

Abstract
Interactions of gas embolism bubbles with endothelial cells, as can occur during decompression events or other forms of intravascular gas entry, are poorly characterized. Endothelial cells respond to microbubble contact via mechanotransduction responses that can lead to cell death or aberrant cellular function. Cultured bovine aortic endothelial cells were individually contacted with microbubbles. Cells were loaded with fluorescent dyes indicating calcium- and nitric oxide signaling and cell viability. A surfactant, Pluronic F-127, and/or albumin were added to the culture media. Control experiments utilized calcium-free media as well as probe-poking in place of microbubble contact. We acquired fluorescence microscopy time-lapse images of cell responses to bubble and probe contact and determined contact effects on cell signaling and cell death. Calcium influx was essential for cell death to occur with bubble contact. Bubble contact stimulated extracellular calcium entry without altering nitric oxide levels unless cell death was provoked. Cell responses were independent of bubble contact duration lasting either one or 30 seconds. Microbubble contact provoked cell death over 7 times more frequently than micropipette poking. Albumin and the surfactant each attenuated the calcium response to bubble contact and also reduced the lethality of microbubble contact by 67.4% and 76.0%, respectively, when used alone, and by 91.2% when used together. This suggests that surface interactions between the bubble or probe interface and plasma- and cell surface-borne macromolecules differentially modulate the mechanism of calcium trafficking such that microbubble contact more substantially induces cell death or aberrant cellular function. The surfactant findings provide a cytoprotective approach to mitigate this form of mechanical injury.
Introduction
The vascular endothelium constitutes an important structural interface between the circulation and the adjoining tissues, playing a critical role in mechanosensing the physical environment of the entirety of the vasculature. Endothelial cells (ECs) respond to different levels of hemodynamic shear stress by releasing vasoconstrictors such as endothelin or platelet-activating factors and vasodilators such as nitric oxide (NO) or prostacyclin to modulate and regulate vascular tone. The endothelium is subject to mechanical interactions, with intravascular gas bubbles occurring during episodes of gas embolism.
Gas bubbles are introduced into the body both accidentally and deliberately, posing a health threat regardless of the gas source. Gas embolism occurs in commercial and recreational divers (1) and is an occupational hazard for hyperbaric chamber workers (2). Gas embolism is also pervasive in clinical medicine in a variety of procedures, including cardiac surgery (3) and numerous other clinical interventions. Intra-arterial gas bubbles can transit the vasculature and deposit into the microcirculation of any end organ. Central nervous system gas embolization has been studied most in relation to cardiac surgery (4–6), but it remains a significant concern with decompression events. Neurological deficits commonly associated with cerebrovascular gas embolism include stroke, impaired consciousness, seizures, and cognitive impairment, all of which limit function (7).
Intravascular bubbles occlude vessels, diminish perfusion, and initiate thrombotic and inflammatory pathways (8–12). A common feature of these pathophysiological events is that they result directly from bubble interactions with endothelial cells and blood elements through adhesion events (13–17) and contact with the gas-liquid interface (18–20). In particular, cellular binding and adhesion phenomena are important in understanding cell-bubble interactions and cellular responses to bubble exposure. Minimizing activation of pathophysiological responses evoked by intravascular bubbles may prevent injury from developing. At present, only hyperbaric oxygen is used and is often not feasible for hours after embolism occurrence (21). A potential method of treatment or prevention is to populate the bubble/blood interface with a chemical constituent to render the interface more biologically inert, shielding biological moieties from encountering an adsorptive surface (22,23). This prevents initiation of pathophysiological processes resulting from adverse molecular interactions between the bubble surface, an adsorbed surface layer and the luminal endothelial surface. Surfactants such as perfluorocarbons, polydimethylsiloxanes and polyols have demonstrated biocompatibility and desired effects on gas embolism (15,17,24,25), inflammation (26–28) and interactions with blood components (19,20,29). Surfactants reduce bubble adhesion to the endothelium (13,14), accelerate bubble clearance from the microcirculation (15–17) and attenuate bubble-associated blood-clotting (19) and platelet-binding interactions in vitro (20).
In this study, we investigated effects of microbubble contact-mediated calcium signaling and cell survival using bovine aortic endothelial cells. We also evaluated cytoprotection conferred by introduction of a surfactant to the liquid medium used during cell exposure to bubbles.
Methods
Cell culture preparation and fluorescence dye-loading
We studied bovine aortic endothelial cells (ECs) based on available historical studies of their mechanotransduction responses and calcium signaling following mechanical perturbation (30). ECs were cultured (passages <18) using Dulbecco’s modified Eagle’s medium (Cambrex, East Rutherford, N.J., USA) supplemented with 10% (v/v) heat-inactivated newborn calf serum (Invitrogen Corporation, Carlsbad, Calif., USA), 1% (v/v) L-glutamine (200 mM/L, Invitrogen) and 2% (v/v) penicillin/streptomycin (10,000U penicillin and 10 mg streptomycin per mL, Cambrex). The cells were seeded onto 8.6 cm2 chambered cover glass (Nalge Nunc International, N.Y., USA) at a density of 1 to 104 cells per chamber. Cells were cultured in 95% oxygen and 5% carbon dioxide cells but were not grown long enough to achieve confluence. Instead they were grown for sufficient time so that cell density on the cover slip permitted only one cell to appear in the microscope view field. This permitted clear spatial definition of single cell responses and eliminated any cell-cell signaling. Cells were studied under room air conditions, which was not expected to influence cell responses.
We obtained three fluorescence dyes, Fluo4/AM (Glycine, N-[4-[6-[(acetyloxy)methoxy]-2,7-difluoro-3-oxo-3H-xanthen-9-yl]-2-[2-[2-[bis[2-(acetyloxy)methoxy]-2-oxoethyl]amino]-5-methylphenoxy]ethoxy]phenyl]-N-[2-[(acetyloxy)methoxy]-2-oxoethyl]-,(acetyloxy)methyl ester; excitation/emission 494/516nm), DAF-FM/DA (3-Amino, 4-aminomethyl-2′,7′-difluorofluorescein diacetate; excitation/emission 495/515 nm) and Propidium Iodide (PI, excitation/emission 535/615 nm) from Molecular Probes (Invitrogen, Eugene, Ore., USA). Fluo4/AM and DAF-FM/DA respond to changes in intracellular Ca2+ (31) and nitric oxide (32–34) respectively. PI binds to nuclear DNA and enables distinction to be made between live and dead cells, since it enters only through a breached cell membrane (35).
