

Abstract
To date, molecular genetic analyses have identified over 500 distinct DNA variants in five disease genes associated with familial Parkinson disease; α-synuclein (SNCA), parkin (PARK2), PTEN-induced putative kinase 1 (PINK1), DJ-1 (PARK7), and Leucine-rich repeat kinase 2 (LRRK2). These genetic variants include ∼82% simple mutations and ∼18% copy number variations. Some mutation subtypes are likely underestimated because only few studies reported extensive mutation analyses of all five genes, by both exonic sequencing and dosage analyses. Here we present an update of all mutations published to date in the literature, systematically organized in a novel mutation database (http://www.molgen.ua.ac.be/PDmutDB). In addition, we address the biological relevance of putative pathogenic mutations. This review emphasizes the need for comprehensive genetic screening of Parkinson patients followed by an insightful study of the functional relevance of observed genetic variants. Moreover, while capturing existing data from the literature it became apparent that several of the five Parkinson genes were also contributing to the genetic etiology of other Lewy Body Diseases and Parkinson-plus syndromes, indicating that mutation screening is recommendable in these patient groups. Hum Mutat 31:763–780, 2010. © 2010 Wiley-Liss, Inc.
Keywords: Parkinson disease, genetic etiology, database, SNCA, PARK2, PINK1, PARK7, LRRK2
Introduction
Parkinson disease (PD) is the second most common progressive neurodegenerative brain disorder. It affects 1 to 2% of the population above 65 years and its prevalence increases to approximately 4% in those above 85 years. As these demographic age groups are growing rapidly due to general aging of the population and increasing lifespans, neurodegenerative diseases will represent an ever-growing social and economic burden for society. Through time, the scientific view on PD etiology has changed dramatically. Due to the observation that only 15 to 20% of PD patients have a clear positive family history of PD, researchers predicted that the majority of the PD patients have a complex etiology, including both a genetic and environmental component. During the last 2 decades, molecular genetic analyses in PD families provided important insights in disease mechanisms underlying PD pathology. Nine genes that contribute to the genetic etiology of familial PD were identified through positional cloning strategies in inherited PD patients and families [Bonifati et al., 2003; Di Fonzo et al., 2009; Kitada et al., 1998; Lautier et al., 2008; Paisan-Ruiz et al., 2004, 2009; Polymeropoulos et al., 1997; Ramirez et al., 2006; Valente et al., 2004a; Zimprich et al., 2004a]. Two more PD genes, UCH-L1 and HTRA2, were identified based on the functional relevance of their corresponding protein to PD pathogenesis [Leroy et al., 1998a, b; Strauss et al., 2005]. Although follow-up genetic studies are inconsistent for some of these genes or conclusive data are still pending, ample evidence for a causal association was obtained for PD with five genes, that is, α-synuclein (SNCA; MIM] 163890), parkin (PARK2; MIM] 602544), PTEN-induced putative kinase 1 (PINK1; MIM] 608309), DJ-1 (PARK7; MIM] 602533), and Leucine-rich repeat kinase 2 (LRRK2; MIM] 609007). Extensive mutation screening of these five causal genes revealed both simple mutations (missense, nonsense, silent, splice site, and untranslated region (UTR) mutations, small insertions and deletions (indels), and copy number variations (CNVs) leading to PD. Approximately 330 confirmed or possible pathogenic mutations in over 1,900 families have been identified so far (Supp. Tables S1-S5; PDmutDB database: http://www.molgen.ua.ac.be/PDmutDB). Possible pathogenic mutations include non-synonymous variants, splice site variants or variants in UTRs that were not observed in control individuals. In this mutation update we present the DNA variants identified so far and elaborate on their clinical and biological relevance. We also discuss the importance of a new publicly available and extensively curated database PDmutDB, and the implications of these analyses for mutation analyses in a diagnostic setting.
Major Genes and Proteins
Autosomal Dominant PD Genes
α-Synuclein
SNCA was the first causal PD gene identified segregating a pathogenic missense mutation—p.Ala53Thr—in a large Italian family (“Contursi”) (MIM 163890) [Polymeropoulos et al., 1996, 1997] (Table 1 and Fig. 1). The 144aa SNCA protein encoded by the three different SNCA transcripts is typically found as a natively unfolded, soluble protein in the cytoplasm or associated with lipid membranes [Davidson et al., 1998] (Table 2). The exact biological function of SNCA in brain is still not fully understood, although there is evidence that implicates SNCA in neurotransmitter release and vesicle turnover at the presynaptic terminals [Abeliovich et al., 2000; Liu et al., 2004].
Table 1.
Overview of the FiMe Major PD Genes
| Mutation spectrum | ||||||||
|---|---|---|---|---|---|---|---|---|
| Gene | MIM number | Inheritance | Position | Gene size | Number of exons | Transcript length | Classic mutations | Copy number variations |
| SNCA | 163890 | AD | 4q21 | 112 kb | 6 | 1,543 bp | Missense (0.9%) | Whole gene duplication and triplication (0.6%) |
| LRRK2 | 609007 | AD | 12q12 | 144 kb | 51 | 9,225 bp | Missense (18.2%) | — |
| PARK2 | 602544 | AR | 6q26 | 1.38 Mb | 12 | 4,073 bp | Nonsense, frameshift (indels and splice site), missense (32.4%) | Single or multiple exon deletions and duplications (15.8%) |
| PINK1 | 608309 | AR | 1p35-36 | 18 kb | 8 | 2,660 bp | Nonsense, framshift (indels), missense (24.7%) | Single or multiple exon deletions; whole gene deletion (1.2%) |
| PARK7 or DJ-1 | 602533 | AR | 1p36 | 34 kb | 7 | 961 bp | Missense (4.4%) | Single or multiple exon deletions and duplications (1.2%) |
(%) Number of (possible) pathogenic mutations for this gene/total number of (possible) pathogenic mutations.
Figure 1.

Representation of SNCA on genomic and transcript level. All three transcripts coding for the same protein SNCA are depicted (t1: NM 001146055.1 /t2: NM_000345.2/t3: NM_007308.2). On transcript level exons are colored alternately.
Table 2.
Features of the Proteins Coded by the Five Major Genes
| Gene | Protein | Number of aa | Functional domains | (Putative) function |
|---|---|---|---|---|
| SNCA | α-synuclein | 144 aa | – | Neurotransmitter release |
| LRRK2 | LRRK2 | 2,527 aa | Ank (ankyrin-like), LRR (leucine rich repeat), Roc (Ras-of-complex proteins), COR (C-terminal of Roc), Kinase, WD40 | – |
| PARK2 | Parkin | 465 aa | UBL (ubiquitin-like), RING1, IBR (in-between-ring), RING2 | Target proteins for degradation, maintenance mitochondrial function |
| PINK1 | PINK1 | 581 aa | Target sequence, kinase | Oxidative stress response, maintenance mitochondrial function |
| PARK7 or DJ-1 | DJ-1 | 189 aa | – | Redox sensor, antioxidant |
Mutations in SNCA are rather rare and explain disease in ∼2.5% of known unrelated affected carriers (see Supp. Tables S1-1 and S1-2 for mutations, PDmutDB for all references: http://www.molgen.ua.ac.be/PDmutDB). Apart from the Italian Contursi family, p.Ala53Thr was also identified in several families of Greek descent [Athanassiadou et al., 1999; Papadimitriou et al., 1999; Polymeropoulos et al., 1996, 1997; Spira et al., 2001]. More recently, p.Ala53Thr was also detected in two other unrelated families from Asia and Sweden [Choi et al., 2008; Ki et al., 2007; Puschmann et al., 2009] as well as in one seemingly sporadic PD patient of Polish origin [Michell et al., 2005]. With only two other missense mutations identified in SNCA—p.Ala30Pro [Kruger et al., 1998] and p.Glu46Lys [Zarranz et al., 2004] (see Supp. Table S1-1)—both also located in the N-terminus of the protein, the missense mutation frequency of SNCA in different populations remains very low. In 2003, a triplication of the wild-type SNCA locus was observed in a large multigenerational family [Singleton et al., 2003], instigating the discovery of SNCA multiplications in several other families with PD and related LBD disorders (see Supp. Table S1-2 for mutations, PDmutDB for all references: http://www.molgen.ua.ac.be/PDmutDB) [Chartier-Harlin et al., 2004; Fuchs et al., 2007; Ibanez et al., 2004, 2009; Ikeuchi et al., 2008; Nishioka et al., 2006, 2009; Nuytemans et al., 2009]. Several of these dosage studies attempted to delineate the boundaries of the multiplicated genomic region identified in families or shared between unrelated carriers. Most SNCA multiplicated regions appeared in different genomic sizes (see Supp. Table S1-2), suggestive of independent mutational events. Few studies, however, reported equally sized duplicated or triplicated regions surrounding SNCA amongst different families or within branches of the same family [Fuchs et al., 2007; Nishioka et al., 2009]. Of interest is that SNCA duplications were also reported in four apparently sporadic PD patients [Ahn et al., 2008; Nishioka et al., 2009; Nuytemans et al., 2009].
Leucine-rich repeat kinase 2 or dardarin
The leucine-rich repeat kinase 2 gene (LRRK2) was the second causal gene linked to autosomal dominant inherited PD (MIM] 609007) [Funayama et al., 2002; Paisan-Ruiz et al., 2004; Zimprich et al., 2004a, 2004b] (Table 1 and Fig. 2). Its transcript contains 51 exons coding for the LRRK2 protein [Paisan-Ruiz et al., 2004] (Table 2). LRRK2 comprises several functional domains suggestive of on the one hand a kinase activity dependent on the GTPase function of the Roc domain and on the other hand a scaffold protein function implied by the multiple protein–protein interaction regions (Fig. 2). Of interest is that LRRK2 was shown to form dimers under physiological conditions [Greggio et al., 2008]. The exact biological function of LRRK2 remains largely unknown, because no physiological substrates have been identified so far.
Figure 2.
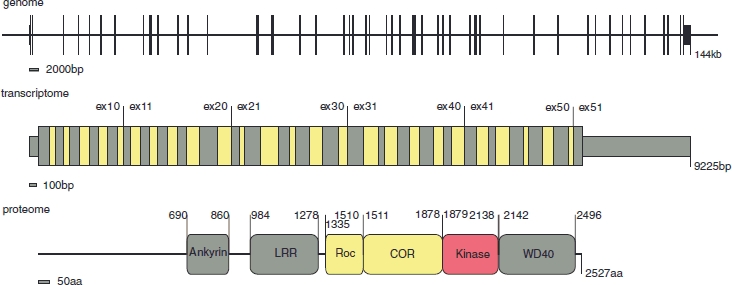
Representation of LRRK2 on genomic and transcript level and the functional domains of the LRRK2 protein. On transcript level exons are colored alternately (NM_198578.2). (LRR: leucine-rich repeat; Roc: Ras-of-complex protein; COR: C-terminal of Roc.)
The first two publications of PD associated mutations in LRRK2 described four different pathogenic missense mutations segregating in families of European and North-American origin [Paisan-Ruiz et al., 2004; Zimprich et al., 2004a]. Subsequent mutation analyses identified about 80 discrete missense mutations in over a 1,000 families and sporadic patients worldwide (see Supp. Table S2 for mutations, PDmutDB for all references: http://www.molgen.ua.ac.be/PDmutDB). This corresponds to about 50% of all reported unrelated carriers of mutations in the five major genes, making LRRK2 the most frequently mutated PD gene so far (Table 3 and PDmutDB: http://www.molgen.ua.ac.be/PDmutDB). The 80 missense mutations are located over the entire LRRK2 protein and affect all predicted functional domains. Some mutations, though, have much higher frequencies than others, for example, p.Gly2019Ser and mutations altering codon Arg1441. Unfortunately, because of the large number of coding exons, only a minority of studies performed mutation analyses of the complete coding region. Most studies focused instead on those exons coding for functional relevant protein domains, namely, Roc, COR, and kinase domains (Fig. 2). Only three studies included dosage analyses aiming at detecting CNVs but did not detect LRRK2 multiplications or deletions [Mata et al., 2005b; Nuytemans et al., 2009; Paisan-Ruiz et al., 2008]. Nonetheless, rare CNVs of LRRK2 or parts thereof cannot be excluded, before more dosage studies have been performed for LRRK2.
Table 3.
Relative Frequencies of Mutation Categories Dependent on Ethnicity and Familial History
| SNCA (%) | LRRK2 (%) | PARK2 (%) | PINK1 (%) | PARK7 (%) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ethnic origin | Classic | CNV | Classic | CNV | Classic | mixed | CNV | Classic | CNV | Classic | CNV | |
| Caucasian | F | 4.13 | 2.07 | 67.36 | 0 | 10.12 | 3.51 | 7.44 | 3.93 | 0.21 | 0.83 | 0.41 |
| S | 0.99 | 0.33 | 52.48 | 0 | 18.15 | 2.97 | 11.88 | 10.89 | 0.33 | 0.99 | 0.66 | |
| Asian | F | 1.01 | 8.08 | 9.09 | 0 | 9010 | 9010 | 42.42 | 17.17 | 0 | 3.03 | 0 |
| S | 0 | 3.13 | 10.42 | 0 | 28.13 | 1.04 | 38.54 | 17.71 | 1.04 | 0 | 0 | |
| Arab | F | 0 | 0 | 88.61 | 0 | 1.27 | 1.27 | 3.80 | 3.80 | 1.27 | 0 | 0 |
| S | 0 | 0 | 97.06 | 0 | 1.47 | 0 | 0.74 | 0 | 0 | 0.74 | 0 | |
| Latin-American | F | 0 | 0 | 57.14 | 0 | 14.29 | 4.76 | 23.81 | 0 | 0 | 0 | 0 |
| S | 0 | 0 | 41.67 | 0 | 41.67 | 0 | 8.33 | 0 | 8.33 | 0 | 0 | |
| Ashkenazi Jews | F | 0 | 0 | 100.00 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| S | 0 | 0 | 98.04 | 0 | 0 | 0 | 0 | 0 | 0 | 1.96 | 0 | |
(%) Number of unrelated mutation carriers with this category of mutation/total number of unrelated mutation carriers (for each ethnicity and familial history). Each row of this table equals 100%.
An important observation is that the LRRK2 mutation frequency is seemingly dependent on the ethnicity of the population analysed. For example, the most frequent mutation with a strong founder effect—p.Gly2019Ser—was reported worldwide with an average frequency of 1% in PD patients [Paisan-Ruiz, 2009]. But, in Arab Berber and Ashkenazi Jewish populations the p.Gly2019Ser frequency was significantly higher (20 and 40%, respectively) [Lesage et al., 2006; Ozelius et al., 2006], whereas in the first comprehensive screening of a Belgian population, p.Gly2019Ser was apparently absent [Nuytemans et al., 2008]. Other codons in LRRK2 are also frequently mutated as a consequence of numerous independent mutational events. The p.Arg1441 codon constitutes a mutation hotspot with three different codon substitutions: p.Arg1441Cys, p.Arg1441Gly, and p.Arg1441His. The relatively high mutation frequencies of these mutations should be approached with some caution though, because underlying founder effects have been reported. The most frequent mutation p.Gly2019Ser is observed on a limited number of haplotypes. Also, p.Arg1441Gly was transmitted from a common founder in the Basque population [Gaig et al., 2006; Gonzalez-Fernandez et al., 2007; Gorostidi et al., 2009; Mata et al., 2005c; Paisan-Ruiz et al., 2004; Simon-Sanchez et al., 2006] while p.Arg1441Cys was observed worldwide on several different founder haplotypes [Di Fonzo et al., 2006a; Gaig et al., 2006; Goldwurm et al., 2005; Gosal et al., 2007; Haugarvoll et al., 2008; Hedrich et al., 2006b; Nuytemans et al., 2008; Pankratz et al., 2006a; Tan et al., 2006a]. Additionally, several missense mutations seemed to be (nearly) private mutations for Asian populations: p.Arg1628-Pro, p.Pro755Leu, and p.Gly2385Arg [An et al., 2008; Di Fonzo et al., 2006b; Farrer et al., 2007; Fung et al., 2006b; Ross et al., 2008; Tan et al., 2007, 2008, 2009; Tomiyama et al., 2008].
In contrast to other PD genes, mutations in LRRK2 have a relatively high frequency of up to 2% in sporadic, late-onset PD patients [Di Fonzo et al., 2005; Gilks et al., 2005; Nichols et al., 2005; Tomiyama et al., 2006]. The high mutation frequency in both familial and sporadic patients makes LRRK2 the most frequently mutated gene of the five major PD genes. Some prudence in interpreting data is warranted though. Some of the missense mutations have also been reported in healthy control individuals, raising questions on the biological role of these rare variants in disease [Meeus et al., 2010]. The highly variable onset ages associated with LRRK2 mutations [Hernandez et al., 2005; Kachergus et al., 2005; Paisan-Ruiz et al., 2005; Zimprich et al., 2004a], the presence of LRRK2 mutations in unaffected individuals [Carmine Belin et al., 2006; Di Fonzo et al., 2006a; Gaig et al., 2006; Hernandez et al., 2005; Kay et al., 2005; Khan et al., 2005b; Latourelle et al., 2008; Nichols et al., 2005; Zimprich et al., 2004a], and the high frequency in sporadic patients render the assessment of pathogenicity of the identified variants extremely difficult as these issues complicate segregation analyses. To date, pathogenicity supported by segregation analyses has only been demonstrated for six LRRK2 mutations (p.Arg1441Cys, p.Arg1441Gly, p.Tyr1699Cys, p.Gly2019Ser, and p.Ile2020Thr).
