication/deletion analysis.11–14
If genetic analysis fails to show evidence of CHM mutations, immunoblot analysis with anti-REP-1 antibody can show the absence of REP-1 protein in peripheral lymphocytes.15 In our patient, given karyotypic evidence of a translocation at the Xq21 CHM gene site, DNA analysis was not needed to explain the phenotype. Anti-REP-1 immunoblot analysis might show absence of the REP-1 protein since selective inactivation of the normal X chromosome was present in the peripheral lymphocyte cell lines. However, if the normal X is active in any non-tested cell lines, the immunoblot analysis may not show an absence of REP-1. Currently, CHM gene analysis is availale for identifying a defined mutation and would be necessary for pre-natal or pre-implantation genetic diagnosis. Immunoblot analysis is only available for research purposes.
CONCLUSION
Our patient with ectodermal dysplasia presented with nyctalopia, vision loss, peripheral visual field loss, with evidence of choroiretinal degeneration on fundus examination, FA, and ERG. Her characteristic facial features, hypotrichosis, dental abnormalities, and presence of hypohidrosis in the setting of her ocular disease is consistent with a diagnosis of hypohidrotic ectodermal dysplasia and choroideremia. Karyotyping confirmed the diagnosis by showing a complex translocation of chromosomes X, 1, and 3 involving loci Xq13 (EDA) and Xq21 (CHM). This translocation combined with a non-random inactivation of the normal X-chromosome resulted in the clinical findings of the two disorders.
ACKNOWLEDGMENTS
Supported in part by the Department of Ophthalmology Research Fund and the Norma Lazar Eye Research Grant of The New York Eye & Ear Infirmary.
Footnotes
Declaration of interest: The authors report no conflict of interest. The authors alone are responsible for the content and writing of the paper.
REFERENCES
- 1.MacDonald IM, Russell L, Chan CC. Choroideremia: New Findings from Ocular Pathology and Review of Recent Literature. Surv Ophthalmol. 2009;54:401–407. doi: 10.1016/j.survophthal.2009.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wald KJ, Hirose T. Ectodermal dysplasia. Ectrodactyly, and Clefting Syndrome and Bilateral Retinal Detachments. Arch Ophthalmol. 1993;111:734. doi: 10.1001/archopht.1993.01090060020009. [DOI] [PubMed] [Google Scholar]
- 3.Hayakawa M, Yanashima K, Kato K, Nakajima A, Yamauchi H. Association of ectodermal dysplasia, ectrodactyly, and macular dystrophy: EEM syndrome. Ophthalmic Pediatr Genet. 1989;10:287–92. doi: 10.3109/13816818909009884. [DOI] [PubMed] [Google Scholar]
- 4.Noble K, Carr R, Siegel I. Fluorescein Angiography of the Hereditary Choroidal Dystrophies. Br J Ophthalmol. 1977;61:43–45. doi: 10.1136/bjo.61.1.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wright JT, Grange DK, Richter MK. [accessed January 1, 2010];Hypohidrotic Ectodermal Dysplasia. Available at: http://www.ncbi.nlm.nih.gov.
- 6. [accessed January 1, 2010];Hypohidrotic Ectodermal Dysplasia. Available at: http://ghr.nlm.nih.gov/condition=hypohidroticectodermaldyspl asia.
- 7.Moloney JB, Blake J, Benham B. Regional Choroidal Atrophy and Alopecia. Acta Ophthalmol. 1988;66:272–276. doi: 10.1111/j.1755-3768.1988.tb04596.x. [DOI] [PubMed] [Google Scholar]
- 8.Alory C, Balch WE. Molecular basis for Rab prenylation. J Cell Biol. 2000;150:89–103. doi: 10.1083/jcb.150.1.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mura M, Sereda C, Jablonski MM. Clinical and functional findings in choroideremia due to complete deletion of the CHM gene. Arch Ophthalmol. 2007;125:1107–1113. doi: 10.1001/archopht.125.8.1107. [DOI] [PubMed] [Google Scholar]
- 10.Schwartz M, Rosenberg T. Prenatal diagnosis of choroideremia. Acta Ophthalmol Scand Suppl. 1996;219:33–36. doi: 10.1111/j.1600-0420.1996.tb00381.x. [DOI] [PubMed] [Google Scholar]
- 11.MacDonald IM, Sereda C, McTaggart K, Mah D. Choroideremia Gene Testing. Expert Rev Mol Diagn. 2004;4:478–484. doi: 10.1586/14737159.4.4.478. [DOI] [PubMed] [Google Scholar]
- 12.van den Hurk JA, Schwartz M, van Bokhoven H, et al. Molecular basis of choroideremia (CHM): mutations involving the Rab escort protein-1 (REP-1) gene. Hum Mutat. 1997;9:110–117. doi: 10.1002/(SICI)1098-1004(1997)9:2<110::AID-HUMU2>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 13.McTaggart KE, Tran M, Mah DY, et al. Mutational analysis of patients with the diagnosis of choroideremia. Hum Mutat. 2002;20:189–196. doi: 10.1002/humu.10114. [DOI] [PubMed] [Google Scholar]
- 14.Hayakawa M, Fujiki K, Hotta Y, et al. Visual impairment and REP-1 gene mutations in Japanese choroideremia patients. Ophthalmic Genet. 1999;20:107–115. doi: 10.1076/opge.20.2.107.2285. [DOI] [PubMed] [Google Scholar]
- 15.MacDonald IM, Mah DY, Ho YK, et al. A practical diagnostic test for choroideremia. Ophthalmology. 1998;105:1637–1640. doi: 10.1016/S0161-6420(98)99031-5. [DOI] [PubMed] [Google Scholar]