Cells were washed twice with a HEPES buffer (Invitrogen) and loaded with Fluo-4/AM or DAF-FM DA for one hour. Fluo-4/AM was prepared by dissolving 50 mg of compound into 100 μl of dimethyl sulfoxide and then diluting the mixture with 9.9 ml of the HEPES buffer to a final concentration of 5.0 mM. DAF-FM DA was prepared by dissolving 50 μg of DAF-FM DA into 20 μl dimethyl sulfoxide, which was then diluted with 19.8 mL of the HEPES buffer for a final concentration of 5.0 μM. After dye loading, cells were washed twice with the HEPES buffer, and the well was loaded with 3 mL of the HEPES buffer. In some experiments EDTA was added to the HEPES buffer used for well loading to exclude the presence of extracellular Ca2+. With regard to cell viability experiments, the well was loaded with 3 mL of 500 nM PI in the HEPES buffer.
Microscopy imaging
Bright field and fluorescence images of cells were obtained with an Olympus IX70 inverted microscope (Olympus America, Inc., Melville, N.Y., USA) equipped with a Chroma PhotoFluor illuminator (Chroma Technology Corporation, Rockingham, Vt., USA) and a 40x magnification objective lens and having a numerical aperture of 0.75. Software scripts were run to alternate shuttering between fluorescence (Fluo-4/AM or DAF-FM/DA) and bright light exposures automatically, with a period of 0.5 seconds using Scanalytics IPLab® (Rockville, Md., USA). Images were obtained with a CCD camera (SensiCam QE®, The Cooke Corporation, Romulus, Mich., USA) connected to a personal computer. Approximately 150–600 images for each wavelength were recorded. Reference experiments showed nominal bleaching to occur with the fluorophores used in these studies, and no observable cell injury was found from long light exposures. A FITC filter set was used to acquire images with Fluo-4/AM and DAF-FM/DA for calcium and NO signaling, respectively. A Texas Red filter set was used to acquire PI images to assess cell viability.
Mechanical cell stimulation conditions
An air microbubble was produced at the submerged tip of a glass micropipette mounted on a nanoliter injector (Nanoject®, Drummond Scientific Company, Broomall, Pa., USA). The injector was held in the stage of a micromanipulator (PPM5000 PiezoPatch® Micromanipulator with joystick control, World Precision Instruments, Fla., USA) at a 40° angle relative to the horizontal line. Micropipette tips were pulled to an outer diameter of 50 μm using a micropipette puller (P-97, Sutter Instrument, Novato, Calif., USA). Tips were beveled to a 40° angle using a grinder (EG-44, Narishige, Tokyo, Japan). To produce a microbubble, the micropipette was backfilled with distilled water followed by aspiration of a small volume of air. The tip was viewed under the microscope and positioned in the liquid phase directly above an endothelial cell. The distance between the tip and cell surface was detected using the microscope z-axis focal plane, and it was adjusted to 80–120 μm. An air microbubble having a 0.2–0.5 nl volume (diameter ~70–100 μm) was injected into the liquid solution and retained on the micropipette tip. The ultramicromanipulator position was subsequently adjusted downward slowly in the z-direction to initiate contact between the microbubble and the cell. Figure 1 shows a schematic representation of our ECs in a chamber with a micropipette tip bearing an air microbubble positioned above a single cell (Figure 1A), while Figures 1B and 1C are light photomicrographs of a pipette tip and a tip with a retained bubble, respectively.
Figure 1.

A) Schematic representation of the micropipette tip with a microbubble positioned above an endothelial cell in the culture chamber. Light microscopy photomicrographs of B) micropipette tip immersed in culture media, and C) a microbubble at the end of the micropipette tip.
To study effects of isolated (single) bubble contact with cells, we performed a preliminary experiment to compare the maximum relative intensities of calcium-signaling responses using Fluo4/AM following individual short (one-second) and long (30-second) contact time bubble exposures. Control experiments were conducted using single micropipette tip poke stimulation of short duration. The micropipette tip for control experiments was pulled to an outer diameter of 5–10 μm and ground to a 40° bevel angle. The pipette tip was positioned under the microscope about 50 μm above a cell and then very slowly lowered using the ultramicromanipulator’s continuous displacement mode (resolution; 0.1 μm, velocity; 0.5 μm/sec). Immediately upon cell contact and activation, the pipette motion was stopped and the tip was quickly elevated off the cell surface.
To help isolate direct bubble surface effects and effects of surface active agents on cell calcium-signaling responses, we conducted experiments with a plain buffer as well as a buffer having either protein [5% w/v bovine serum albumin (BSA; Sigma-Aldrich)], surfactant [0.1% w/v Pluronic F-127 (PF127, Invitrogen)] or both protein and surfactant added. In this concentration BSA rapidly (<1 second) adsorbs to, and saturates, the bubble surface (36). Pluronic F-127, a polymer of polyoxyethylene and polyoxypropylene, competes with proteins for bubble surface occupancy, alters the size and distribution of bubbles within the microcirculation, and decreases the adhesion of bubbles to the endothelium (14–16,37). These various conditions provide interfacial surface layers that can have interactions with cells different from those promoted by cell contact with a clean interface. For these experiments we made single short-duration microbubble contact, and for comparison we conducted control micropipette tip contact experiments in a plain HEPES buffer. Some experiments were also conducted with calcium chelation from the extracellular medium to identify specific effects of intracellular calcium release and entry of extracellular calcium as the source of intracellular calcium signaling generated by microbubble and micropipette contact.