Autosomal recessive PD genes
PARK2 or parkin
The first of three recessive PD genes identified is PARK2 (MIM 602544), which was linked with disease in a nuclear Japanese consanguineous family [Kitada et al., 1998] (Table 1 and Fig. 3). PARK2 spans approximately 1.38 Mb and encodes the protein parkin. The 456 amino acid protein harbors four major functional domains corresponding to its function as an E3 ubiquitin ligase (Table 2) [Imai et al., 2000; Shimura et al., 2000; Zhang et al., 2000]. Its role in the ubiquitin proteasome system (UPS) comprises of tagging dysfunctional or excessive proteins for degradation. Further, it was shown that under physiological conditions parkin is involved in mitochondrial maintenance [Deng et al., 2008a; Exner et al., 2007; Park et al., 2009; Poole et al., 2008; Weihofen et al., 2009] and might induce subsequent autophagy of dysfunctional mitochondria [Narendra et al., 2008, 2009].
Figure 3.
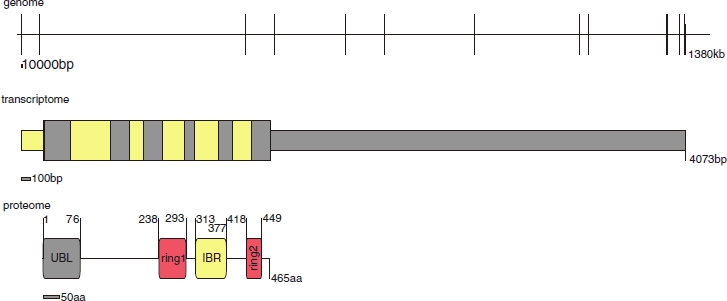
Representation of PARK2 on genomic and transcript level and the functional domains of the parkin protein. On transcript level exons are colored alternately (NM_004562.2). (UBL: ubiquitin-like; IBR: in-between-ring.)
The first mutation reports indicated a wide spectrum of loss-of-function mutations in PARK2 including simple mutations like nonsense, missense and splice site mutations, indels, as well as CNVs of the promoter region and single or multiple exons (Table 2) [Hattori et al., 1998a, b; Kitada et al., 1998]. PARK2 mutations were identified spread across the entire gene in either homozygous, compound heterozygous or heterozygous state in familial and sporadic patients from different ethnicities (see Supp. Table S3-1 and S3-2 for mutations, PDmutDB for all references: http://www.molgen.ua.ac.be/PDmutDB). Heterozygous PARK2 variants have also been observed in healthy control individuals, making assessment of pathogenicity for these variants quite complex. Approximately 40% of unrelated mutation carriers were reported to harbor a mutation in PARK2 (Table 3 and PDmutDB: http://www.molgen.ua.ac.be/PDmutDB). Of these, close to 8% carry both a simple mutation as a CNV, whereas carriers of only simple mutations or CNVs are almost equally common (43.8% vs. 47.9%). Investigation of the haplotypes on which frequent PARK2 mutations reside, showed that most CNVs are independent events, whereas point mutations were more commonly transmitted from common founders [Periquet et al., 2001]. This suggests that the high mutation frequency in PARK2 is only partly due to small founder effects.
P-TEN-induced putative kinase 1
Homozygosity mapping in PARK2 negative European families led to the identification of the second autosomal recessive gene, P-TEN induced putative kinase 1 (PINK1; MIM] 608309) [Valente et al., 2001, 2002, 2004a] (Table 1 and Fig. 4). The PINK1 protein is a putative serine/threonine kinase involved in mitochondrial response to cellular and oxidative stress [Valente et al., 2004a] (Table 2). This response is likely mediated by regulation of the calcium efflux, influencing processes such as mitochondrial trafficking [Wang and Schwarz, 2009; Weihofen et al., 2009], ROS formation, mitochondrial respiration efficacy [Liu et al., 2009], and opening of the mitochondrial permeability transition pore [Gandhi et al., 2009] as well as by interaction with cell death inhibitors and chaperones [Plun-Favreau et al., 2007; Pridgeon et al., 2007; Wang et al., 2007]. In addition, PINK1 is an important player in the alleged PINK1/parkin pathway, regulating mitochondrial morphology and functionality in response to stressors [Deng et al., 2008a; Exner et al., 2007; Park et al., 2009; Poole et al., 2008; Weihofen et al., 2009].
Figure 4.
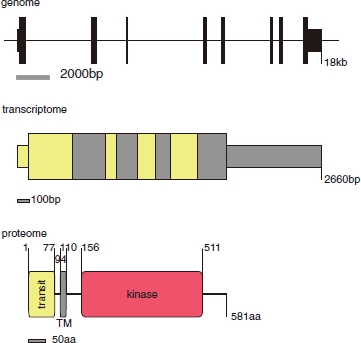
Representation of PINK1 on genomic and transcript level and the functional domains of the PINK1 protein. On transcript level exons are colored alternately (NM_032409.2). (TM: transmembranair.)
The PINK1 mutation spectrum involves nonsense and missense mutations, indels, and whole-gene or single/multiple exon CNVs (Table 2) located across the entire gene. Mutation analyses in familial as well as sporadic patients identified homozygous and compound heterozygous mutations (see Supp. Table S4-1 for mutations, PDmutDB for all references: http://www.molgen.ua.ac.be/PDmutDB). Approximately 6.5% of known mutation carriers carry a mutation in PINK1 (Table 3). Again, many putative pathogenic mutations were also observed in heterozygous state in familial and sporadic patients as well as in healthy control individuals [Abou-Sleiman et al., 2006; Bonifati et al., 2005; Brooks et al., 2009; Choi et al., 2008; Djarmati et al., 2006; Fung et al., 2006a; Healy et al., 2004; Klein et al., 2005; Kumazawa et al., 2008; Mellick et al., 2009; Nuytemans et al., 2009; Rogaeva et al., 2004; Tan et al., 2005, 2006b; Valente et al., 2004b; Weng et al., 2007]. With the current available mutation data, it seems that CNVs in PINK1 are less common than simple loss-of-function mutations (see Supp. Table S4-2). But at this stage we cannot exclude that this observation represents an ascertainment bias because many studies did not perform PINK1 dosage analyses and therefore might have missed CNVs in their patient groups.
PARK7 or DJ-1
The third autosomal recessive PD gene, PARK7 (or DJ-1; MIM 602533) was identified by homozygosity mapping in an extended Dutch family with multiple consanguinity loops [Bonifati et al., 2003; van Duijn et al., 2001] (Table 1 and Fig. 5). The DJ-1 protein was found to be H2O2 responsive suggesting that DJ-1 represents a sensor for oxidative stress, for example, dopamine toxicity [Lev et al., 2009], and acts as an antioxidant [Mitsumoto and Nakagawa, 2001] (Table 2). It was further hypothesized that DJ-1 could be part of a novel E3 ligase complex together with parkin and PINK1 [Xiong et al., 2009].
Figure 5.
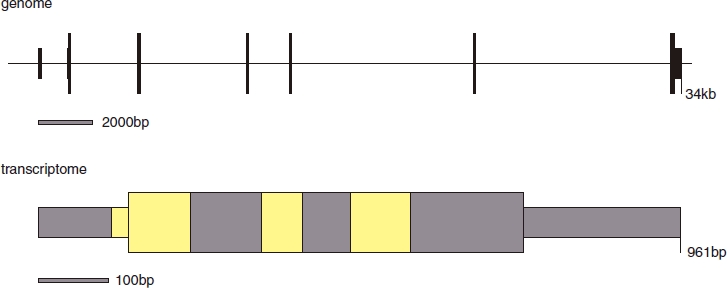
Representation of PARK7 on genomic and transcript level. On transcript level exons are colored alternately (NM_007262.4).
Mutation analyses identified homozygous, compound heterozygous as well as heterozygous [Bonifati et al., 2003; Clark et al., 2004; Hague et al., 2003; Hedrich et al., 2004a; Nuytemans et al., 2009] missense mutations and CNVs in patients (see Supp. Tables S5-1 and S5-2 for mutations, PDmutDB for all references: http://www.molgen.ua.ac.be/PDmutDB). Also for PARK7, heterozygous variants were observed in control individuals. Mutations in PARK7 are reported near 1% of all known mutation carriers (Table 3). Current mutation data indicates that CNVs in PARK7 are less frequent than simple mutations. But, because of the rarity of mutations in PARK7, most studies have not analysed their PD patient groups, making it highly likely that putative pathogenic mutations have been missed and that the current mutation frequency of PARK7 is an underestimate.
Clinical Implications
Clinical features of PD patients typically include tremor, bradykinesia, rigidity, good levodopa response, and/or postural instability. Interestingly, PD is part of a wide Lewy Body Diseases (LBD) spectrum made up by closely related clinical phenotypes characterized by variable manifestation of parkinsonism and dementia (PD, PD with dementia [PDD], Dementia with LB (DLB), LB variant of Alzheimer's disease [AD] and AD). On the other hand, parkinsonism can be accompanied by additional atypical features defining the parkinson-plus syndromes, like multiple system atrophy (MSA; dysautonomia and/or cerebellar signs), progressive supranuclear palsy (PSP; impaired vertical eye movements and prominent postural instability) and corticobasal degeneration (CBD; apraxia). The clinical features reported in literature are mostly typical for disorders of the LBD spectrum. In some cases, however, more atypical features indicative of other related diseases, such as the Parkinson-plus syndromes were observed. This indicates there is a high variability in phenotypes associated by mutations in SNCA, LRRK2, PARK2, PINK1, and PARK7.
Here we summarize typical and atypical presentations of specific mutation groups and discuss some of its implications. This and more detailed information on familial, individual, and clinical data can be found in the newly constructed and publicly available PDmutDB database (http://www.molgen.ua.ac.be/PDmutDB).
SNCA is the only one of the five genes in which an obvious correlation can be made between distinct missense mutations or distinct CNVs and the resulting different phenotypes. The majority of the familial PD patients carrying the SNCA missense mutations p.Ala53Thr or p.Ala30Pro typically present with bradykinesia and rigidity at an early onset age (< 55years) [Bostantjopoulou et al., 2001; Ki et al., 2007; Kruger et al., 1998; Papapetropoulos et al., 2001, 2003; Puschmann et al., 2009; Spira et al., 2001]. The sporadic Polish patient carrying p.Ala53Thr, however, showed typical PD features, that is, late onset at 74 years, rigidity, progressive bradykinesia, and mild tremor [Michell et al., 2005]. Also, clinical features in carriers of the third SNCA missense mutation p.Glu46Lys are atypical in such that these carriers present with symptoms at later age and suffer from dementia within several years after PD onset [Zarranz et al., 2004]. Brain pathology in one p.Glu46Lys carrier showed diffuse LB consistent with a diagnosis of DLB confirming the atypical clinical presentation [Zarranz et al., 2004]. Also, patients carrying SNCA multiplications present with atypical forms of the disease. A direct correlation between phenotype and number of SNCA copies was consistently observed among different studies. Most duplication carriers present with late-onset parkinsonism [Chartier-Harlin et al., 2004; Fuchs et al., 2007; Ibanez et al., 2004, 2009; Nishioka et al., 2006, 2009], which can be accompanied by a later onset cognitive decline (PDD) [Nishioka et al., 2006, 2009; Nuytemans et al., 2009]. Triplication carriers however seem to be more severely affected suffering from a more aggressive form of dementia despite their shorter disease duration (DLB) [Farrer et al., 2004; Ibanez et al., 2009; Singleton et al., 2003]. Also, asymptomatic carriers have been reported in families of both seemingly sporadic and familial PD patients [Ahn et al., 2008; Ibanez et al., 2009; Nishioka et al., 2006, 2009]. Only few of these carriers have exceeded the onset age of the proband [Ibanez et al., 2009; Nishioka et al., 2009], indicating variable onset ages or reduced penetrance for this mutation. When considering all unaffected duplication carriers that are older than the average onset age of the affected carriers as true asymptomatic individuals a crude estimate of 85% penetrance could be obtained from the information in PDmutDB (http://www.molgen.ua.ac.be/PDmutDB). Interestingly, one study describing both duplication and triplication of SNCA in two separate branches of the same family, also reported clinical features reminiscent of MSA (orthostatic hypotension and poor levodopa response) in both branches [Fuchs et al., 2007].
Typically, patients carrying LRRK2 missense mutations present with clinical features similar to those of idiopathic PD, that is, asymmetrical late onset, bradykinesia, rigidity, tremor, and good l-dopa response. The incidence of tremor, however, seems to be elevated in LRRK2 carriers indicating that LRRK2 mutations most likely lead to tremor-dominant disease [Haugarvoll et al., 2008; Nuytemans et al., 2008; Paisan-Ruiz et al., 2004]. On the other hand, isolated studies have also reported LRRK2 mutations in carriers with a clinical diagnosis of sporadic PD with late-onset AD as well as CBD, PSP, or frontotemporal dementia (FTD) [Chen-Plotkin et al., 2008; Santos-Reboucas et al., 2008; Spanaki et al., 2006].
Clinical features of PARK2 homozygous mutation carriers are generally indistinguishable from those of idiopathic PD patients with the exception of a clear drop in onset age. Typically PARK2 patients present with disease onset before the age of 50 years and a slow disease progression [Abbas et al., 1999; Khan et al., 2005a; Lucking et al., 2000]. Although they respond well to levodopa treatment they are more likely to develop treatment-induced motor complications earlier in the treatment [Deng et al., 2008b; Khan et al., 2005a; Lucking et al., 2000]. Further, PARK2 mutations were also identified in patients with a clinical diagnosis of PSP, PD plus essential tremor (ET), as well as ET and restless legs syndrome (RLS) [Adel et al., 2006; Deng et al., 2007; Limousin et al., 2009; Pellecchia et al., 2007; Pigullo et al., 2004; Sanchez et al., 2002].
Homozygous PINK1 mutation carriers are clinically indistinguishable from homozygous PARK2 mutation carriers [Bentivoglio et al., 2001; Valente et al., 2004a]. Although rare, PINK1 mutations were also associated with late-onset PD, RLS with parkinsonism, and dopa-responsive dystonia [Gelmetti et al., 2008; Leutenegger et al., 2006; Tan et al., 2005, 2006b]. Further, a few PINK1 homozygous mutation carriers also presented with cognitive and psychiatric problems in addition to parkinsonism [Ephraty et al., 2007; Reetz et al., 2008; Savettieri et al., 2008].
Clinical features of carriers with a homozygous mutation in the recessive PARK7 gene are also similar to those of homozygous PARK2 and PINK1 carriers [Bonifati et al., 2003]. Also here, clinical heterogeneity with a wide range of clinical phenotypes among unrelated and related carriers was reported. For example, in one family segregating two distinct homozygous variations were diagnosed with early onset parkinsonism, dementia, and amyo-trophic lateral sclerosis (ALS) [Annesi et al., 2005]. In addition, the initially reported 14 kb deletion of the 50 region of PARK7 linked to typical PD was also observed heterozygously in two dementia patients without signs of parkinsonism [Arias et al., 2004].
The available clinical data showed us that mutations in these five PD genes are not only present in patients but also in patients diagnosed with related disorders. Some clinical features are known to overlap between these disorders, so clinical diagnoses may not always be accurate or different disorders might share a common etiology. In both cases, it might be worthwhile screening for mutations in “PD-associated-genes” in larger groups of patients with clinical diagnoses related to PD to further explore the genotype-phenotype correlations. Alternatively, no information was provided on mutation analyses of additional genes so other currently unknown mutations might still explain this range of clinical features for these patients.