To determine effects of microbubble contact on NO production, we conducted short-duration microbubble and control micropipette contact experiments using ECs loaded with DAF-FM/DA. Experimental conditions included use of the same plain, protein- and surfactant-containing buffers described above and buffers to which the NO substrate L-Arginine (Sigma-Aldrich, St. Louis, Mo., USA) had been added at 1mM concentration. We continuously observed ECs for 60 minutes after contact. To confirm that the DAF-FM/DA dye worked well under these conditions, we added 0.1 μM Carbachol (Sigma-Aldrich), which stimulates EC NO production, 0.1 μM Carbachol + 1 mM L-NAME (Sigma-Aldrich), an inhibitor of NO synthase, or 10 μM adenosine triphosphatase (ATP), which is an activator of calcium signal pathways, to the extracellular media. The measured resultant relative intensity of the NO signal remained stable in control cells and increased in the Carbachol and ATP groups, with L-NAME inhibiting NO production, as shown in Figure 2. Thus, we are assured that the DAF-FM/DA dye performed adequately in the experiments.
Figure 2.

Fluorescence response of endothelial cells loaded with Fluo4/AM for calcium detection during A) control time period; B) with 0.2 mM Carbachol exposure; C) with 10 μM ATP exposure and D) with 0.2 mM Carbachol plus 100 mM L-NAME exposure.
To assess cell viability following microbubble contact we performed both short duration bubble and control micropipette contact experiments on ECs loaded with PI. Buffers used the same as those used in the previous studies. Viability assessment was conducted 15 minutes after the contact was initiated. The PI dye was considered to be positive (i.e., cell death had occurred) if the mean relative intensity (F/F0) in nucleus was greater than or equal to 5.
During decompression, showers of bubbles may continue to occur, resulting in repeated or successive gaseous emboli passage through the vasculature. To study effects of repeated bubble contact with cells, we compared the maximum relative intensities of both calcium and NO-signaling responses and evidence of cell viability following two short contact time bubble or control pipette exposures. Based on results of the preliminary contact time experiment, only short-duration stimuli were used. The separation time between the first and second bubble or pipette contact was 15 minutes in all cases. For these experiments we compared the relative maximum signal intensity following the second exposure to that response generated by the first (baseline) exposure. Buffer conditions were as described earlier.
Data presentation and statistics
Recorded microscopy images were compiled into a time-lapse video format for off-line data analysis. Within each time-lapse video, a region of interest (ROI) of a targeted single cell was selected to measure localized fluorescence intensity as a function of time (IPLab® scripts by BioVision Technologies, Exton, Pa., USA). For experiments with Fluo4/AM and DAF-FM/DA, a 5 μm diameter perinuclear ROI in the cytoplasm was chosen within each cell. For PI studies, the cell nucleus was selected as the ROI. Relative fluorescence intensity data F/F0 were derived in which F was the time-dependent intensity in the specific region of interest and F0 was the baseline intensity at time zero. Ratios were exported to Microsoft Excel spreadsheets for statistical analysis and graphing.
For all the analysis we excluded all cases in which the EC cell membrane was immediately broken by bubble or probe contact. Results for all included experiments are presented as mean ± standard deviation. The paired Student’s t-Test was used to determine the significance of the comparison between the peak fluorescence response following successive microbubble contacts. Group variances were examined using the F-Test (for two group comparisons) or the Bartlett Test (for more than two group comparisons). For equal variances statistical significance was established using the unpaired Student’s t-Test about two group comparisons. For two or more group comparisons, one-factor analysis of variance (ANOVA) followed by Bonferroni’s Multiple Comparison Test was used. For unequal variances, the t-Test for unequal variances was used. The Chi-square (χ2) Test for independence was used for the PI cell viability analysis. P < 0.05 was considered statistically significant.
Results
Both microbubble and micropipette contact activated EC calcium-signaling pathways and elicited a wave of calcium progressing through the cell. Figure 3 shows fluorescence microscopy images of an endothelial cell loaded with Fluo4/AM before and after microbubble contact. Figure 3A displays the cell prior to contact. The cytoplasm appears dark and almost homogenous. Figure 3B was acquired a few seconds after microbubble contact. The increase in fluorescence intensity indicates an increase in calcium concentration in the small area touched by the microbubble. Figure 3C, obtained ~10 seconds after microbubble contact, demonstrates that the fluorescence intensity has increased throughout the cytoplasm as the calcium wave has attained near-maximal intensity. A filamentous construction localizing the calcium is evident in the bright regions of the cell’s interior. Figure 3D showing the cell ~120 seconds after microbubble contact illustrates that intracellular calcium has been taken up into storage organelles, but the intracellular concentration profile has not returned to the pre-contact baseline. Figure 3E graphically demonstrates the calcium wave determined using the prescribed ROI, with the four successive time points indicated on the plot corresponding to Figure 3A–D. The calcium flux represented in Figure 3 was absent in experiments conducted without calcium present in the extracellular medium, confirming that the calcium wave observed is due to extracellular calcium influx into cells following cell stimulation by either micropipette or microbubble contact.
Figure 3.
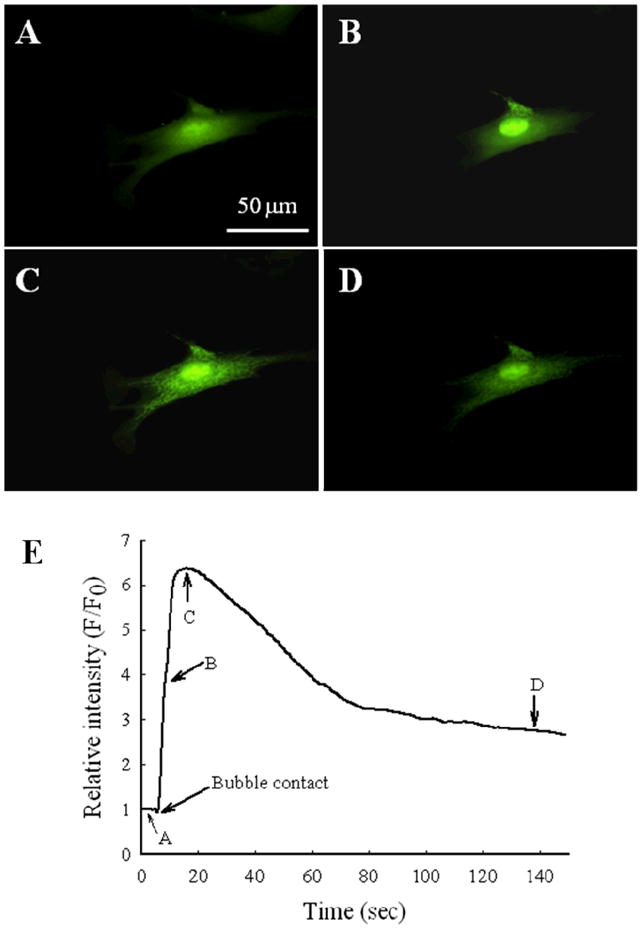
Examplar fluorescence microscopy images of an endothelial cell loaded with Fluo4/AM for calcium detection. Images were acquired A) prior to microbubble contact; B) shortly after microbubble contact; C) ~10 sec after microbubble contact; and D) ~120 sec after the bubble contact. E) The time-lapse history of the relative fluorescence intensity within the region of interest. Time points 1–4 correspond to images in panels A–D, respectively.