When discussing genotype-phenotype correlations, one needs to take into account that at times it can be difficult to comprehend the clinical implications of some genetic variants. Although homozygous mutations in the recessive genes have a penetrance of 100% with only two carriers older than the onset age of affected relatives reported in literature (PARK2 p.Trp74fsCysX8 [Pineda-Trujillo et al., 2001] and PARK7 p.Glu64Asp [Hering et al., 2004]), the effect of heterozygous mutations is far less clear. The presence of these mutations in PARK2, PINK1, and PARK7 has instigated a debate on the role of heterozygous recessive mutations as risk factors for disease. In many studies the prevalence of these heterozygous rare variants is (significantly) higher in patients than in control individuals (PARK2: [Brooks et al., 2009; Clark et al., 2006; Lesage et al., 2008; Nuytemans et al., 2009; Sun et al., 2006]/PINK1: [Abou-Sleiman et al., 2006; Bonifati et al., 2005; Brooks et al., 2009; Marongiu et al., 2008; Rogaeva et al., 2004; Valente et al., 2004b]), implying that the presence of a heterozygous recessive mutation might increase the carrier's susceptibility to develop PD. In addition, several families reported affected heterozygous family members of a homozygous proband creating a false impression of dominant inheritance and indicating a possible predisposition of PARK2 or PINK1 variants to PD (PARK2: [Maruyama et al., 2000; Munhoz et al., 2004; Tan et al., 2003]/PINK1: [Criscuolo et al., 2006; Djarmati et al., 2006; Hedrich et al., 2006a; Ibanez et al., 2006]). Investigation of clinical features in patients with digenic combinations of heterozygous mutations might provide us with more insight in the effects of these variants (Table 4). For example, the clinical presentation and onset of PD does not differ between patients carrying a heterozygous LRRK2 mutation and patients carrying a digenic combination of LRRK2 and PARK2 mutations [Bras et al., 2008; Ferreira et al., 2007; Gao et al., 2009; Illarioshkin et al., 2007; Lesage et al., 2006; Marras et al., 2010]. Illiaroshkin and coworkers, though, reported early occurrence of dyskinesias during treatment, more common in PARK2 mutation carriers, in a LRRK2/PARK2 digenic mutation carrier [Illarioshkin et al., 2007]. Reports of carriers with digenic mutations of two recessive genes are rare, mostly because many mutation studies reported so far have not analyzed all five PD genes. One study describing patients carrying a single heterozygous PINK1 mutation on top of a homozygous PARK2 mutation indicated, nevertheless, that these patients present with a significant earlier onset age than patients carrying only PARK2 mutations [Funayama et al., 2008]. This suggested that heterozygous PINK1 mutations might indeed effect the development of PD, although more research into their biological role is warranted.
Table 4.
Relative Frequencies of Homozygotes or Compound Heterozygotes and Digenic Combinations Dependent on Ethnicity and Familial History
| Ethnic origin | Homozygotes (%) | Compound heterozygotes (%) | Digenic combinations (%) | ||||
|---|---|---|---|---|---|---|---|
| Caucasian | F | 10.33 | LRRK2, PARK2, PINK1, and DJ-1 | 8.06 | LRRK2, PARK2, and PINK1 | 0.20 | LRRK2-PARK2 |
| S | 8.58 | PARK2, PINK1, and DJ-1 | 6.60 | LRRK2, PARK2, and PINK1 | 1.65 | LRRK2-PARK2 | |
| Asian | F | 41.41 | SNCA, PARK2, PINK1, and DJ-1 | 22.22 | PARK2 and PINK1 | 1.01 | PINK1-DJ-1 |
| S | 38.54 | PARK2, and PINK1 | 6.25 | PARK2 | 0 | ||
| Arab | F | 50.63 | LRRK2, PARK2, and PINK1 | 1.27 | PARK2 | 0 | |
| S | 13.97 | LRRK2, PARK2, and DJ-1 | 0 | 0 | |||
| Latin-American | F | 23.81 | PARK2 | 14.29 | PARK2 | 0 | |
| S | 16.67 | LRRK2 and PINK1 | 25.00 | PARK2 | 0 |
(%) Number of unrelated mutation carriers with this category of mutation/total number of unrelated mutation carriers (for each ethnicity and familial history).
Biological and Pathological Relevance
The pathology in PD brain generally consists of progressive neuronal depigmentation and dopaminergic cell loss in the substantia nigra, accompanied by presence of LB in the residual neurons (Table 5). Interestingly, the LB are common to all disorders in the LBD spectrum, although their location in the patient's brain can help specify the exact disorder. In nondemented PD patients the LB are usually confined to the brainstem, whereas more widespread cortical LB point to PDD or DLB. It is not fully understood yet how mutations in the causal PD genes might cause such pathology. Because SNCA is the main constituent of LB [Baba et al., 1998], many studies have tried elucidating the biological processes that trigger SNCA aggregation. Direct investigation of SNCA itself has provided evidence that mutant SNCA has a greater tendency to acquire a misfolded conformation [Conway et al., 2000; Cookson, 2005; Kazantsev and Kolchinsky, 2008], stabilized by oligomerisation [Uversky et al., 2001a, b]. But overexpression of wild-type SNCA produces the same effect by triggering a shift from natively unfolded SNCA to small oligomers due to concentration burden [Kazantsev et al., 2008; Uversky et al., 2001b]. Aggregation of SNCA has been shown to be neurotoxic for the cell through the formation of intermediate aggregates called protofibrils [Conway et al., 2000; Spillantini et al., 1998]. Because of their conformation these protofibrils can bind lipid membranes and cause membrane permeabilization. It is suspected that LB sequester these protofibrils as part of a defense mechanism of the cell against toxic effects [Bodner et al., 2006; Kazantsev and Kolchinsky, 2008]. Although a few studies reported the presence of LRRK2 in ubiquitin-positive inclusion bodies [Greggio et al., 2006; Perry et al., 2008], it is generally perceived that LRRK2 does not reside in LB in affected brains. Interestingly though, associated LRRK2 pathology comprises variable lesions; (diffuse) LBs and/or PSP-like tau aggregation or none of the above [Zimprich et al., 2004a], suggesting that LRRK2 dysfunction might be an upstream event in neurodegeneration and causing disturbances in different pathways. The biological function of LRRK2, however, is still largely unknown. Mutations in the kinase domain of LRRK2 (i.e., p.Gly2019Ser and p.Ile2020Thr) were reported to increase kinase activity [Anand et al., 2009; Gloeckner et al., 2006; Greggio et al., 2006; Guo et al., 2007; Imai et al., 2008; West et al., 2005, 2007], but these results were based on autophosphorylation or phosphorylation of heterologous substrates, warranting caution in interpreting these data. The mutations in the Roc domain, the GTPase regulating the kinase domain, are suspected to impair the function of the GTPase, therefore inducing sustained kinase activity of LRRK2 [Guo et al., 2007; Lewis et al., 2007; Li et al., 2007]. Furthermore, mutations like the substitutions at codon p.Arg1441 and p.Arg1442Pro are located at key positions for the formation of functional LRRK2 dimers; possibly also resulting in a decreased GTPase activity [Gotthardt et al., 2008]. As the exact functions of the other domains in LRRK2 in relation to kinase activity are unclear, it is difficult to assess the impact of mutations in these domains.
Table 5.
Overview of Pathology Associated with Mutations in the Five Different PD Genes
| Gene | Pathology | Reference(s) |
|---|---|---|
| SNCA | Typical LB disease | [Spira et al., 2001] |
| Brainstem and cortical LB and neuritic staining | [Farrer et al., 2004; Fuchs et al., 2007; Gwinn-Hardy et al., 2000; Ikeuchi et al., 2008; Obi et al., 2008; Wakabayashi et al., 1998; Waters and Miller, 1994; Zarranz et al., 2004] | |
| LRRK2 | Typical LB disease | [Giasson et al., 2006; Giordana et al., 2007; Papapetropoulos et al., 2006] |
| Tau-positive pathology without LB | [Gaig et al., 2007; Rajput et al., 2006; Zimprich et al., 2004a] | |
| Nigral degeneration, with neither LB nor NFT | [Dachsel et al., 2007; Gaig et al., 2008; Giasson et al., 2006] | |
| PARK2 | Loss of dopaminergic neurons in SN and LC without LB or NFT pathology | [Gouider-Khouja et al., 2003; Hayashi et al., 2000; Kitada et al., 1998; Sasaki et al., 2004] |
| Typical LB disease | [Pramstaller et al., 2005] | |
| PINK1 | Typical LB disease | [Samaranch et al., 2010] |
| PARK7 or DJ-1 | Remains to be determined |
LB, lewy body; NFT, neurofibrillary tangles; SN, substantia nigra, LC, locus ceruleus.
The proteins encoded by the recessive PD genes are all involved in the cell's response mechanism to cellular and oxidative stress, implying cell dysfunction or increased vulnerability to neurode-generation in patients carrying mutations in these genes. Mutations in PARK2 were reported to impair the E3 ubiquitin ligase activity of parkin [Shimura et al., 2000], which resulted in insufficient protein clearance and the subsequent formation of protein aggregates. On the other hand, PINK1 mutations were shown to interfere with its protein stability and kinase activity [Sim et al., 2006], possible causing disrupted mitochondrial trafficking [Wang and Schwarz, 2009; Weihofen et al., 2009], reduced performance of the electron transport complexes [Gandhi et al., 2009; Liu et al., 2009] and elevated ROS formation [Gandhi et al., 2009] due to disturbed calcium homestasis, as well as activation of cell death proteins [Plun-Favreau et al., 2007; Pridgeon et al., 2007; Wang et al., 2007]. Together parkin and PINK1 are thought to be involved in the same pathway upstream of the mitochondrial fission/fusion machinery and mutations in both have been shown to result in an increase of mitochondrial fission in mammalian cells [Exner et al., 2007; Weihofen et al., 2009]. In addition, parkin was shown to be recruited to dysfunctional mitochondria pointing toward a possible role of parkin in the induction of mitophagy [Narendra et al., 2008, 2009]. Mutations in PARK2 might impair this function and eventually result in increased cellular toxicity. This hypothesis was supported by a parkin null Drosophila model, which showed mitochondrial defects and elevated oxidative stress rather than UPS impairment [Greene et al., 2003; Pesah et al., 2004], implying that parkin's involvement in mitochondria might be its primary activity. Nonetheless, more studies are needed to investigate the contribution of parkin to this pathogenic pathway.
In light of this, it seems plausible that digenic combinations of heterozygous mutations in PARK2 and PINK1 could be sufficient to cause disease as they might enhance each other's pathogenic effect by concomitant partial loss of function of two important enzymes active in the same pathway. Further, mutations in PARK7 were suspected to contribute to neuronal death through loss of antioxidant activity of DJ-1 and subsequent increase in oxidative stress of the cell [Moore et al., 2003; Ramsey and Giasson, 2008; Taira et al., 2004]. It is not clear yet how the PARK7 mutations can lead to this impaired functionality. As concomitant deficits in mitochondrial function and UPS activity have been observed, one might suspect a feedback loop between both cellular processes ultimately resulting in cell death and protein excess and aggregation.
Diagnostic Relevance
The past decade has been very exciting for molecular genetic research of PD. Genetic variants in at least 11 genes have been associated with increased risk for PD, and study of the corresponding proteins has been critical for our knowledge of the disease mechanisms underlying PD pathogenesis. For five genes there is extensive evidence of causality but screening all five of them for diagnostic or research purposes is a laborious undertaking. Therefore, mutation studies have been often restricted to sequence analyses of the two most frequently mutated genes—LRRK2 and parkin—and sometimes even further restricted to sequences coding for functional domains within these genes. Therefore, we have incomplete data to calculate the precise contribution of mutations in different PD genes together with an underestimation of more complex mutations like CNVs.
Ideally mutation analyses of the five major PD genes should include both sequence analysis and dosage analyses to detect CNVs. We investigated 310 Flanders-Belgian patients [Nuytemans et al., 2009], and showed high frequencies of heterozygous variants in PARK2 (9.0%) and LRRK2 (6.1%) and low contributions for SNCA, PINK1, and PARK7 mutations (0.3, 0.3, and 0.6%, respectively). In contrast to other populations, we did not observe the most frequent mutation in LRRK2, p.Gly2019Ser.
It is difficult to compare mutation frequencies between patient groups of different ethnic background because even the more recent and extensive mutation analyses in Brazilian, Dutch, Korean, Australian, or Portugese patient groups [Aguiar et al., 2008; Bras et al., 2008; Camargos et al., 2009; Choi et al., 2008; Macedo et al., 2009; Mellick et al., 2009] employed different study setups (selection of patients, genes of interest, domains of interest, etc.). Here, we provided a comprehensive presentation of the mutation frequencies, based on the published studies (Tables 3 and 4 PDmutDB: http://www.molgen.ua.ac.be/PDmutDB). When analyzing these data it became clear that, as in our Flanders-Belgian study, the contributions of SNCA, PINK1, and PARK7 are relatively low. Remarkably, the mutation burden of PINK1 was increased almost twofold in Asian patient groups. LRRK2 remains the most frequently mutated gene, even when heterozygous PARK2 mutation carriers were included in the equation. These data reinforced the guidelines on molecular diagnosis of PD that were proposed by the European Federation of Neurological Sciences (EFNS) [Harbo et al., 2009]. It is important to stress that the frequency data depicted here were extracted from reported studies only, and therefore is likely biased because SNCA, PINK1, and PARK7 analyses were often incomplete or even absent.
Influences of ethnicity on mutation frequencies as well as founder effects have been documented for several PD genes. Consequently, only a complete mutation analysis of these genes will allow the identification of all relevant mutations both for the individual patient as well as a population of interest. In addition, it is important to go on with the genetic characterization of patients even if they have been shown to carry a mutation in one gene, because unexpected digenic combinations might explain some atypical clinical presentations of individual PD patients. Despite the fact that not all studies implemented CNV analyses the observed CNV frequencies are higher than expected, implying that gene dosage is a major feature of the genetic etiology of PD. For PARK2, for instance, in approximately 50% of mutation carriers, deletions or duplications of (single) exons were identified. For SNCA, multiplications were observed not only in familial (∼88%) but also in seemingly sporadic patients (∼12%), resulting in higher CNV frequencies than originally anticipated. These data indicate that dosage analysis should be considered in all mutation screenings. On the other hand, when performing extensive mutation analyses, problems with pathogenicity assessment can occur for some types of mutations. For example, genetic variants appearing in LRRK2 domains with unclear biological function or heterozygous PARK2 mutations. The current efforts aiming at developing novel functional assays should be helpful in obtaining sufficient evidence to support a pathogenic role—and thus clinical implication—of individual mutations in the near future.
A PD Mutation Database (PDmutDB)
A huge amount of information on genetic variability in SNCA, PARK2, PINK1, PARK7, and LRRK2 and corresponding clinical phenotypes is present in the scientific literature, though, contained within numerous articles published over the last 2 decades. In addition, the data provided is often incomplete, fragmented, or sometimes even hard to interpret because, for example, clinical and genetic data of one family or group of patients are reported in separate articles and/or in different formats. Some of the current mutation databases do not systematically provide information on clinical features, familial history, and so on, whereas others are maintained by the goodwill of the researchers themselves, and consequently, are often incomplete or not up to date. Therefore, we decided to construct a novel PD mutation database, called PDmutDB (http://www.molgen.ua.ac.be/PDmutDB). This database will be publicly available and will hold information of reported variants with correct nomenclature and references to original studies. To allow for genotype-phenotype correlations, we added detailed familial and clinical data. Importantly, all informative family members are linked to their individual clinical features and identified variants in multiple genes with indication of zygosity. Also, we make an effort to provide an indication of pathogenicity for each variant, whenever sufficient data are available.
Data from new publications will be included in the database whenever they contain sufficient genetic information to correctly link each individual to the respective mutations. Individual researchers can submit genetic and/or clinical information using an additional file when their publications do not permit excessive tables. Contact: PDmutDB@molgen.vib-ua.be.
Conclusion and Future Prospects
During the last 2 decades molecular genetic research has lead to the identification of five important PD genes bearing approximately 500 different DNA variants. These variants make up a wide mutation spectrum including different simple mutations as well as genomic rearrangements. Gathering this information from literature is very laborious because it is scattered across many publications and different studies employ different study designs. Here we present a novel publicly available mutation database PDmutDB (http://www.molgen.ua.ac.be/PDmutDB). Next to the systematic organization of all DNA variants, this database provides information on family history, clinical features, and mutation zygosity. At this time, data on approximately 1,900 families of sporadic and familial patients are available. Data meta-analysis indicated both high genetic and clinical heterogeneity among mutation carriers. Mutations have been identified in patients with PD but also clinically related disorders such as LBD and Parkinson-plus syndromes. These data underline the complex genetic nature of these neurodegenerative diseases linking them together in spectrum disorders. As only few studies have included patients with PD-related disorders in their mutation analyses, the exact contribution of PD genes to the etiology of other neurodegenerative diseases remains unclear. Even for PD itself, this assessment is not straightforward as most mutation reports present fragmented data on only one or few PD genes. Because more recent data support that dosage plays a major role in PD pathogenesis (e.g., higher frequency of multiplications for SNCA than missense mutations and significant contribution of dosage effects in recessive genes), a lot of attention is drawn to gene expression regulation. Noncoding sequence variants in the promoter and UTR regions of PD genes are all potential disease associated variants. Indeed, variants in the SNCA promoter and UTR regions (both 5′ and 3′) were reported to be associated with increased risk for PD [Brighina et al., 2008; Farrer et al., 2001b; Hadjigeorgiou et al., 2006; Izumi et al., 2001; Kay et al., 2008; Maraganore et al., 2006; Mizuta et al., 2006; Mueller et al., 2005; Myhre et al., 2008b; Pals et al., 2004b; Parsian et al., 2007; Ross et al., 2007; Tan et al., 2004; Winkler et al., 2007]. Moreover, recent genome-wide association studies (GWAS) in PD patient and control groups confirmed the SNCA region as a major player in PD susceptibility [Pankratz et al., 2009; Satake et al., 2009; Simon-Sanchez et al., 2009]. Also, variants located upstream of LRRK2 were identified to be associated with increased risk for PD, suggesting that variants causing transcriptional upregulation of LRRK2 might be part of PD etiology [Satake et al., 2009; Simon-Sanchez et al., 2009]. Given that not all is known about the cell's transcription and translation mechanisms, variants detected in previously unexplored genomic regions might turn out to represent a novel group of pathogenic variations. It is clear that the research field should keep an open mind when performing mutation analyses and interpreting its results as exemplified by the variants in the promoter and UTR regions that were previously overlooked. Unfortunately, it is difficult to assess the pathogenic nature of these new subtypes of genetic variants without relevant functional assays. This concern already exists for several other groups of mutations in known PD genes. For example, we do not have unambiguous information on the actual involvement of PARK2 heterozygotes in PD pathogenicity or on the implications of mutations in LRRK2 regions with less known functionality. Even the current functional analyses of putative pathogenicity of LRRK2 mutations might be misleading because no physiological substrate has yet been identified. These concerns signify the urgent need of the molecular genetics field to invest more time and efforts in the development of relevant functional assays.
For the novel PDmutDB database to be usable in the broader research field, data on other genes associated with PD will be added in the near future. Functional data will also be included in the database when these data become available. This way we strive to develop a valuable, easy accessible, and up-to-date instrument for future research or diagnostic purposes.
In conclusion, it is clear that our knowledge on underlying genetics of PD gathered in the last 2 decades has provided researchers with incredible amounts of information on the different biological pathways involved in the pathogenesis of PD. There are still a large number of unanswered questions residing among the few solved mysteries, which will need further attention to fully understand PD in all its facets.
Acknowledgments
The research in the authors' research group was supported in part by by the Special Research Fund of the University of Antwerp, the Fund for Scientific Research Flanders (FWO-F), the Institute for Science and Technology-Flanders (IWT-F), the Foundation for Alzheimer Research (SAO/FRMA), and a Methusalem excellence grant of the Flemish Government; K.N. was holder of a PhD fellowship of the IWT-F and J.T. holds a postdoctoral fellowships of the FWO-V.
Supplementary material
References
- Abbas N, Lucking CB, Ricard S, Durr A, Bonifati V, De MG, Bouley S, Vaughan JR, Gasser T, Marconi R, Broussolle E, Brefel-Courbon C, Harhangi BS, Oostra BA, Fabrizio E, Bohme GA, Pradier L, Wood NW, Filla A, Meco G, Denefle P, Agid Y, Brice A. A wide variety of mutations in the parkin gene are responsible for autosomal recessive parkinsonism in Europe. French Parkinson's Disease Genetics Study Group and the European Consortium on Genetic Susceptibility in Parkinson's Disease. Hum Mol Genet. 1999;8:567–574. doi: 10.1093/hmg/8.4.567. [DOI] [PubMed] [Google Scholar]
- Abeliovich A, Schmitz Y, Farinas I, Choi-Lundberg D, Ho WH, Castillo PE, Shinsky N, Verdugo JM, Armanini M, Ryan A, Hynes M, Phillips H, Sulzer D, Rosenthal A. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron. 2000;25:239–252. doi: 10.1016/s0896-6273(00)80886-7. [DOI] [PubMed] [Google Scholar]
- Abou-Sleiman PM, Healy DG, Quinn N, Lees AJ, Wood NW. The role of pathogenic DJ-1 mutations in Parkinson's disease. Ann Neurol. 2003;54:283–286. doi: 10.1002/ana.10675. [DOI] [PubMed] [Google Scholar]
- Abou-Sleiman PM, Muqit MM, McDonald NQ, Yang YX, Gandhi S, Healy DG, Harvey K, Harvey RJ, Deas E, Bhatia K, Quinn N, Lees A, Latchman DS, Wood NW. A heterozygous effect for PINK1 mutations in Parkinson's disease? Ann Neurol. 2006;60:414–419. doi: 10.1002/ana.20960. [DOI] [PubMed] [Google Scholar]
- Adel S, Djarmati A, Kabakci K, Pichler I, Eskelson C, Lohnau T, Kock N, Hagenah J, Hedrich K, Schwinger E, Kramer PL, Pramstaller PP, Klein C. Co-occurrence of restless legs syndrome and Parkin mutations in two families. Mov Disord. 2006;21:258–263. doi: 10.1002/mds.20690. [DOI] [PubMed] [Google Scholar]
- Aguiar PC, Lessa PS, Godeiro C, Jr, Barsottini O, Felicio AC, Borges V, Silva SM, Saba RA, Ferraz HB, Moreira-Filho CA, Andrade LA. Genetic and environmental findings in early-onset Parkinson's disease Brazilian patients. Mov Disord. 2008;23:1228–1233. doi: 10.1002/mds.22032. [DOI] [PubMed] [Google Scholar]
- Ahn TB, Kim SY, Kim JY, Park SS, Lee DS, Min HJ, Kim YK, Kim SE, Kim JM, Kim HJ, Cho J, Jeon BS. alpha-Synuclein gene duplication is present in sporadic Parkinson disease. Neurology. 2008;70:43–49. doi: 10.1212/01.wnl.0000271080.53272.c7. [DOI] [PubMed] [Google Scholar]
- Alvarez V, Guisasola LM, Moreira VG, Lahoz CH, Coto E. Early-onset Parkinson's disease associated with a new parkin mutation in a Spanish family. Neurosci Lett. 2001;313:108–110. doi: 10.1016/s0304-3940(01)02235-2. [DOI] [PubMed] [Google Scholar]
- An XK, Peng R, Li T, Burgunder JM, Wu Y, Chen WJ, Zhang JH, Wang YC, Xu YM, Gou YR, Yuan GG, Zhang ZJ. LRRK2 Gly2385Arg variant is a risk factor of Parkinson's disease among Han-Chinese from mainland China. Eur J Neurol. 2008;15:301–305. doi: 10.1111/j.1468-1331.2007.02052.x. [DOI] [PubMed] [Google Scholar]
- Anand VS, Reichling LJ, Lipinski K, Stochaj W, Duan W, Kelleher K, Pungaliya P, Brown EL, Reinhart PH, Somberg R, Hirst WD, Riddle SM, Braithwaite SP. Investigation of leucine-rich repeat kinase 2: enzymological properties and novel assays. FEBS J. 2009;276:466–478. doi: 10.1111/j.1742-4658.2008.06789.x. [DOI] [PubMed] [Google Scholar]
- Annesi F, Rocca EF, Ciro Candiano I, Carrideo S, Tarantino P, Provenzano G, Civitelli D, De Marco EV, Quattrone A, Annesi G. Novel human pathological mutations. Gene symbol: PARK2. Disease: Parkinson's disease. Hum Genet. 2007;122:415. [PubMed] [Google Scholar]
- Annesi G, Savettieri G, Pugliese P, D'Amelio M, Tarantino P, Ragonese P, La Bella V, Piccoli T, Civitelli D, Annesi F, Fierro B, Piccoli F, Arabia G, Caracciolo M, Ciro Candiano I, Quattrone A. DJ-1 mutations and parkinsonism-dementia-amyotrophic lateral sclerosis complex. Ann Neurol. 2005;58:803–807. doi: 10.1002/ana.20666. [DOI] [PubMed] [Google Scholar]
- Arias VA, Sleegers K, Dekker MC, van Gool WA, van Swieten JC, Aulchenko YS, Oostra BA, van Duijn CM. A deletion in DJ-1 and the risk of dementia—a population-based survey. Neurosci Lett. 2004;372:196–199. doi: 10.1016/j.neulet.2004.09.040. [DOI] [PubMed] [Google Scholar]
- Athanassiadou A, Voutsinas G, Psiouri L, Leroy E, Polymeropoulos MH, Ilias A, Maniatis GM, Papapetropoulos T. Genetic analysis of families with Parkinson disease that carry the Ala53Thr mutation in the gene encoding alpha-synuclein. Am J Hum Genet. 1999;65:555–558. doi: 10.1086/302486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba M, Nakajo S, Tu PH, Tomita T, Nakaya K, Lee VM, Trojanowski JQ, Iwatsubo T. Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson's disease and dementia with Lewy bodies. Am J Pathol. 1998;152:879–884. [PMC free article] [PubMed] [Google Scholar]
- Bardien S, Keyser R, Yako Y, Lombard D, Carr J. Molecular analysis of the parkin gene in South African patients diagnosed with Parkinson's disease. Parkinsonism Relat Disord. 2009;15:116–121. doi: 10.1016/j.parkreldis.2008.04.005. [DOI] [PubMed] [Google Scholar]
- Bayrakli F, Bilguvar K, Mason CE, DiLuna ML, Bayri Y, Gungor L, Terzi M, Mane SM, Lifton RP, State MW, Gunel M. Rapid identification of disease-causing mutations using copy number analysis within linkage intervals. Hum Mutat. 2007;28:1236–1240. doi: 10.1002/humu.20592. [DOI] [PubMed] [Google Scholar]
- Bentivoglio AR, Cortelli P, Valente EM, Ialongo T, Ferraris A, Elia A, Montagna P, Albanese A. Phenotypic characterisation of autosomal recessive PARK6-linked parkinsonism in three unrelated Italian families. Mov Disord. 2001;16:999–1006. doi: 10.1002/mds.10034. [DOI] [PubMed] [Google Scholar]
- Berg D, Schweitzer K, Leitner P, Zimprich A, Lichtner P, Belcredi P, Brussel T, Schulte C, Maass S, Nagele T. Type and frequency of mutations in the LRRK2 gene in familial and sporadic Parkinson's disease. Brain. 2005;128:3000–3011. doi: 10.1093/brain/awh666. [DOI] [PubMed] [Google Scholar]
- Bertoli-Avella AM, Giroud-Benitez JL, Akyol A, Barbosa E, Schaap O, van der Linde HC, Martignoni E, Lopiano L, Lamberti P, Fincati E. Novel parkin mutations detected in patients with early-onset Parkinson's disease. Mov Disord. 2005;20:424–431. doi: 10.1002/mds.20343. [DOI] [PubMed] [Google Scholar]
- Biswas A, Gupta A, Naiya T, Das G, Neogi R, Datta S, Mukherjee S, Das SK, Ray K, Ray J. Molecular pathogenesis of Parkinson's disease: identification of mutations in the Parkin gene in Indian patients. Parkinsonism Relat Disord. 2006;12:420–426. doi: 10.1016/j.parkreldis.2006.04.005. [DOI] [PubMed] [Google Scholar]
- Bodner RA, Housman DE, Kazantsev AG. New directions for neurodegenerative disease therapy: using chemical compounds to boost the formation of mutant protein inclusions. Cell Cycle. 2006;5:1477–1480. doi: 10.4161/cc.5.14.2929. [DOI] [PubMed] [Google Scholar]
- Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ, Krieger E, Dekker MC, Squitieri F, Ibanez P, Joosse M, van Dongen JW, Vanacore N, van Swieten JC, Brice A, Meco G, van Duijn CM, Oostra BA, Heutink P. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003;299:256–259. doi: 10.1126/science.1077209. [DOI] [PubMed] [Google Scholar]
- Bonifati V, Rohe CF, Breedveld GJ, Fabrizio E, De MM, Tassorelli C, Tavella A, Marconi R, Nicholl DJ, Chien HF, Fincati E, Abbruzzese G, Marini P. Early-onset parkinsonism associated with PINK1 mutations: frequency, genotypes, and phenotypes. Neurology. 2005;65:87–95. doi: 10.1212/01.wnl.0000167546.39375.82. [DOI] [PubMed] [Google Scholar]
- Bostantjopoulou S, Katsarou Z, Papadimitriou A, Veletza V, Hatzigeorgiou G, Lees A. Clinical features of parkinsonian patients with the alpha-synuclein (G209A) mutation. Mov Disord. 2001;16:1007–1013. doi: 10.1002/mds.1221. [DOI] [PubMed] [Google Scholar]
- Bras J, Guerreiro R, Ribeiro M, Morgadinho A, Januario C, Dias M, Calado A, Semedo C, Oliveira C, Hardy J, Singleton A. Analysis of Parkinson disease patients from Portugal for mutations in SNCA, PRKN, PINK1 and LRRK2. BMC Neurol. 2008;8:1. doi: 10.1186/1471-2377-8-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brighina L, Frigerio R, Schneider NK, Lesnick TG, de Andrade M, Cunningham JM, Farrer MJ, Lincoln SJ, Checkoway H, Rocca WA, Maraganore DM. Alpha-synuclein, pesticides, and Parkinson disease: a case–control study. Neurology. 2008;70:1461–1469. doi: 10.1212/01.wnl.0000304049.31377.f2. [DOI] [PubMed] [Google Scholar]
- Brooks J, Ding J, Simon-Sanchez J, Paisan-Ruiz C, Singleton AB, Scholz SW. Parkin and PINK1 mutations in early-onset Parkinson's disease: comprehensive screening in publicly available cases and control. J Med Genet. 2009;46:375–381. doi: 10.1136/jmg.2008.063917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruggemann N, Mitterer M, Lanthaler AJ, Djarmati A, Hagenah J, Wiegers K, Winkler S, Pawlack H, Lohnau T, Pramstaller PP, Klein C, Lohmann K. Frequency of heterozygous Parkin mutations in healthy subjects: need for careful prospective follow-up examination of mutation carriers. Parkinsonism Relat Disord. 2009;15:425–429. doi: 10.1016/j.parkreldis.2008.11.014. [DOI] [PubMed] [Google Scholar]
- Camargos ST, Dornas LO, Momeni P, Lees A, Hardy J, Singleton A, Cardoso F. Familial Parkinsonism and early onset Parkinson's disease in a Brazilian movement disorders clinic: phenotypic characterization and frequency of SNCA, PRKN, PINK1, and LRRK2 mutations. Mov Disord. 2009;24:662–666. doi: 10.1002/mds.22365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmine Belin A, Westerlund M, Sydow O, Lundstromer K, Hakansson A, Nissbrandt H, Olson L, Galter D. Leucine-rich repeat kinase 2 (LRRK2) mutations in a Swedish Parkinson cohort and a healthy nonagenarian. Mov Disord. 2006;21:1731–1734. doi: 10.1002/mds.21016. [DOI] [PubMed] [Google Scholar]
- Cazeneuve C, San C, Ibrahim SA, Mukhtar MM, Kheir MM, Leguern E, Brice A, Salih MA. A new complex homozygous large rearrangement of the PINK1 gene in a Sudanese family with early onset Parkinson's disease. Neurogenetics. 2009;10:265–270. doi: 10.1007/s10048-009-0174-4. [DOI] [PubMed] [Google Scholar]
- Chan DK, Mok V, Ng PW, Yeung J, Kwok JB, Fang ZM, Clarke R, Wong L, Schofield PR, Hattori N. PARK2 mutations and clinical features in a Chinese population with early-onset Parkinson's disease. J Neural Transm. 2008;115:715–719. doi: 10.1007/s00702-007-0011-6. [DOI] [PubMed] [Google Scholar]
- Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, Levecque C, Larvor L, Andrieux J, Hulihan M, Waucquier N, Defebvre L, Amouyel P, Farrer M, Destee A. Alpha-synuclein locus duplication as a cause of familial Parkinson's disease. Lancet. 2004;364:1167–1169. doi: 10.1016/S0140-6736(04)17103-1. [DOI] [PubMed] [Google Scholar]
- Chaudhary S, Behari M, Dihana M, Swaminath PV, Govindappa ST, Jayaram S, Goyal V, Maitra A, Muthane UB, Juyal RC, Thelma BK. Parkin mutations in familial and sporadic Parkinson's disease among Indians. Parkinsonism Relat Disord. 2006;12:239–245. doi: 10.1016/j.parkreldis.2005.12.004. [DOI] [PubMed] [Google Scholar]
- Chen R, Gosavi NS, Langston JW, Chan P. Parkin mutations are rare in patients with young-onset parkinsonism in a US population. Parkinsonism Relat Disord. 2003;9:309–312. doi: 10.1016/s1353-8020(03)00018-x. [DOI] [PubMed] [Google Scholar]
- Chen-Plotkin AS, Yuan W, Anderson C, McCarty WE, Hurtig HI, Clark CM, Miller BL, Lee VM, Trojanowski JQ, Grossman M, Van Deerlin V. Corticobasal syndrome and primary progressive aphasia as manifestations of LRRK2 gene mutations. Neurology. 2008;70:521–527. doi: 10.1212/01.WNL.0000280574.17166.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chishti MA, Bohlega S, Ahmed M, Loualich A, Carroll P, Sato C, St. George-Hyslop P, Westaway D, Rogaeva E. T313M PINK1 mutation in an extended highly consanguineous Saudi family with early-onset Parkinson disease. Arch Neurol. 2006;63:1483–1485. doi: 10.1001/archneur.63.10.1483. [DOI] [PubMed] [Google Scholar]
- Choi JM, Woo MS, Ma HI, Kang SY, Sung YH, Yong SW, Chung SJ, Kim JS, Shin HW, Lyoo CH, Lee PH, Baik JS, Kim SJ, Park MY, Sohn YH, Kim JH, Kim JW, Lee MS, Lee MC, Kim DH, Kim YJ. Analysis of PARK genes in a Korean cohort of early-onset Parkinson disease. Neurogenetics. 2008;9:263–269. doi: 10.1007/s10048-008-0138-0. [DOI] [PubMed] [Google Scholar]