The results for the basic experiment of single EC stimulation by probe poking or microbubble contact using a HEPES buffer are shown in Figure 4. The maximum relative intensity of the calcium response was not different between the short- and long-duration microbubble contact groups, and these were not different from the result obtained for micropipette contact. Based on these findings, only short-duration stimuli were used in subsequent investigation.
Figure 4.

Maximum relative fluorescence intensity for calcium signaling following microbubble and micropipette contact. Stimuli were short duration (n = 34), long duration (n = 19) microbubble contact and micropipette contact (n = 24).
The presence of both BSA and PF127, either individually or added together in the buffer, did influence calcium signaling following microbubble, but not micropipette, contact. Figure 5 shows results of the maximum fluorescence intensity elicited from ECs loaded with Fluo4/AM following short-duration microbubble and pipette stimulation in the HEPES buffer and in the buffer with BSA, PF127 and both compounds present. With regard to bubble stimulation, the maximum relative intensity was significantly less with either the surfactant or the protein present by themselves or together (P < 0.01 for all cases). There were no significant differences in the results obtained for micropipette stimulation for the various cell media conditions. There were no differences between the maximal intensities after bubble and pipette stimulations for identical cell media conditions.
Figure 5.

Maximum relative fluorescence intensity for endothelial cell calcium signaling following microbubble or micropipette contact in plain HEPES buffer and buffer with BSA, PF-127 or both added (n = 24 per group). * P < 0.05 compared to maximum relative fluorescence intensity following stimulation of cells in plain buffer.
The lethality of bubble contact was highly dependent on the cell media conditions. Without calcium present in the extracellular media, bubble contact did not elicit cell death, indicating that calcium entry is an essential component of the mechanism of bubble-induced injury. In the presence of extracellular calcium, cell viability after microbubble contact depended on the presence of protein and/or surfactant in the culture media, as shown in Figure 6. A large fraction (38.3%) of cells undergoing microbubble contact in the HEPES buffer became PI-positive. This was reduced by 67.4% to 12.5% (P < 0.05) becoming PI-positive if BSA were added to the cell media, reduced by 76.0% to 9.2% (P < 0.05) becoming PI-positive if PF127 were added to the cell media and reduced by 91.2% to 3.4% (P < 0.05) becoming PI-positive if both BSA and PF127 were added to the cell media. By comparison, 5.3% of cells became PI-positive following micropipette contact in the HEPES buffer.
Figure 6.

Effect of PF-127 and 5% BSA and microbubble or micropipette contact on endothelial cell viability using Propidium Iodide to assess cell death (n ≥ 40 for all groups). * P < 0.01 compared to microbubble contact in plain HEPES buffer.
Within the 60-minute observation window if a cell died, we observed the appearance of a spike (~55% increase over baseline) and then an abrupt decline (~ 60% decrease below baseline) in the associated fluorescence signal for ECs loaded with DAF-FM/DA. An example for a single sentinel event is shown in Figure 7. However, if cells did not die, no change in the NO levels was detected using DAF-FM/DA. This result was independent of whether or not L-Arg was present in the buffer.
Figure 7.

Relative fluorescence intensity for nitric oxide signaling following microbubble contact, as associated with occurrence of cell death.
In Figures 8 and 9 the group means and standard deviations are shown for the normalized second measurements of maximum relative intensities for calcium- and nitric oxide signaling, respectively, generated by successive bubble contacts. The normalization scheme used for calcium signaling was to calculate the ratio of the maximum fluorescence achieved with the second and first stimuli, using the first maximum response as the baseline. The normalization scheme used for nitric oxide signaling was to measure the mean fluorescence during a 60-second time period that began 30 seconds after bubble contact, and then to calculate the ratio of the mean fluorescence achieved with the second and first stimuli, using the first signaling response as the baseline. The maximum relative intensity following the second bubble contact was significantly lower than that elicited by the first bubble contact for all four of the experimental conditions (P <0.05 in all cases), whereas the signaling from the second pipette stimulus was no different from the initial response. Both the calcium- and nitric oxide-signaling responses to successive microbubble contacts were more robust and better preserved with PF127 present, regardless of whether BSA was included in the buffer (P <0.05 for both cases). The addition of BSA to the buffer did not alter the latent response from that achieved using the buffer alone.
Figure 8.

Maximum relative fluorescence intensity for endothelial cell nitric oxide signaling following repeated microbubble contact (n = 16 per group). * P < 0.05 compared to maximum relative fluorescence intensity following first stimulus. ** P < 0.05 compared to maximum relative fluorescence elicited for cells in plain buffer.
Figure 9.

Maximum relative fluorescence intensity for endothelial cell calcium signaling following repeated microbubble contact (n = 16 per group). * P < 0.05 compared to maximum relative fluorescence intensity following first stimulus. ** P < 0.05 compared to maximum relative fluorescence elicited for cells in plain buffer.
Discussion
This is the first study demonstrating that microbubble contact activates isolated cultured endothelial cell calcium-signaling pathways and provokes cell death in vitro. Endothelial cells regulate a variety of physiological functions, including blood-tissue permeability, blood coagulation, angiogenesis and vessel repair, vascular tone and some immune responses (38). Many aspects of endothelial cell function depend highly on modulation of the intracellular Ca2+ concentration, whose regulation is important for transmission of cell-surface activation signals. Intracellular Ca2+ concentration is increased and the endothelium is activated by mechanical stimulation, including shear stress (39) and physical perturbation (30), as well as by hypoxia (40). Large alterations in calcium homeostasis can signal apoptosis or necrosis, with various signaling systems becoming overwhelmed by massive calcium release (41). Without causing cell death, the endothelial cell response to bubbles can induce temporary dysfunction, leading to sufficiently aberrant physiology as to effect organ function. This can depend on chemo- and mechanotransduction beginning with ECs sensing the luminal environment, including temporal aspects of the underlying shear stress profile. Recently we have shown that gas bubble passage through the microcirculation induces travel of a biphasic shear stress wave axially along the cell membrane (18,42–45). The shear stress rapidly switches direction, leading to fast force dynamics that can markedly compress and then stretch the cell membrane locally. This work further indicates that the presence of a surfactant markedly attenuates the magnitude of the shear stress gradients and therefore should reduce the mechanical stimulation cells endure (18). This is consistent with the observations we have made in the present experiments as well as our prior in vivo findings (15,17,20,46).
As intracellular Ca2+ levels rise, calmodulin binding increases. The Ca2+-calmodulin complex activates calmodulin kinase, which phosphorylates proteins (38) into their active forms. Phosphorylation activates endothelial nitric oxide synthase (eNOS) (47), which increases NO synthesis to produce vasodilation. We regularly observed a prolonged hyperemic phase of vasodilation and loss of vessel tone regulation following bubble passage through the arteriolar microcirculation (15–17,46). This indicates an embolism-induced alteration in vessel reactivity and may result from activation of Ca2+ - and NO-dependent pathways by bubble transit through the vessel that is not discernible from these experiments with contact stimulation of single cells.
While the activation of ECs by microbubble contact appears to be very similar to that provoked by micropipette tip contact, there are important differences manifested by effects of a surfactant and/or the protein added to the media and on induction of cell death. The visual phenomena associated with the fluorescence indicator of calcium fluxes Fluo4/AM appear characteristically similar for both microbubble and micropipette contact. Beginning at the location of either bubble or pipette contact, the calcium wave proceeded to spread throughout the cytoplasm. The initial brightness within the cytoplasm appeared at first to increase in intensity homogeneously; then bright areas dimmed and discrete, heterogeneous bright regions appeared throughout the cytoplasm. Anticipating that these regions would be mainly endoplasmic reticulum and mitochondria into which Ca2+ reuptake and storage primarily occur (38), we conducted additional experiments using fluorescence trackers specific for these particular organelles. Overlaying images from these double dye-loading experiments enabled us to identify both the endoplasmic reticulum and mitochondria as having structures and locations similar to those of the bright elements present with the Fluo4/AM dye (data not shown). Based on the studies utilizing chemical chelation of calcium from the HEPES buffer, the calcium wave dynamics and any associated linkage to provocation of cell death following microbubble contact are heavily dependent on calcium influx into the cell. The mechanism of calcium entry and subsequent calcium trafficking following bubble contact is likely different from that occurring after probe contact, given that the magnitudes of the calcium fluxes are so similar yet cell death rates are significantly different without the addition of cytoprotective elements including the surfactant.
For these studies we adopted the maximum relative fluorescence intensity with Fluo4/AM as our measure of the degree of activation of the EC calcium-signaling pathway. We limited cell-cell signaling by using cultured cells that had not achieved confluence. Additionally, we were extremely careful in monitoring for the morphological predictors of impending cell demise, such as bleb formations, during all of our experiments. We discarded the cells if such predictors were present and did not conduct experiments with cells out of the incubator beyond three to five hours. Nonetheless, these considerations do not make up for inherent experimental limitations present in our method, including the use of bovine, nonconfluent ECs in vitro, rather than confluent human cells in vivo, the absence of physiological temperature, oxygen tension, exposure to blood and the lack of blood pressure and shear stress exposures expected within the vasculature.
One feature of gas embolism is that bubbles can pass through a vessel or lodge in the vasculature and remain in contact with the endothelium. Thus, to determine the effect of bubble contact duration on endothelial cell response, we studied both a very short, or transient, contact and a long-duration contact. With single-contact stimulation we found that bubble-contact duration had no significant effect on the maximum relative intensity of the calcium wave elicited, although even short-term bubble contact was frequently lethal to cells. This was most likely from calcium overload produced by a lethal stimulus not mimicked by micropipette probe contact. Nontheless, sublethal stimuli may be repeated by multiple embolic events physiologically, and this may have the cumulative effect of producing a lethal injury. In rat cremaster intravital microscopy studies of intravascular gas embolism (15,17,20,46), we have often witnessed repeated bubble passage through a single vessel accompanied by alterations in vasomotor tone regulatory function.
Our experiments of sequential stimuli were performed to determine the effect of repeated, otherwise sublethal, bubble contacts with an endothelial cell in such cases. While we generally found the maximum relative intensity of the calcium and nitric oxide signals produced by the repeated bubble contact was so commonly lethal with only the plain buffer or buffer with BSA added, it was also a challenge to complete sufficient non-lethal cases to assess statistical results. This occurred despite a single bubble contact not being instantly lethal. We determined that the baseline absolute fluorescence intensity values just prior to the second cell contact were not significantly different from those before the initial contact within any of the double-contact groups (P <0.05 by paired t-Test, data not shown). This indicates that the intracellular calcium concentration within the ROI had already decreased to its pre-stimulus baseline level when the second contact occurred, although the response to the second stimulus was muted. The mechanism of this desensitization is not clear, but Sigurdson et al., (30) have previously reported the occurrence of desensitization in calcium-signaling pathway activation following mechanical perturbation. Its association with cell lethality is heretofore unknown.