- Chung SJ, Park HK, Ki CS, Kim MJ, Lee MC. Marked diurnal fluctuation and rest benefit in a patient with parkin mutation. Mov Disord. 2008;23:624–626. doi: 10.1002/mds.21951. [DOI] [PubMed] [Google Scholar]
- Ciro Candiano I, Annesi F, Rocca EF, Carrideo S, Tarantino P, Provenzano G, Civitelli D, De Marco EV, Quattrone A, Annesi G. Novel human pathological mutations. Gene symbol: PARK2. Disease: Parkinson's disease. Hum Genet. 2007;122:416. [PubMed] [Google Scholar]
- Clarimon J, Johnson J, Dogu O, Horta W, Khan N, Lees AJ, Hardy J, Singleton A. Defining the ends of Parkin exon 4 deletions in two different families with Parkinson's disease. Am J Med Genet B Neuropsychiatr Genet. 2005;133B:120–123. doi: 10.1002/ajmg.b.30119. [DOI] [PubMed] [Google Scholar]
- Clarimon J, Pagonabarraga J, Paisan-Ruiz C, Campolongo A, Pascual-Sedano B, Marti-Masso JF, Singleton AB, Kulisevsky J. Tremor dominant parkinsonism: Clinical description and LRRK2 mutation screening. Mov Disord. 2008;23:518–523. doi: 10.1002/mds.21771. [DOI] [PubMed] [Google Scholar]
- Clark LN, Afridi S, Karlins E, Wang Y, Mejia-Santana H, Harris J, Louis ED, Cote LJ, Andrews H, Fahn S, Waters C, Ford B, Frucht S, Ottman R, Marder K. Case–control study of the parkin gene in early-onset Parkinson disease. Arch Neurol. 2006;63:548–552. doi: 10.1001/archneur.63.4.548. [DOI] [PubMed] [Google Scholar]
- Clark LN, Afridi S, Mejia-Santana H, Harris J, Louis ED, Cote LJ, Andrews H, Singleton A, Wavrant De-Vrieze F, Hardy J, Mayeux R, Fahn S, Waters C, Ford B, Frucht S, Ottman R, Marder K. Analysis of an early-onset Parkinson's disease cohort for DJ-1 mutations. Mov Disord. 2004;19:796–800. doi: 10.1002/mds.20131. [DOI] [PubMed] [Google Scholar]
- Conway KA, Lee SJ, Rochet JC, Ding TT, Williamson RE, Lansbury PT., Jr Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson's disease: implications for pathogenesis and therapy. Proc Natl Acad Sci USA. 2000;97:571–576. doi: 10.1073/pnas.97.2.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cookson MR. The biochemistry of Parkinson's disease. Annu Rev Biochem. 2005;74:29–52. doi: 10.1146/annurev.biochem.74.082803.133400. [DOI] [PubMed] [Google Scholar]
- Criscuolo C, Volpe G, De Rosa A, Varrone A, Marongiu R, Mancini P, Salvatore E, Dallapiccola B, Filla A, Valente EM, De Marco G. PINK1 homozygous W437X mutation in a patient with apparent dominant transmission of parkinsonism. Mov Disord. 2006;21:1265–1267. doi: 10.1002/mds.20933. [DOI] [PubMed] [Google Scholar]
- Dachsel JC, Mata IF, Ross OA, Taylor JP, Lincoln SJ, Hinkle KM, Huerta C, Ribacoba R, Blazquez M, Alvarez V, Farrer MJ. Digenic parkinsonism: investigation of the synergistic effects of PRKN and LRRK2. Neurosci Lett. 2006;410:80–84. doi: 10.1016/j.neulet.2006.06.068. [DOI] [PubMed] [Google Scholar]
- Dachsel JC, Ross OA, Mata IF, Kachergus J, Toft M, Cannon A, Baker M, Adamson J, Hutton M, Dickson DW, Farrer MJ. Lrrk2 G2019S substitution in frontotemporal lobar degeneration with ubiquitin-immunoreactive neuronal inclusions. Acta Neuropathol. 2007;113:601–606. doi: 10.1007/s00401-006-0178-1. [DOI] [PubMed] [Google Scholar]
- Davidson WS, Jonas A, Clayton DF, George JM. Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J Biol Chem. 1998;273:9443–9449. doi: 10.1074/jbc.273.16.9443. [DOI] [PubMed] [Google Scholar]
- Deng H, Dodson MW, Huang H, Guo M. The Parkinson's disease genes pink1 and parkin promote mitochondrial fission and/or inhibit fusion in Drosophila. Proc Natl Acad Sci USA. 2008a;105:14503–14508. doi: 10.1073/pnas.0803998105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng H, Le W, Shahed J, Xie W, Jankovic J. Mutation analysis of the parkin and PINK1 genes in American Caucasian early-onset Parkinson disease families. Neurosci Lett. 2008b;430:18–22. doi: 10.1016/j.neulet.2007.10.018. [DOI] [PubMed] [Google Scholar]
- Deng H, Le WD, Hunter CB, Mejia N, Xie WJ, Jankovic J. A family with Parkinson disease, essential tremor, bell palsy, and parkin mutations. Arch Neurol. 2007;64:421–424. doi: 10.1001/archneur.64.3.421. [DOI] [PubMed] [Google Scholar]
- Di Fonzo A, Dekker MC, Montagna P, Baruzzi A, Yonova EH, Correia GL, Szczerbinska A, Zhao T, Dubbel-Hulsman LO, Wouters CH, de Graaff E, Oyen WJ, Simons EJ, Breedveld GJ, Oostra BA, Horstink MW, Bonifati V. FBXO7 mutations cause autosomal recessive, early-onset parkinsonian-pyramidal syndrome. Neurology. 2009;72:240–245. doi: 10.1212/01.wnl.0000338144.10967.2b. [DOI] [PubMed] [Google Scholar]
- Di Fonzo A, Rohe CF, Ferreira J, Chien HF, Vacca L, Stocchi F, Guedes L, Fabrizio E, Manfredi M, Vanacore N, Goldwurm S, Breedveld G, Sampaio C, Meco G, Barbosa E, Oostra BA, Bonifati V. A frequent LRRK2 gene mutation associated with autosomal dominant Parkinson's disease. Lancet. 2005;365:412–415. doi: 10.1016/S0140-6736(05)17829-5. [DOI] [PubMed] [Google Scholar]
- Di Fonzo A, Tassorelli C, De MM, Chien HF, Ferreira J, Rohe CF, Riboldazzi G, Antonini A, Albani G, Mauro A, Marconi R, Abbruzzese G, Lopiano L, Fincati E, Guidi M, Marini P, Stocchi F, Onofrj M, Toni V, Tinazzi M, Fabbrini G, Lamberti P, Vanacore N, Meco G, Leitner P, Uitti RJ, Wszolek ZK, Gasser T, Simons EJ, Breedveld GJ, Goldwurm S, Pezzoli G, Sampaio C, Barbosa E, Martignoni E, Oostra BA, Bonifati V. Comprehensive analysis of the LRRK2 gene in sixty families with Parkinson's disease. Eur J Hum Genet. 2006a;14:322–331. doi: 10.1038/sj.ejhg.5201539. [DOI] [PubMed] [Google Scholar]
- Di Fonzo A, Wu-Chou YH, Lu CS, van Doeselaar M, Simons EJ, Rohe CF, Chang HC, Chen RS, Weng YH, Vanacore N, Breedveld GJ, Oostra BA, Bonifati V. A common missense variant in the LRRK2 gene, Gly2385Arg, associated with Parkinson's disease risk in Taiwan. Neurogenetics. 2006b;7:133–138. doi: 10.1007/s10048-006-0041-5. [DOI] [PubMed] [Google Scholar]
- Djarmati A, Hedrich K, Svetel M, Lohnau T, Schwinger E, Romac S, Pramstaller PP, Kostic V, Klein C. Heterozygous PINK1 mutations: a susceptibility factor for Parkinson disease? Mov Disord. 2006;21:1526–1530. doi: 10.1002/mds.20977. [DOI] [PubMed] [Google Scholar]
- Djarmati A, Hedrich K, Svetel M, Schafer N, Juric V, Vukosavic S, Hering R, Riess O, Romac S, Klein C, Kostic V. Detection of Parkin (PARK2) and DJ1 (PARK7) mutations in early-onset Parkinson disease: Parkin mutation frequency depends on ethnic origin of patients. Hum Mutat. 2004;23:525. doi: 10.1002/humu.9240. [DOI] [PubMed] [Google Scholar]
- Ephraty L, Porat O, Israeli D, Cohen OS, Tunkel O, Yael S, Hatano Y, Hattori N, Hassin-Baer S. Neuropsychiatric and cognitive features in autosomal-recessive early parkinsonism due to PINK1 mutations. Mov Disord. 2007;22:566–569. doi: 10.1002/mds.21319. [DOI] [PubMed] [Google Scholar]
- Exner N, Treske B, Paquet D, Holmstrom K, Schiesling C, Gispert S, Carballo-Carbajal I, Berg D, Hoepken HH, Gasser T, Kruger R, Winklhofer KF, Vogel F, Reichert AS, Auburger G, Kahle PJ, Schmid B, Haass C. Loss-of-function of human PINK1 results in mitochondrial pathology and can be rescued by parkin. J Neurosci. 2007;27:12413–12418. doi: 10.1523/JNEUROSCI.0719-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrer M, Chan P, Chen R, Tan L, Lincoln S, Hernandez D, Forno L, Gwinn-Hardy K, Petrucelli L, Hussey J, Singleton A, Tanner C, Hardy J, Langston JW. Lewy bodies and parkinsonism in families with parkin mutations. Ann Neurol. 2001a;50:293–300. doi: 10.1002/ana.1132. [DOI] [PubMed] [Google Scholar]
- Farrer M, Kachergus J, Forno L, Lincoln S, Wang DS, Hulihan M, Maraganore D, Gwinn-Hardy K, Wszolek Z, Dickson D, Langston JW. Comparison of kindreds with parkinsonism and alpha-synuclein genomic multiplications. Ann Neurol. 2004;55:174–179. doi: 10.1002/ana.10846. [DOI] [PubMed] [Google Scholar]
- Farrer M, Maraganore DM, Lockhart P, Singleton A, Lesnick TG, de Andrade M, West A, de Silva R, Hardy J, Hernandez D. alpha-Synuclein gene haplotypes are associated with Parkinson's disease. Hum Mol Genet. 2001b;10:1847–1851. doi: 10.1093/hmg/10.17.1847. [DOI] [PubMed] [Google Scholar]
- Farrer M, Stone J, Mata IF, Lincoln S, Kachergus J, Hulihan M, Strain KJ, Maraganore DM. LRRK2 mutations in Parkinson disease. Neurology. 2005;65:738–740. doi: 10.1212/01.wnl.0000169023.51764.b0. [DOI] [PubMed] [Google Scholar]
- Farrer MJ, Stone JT, Lin CH, Dachsel JC, Hulihan MM, Haugarvoll K, Ross OA, Wu RM. Lrrk2 G2385R is an ancestral risk factor for Parkinson's disease in Asia. Parkinsonism Relat Disord. 2007;13:89–92. doi: 10.1016/j.parkreldis.2006.12.001. [DOI] [PubMed] [Google Scholar]
- Ferreira JJ, Guedes LC, Rosa MM, Coelho M, van DM, Schweiger D, Di FA, Oostra BA, Sampaio C, Bonifati V. High prevalence of LRRK2 mutations in familial and sporadic Parkinson's disease in Portugal. Mov Disord. 2007;22:1194–1201. doi: 10.1002/mds.21525. [DOI] [PubMed] [Google Scholar]
- Foroud T, Uniacke SK, Liu L, Pankratz N, Rudolph A, Halter C, Shults C, Marder K, Conneally PM, Nichols WC. Heterozygosity for a mutation in the parkin gene leads to later onset Parkinson disease. Neurology. 2003;60:796–801. doi: 10.1212/01.wnl.0000049470.00180.07. [DOI] [PubMed] [Google Scholar]
- Fuchs J, Nilsson C, Kachergus J, Munz M, Larsson EM, Schule B, Langston JW, Middleton FA, Ross OA, Hulihan M, Gasser T, Farrer MJ. Phenotypic variation in a large Swedish pedigree due to SNCA duplication and triplication. Neurology. 2007;68:916–922. doi: 10.1212/01.wnl.0000254458.17630.c5. [DOI] [PubMed] [Google Scholar]
- Funayama M, Hasegawa K, Kowa H, Saito M, Tsuji S, Obata F. A new locus for Parkinson's disease (PARK8) maps to chromosome 12p11.2–q13.1. Ann Neurol. 2002;51:296–301. doi: 10.1002/ana.10113. [DOI] [PubMed] [Google Scholar]
- Funayama M, Li Y, Tsoi TH, Lam CW, Ohi T, Yazawa S, Uyama E, Djaldetti R, Melamed E, Yoshino H, Imamichi Y, Takashima H, Nishioka K, Sato K, Tomiyama H, Kubo S, Mizuno Y, Hattori N. Familial Parkinsonism with digenic parkin and PINK1 mutations. Mov Disord. 2008;23:1461–1465. doi: 10.1002/mds.22143. [DOI] [PubMed] [Google Scholar]
- Fung HC, Chen CM, Hardy J, Singleton AB, Lee-Chen GJ, Wu YR. Analysis of the PINK1 gene in a cohort of patients with sporadic early-onset parkinsonism in Taiwan. Neurosci Lett. 2006a;394:33–36. doi: 10.1016/j.neulet.2005.10.005. [DOI] [PubMed] [Google Scholar]
- Fung HC, Chen CM, Hardy J, Singleton AB, Wu YR. A common genetic factor for Parkinson disease in ethnic Chinese population in Taiwan. BMC Neurol. 2006b;6:47. doi: 10.1186/1471-2377-6-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaig C, Ezquerra M, Marti MJ, Munoz E, Valldeoriola F, Tolosa E. LRRK2 mutations in Spanish patients with Parkinson disease: frequency, clinical features, and incomplete penetrance. Arch Neurol. 2006;63:377–382. doi: 10.1001/archneur.63.3.377. [DOI] [PubMed] [Google Scholar]
- Gaig C, Ezquerra M, Marti MJ, Valldeoriola F, Munoz E, Llado A, Rey MJ, Cardozo A, Molinuevo JL, Tolosa E. Screening for the LRRK2 G2019S and codon-1441 mutations in a pathological series of parkinsonian syndromes and frontotemporal lobar degeneration. J Neurol Sci. 2008;270:94–98. doi: 10.1016/j.jns.2008.02.010. [DOI] [PubMed] [Google Scholar]
- Gaig C, Marti MJ, Ezquerra M, Rey MJ, Cardozo A, Tolosa E. G2019S LRRK2 mutation causing Parkinson's disease without Lewy bodies. J Neurol Neurosurg Psychiatry. 2007;78:626–628. doi: 10.1136/jnnp.2006.107904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi S, Wood-Kaczmar A, Yao Z, Plun-Favreau H, Deas E, Klupsch K, Downward J, Latchman DS, Tabrizi SJ, Wood NW, Duchen MR, Abramov AY. PINK1-associated Parkinson's disease is caused by neuronal vulnerability to calcium-induced cell death. Mol Cell. 2009;33:627–638. doi: 10.1016/j.molcel.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L, Gomez-Garre P, az-Corrales FJ, Carrillo F, Carballo M, Palomino A, az-Martin J, Mejias R, Vime PJ, Lopez-Barneo J, Mir P. Prevalence and clinical features of LRRK2 mutations in patients with Parkinson's disease in southern Spain. Eur J Neurol. 2009 doi: 10.1111/j.1468-1331.2009.02620.x. [DOI] [PubMed] [Google Scholar]
- Gelmetti V, Ferraris A, Brusa L, Romano F, Lombardi F, Barzaghi C, Stanzione P, Garavaglia B, Dallapiccola B, Valente EM. Late onset sporadic Parkinson's disease caused by PINK1 mutations: clinical and functional study. Mov Disord. 2008;23:881–885. doi: 10.1002/mds.21960. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Covy JP, Bonini NM, Hurtig HI, Farrer MJ, Trojanowski JQ, Van DV. Biochemical and pathological characterization of Lrrk2. Ann Neurol. 2006;59:315–322. doi: 10.1002/ana.20791. [DOI] [PubMed] [Google Scholar]
- Gilks WP, bou-Sleiman PM, Gandhi S, Jain S, Singleton A, Lees AJ, Shaw K, Bhatia KP, Bonifati V, Quinn NP, Lynch J, Healy DG, Holton JL, Revesz T, Wood NW. A common LRRK2 mutation in idiopathic Parkinson's disease. Lancet. 2005;365:415–416. doi: 10.1016/S0140-6736(05)17830-1. [DOI] [PubMed] [Google Scholar]
- Giordana MT, D'Agostino C, Albani G, Mauro A, Di FA, Antonini A, Bonifati V. Neuropathology of Parkinson's disease associated with the LRRK2 Ile1371Val mutation. Mov Disord. 2007;22:275–278. doi: 10.1002/mds.21281. [DOI] [PubMed] [Google Scholar]
- Gloeckner CJ, Kinkl N, Schumacher A, Braun RJ, O'Neill E, Meitinger T, Kolch W, Prokisch H, Ueffing M. The Parkinson disease causing LRRK2 mutation I2020T is associated with increased kinase activity. Hum Mol Genet. 2006;15:223–232. doi: 10.1093/hmg/ddi439. [DOI] [PubMed] [Google Scholar]
- Godeiro-Junior C, de Carvalho-Aguiar PM, Felicio AC, Barsottini OG, Silva SM, Borges V, Andrade LA, Ferraz HB. PINK1 mutations in a Brazilian cohort of early-onset Parkinson's disease patients. Mov Disord. 2009 doi: 10.1002/mds.22685. [DOI] [PubMed] [Google Scholar]
- Goldwurm S, Di Fonzo A, Simons EJ, Rohe CF, Zini M, Canesi M, Tesei S, Zecchinelli A, Antonini A, Mariani C, Meucci N, Sacilotto G, Sironi F, Salani G, Ferreira J, Chien HF, Fabrizio E, Vanacore N, Dalla LA, Stocchi F, Diroma C, Lamberti P, Sampaio C, Meco G, Barbosa E, Bertoli-Avella AM, Breedveld GJ, Oostra BA, Pezzoli G, Bonifati V. The G6055A (G2019S) mutation in LRRK2 is frequent in both early and late onset Parkinson's disease and originates from a common ancestor. J Med Genet. 2005;42:e65. doi: 10.1136/jmg.2005.035568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Fernandez MC, Lezcano E, Ross OA, Gomez-Esteban JC, Gomez-Busto F, Velasco F, varez-Alvarez M, Rodriguez-Martinez MB, Ciordia R, Zarranz JJ, Farrer MJ, Mata IF, de Pancorbo MM. Lrrk2-associated parkinsonism is a major cause of disease in Northern Spain. Parkinsonism Relat Disord. 2007;13:509–515. doi: 10.1016/j.parkreldis.2007.04.003. [DOI] [PubMed] [Google Scholar]
- Gorostidi A, Ruiz-Martinez J, de Munain AL, Alzualde A, Masso JF. LRRK2 G2019S and R1441G mutations associated with Parkinson's disease are common in the Basque Country, but relative prevalence is determined by ethnicity. Neurogenetics. 2009;10:157–159. doi: 10.1007/s10048-008-0162-0. [DOI] [PubMed] [Google Scholar]
- Gosal D, Lynch T, Ross OA, Haugarvoll K, Farrer MJ, Gibson JM. Global distribution and reduced penetrance: Lrrk2 R1441C in an Irish Parkinson's disease kindred. Mov Disord. 2007;22:291–292. doi: 10.1002/mds.21200. [DOI] [PubMed] [Google Scholar]
- Gotthardt K, Weyand M, Kortholt A, Van Haastert PJ, Wittinghofer A. Structure of the Roc-COR domain tandem of C. tepidum, a prokaryotic homologue of the human LRRK2 Parkinson kinase. EMBO J. 2008;27:2352. doi: 10.1038/emboj.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouider-Khouja N, Larnaout A, Amouri R, Sfar S, Belal S, Ben HC, Ben HM, Hattori N, Mizuno Y, Hentati F. Autosomal recessive parkinsonism linked to parkin gene in a Tunisian family. Clinical, genetic and pathological study. Parkinsonism.Relat Disord. 2003;9:247–251. doi: 10.1016/s1353-8020(03)00016-6. [DOI] [PubMed] [Google Scholar]
- Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci USA. 2003;100:4078–4083. doi: 10.1073/pnas.0737556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greggio E, Jain S, Kingsbury A, Bandopadhyay R, Lewis P, Kaganovich A, van der Brug MP, Beilina A, Blackinton J, Thomas KJ, Ahmad R, Miller DW, Kesavapany S, Singleton A, Lees A, Harvey RJ, Harvey K, Cookson MR. Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol Dis. 2006;23:329–341. doi: 10.1016/j.nbd.2006.04.001. [DOI] [PubMed] [Google Scholar]
- Greggio E, Zambrano I, Kaganovich A, Beilina A, Taymans JM, Daniels V, Lewis P, Jain S, Ding J, Syed A, Thomas KJ, Baekelandt V, Cookson MR. The Parkinson disease-associated leucine-rich repeat kinase 2 (LRRK2) is a dimer that undergoes intramolecular autophosphorylation. J Biol Chem. 2008;283:16906–16914. doi: 10.1074/jbc.M708718200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JF, Xiao B, Liao B, Zhang XW, Nie LL, Zhang YH, Shen L, Jiang H, Xia K, Pan Q, Yan XX, Tang BS. Mutation analysis of Parkin, PINK1, DJ-1 and ATP13A2 genes in Chinese patients with autosomal recessive early-onset Parkinsonism. Mov Disord. 2008;23:2074–2079. doi: 10.1002/mds.22156. [DOI] [PubMed] [Google Scholar]
- Guo L, Gandhi PN, Wang W, Petersen RB, Wilson-Delfosse AL, Chen SG. The Parkinson's disease-associated protein, leucine-rich repeat kinase 2 (LRRK2), is an authentic GTPase that stimulates kinase activity. Exp Cell Res. 2007;313:3658–3670. doi: 10.1016/j.yexcr.2007.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwinn-Hardy K, Mehta ND, Farrer M, Maraganore D, Muenter M, Yen SH, Hardy J, Dickson DW. Distinctive neuropathology revealed by alpha-synuclein antibodies in hereditary parkinsonism and dementia linked to chromosome 4p. Acta Neuropathol. 2000;99:663–672. doi: 10.1007/s004010051177. [DOI] [PubMed] [Google Scholar]
- Hadjigeorgiou GM, Xiromerisiou G, Gourbali V, Aggelakis K, Scarmeas N, Papadimitriou A, Singleton A. Association of alpha-synuclein Rep1 polymorphism and Parkinson's disease: influence of Rep1 on age at onset. Mov Disord. 2006;21:534–539. doi: 10.1002/mds.20752. [DOI] [PubMed] [Google Scholar]
- Hague S, Rogaeva E, Hernandez D, Gulick C, Singleton A, Hanson M, Johnson J, Weiser R, Gallardo M, Ravina B, Gwinn-Hardy K, Crawley A, PH SG-H, Lang AE, Heutink P, Bonifati V, Hardy J, Singleton A. Early-onset Parkinson's disease caused by a compound heterozygous DJ-1 mutation. Ann Neurol. 2003;54:271–274. doi: 10.1002/ana.10663. [DOI] [PubMed] [Google Scholar]
- Harbo HF, Finsterer J, Baets J, Van BC, Di DS, Fontaine B, De JP, Lossos A, Lynch T, Mariotti C, Schols L, Spinazzola A, Szolnoki Z, Tabrizi SJ, Tallaksen C, Zeviani M, Burgunder JM, Gasser T. EFNS guidelines on the molecular diagnosis of neurogenetic disorders: general issues, Huntington's disease, Parkinson's disease and dystonias. Eur J Neurol. 2009;16:777–785. doi: 10.1111/j.1468-1331.2009.02646.x. [DOI] [PubMed] [Google Scholar]
- Hatano Y, Li Y, Sato K, Asakawa S, Yamamura Y, Tomiyama H, Yoshino H, Asahina M, Kobayashi S, Hassin-Baer S, Lu CS, Ng AR, Rosales RL, Shimizu N, Toda T, Mizuno Y, Hattori N. Novel PINK1 mutations in early-onset parkinsonism. Ann Neurol. 2004;56:424–427. doi: 10.1002/ana.20251. [DOI] [PubMed] [Google Scholar]
- Hattori N, Kitada T, Matsumine H, Asakawa S, Yamamura Y, Yoshino H, Kobayashi T, Yokochi M, Wang M, Yoritaka A, Kondo T, Kuzuhara S, aNakamura S, Shimizu N, Mizuno Y. Molecular genetic analysis of a novel Parkin gene in Japanese families with autosomal recessive juvenile parkinsonism: evidence for variable homozygous deletions in the Parkin gene in affected individuals. Ann Neurol. 1998a;44:935–941. doi: 10.1002/ana.410440612. [DOI] [PubMed] [Google Scholar]
- Hattori N, Matsumine H, Asakawa S, Kitada T, Yoshino H, Elibol B, Brookes AJ, Yamamura Y, Kobayashi T, Wang M, Yoritaka A, Minoshima S, Shimizu N, Mizuno Y. Point mutations (Thr240Arg and Gln311Stop) [correction of Thr240Arg and Ala311Stop] in the Parkin gene. Biochem Biophys Res Commun. 1998b;249:754–758. doi: 10.1006/bbrc.1998.9134. [DOI] [PubMed] [Google Scholar]
- Haubenberger D, Bonelli S, Hotzy C, Leitner P, Lichtner P, Samal D, Katzenschlager R, Djamshidian A, Brucke T, Steffelbauer M, Bancher C, Grossmann J, Ransmayr G, Strom TM, Meitinger T, Gasser T, Auff E, Zimprich A. A novel LRRK2 mutation in an Austrian cohort of patients with Parkinson's disease. Mov Disord. 2007;22:1640–1643. doi: 10.1002/mds.21568. [DOI] [PubMed] [Google Scholar]
- Haugarvoll K, Rademakers R, Kachergus JM, Nuytemans K, Ross OA, Gibson JM, Tan EK. Lrrk2 R1441C parkinsonism is clinically similar to sporadic Parkinson disease. Neurology. 2008;70:1456–1460. doi: 10.1212/01.wnl.0000304044.22253.03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi S, Wakabayashi K, Ishikawa A, Nagai H, Saito M, Maruyama M, Takahashi T, Ozawa T, Tsuji S, Takahashi H. An autopsy case of autosomal-recessive juvenile parkinsonism with a homozygous exon 4 deletion in the parkin gene. Mov Disord. 2000;15:884–888. doi: 10.1002/1531-8257(200009)15:5<884::aid-mds1019>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- Healy DG, bou-Sleiman PM, Gibson JM, Ross OA, Jain S, Gandhi S, Gosal D, Muqit MM, Wood NW, Lynch T. PINK1 (PARK6) associated Parkinson disease in Ireland. Neurology. 2004;63:1486–1488. doi: 10.1212/01.wnl.0000142089.38301.8e. [DOI] [PubMed] [Google Scholar]
- Hedrich K, Djarmati A, Schafer N, Hering R, Wellenbrock C, Weiss PH, Hilker R, Vieregge P, Ozelius LJ, Heutink P, Bonifati V, Schwinger E, Lang AE, Noth J, Bressman SB, Pramstaller PP, Riess O, Klein C. DJ-1 (PARK7) mutations are less frequent than Parkin (PARK2) mutations in early-onset Parkinson disease. Neurology. 2004a;62:389–394. doi: 10.1212/01.wnl.0000113022.51739.88. [DOI] [PubMed] [Google Scholar]
- Hedrich K, Eskelson C, Wilmot B, Marder K, Harris J, Garrels J, Meija-Santana H, Vieregge P, Jacobs H, Bressman SB, Lang AE, Kann M, Abbruzzese G, Martinelli P, Schwinger E, Ozelius LJ, Pramstaller PP, Klein C, Kramer P. Distribution, type, and origin of Parkin mutations: review and case studies. Mov Disord. 2004b;19:1146–1157. doi: 10.1002/mds.20234. [DOI] [PubMed] [Google Scholar]
- Hedrich K, Hagenah J, Djarmati A, Hiller A, Lohnau T, Lasek K, Grunewald A, Hilker R, Steinlechner S, Boston H, Kock N, Schneider-Gold C, Kress W, Siebner H, Binkofski F, Lencer R, Munchau A, Klein C. Clinical spectrum of homozygous and heterozygous PINK1 mutations in a large German family with Parkinson disease: role of a single hit? Arch Neurol. 2006a;63:833–838. doi: 10.1001/archneur.63.6.833. [DOI] [PubMed] [Google Scholar]
- Hedrich K, Kann M, Lanthaler AJ, Dalski A, Eskelson C, Landt O, Schwinger E, Vieregge P, Lang AE, Breakefield XO, Ozelius LJ, Pramstaller PP, Klein C. The importance of gene dosage studies: mutational analysis of the parkin gene in early-onset parkinsonism. Hum Mol Genet. 2001;10:1649–1656. doi: 10.1093/hmg/10.16.1649. [DOI] [PubMed] [Google Scholar]
- Hedrich K, Marder K, Harris J, Kann M, Lynch T, Meija-Santana H, Pramstaller PP, Schwinger E, Bressman SB, Fahn S, Klein C. Evaluation of 50 probands with early-onset Parkinson's disease for Parkin mutations. Neurology. 2002;58:1239–1246. doi: 10.1212/wnl.58.8.1239. [DOI] [PubMed] [Google Scholar]
- Hedrich K, Winkler S, Hagenah J, Kabakci K, Kasten M, Schwinger E, Volkmann J, Pramstaller PP, Kostic V, Vieregge P, Klein C. Recurrent LRRK2 (Park8) mutations in early-onset Parkinson's disease. Mov Disord. 2006b;21:1506–1510. doi: 10.1002/mds.20990. [DOI] [PubMed] [Google Scholar]
- Hering R, Strauss KM, Tao X, Bauer A, Woitalla D, Mietz EM, Petrovic S, Bauer P, Schaible W, Muller T, Schols L, Klein C, Berg D, Meyer PT, Schulz JB, Wollnik B, Tong L, Kruger R, Riess O. Novel homozygous p.E64D mutation in DJ1 in early onset Parkinson disease (PARK7) Hum Mutat. 2004;24:321–329. doi: 10.1002/humu.20089. [DOI] [PubMed] [Google Scholar]
- Hernandez D, Paisan RC, Crawley A, Malkani R, Werner J, Gwinn-Hardy K, Dickson D, Wavrant DF, Hardy J, Singleton A. The dardarin G 2019 S mutation is a common cause of Parkinson's disease but not other neurodegenerative diseases. Neurosci Lett. 2005;389:137–139. doi: 10.1016/j.neulet.2005.07.044. [DOI] [PubMed] [Google Scholar]
- Hertz JM, Ostergaard K, Juncker I, Pedersen S, Romstad A, Moller LB, Guttler F, Dupont E. Low frequency of Parkin, Tyrosine Hydroxylase, and GTP Cyclohydrolase I gene mutations in a Danish population of early-onset Parkinson's Disease. Eur J Neurol. 2006;13:385–390. doi: 10.1111/j.1468-1331.2006.01249.x. [DOI] [PubMed] [Google Scholar]
- Hoenicka J, Vidal L, Morales B, Ampuero I, Jimenez-Jimenez FJ, Berciano J, del ST, Jimenez A, Ruiz PG, de Yebenes JG. Molecular findings in familial Parkinson disease in Spain. Arch Neurol. 2002;59:966–970. doi: 10.1001/archneur.59.6.966. [DOI] [PubMed] [Google Scholar]
- Huang Y, Halliday GM, Vandebona H, Mellick GD, Mastaglia F, Stevens J, Kwok J, Garlepp M, Silburn PA, Horne MK, Kotschet K, Venn A, Rowe DB, Rubio JP, Sue CM. Prevalence and clinical features of common LRRK2 mutations in Australians with Parkinson's disease. Mov Disord. 2007;22:982–989. doi: 10.1002/mds.21477. [DOI] [PubMed] [Google Scholar]
- Ibanez P, Bonnet AM, Debarges B, Lohmann E, Tison F, Pollak P, Agid Y, Durr A, Brice A. Causal relation between alpha-synuclein gene duplication and familial Parkinson's disease. Lancet. 2004;364:1169–1171. doi: 10.1016/S0140-6736(04)17104-3. [DOI] [PubMed] [Google Scholar]
- Ibanez P, Lesage S, Janin S, Lohmann E, Durif F, Destee A, Bonnet AM, Brefel-Courbon C, Heath S, Zelenika D, Agid Y, Durr A, Brice A. Alpha-synuclein gene rearrangements in dominantly inherited parkinsonism: frequency, phenotype, and mechanisms. Arch Neurol. 2009;66:102–108. doi: 10.1001/archneurol.2008.555. [DOI] [PubMed] [Google Scholar]
- Ibanez P, Lesage S, Lohmann E, Thobois S, De MG, Borg M, Agid Y, Durr A, Brice A. Mutational analysis of the PINK1 gene in early-onset parkinsonism in Europe and North Africa. Brain. 2006;129:686–694. doi: 10.1093/brain/awl005. [DOI] [PubMed] [Google Scholar]
- Ikeuchi T, Kakita A, Shiga A, Kasuga K, Kaneko H, Tan CF, Idezuka J, Wakabayashi K, Onodera O, Iwatsubo T, Nishizawa M, Takahashi H, Ishikawa A. Patients homozygous and heterozygous for SNCA duplication in a family with parkinsonism and dementia. Arch Neurol. 2008;65:514–519. doi: 10.1001/archneur.65.4.514. [DOI] [PubMed] [Google Scholar]
- Illarioshkin SN, Periquet M, Rawal N, Lucking CB, Zagorovskaya TB, Slominsky PA, Miloserdova OV, Markova ED, Limborska SA, Ivanova-Smolenskaya IA, Brice A. Mutation analysis of the parkin gene in Russian families with autosomal recessive juvenile parkinsonism. Mov Disord. 2003;18:914–919. doi: 10.1002/mds.10467. [DOI] [PubMed] [Google Scholar]
- Illarioshkin SN, Shadrina MI, Slominsky PA, Bespalova EV, Zagorovskaya TB, Bagyeva GK, Markova ED, Limborska SA, Ivanova-Smolenskaya IA. A common leucine-rich repeat kinase 2 gene mutation in familial and sporadic Parkinson's disease in Russia. Eur J Neurol. 2007;14:413–417. doi: 10.1111/j.1468-1331.2007.01685.x. [DOI] [PubMed] [Google Scholar]