We have hypothesized that surface interactions between the bubble interface and plasma-and cell surface-borne macromolecules activate specific biochemical pathways which lead to cell death or aberrant cellular function, and these studies contribute to our understanding of these phenomena. Blood-borne macromolecules, particularly proteins, adsorb rapidly onto gas–liquid interfaces (48,49), where they have mechanical implications for producing adhesion between bubble and cell surfaces (13,46,50). The addition of either PF-127 or albumin (or both) to the culture media attenuated calcium signaling responses and rendered microbubble contact less lethal. In the absence of calcium in the extracellular media, bubble contact could not be considered lethal. These findings strongly suggest that calcium entry into the cell is essential for the provocation of cellular demise, and that the gas–liquid interface plays a key role in eliciting the calcium-signaling response. These findings amplify previous results showing that surface-active agents attenuate gas-embolism induced endothelial injury in excised vessels (14) by demonstrating a protective effect of surface adsorption of large-molecule agents at the level of the individual cell. While the detailed mechanism of interaction between the bubble and cell surfaces and the resultant cell response has not been established, these new findings show the bubble interface to be an important mediator of the endothelial cell pathophysiology in response to gas embolism. Manipulation of the interface by the addition of the surfactant confers distinct protection from cell death and alters the cell response to contact with an embolism bubble.
We consider that microbubble contact provides a unique stimulus that induces normal and shear stresses through liquid motion. Surface binding interactions between the cell and bubble may readily be affected by competition between adsorbed proteins and macromolecules on the endothelial cell surface to occupy the bubble interface. Bubble contact effects on cell viability may result from bubble retraction from the cell surface, causing tugging or tearing free of macromolecules bridging from the cell surface to the bubble interface. We acknowledge that direct comparison with in vivo events will depend on the state of the glycocalyx, which may not have the same structure or thickness as that present on our cultured cells.
We anticipated that the NO pathway would be activated following activation of the calcium-signaling pathway (47), yet we did not detect any such obvious occurrence except in those cases in which the cell died. The reason is not clear, but it is possible that the NO pathway is easily affected by many other factors precisely because of its downstream location. While cells appeared to be healthy morphologically after bubble contact, intracellular function was clearly altered by the bubble contact. Evidence for this includes our findings that repeated bubble contact was highly lethal with a plain buffer or a buffer with BSA alone, whereas single contacts were not, and that a less intense calcium wave was always elicited by a second bubble contact. We did confirm that the DAF/FM DA dye detected physiologically produced NO. Although ATP is a strong activator of calcium pathway, the associated increase in relative intensity of DAF/FM DA we observed was about 40%. Mizuno et al., (51) showed that EC intracellular calcium concentration [Ca2+]i elevation regulates NO production, but the peak [Ca2+]i elevation- or the integrated [Ca2+]i elevation-NO production relationships varies depending on the type of agonists. Thus it is also possible that a microbubble stimulation produced [Ca2+]i elevation without any accompanying significant elevation of NO production. However, we were able to detect changes in NO when the stimulus was lethal to cells.
We conclude that microbubble contact activated EC calcium-signaling via mechanotransduction events mediated by the gas liquid interface. Microbubble contact readily killed cells in the presence of extracellular calcium, and this was markedly attenuated by the presence of the surfactant in the buffer. Surface interactions between the bubble interface and the cell surface are therefore implicated in cell death and aberrant cellular function produced by microbubble contact. Targeting the mechanism of bubble-induced endothelial cell injury in advance of, or during decompression events provides a potential pharmacological approach to preventive therapy for gas embolism.
Acknowledgments
This work was supported by ONR grant N00014-08-1-0436 and NIH grant R01 HL067986.
References
- 1.Smerz RW. Concomitant cerebral and coronary arterial gas emboli in a sport diver: a case report. Hawaii Med J. 2005;64:12–3. [PubMed] [Google Scholar]
- 2.Risberg J, Englund M, Aanderud L, Eftedal O, Flook V, Thorsen E. Venous gas embolism in chamber attendants after hyperbaric exposure. Undersea Hyperb Med. 2004;31:417–29. [PubMed] [Google Scholar]
- 3.Abu-Omar Y, Balacumaraswami L, Pigott DW, Matthews PM, Taggart DP. Solid and gaseous cerebral microembolization during off-pump, on-pump, and open cardiac surgery procedures. J Thorac Cardiovasc Surg. 2004;127:1759–65. doi: 10.1016/j.jtcvs.2003.09.048. [DOI] [PubMed] [Google Scholar]