- Imai Y, Gehrke S, Wang HQ, Takahashi R, Hasegawa K, Oota E, Lu B. Phosphorylation of 4E-BP by LRRK2 affects the maintenance of dopaminergic neurons in Drosophila. EMBO J. 2008;27:2432–2443. doi: 10.1038/emboj.2008.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai Y, Soda M, Takahashi R. Parkin suppresses unfolded protein stress-induced cell death through its E3 ubiquitin-protein ligase activity. J Biol Chem. 2000;275:35661–35664. doi: 10.1074/jbc.C000447200. [DOI] [PubMed] [Google Scholar]
- Izumi Y, Morino H, Oda M, Maruyama H, Udaka F, Kameyama M, Nakamura S, Kawakami H. Genetic studies in Parkinson's disease with an alpha-synuclein/NACP gene polymorphism in Japan. Neurosci Lett. 2001;300:125–127. doi: 10.1016/s0304-3940(01)01557-9. [DOI] [PubMed] [Google Scholar]
- Johnson J, Paisan-Ruiz C, Lopez G, Crews C, Britton A, Malkani R, Evans EW, Inerney-Leo A, Jain S, Nussbaum RL, Foote KD, Mandel RJ, Crawley A, Reimsnider S, Fernandez HH, Okun MS, Gwinn-Hardy K, Singleton AB. Comprehensive screening of a North American Parkinson's disease cohort for LRRK2 mutation. Neurodegener Dis. 2007;4:386–391. doi: 10.1159/000105160. [DOI] [PubMed] [Google Scholar]
- Kachergus J, Mata IF, Hulihan M, Taylor JP, Lincoln S, Aasly J, Gibson JM, Ross OA, Lynch T, Wiley J, Payami H, Nutt J, Maraganore DM, Czyzewski K, Styczynska M, Wszolek ZK, Farrer MJ, Toft M. Identification of a novel LRRK2 mutation linked to autosomal dominant parkinsonism: evidence of a common founder across European populations. Am J Hum Genet. 2005;76:672–680. doi: 10.1086/429256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kann M, Jacobs H, Mohrmann K, Schumacher K, Hedrich K, Garrels J, Wiegers K, Schwinger E, Pramstaller PP, Breakefield XO, Ozelius LJ, Vieregge P, Klein C. Role of parkin mutations in 111 community-based patients with early-onset parkinsonism. Ann Neurol. 2002;51:621–625. doi: 10.1002/ana.10179. [DOI] [PubMed] [Google Scholar]
- Kay DM, Factor SA, Samii A, Higgins DS, Griffith A, Roberts JW, Leis BC, Nutt JG, Montimurro JS, Keefe RG, Atkins AJ, Yearout D, Zabetian CP, Payami H. Genetic association between alpha-synuclein and idiopathic Parkinson's disease. Am J Med Genet B Neuropsychiatr Genet. 2008;147B:1222–1230. doi: 10.1002/ajmg.b.30758. [DOI] [PubMed] [Google Scholar]
- Kay DM, Kramer P, Higgins D, Zabetian CP, Payami H. Escaping Parkinson's disease: a neurologically healthy octogenarian with the LRRK2 G2019S mutation. Mov Disord. 2005;20:1077–1078. doi: 10.1002/mds.20618. [DOI] [PubMed] [Google Scholar]
- Kay DM, Moran D, Moses L, Poorkaj P, Zabetian CP, Nutt J, Factor SA, Yu CE, Montimurro JS, Keefe RG, Schellenberg GD, Payami H. Heterozygous parkin point mutations are as common in control subjects as in Parkinson's patients. Ann Neurol. 2007;61:47–54. doi: 10.1002/ana.21039. [DOI] [PubMed] [Google Scholar]
- Kazantsev AG, Kolchinsky AM. Central role of alpha-synuclein oligomers in neurodegeneration in Parkinson disease. Arch Neurol. 2008;65:1577–1581. doi: 10.1001/archneur.65.12.1577. [DOI] [PubMed] [Google Scholar]
- Khan NL, Horta W, Eunson L, Graham E, Johnson JO, Chang S, Davis M, Singleton A, Wood NW, Lees AJ. Parkin disease in a Brazilian kindred: manifesting heterozygotes and clinical follow-up over 10 years. Mov Disord. 2005a;20:479–484. doi: 10.1002/mds.20335. [DOI] [PubMed] [Google Scholar]
- Khan NL, Jain S, Lynch JM, Pavese N, bou-Sleiman P, Holton JL, Healy DG, Gilks WP, Sweeney MG, Ganguly M, Gibbons V, Gandhi S, Vaughan J, Eunson LH, Katzenschlager R, Gayton J, Lennox G, Revesz T, Nicholl D, Bhatia KP, Quinn N, Brooks D, Lees AJ, Davis MB, Piccini P, Singleton AB, Wood NW. Mutations in the gene LRRK2 encoding dardarin (PARK8) cause familial Parkinson's disease: clinical, pathological, olfactory and functional imaging and genetic data. Brain. 2005b;128:2786–2796. doi: 10.1093/brain/awh667. [DOI] [PubMed] [Google Scholar]
- Ki CS, Stavrou EF, Davanos N, Lee WY, Chung EJ, Kim JY, Athanassiadou A. The Ala53Thr mutation in the alpha-synuclein gene in a Korean family with Parkinson disease. Clin Genet. 2007;71:471–473. doi: 10.1111/j.1399-0004.2007.00781.x. [DOI] [PubMed] [Google Scholar]
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- Klein C, Djarmati A, Hedrich K, Schafer N, Scaglione C, Marchese R, Kock N, Schule B, Hiller A, Lohnau T, Winkler S, Wiegers K, Hering R, Bauer P, Riess O, Abbruzzese G, Martinelli P, Pramstaller PP. PINK1, Parkin, and DJ-1 mutations in Italian patients with early-onset parkinsonism. Eur J Hum Genet. 2005;13:1086–1093. doi: 10.1038/sj.ejhg.5201455. [DOI] [PubMed] [Google Scholar]
- Klein C, Pramstaller PP, Kis B, Page CC, Kann M, Leung J, Woodward H, Castellan CC, Scherer M, Vieregge P, Breakefield XO, Kramer PL, Ozelius LJ. Parkin deletions in a family with adult-onset, tremor-dominant parkinsonism: expanding the phenotype. Ann Neurol. 2000;48:65–71. [PubMed] [Google Scholar]
- Kobayashi T, Wang M, Hattori N, Matsumine H, Kondo T, Mizuno Y. Exonic deletion mutations of the Parkin gene among sporadic patients with Parkinson's disease. Parkinsonism Relat Disord. 2000;6:129–131. doi: 10.1016/s1353-8020(00)00006-7. [DOI] [PubMed] [Google Scholar]
- Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Riess O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat Genet. 1998;18:106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- Kumazawa R, Tomiyama H, Li Y, Imamichi Y, Funayama M, Yoshino H, Yokochi F, Fukusako T, Takehisa Y, Kashihara K, Kondo T, Elibol B, Bostantjopoulou S, Toda T, Takahashi H, Yoshii F, Mizuno Y, Hattori N. Mutation analysis of the PINK1 gene in 391 patients with Parkinson disease. Arch Neurol. 2008;65:802–808. doi: 10.1001/archneur.65.6.802. [DOI] [PubMed] [Google Scholar]
- Latourelle JC, Sun M, Lew MF, Suchowersky O, Klein C, Golbe LI, Mark MH, Growdon JH, Wooten GF, Watts RL, Guttman M, Racette BA, Perlmutter JS, Ahmed A, Shill HA, Singer C, Goldwurm S, Pezzoli G, Zini M, Saint-Hilaire MH, Hendricks AE, Williamson S, Nagle MW, Wilk JB, Massood T, Huskey KW, Laramie JM, DeStefano AL, Baker KB, Itin I, Litvan I, Nicholson G, Corbett A, Nance M, Drasby E, Isaacson S, Burn DJ, Chinnery PF, Pramstaller PP, Al-hinti J, Moller AT, Ostergaard K, Sherman SJ, Roxburgh R, Snow B, Slevin JT, Cambi F, Gusella JF, Myers RH. The Gly2019Ser mutation in LRRK2 is not fully penetrant in familial Parkinson's disease: the GenePD study. BMC Med. 2008;6:32. doi: 10.1186/1741-7015-6-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lautier C, Goldwurm S, Durr A, Giovannone B, Tsiaras WG, Pezzoli G, Brice A, Smith RJ. Mutations in the GIGYF2 (TNRC15) gene at the PARK11 locus in familial Parkinson disease. Am J Hum Genet. 2008;82:822–833. doi: 10.1016/j.ajhg.2008.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MJ, Mata IF, Lin CH, Tzen KY, Lincoln SJ, Bounds R, Lockhart PJ, Hulihan MM, Farrer MJ, Wu RM. Genotype–phenotype correlates in Taiwanese patients with early-onset recessive Parkinsonism. Mov Disord. 2009;24:104–108. doi: 10.1002/mds.22093. [DOI] [PubMed] [Google Scholar]
- Leroy E, Anastasopoulos D, Konitsiotis S, Lavedan C, Polymeropoulos MH. Deletions in the Parkin gene and genetic heterogeneity in a Greek family with early onset Parkinson's disease. Hum Genet. 1998a;103:424–427. doi: 10.1007/s004390050845. [DOI] [PubMed] [Google Scholar]
- Leroy E, Boyer R, Auburger G, Leube B, Ulm G, Mezey E, Harta G, Brownstein MJ, Jonnalagada S, Chernova T, Dehejia A, Lavedan C, Gasser T, Steinbach PJ, Wilkinson KD, Polymeropoulos MH. The ubiquitin pathway in Parkinson's disease. Nature. 1998b;395:451–452. doi: 10.1038/26652. [DOI] [PubMed] [Google Scholar]
- Lesage S, Condroyer C, Lannuzel A, Lohmann E, Troiano A, Tison F, Damier P, Thobois S, Ouvrard-Hernandez AM, Rivaud-Pechoux S, Brefel-Courbon C, Destee A, Tranchant C, Romana M, Leclere L, Durr A, Brice A. Molecular analyses of the LRRK2 gene in European and North-African autosomal dominant Parkinson's disease. J Med Genet. 2009;46:458–464. doi: 10.1136/jmg.2008.062612. [DOI] [PubMed] [Google Scholar]
- Lesage S, Durr A, Tazir M, Lohmann E, Leutenegger AL, Janin S, Pollak P, Brice A. LRRK2 G2019S as a cause of Parkinson's disease in North African Arabs. N Engl J Med. 2006;354:422–423. doi: 10.1056/NEJMc055540. [DOI] [PubMed] [Google Scholar]
- Lesage S, Janin S, Lohmann E, Leutenegger AL, Leclere L, Viallet F, Pollak P, Durif F, Thobois S, Layet V, Vidailhet M, Agid Y, Durr A, Brice A, Bonnet AM, Borg M, Broussolle E, Damier P, Destee A, Martinez M, Penet C, Rasco O, Tison F, Tranchan C, Verin M. LRRK2 exon 41 mutations in sporadic Parkinson disease in Europeans. Arch Neurol. 2007a;64:425–430. doi: 10.1001/archneur.64.3.425. [DOI] [PubMed] [Google Scholar]
- Lesage S, Lohmann E, Tison F, Durif F, Durr A, Brice A. Rare heterozygous parkin variants in French early-onset Parkinson disease patients and controls. J Med Genet. 2008;45:43–46. doi: 10.1136/jmg.2007.051854. [DOI] [PubMed] [Google Scholar]
- Lesage S, Magali P, Lohmann E, Lacomblez L, Teive H, Janin S, Cousin PY, Durr A, Brice A. Deletion of the parkin and PACRG gene promoter in early-onset parkinsonism. Hum Mutat. 2007b;28:27–32. doi: 10.1002/humu.20436. [DOI] [PubMed] [Google Scholar]
- Leutenegger AL, Salih MA, Ibanez P, Mukhtar MM, Lesage S, Arabi A, Lohmann E, Durr A, Ahmed AE, Brice A. Juvenile-onset Parkinsonism as a result of the first mutation in the adenosine triphosphate orientation domain of PINK1. Arch Neurol. 2006;63:1257–1261. doi: 10.1001/archneur.63.9.1257. [DOI] [PubMed] [Google Scholar]
- Lev N, Ickowicz D, Barhum Y, Lev S, Melamed E, Offen D. DJ-1 protects against dopamine toxicity. J Neural Transm. 2009;116:151–160. doi: 10.1007/s00702-008-0134-4. [DOI] [PubMed] [Google Scholar]
- Lewis PA, Greggio E, Beilina A, Jain S, Baker A, Cookson MR. The R1441C mutation of LRRK2 disrupts GTP hydrolysis. Biochem Biophys Res Commun. 2007;357:668–671. doi: 10.1016/j.bbrc.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Kitami T, Wang M, Mizuno Y, Hattori N. Geographic and ethnic differences in frequencies of two polymorphisms (D/N394 and L/I272) of the parkin gene in sporadic Parkinson's disease. Parkinsonism.Relat Disord. 2005a;11:485–491. doi: 10.1016/j.parkreldis.2005.06.006. [DOI] [PubMed] [Google Scholar]
- Li X, Tan YC, Poulose S, Olanow CW, Huang XY, Yue Z. Leucine-rich repeat kinase 2 (LRRK2)/PARK8 possesses GTPase activity that is altered in familial Parkinson's disease R1441C/G mutants. J Neurochem. 2007;103:238–247. doi: 10.1111/j.1471-4159.2007.04743.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Tomiyama H, Sato K, Hatano Y, Yoshino H, Atsumi M, Kitaguchi M, Sasaki S, Kawaguchi S, Miyajima H, Toda T, Mizuno Y, Hattori N. Clinicogenetic study of PINK1 mutations in autosomal recessive early-onset parkinsonism. Neurology. 2005b;64:1955–1957. doi: 10.1212/01.WNL.0000164009.36740.4E. [DOI] [PubMed] [Google Scholar]
- Limousin N, Konofal E, Karroum E, Lohmann E, Theodorou I, Durr A, Arnulf I. Restless legs syndrome, rapid eye movement sleep behavior disorder, and hypersomnia in patients with two parkin mutations. Mov Disord. 2009;24:1970–1976. doi: 10.1002/mds.22711. [DOI] [PubMed] [Google Scholar]
- Liu S, Ninan I, Antonova I, Battaglia F, Trinchese F, Narasanna A, Kolodilov N, Dauer W, Hawkins RD, Arancio O. alpha-Synuclein produces a long-lasting increase in neurotransmitter release. EMBO J. 2004;23:4506–4516. doi: 10.1038/sj.emboj.7600451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Vives-Bauza C, Acin P, Yamamoto A, Tan Y, Li Y, Magrane J, Stavarache MA, Shaffer S, Chang S, Kaplitt MG, Huang XY, Beal MF, Manfredi G, Li C. PINK1 defect causes mitochondrial dysfunction, proteasomal deficit and alpha-synuclein aggregation in cell culture models of Parkinson's disease. PLoS ONE. 2009;4:e4597. doi: 10.1371/journal.pone.0004597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucking CB, Abbas N, Durr A, Bonifati V, Bonnet AM, de BT, De MG, Wood NW, Agid Y, Brice A. Homozygous deletions in parkin gene in European and North African families with autosomal recessive juvenile parkinsonism. The European Consortium on Genetic Susceptibility in Parkinson's Disease and the French Parkinson's Disease Genetics Study Group. Lancet. 1998;352:1355–1356. doi: 10.1016/s0140-6736(05)60746-5. [DOI] [PubMed] [Google Scholar]
- Lucking CB, Durr A, Bonifati V, Vaughan J, De MG, Gasser T, Harhangi BS, Meco G, Denefle P, Wood NW, Agid Y, Brice A. Association between early-onset Parkinson's disease and mutations in the parkin gene. N Engl J Med. 2000;342:1560–1567. doi: 10.1056/NEJM200005253422103. [DOI] [PubMed] [Google Scholar]
- Macedo MG, Verbaan D, Fang Y, van Rooden SM, Visser M, Anar B, Uras A, Groen JL, Rizzu P, van Hilten JJ, Heutink P. Genotypic and phenotypic characteristics of Dutch patients with early onset Parkinson's disease. Mov Disord. 2009;24:196–203. doi: 10.1002/mds.22287. [DOI] [PubMed] [Google Scholar]
- Madegowda RH, Kishore A, Anand A. Mutational screening of the parkin gene among South Indians with early onset Parkinson's disease. J Neurol Neurosurg Psychiatry. 2005;76:1588–1590. doi: 10.1136/jnnp.2004.046888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maraganore DM, de AM, Elbaz A, Farrer MJ, Ioannidis JP, Kruger R, Rocca WA, Schneider NK, Lesnick TG, Lincoln SJ, Hulihan MM, Aasly JO. Collaborative analysis of alpha-synuclein gene promoter variability and Parkinson disease. JAMA. 2006;296:661–670. doi: 10.1001/jama.296.6.661. [DOI] [PubMed] [Google Scholar]