- 4.Moody DM, Bell MA, Challa VR, Johnston WE, Prough DS. Brain microemboli during cardiac surgery or aortography. Ann Neurol. 1990;28:477–86. doi: 10.1002/ana.410280403. [DOI] [PubMed] [Google Scholar]
- 5.Challa VR, Moody DM, Troost BT. Brain embolic phenomena associated with cardiopulmonary bypass. J Neurol Sci. 1993;117:224–31. doi: 10.1016/0022-510x(93)90177-z. [DOI] [PubMed] [Google Scholar]
- 6.Mangano CM. Scuds, scads, and other air-borne hazards. Anesthesiology. 1997;87:476–8. doi: 10.1097/00000542-199709000-00004. [DOI] [PubMed] [Google Scholar]
- 7.Cheung AT, Stecker MM. Neurologic complications of cardiac surgery. Progress in Anesthesiology. 1998;12:3–20. [Google Scholar]
- 8.Albertine KH, Wiener-Kronish JP, Koike K, Staub NC. Quantification of damage by air emboli to lung microvessels in anesthetized sheep. J Appl Physiol. 1984;57:1360–8. doi: 10.1152/jappl.1984.57.5.1360. [DOI] [PubMed] [Google Scholar]
- 9.Clark RE, Brillman J, Davis DA, Lovell MR, Price JR, Magovern GJ. Microemboli during coronary artery bypass grafting. J Thorac Cardiovasc Surg. 1995;109:249–58. doi: 10.1016/S0022-5223(95)70386-1. [DOI] [PubMed] [Google Scholar]
- 10.Philp RB, Inwood MJ, Warren BA. Interactions between gas bubbles and components of the blood: implications in decompression sickness. Aerospace Med. 1972;43:946–53. [PubMed] [Google Scholar]
- 11.Warren BA, Philp RB, Inwood MJ. The ultrastructural morphology of air embolism: Platelet adhesion to the interface and endothelial damage. Br J Exp Path. 1973;54:163–72. [PMC free article] [PubMed] [Google Scholar]
- 12.Muth CM, Shank ES. Gas embolism. N Engl J Med. 2000;342:476–82. doi: 10.1056/NEJM200002173420706. [DOI] [PubMed] [Google Scholar]
- 13.Suzuki A, Eckmann DM. Embolism bubble adhesion force in excised perfused microvessels. Anesthesiology. 2003;99:400–8. doi: 10.1097/00000542-200308000-00022. [DOI] [PubMed] [Google Scholar]
- 14.Suzuki A, Armstead SC, Eckmann DM. Surfactant reduction in embolism bubble adhesion and endothelial damage. Anesthesiology. 2004;101:97–103. doi: 10.1097/00000542-200407000-00016. [DOI] [PubMed] [Google Scholar]
- 15.Eckmann DM, Lomivorotov VN. Microvascular gas embolization clearance following perfluorocarbon administration. J Appl Physiol. 2003;94:860–8. doi: 10.1152/japplphysiol.00719.2002. [DOI] [PubMed] [Google Scholar]
- 16.Eckmann DM, Kobayashi S, Li M. Microvascular embolization following polidocanol microfoam sclerosant administration. Dermatol Surg. 2005;31:636–43. doi: 10.1111/j.1524-4725.2005.31605. [DOI] [PubMed] [Google Scholar]
- 17.Branger AB, Eckmann DM. Accelerated arteriolar gas embolism reabsorption by an exogenous surfactant. Anesthesiology. 2002;96:971–9. doi: 10.1097/00000542-200204000-00027. [DOI] [PubMed] [Google Scholar]
- 18.Swaminathan TN, Ayyaswamy PS, Mukundakrishnan K, Eckmann DM. Effect of a soluble surfactant on a finite sized bubble motion in a blood vessel. J Fluid Mech. 2010;642:509–39. doi: 10.1017/S0022112009992692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eckmann DM, Diamond SL. Surfactants attenuate gas embolism-induced thrombin production. Anesthesiology. 2004;100:77–84. doi: 10.1097/00000542-200401000-00015. [DOI] [PubMed] [Google Scholar]
- 20.Eckmann DM, Armstead SC, Mardini F. Surfactant reduces platelet-bubble and platelet-platelet binding induced by in vitro air embolism. Anesthesiology. 2005;103:1204–10. doi: 10.1097/00000542-200512000-00015. [DOI] [PubMed] [Google Scholar]
- 21.Moon RE. Bubbles in the brain: what to do for arterial gas embolism? Crit Care Med. 2005;33:909–10. doi: 10.1097/01.ccm.0000159726.47336.f8. [DOI] [PubMed] [Google Scholar]
- 22.Miller R, Fainerman VB, Makievski AV, Krägel J, Grigoriev DO, Kazakov VN, Sinyachenko OV. Dynamics of protein and mixed protein/surfactant adsorption layers at the water/fluid interface. Adv Colloid Interface Sci. 2000;86:39–82. doi: 10.1016/s0001-8686(00)00032-4. [DOI] [PubMed] [Google Scholar]
- 23.Kragel J, Wustneck R, Husband F, Wilde PJ, Makievski AV, Grigoriev DO, Li JB. Properties of mixed protein/surfactant adsorption layers. Colloids Surf B Biointerfaces. 1999;12:399–407. [Google Scholar]
- 24.Cavanagh DP, Eckmann DM. The effects of a soluble surfactant on the interfacial dynamics of stationary bubbles in inclined tubes. J Fluid Mech. 2002;469:369–400. [Google Scholar]
- 25.Eckmann DM, Cavanagh DP. Bubble detachment by diffusion-controlled surfactant adsorption. Colloids Surf A. 2003;227:21–33. [Google Scholar]
- 26.Edwards CM, Lowe H, Trabelsi H, Lucas P, Cambon A. Novel fluorinated surfactants for perfluorochemical emulsification: biocompatibility assessments of glycosidic and polyol derivatives. Art Cells, Blood Subs and Immob Biotech. 1997;25:327–33. doi: 10.3109/10731199709118922. [DOI] [PubMed] [Google Scholar]
- 27.Kalman PG, McCullough DA, Ward C. Evacuation of microscopic air bubbles from dacron reduces complement activation and platelet aggregation. J Vasc Surg. 1990;11:591–8. [PubMed] [Google Scholar]
- 28.Paustian PW, McPherson JC, 3d, Haase RR, Runner RR, Plowman KM, Ward DF, Nguyen TH, McPherson JCJ. Intravenous Pluronic F-127 in early burn wound treatment in rats. Burns. 1993;19:187–91. doi: 10.1016/0305-4179(93)90146-y. [DOI] [PubMed] [Google Scholar]
- 29.Edwards CM, Heptinstall S, Lowe KC. F-68 inhibits agonist-induced platelet aggregation in human whole blood in vitro. Art Cells, Blood Subs and Immob Biotech. 1998;26:441–7. doi: 10.3109/10731199809117464. [DOI] [PubMed] [Google Scholar]
- 30.Sigurdson WJ, Sachs F, Diamond SL. Mechanical perturbation of cultured human endothelial cells causes rapid increases of intracellular calcium. Am J Physiol. 1993;264:H1745–H1752. doi: 10.1152/ajpheart.1993.264.6.H1745. [DOI] [PubMed] [Google Scholar]
- 31.Scheenen WJJM, Hofer AM, Pozzan T. In: Intracellular measurement of calcium using fluorescent probes, Cell Biology: a laboratory handbook. 2. Celis JE, editor. New York: Academic Press; 1998. pp. 363–74. [Google Scholar]
- 32.Kojima H, Nakatsubo N, Kikuchi K, Urano Y, Higuchi T, Tanaka J, Kudo Y, Nagano T. Direct evidence of NO production in rat hippocampus and cortex using a new fluorescent indicator: DAF-2 DA. Neuroreport. 1998;9:3345–8. doi: 10.1097/00001756-199810260-00001. [DOI] [PubMed] [Google Scholar]
- 33.Itoh Y, Ma FH, Hoshi H, Oka M, Noda K, Ukai Y, Kojima H, Nagano T, Toda N. Determination and bioimaging method for nitric oxide in biological specimens by diaminofluorescein fluorometry. Anal Biochem. 2000;287:203–9. doi: 10.1006/abio.2000.4859. [DOI] [PubMed] [Google Scholar]
- 34.Suzuki N, Kojima H, Urano Y, Kikuchi K, Hirata Y, Nagano T. Orthogonality of calcium concentration and ability of 4,5-diaminofluorescein to detect NO. J Biol Chem. 2002;277:47–9. doi: 10.1074/jbc.M108195200. [DOI] [PubMed] [Google Scholar]
- 35.Wisnoskey BJ, Estacion M, Schilling WP. Maitotoxin-induced cell death cascade in bovine aortic endothelial cells: divalent cation specificity and selectivity. Am J Physiol Cell Physiol. 2004;287:C345–C356. doi: 10.1152/ajpcell.00473.2003. [DOI] [PubMed] [Google Scholar]
- 36.Hansen FK, Myrvold R. The kinetics of albumin adsorption to the air/water interface measured by automatic axisymmetric drop shape analysis. J Colloid Interface Sci. 1995;176:408–17. doi: 10.1006/jcis.1998.5745. [DOI] [PubMed] [Google Scholar]
- 37.Eckmann DM, Zhang J, Lampe J, Ayyaswamy PS. Surfactant based intervention to eliminate gas embolism during long duration space based activity. Ann NY Acad Sci. 2006;1077:256–69. doi: 10.1196/annals.1362.039. [DOI] [PubMed] [Google Scholar]
- 38.Tran QK, Ohashi K, Watanabe H. Calcium signalling in endothelial cells. Cardiovasc Res. 2000;48:13–22. doi: 10.1016/s0008-6363(00)00172-3. [DOI] [PubMed] [Google Scholar]
- 39.Ando J, Komatsuda T, Kamiya A. Cytoplasmic calcium response to fluid shear stress in cultured vascular endothelial cells. In Vitro Cell Dev Biol. 1988;24:871–7. doi: 10.1007/BF02623896. [DOI] [PubMed] [Google Scholar]
- 40.Berna N, Arnould T, Remacle J, Michiels C. Hypoxia-induced increase in intracellular calcium concentration in endothelial cells: role of the Na(+)-glucose cotransporter. J Cell Biochem. 2001;84:115–31. doi: 10.1002/jcb.1271. [DOI] [PubMed] [Google Scholar]
- 41.Orrenius S, Zhivotovsky B, Nicotera P. Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol. 2003;4:552–65. doi: 10.1038/nrm1150. [DOI] [PubMed] [Google Scholar]
- 42.Mukundakrishnan K, Quan SP, Eckmann DM, Ayyaswamy PS. Numerical study of wall effects on buoyant gas-bubble rise in a liquid-filled finite cylinder. Phys Rev E. 2007;76:036308, 1–15. doi: 10.1103/PhysRevE.76.036308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mukundakrishnan K, Ayyaswamy PS, Eckmann DM. Finite-sized gas bubble motion in a blood vessel: Non-Newtonian effects. Phys Rev E. 2008;78:036303, 1–15. doi: 10.1103/PhysRevE.78.036303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mukundakrishnan K, Eckmann DM, Ayyaswamy PS. Bubble Motion through a Generalized Power-Law Fluid Flowing in a Vertical Tube. Ann NY Acad Sci. 2009;1161:256–67. doi: 10.1111/j.1749-6632.2009.04089.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mukundakrishnan K, Ayyaswamy PS, Eckmann DM. Bubble Motion in a Blood Vessel: Shear Stress Induced Endothelial Cell Injury. J Biomech Eng. 2009;131:074516, 1–5. doi: 10.1115/1.3153310. [DOI] [PubMed] [Google Scholar]
- 46.Branger AB, Eckmann DM. Theoretical and experimental intravascular gas embolism absorption dynamics. J Appl Physiol. 1999;87:1287–95. doi: 10.1152/jappl.1999.87.4.1287. [DOI] [PubMed] [Google Scholar]
- 47.Schneider JC, El Kebir D, Chereau C, Lanone S, Huang XL, Buys Roessingh AS, Mercier JC, Dall’Ava-Santucci J, Dinh-Xuan AT. Involvement of Ca2+/calmodulin-dependent protein kinase II in endothelial NO production and endothelium-dependent relaxation. Am J Physiol Heart Circ Physiol. 2003;284:H2311–H2319. doi: 10.1152/ajpheart.00932.2001. [DOI] [PubMed] [Google Scholar]
- 48.Porter MR. Use of Surfactant Theory. London: Blackie Academic & Professional; 1994. pp. 26–92. [Google Scholar]
- 49.Rosen MJ. Characteristic Features of Surfactants. New York: John Wiley & Sons; 1978. pp. 1–24. [Google Scholar]
- 50.Lindner JR, Ismail S, Spotnitz WD, Skyba DM, Jayaweera AR, Kaul S. Albumin microbubble persistence during myocardial contrast echocardiography is associated with microvascular endothelial glycocalyx damage. Circulation. 1998;98:2187–94. doi: 10.1161/01.cir.98.20.2187. [DOI] [PubMed] [Google Scholar]
- 51.Mizuno O, Kobayashi S, Hirano K, Nishimura J, Kubo C, Kanaide H. Stimulus-specific alteration of the relationship between cytosolic Ca(2+) transients and nitric oxide production in endothelial cells ex vivo. Br J Pharmacol. 2000;130:1140–6. doi: 10.1038/sj.bjp.0703420. [DOI] [PMC free article] [PubMed] [Google Scholar]