- Marongiu R, Brancati F, Antonini A, Ialongo T, Ceccarini C, Scarciolla O, Capalbo A, Benti R, Pezzoli G, Dallapiccola B, Goldwurm S, Valente EM. Whole gene deletion and splicing mutations expand the PINK1 genotypic spectrum. Hum Mutat. 2007;28:98. doi: 10.1002/humu.9472. [DOI] [PubMed] [Google Scholar]
- Marongiu R, Ferraris A, Ialongo T, Michiorri S, Soleti F, Ferrari F, Elia AE, Ghezzi D. PINK1 heterozygous rare variants: prevalence, significance and phenotypic spectrum. Hum Mutat. 2008;29:565. doi: 10.1002/humu.20719. [DOI] [PubMed] [Google Scholar]
- Marras C, Klein C, Lang AE, Wakutani Y, Moreno D, Sato C, Yip E, Munhoz RP, Lohmann K, Djarmati A, Bi A, Rogaeva E. LRRK2 and Parkin mutations in a family with parkinsonism—Lack of genotype–phenotype correlation. Neurobiol Aging. 2010;31:721–722. doi: 10.1016/j.neurobiolaging.2008.05.030. [DOI] [PubMed] [Google Scholar]
- Maruyama M, Ikeuchi T, Saito M, Ishikawa A, Yuasa T, Tanaka H, Hayashi S, Wakabayashi K, Takahashi H, Tsuji S. Novel mutations, pseudo-dominant inheritance, and possible familial affects in patients with autosomal recessive juvenile parkinsonism. Ann Neurol. 2000;48:245–250. [PubMed] [Google Scholar]
- Mata IF, Alvarez V, Coto E, Blazquez M, Guisasola LM, Salvador C, Kachergus JM, Lincoln SJ, Farrer M. Homozygous partial genomic triplication of the parkin gene in early-onset parkinsonism. Neurosci Lett. 2005a;380:257–259. doi: 10.1016/j.neulet.2005.01.051. [DOI] [PubMed] [Google Scholar]
- Mata IF, Kachergus JM, Taylor JP, Lincoln S, Aasly J, Lynch T, Hulihan MM, Cobb SA, Wu RM, Lu CS, Lahoz C, Wszolek ZK, Farrer MJ. Lrrk2 pathogenic substitutions in Parkinson's disease. Neurogenetics. 2005b;6:171–177. doi: 10.1007/s10048-005-0005-1. [DOI] [PubMed] [Google Scholar]
- Mata IF, Taylor JP, Kachergus J, Hulihan M, Huerta C, Lahoz C, Blazquez M, Guisasola LM, Salvador C, Ribacoba R, Martinez C, Farrer M, Alvarez V. LRRK2 R1441G in Spanish patients with Parkinson's disease. Neurosci Lett. 2005c;382:309–311. doi: 10.1016/j.neulet.2005.03.033. [DOI] [PubMed] [Google Scholar]
- Meeus B, Theuns J, Van Broeckhoven C. GIGYF2 in Parkinson's disease: innocent until proven otherwise. Neurobiol Aging. 2010 (in press) [Google Scholar]
- Mellick GD, Siebert GA, Funayama M, Buchanan DD, Li Y, Imamichi Y, Yoshino H, Silburn PA, Hattori N. Screening PARK genes for mutations in early-onset Parkinson's disease patients from Queensland, Australia. Parkinsonism Relat Disord. 2009;15:105–109. doi: 10.1016/j.parkreldis.2007.11.016. [DOI] [PubMed] [Google Scholar]
- Michell AW, Barker RA, Raha SK, Raha-Chowdhury R. A case of late onset sporadic Parkinson's disease with an A53T mutation in alpha-synuclein. J Neurol Neurosurg Psychiatry. 2005;76:596–597. doi: 10.1136/jnnp.2004.046425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsumoto A, Nakagawa Y. DJ-1 is an indicator for endogenous reactive oxygen species elicited by endotoxin. Free Radic Res. 2001;35:885–893. doi: 10.1080/10715760100301381. [DOI] [PubMed] [Google Scholar]
- Mizuta I, Satake W, Nakabayashi Y, Ito C, Suzuki S, Momose Y, Nagai Y, Oka A, Inoko H, Fukae J, Saito Y, Sawabe M, Murayama S, Yamamoto M, Hattori N, Murata M, Toda T. Multiple candidate gene analysis identifies alpha-synuclein as a susceptibility gene for sporadic Parkinson's disease. Hum Mol Genet. 2006;15:1151–1158. doi: 10.1093/hmg/ddl030. [DOI] [PubMed] [Google Scholar]
- Moore DJ, Zhang L, Dawson TM, Dawson VL. A missense mutation (L166P) in DJ-1, linked to familial Parkinson's disease, confers reduced protein stability and impairs homo-oligomerization. J Neurochem. 2003;87:1558–1567. doi: 10.1111/j.1471-4159.2003.02265.x. [DOI] [PubMed] [Google Scholar]
- Moro E, Volkmann J, Konig IR, Winkler S, Hiller A, Hassin-Baer S, Herzog J, Schnitzler A, Lohmann K, Pinsker MO, Voges J, Djarmatic A, Seibler P, Lozano AM, Rogaeva E, Lang AE, Deuschl G, Klein C. Bilateral subthalamic stimulation in Parkin and PINK1 parkinsonism. Neurology. 2008;70:1186–1191. doi: 10.1212/01.wnl.0000307748.11216.03. [DOI] [PubMed] [Google Scholar]
- Mueller JC, Fuchs J, Hofer A, Zimprich A, Lichtner P, Illig T, Berg D, Wullner U, Meitinger T, Gasser T. Multiple regions of alpha-synuclein are associated with Parkinson's disease. Ann Neurol. 2005;57:535–541. doi: 10.1002/ana.20438. [DOI] [PubMed] [Google Scholar]
- Munhoz RP, Sa DS, Rogaeva E, Salehi-Rad S, Sato C, Medeiros H, Farrer M, Lang AE. Clinical findings in a large family with a parkin ex3delta40 mutation. Arch Neurol. 2004;61:701–704. doi: 10.1001/archneur.61.5.701. [DOI] [PubMed] [Google Scholar]
- Munoz E, Pastor P, Marti MJ, Oliva R, Tolosa E. A new mutation in the parkin gene in a patient with atypical autosomal recessive juvenile parkinsonism. Neurosci Lett. 2000;289:66–68. doi: 10.1016/s0304-3940(00)01248-9. [DOI] [PubMed] [Google Scholar]
- Munoz E, Tolosa E, Pastor P, Marti MJ, Valldeoriola F, Campdelacreu J, Oliva R. Relative high frequency of the c.255delA parkin gene mutation in Spanish patients with autosomal recessive parkinsonism. J Neurol Neurosurg Psychiatry. 2002;73:582–584. doi: 10.1136/jnnp.73.5.582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myhre R, Steinkjer S, Stormyr A, Nilsen GL, Abu ZH, Horany K, Nusier MK, Klungland H. Significance of the parkin and PINK1 gene in Jordanian families with incidences of young-onset and juvenile parkinsonism. BMC Neurol. 2008a;8:47. doi: 10.1186/1471-2377-8-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myhre R, Toft M, Kachergus J, Hulihan MM, Aasly JO, Klungland H, Farrer MJ. Multiple alpha-synuclein gene polymorphisms are associated with Parkinson's disease in a Norwegian population. Acta Neurol Scand. 2008b;118:320–327. doi: 10.1111/j.1600-0404.2008.01019.x. [DOI] [PubMed] [Google Scholar]
- Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin-induced mitophagy in the pathogenesis of Parkinson disease. Autophagy. 2009;5:706–708. doi: 10.4161/auto.5.5.8505. [DOI] [PubMed] [Google Scholar]
- Nichols WC, Elsaesser VE, Pankratz N, Pauciulo MW, Marek DK, Halter CA, Rudolph A, Shults CW, Foroud T. LRRK2 mutation analysis in Parkinson disease families with evidence of linkage to PARK8. Neurology. 2007;69:1737–1744. doi: 10.1212/01.wnl.0000278115.50741.4e. [DOI] [PubMed] [Google Scholar]
- Nichols WC, Pankratz N, Hernandez D, Paisan-Ruiz C, Jain S, Halter CA, Michaels VE, Reed T, Rudolph A, Shults CW, Singleton A, Foroud T. Genetic screening for a single common LRRK2 mutation in familial Parkinson's disease. Lancet. 2005;365:410–412. doi: 10.1016/S0140-6736(05)17828-3. [DOI] [PubMed] [Google Scholar]
- Nichols WC, Pankratz N, Uniacke SK, Pauciulo MW, Halter C, Rudolph A, Conneally PM, Foroud T. Linkage stratification and mutation analysis at the Parkin locus identifies mutation positive Parkinson's disease families. J Med Genet. 2002;39:489–492. doi: 10.1136/jmg.39.7.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishioka K, Hayashi S, Farrer MJ, Singleton AB, Yoshino H, Imai H, Kitami T, Sato K, Kuroda R, Tomiyama H, Mizoguchi K, Murata M, Toda T, Imoto I, Inazawa J, Mizuno Y, Hattori N. Clinical heterogeneity of alpha-synuclein gene duplication in Parkinson's disease. Ann Neurol. 2006;59:298–309. doi: 10.1002/ana.20753. [DOI] [PubMed] [Google Scholar]
- Nishioka K, Ross OA, Ishii K, Kachergus JM, Ishiwata K, Kitagawa M, Kono S, Obi T, Mizoguchi K, Inoue Y, Imai H, Takanashi M, Mizuno Y, Farrer MJ, Hattori N. Expanding the clinical phenotype of SNCA duplication carriers. Mov Disord. 2009;24:1811–1819. doi: 10.1002/mds.22682. [DOI] [PubMed] [Google Scholar]
- Nisipeanu P, Inzelberg R, Abo MS, Carasso RL, Blumen SC, Zhang J, Matsumine H, Hattori N, Mizuno Y. Parkin gene causing benign autosomal recessive juvenile parkinsonism. Neurology. 2001;56:1573–1575. doi: 10.1212/wnl.56.11.1573. [DOI] [PubMed] [Google Scholar]
- Nuytemans K, Meeus B, Crosiers D, Brouwers N, Goossens D, Engelborghs S, Pals P, Pickut B, Van den BM, Corsmit E, Cras P, De Deyn PP, Del-Favero J, Van BC, Theuns J. Relative contribution of simple mutations vs. copy number variations in five Parkinson disease genes in the Belgian population. Hum Mutat. 2009;30:1054–1061. doi: 10.1002/humu.21007. [DOI] [PubMed] [Google Scholar]
- Nuytemans K, Rademakers R, Theuns J, Pals P, Engelborghs S, Pickut B, de PT, Peeters K, Mattheijssens M, Van den BM, Cras P, De Deyn PP, Van BC. Founder mutation p.R1441C in the leucine-rich repeat kinase 2 gene in Belgian Parkinson's disease patients. Eur J Hum Genet. 2008;16:471–479. doi: 10.1038/sj.ejhg.5201986. [DOI] [PubMed] [Google Scholar]
- Obi T, Nishioka K, Ross OA, Terada T, Yamazaki K, Sugiura A, Takanashi M, Mizoguchi K, Mori H, Mizuno Y, Hattori N. Clinicopathologic study of a SNCA gene duplication patient with Parkinson disease and dementia. Neurology. 2008;70:238–241. doi: 10.1212/01.wnl.0000299387.59159.db. [DOI] [PubMed] [Google Scholar]
- Okubadejo N, Britton A, Crews C, Akinyemi R, Hardy J, Singleton A, Bras J. Analysis of Nigerians with apparently sporadic Parkinson disease for mutations in LRRK2, PRKN and ATXN3. PLoS ONE. 2008;3:e3421. doi: 10.1371/journal.pone.0003421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira SA, Scott WK, Martin ER, Nance MA, Watts RL, Hubble JP, Koller WC, Pahwa R. Parkin mutations and susceptibility alleles in late-onset Parkinson's disease. Ann Neurol. 2003;53:624–629. doi: 10.1002/ana.10524. [DOI] [PubMed] [Google Scholar]
- Orr-Urtreger A, Shifrin C, Rozovski U, Rosner S, Bercovich D, Gurevich T, Yagev-More H, Bar-Shira A, Giladi N. The LRRK2 G2019S mutation in Ashkenazi Jews with Parkinson disease: is there a gender effect? Neurology. 2007;69:1595–1602. doi: 10.1212/01.wnl.0000277637.33328.d8. [DOI] [PubMed] [Google Scholar]
- Ozelius LJ, Senthil G, Saunders-Pullman R, Ohmann E, Deligtisch A, Tagliati M, Hunt AL, Klein C, Henick B, Hailpern SM, Lipton RB, Soto-Valencia J, Risch N, Bressman SB. LRRK2 G2019S as a cause of Parkinson's disease in Ashkenazi Jews. N Engl J Med. 2006;354:424–425. doi: 10.1056/NEJMc055509. [DOI] [PubMed] [Google Scholar]
- Paisan-Ruiz C. LRRK2 gene variation and its contribution to Parkinson disease. Hum Mutat. 2009;30:1153–1160. doi: 10.1002/humu.21038. [DOI] [PubMed] [Google Scholar]
- Paisan-Ruiz C, Bhatia KP, Li A, Hernandez D, Davis M, Wood NW, Hardy J, Houlden H, Singleton A, Schneider SA. Characterization of PLA2G6 as a locus for dystonia-parkinsonism. Ann Neurol. 2009;65:19–23. doi: 10.1002/ana.21415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paisan-Ruiz C, Jain S, Evans EW, Gilks WP, Simon J, van der BM, Lopez de MA, Aparicio S, Gil AM, Khan N, Johnson J, Martinez JR, Nicholl D, Carrera IM, Pena AS, de SR, Lees A, Marti-Masso JF, Perez-Tur J, Wood NW, Singleton AB. Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron. 2004;44:595–600. doi: 10.1016/j.neuron.2004.10.023. [DOI] [PubMed] [Google Scholar]
- Paisan-Ruiz C, Lang AE, Kawarai T, Sato C, Salehi-Rad S, Fisman GK, Al-Khairallah T, St George-Hyslop P, Singleton A, Rogaeva E. LRRK2 gene in Parkinson disease: mutation analysis and case control association study. Neurology. 2005;65:696–700. doi: 10.1212/01.wnl.0000167552.79769.b3. [DOI] [PubMed] [Google Scholar]
- Paisan-Ruiz C, Nath P, Washecka N, Gibbs JR, Singleton AB. Comprehensive analysis of LRRK2 in publicly available Parkinson's disease cases and neurologically normal controls. Hum Mutat. 2008;29:485–490. doi: 10.1002/humu.20668. [DOI] [PubMed] [Google Scholar]
- Pals P, Lincoln S, Manning J, Heckman M, Skipper L, Hulihan M, Van den Broeck M, De Pooter T, Cras P, Crook J, Van Broeckhoven C, Farrer MJ. alpha-Synuclein promoter confers susceptibility to Parkinson's disease. Ann Neurol. 2004a;56:591–595. doi: 10.1002/ana.20268. [DOI] [PubMed] [Google Scholar]
- Pals P, Lincoln S, Manning J, Heckman M, Skipper L, Hulihan M, Van den BM, de Pooter T, Cras P, Crook J, Van BC, Farrer MJ. alpha-Synuclein promoter confers susceptibility to Parkinson's disease. Ann Neurol. 2004b;56:591–595. doi: 10.1002/ana.20268. [DOI] [PubMed] [Google Scholar]
- Pankratz N, Pauciulo MW, Elsaesser VE, Marek DK, Halter CA, Rudolph A, Shults CW, Foroud T, Nichols WC. Mutations in LRRK2 other than G2019S are rare in a north American-based sample of familial Parkinson's disease. Mov Disord. 2006a;21:2257–2260. doi: 10.1002/mds.21162. [DOI] [PubMed] [Google Scholar]
- Pankratz N, Pauciulo MW, Elsaesser VE, Marek DK, Halter CA, Wojcieszek J, Rudolph A, Shults CW, Foroud T, Nichols WC. Mutations in DJ-1 are rare in familial Parkinson disease. Neurosci Lett. 2006b;408:209–213. doi: 10.1016/j.neulet.2006.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankratz N, Wilk JB, Latourelle JC, DeStefano AL, Halter C, Pugh EW, Doheny KF, Gusella JF, Nichols WC, Foroud T, Myers RH. Genomewide association study for susceptibility genes contributing to familial Parkinson disease. Hum Genet. 2009;124:593–605. doi: 10.1007/s00439-008-0582-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadimitriou A, Veletza V, Hadjigeorgiou GM, Patrikiou A, Hirano M, Anastasopoulos I. Mutated alpha-synuclein gene in two Greek kindreds with familial PD: incomplete penetrance? Neurology. 1999;52:651–654. doi: 10.1212/wnl.52.3.651. [DOI] [PubMed] [Google Scholar]
- Papapetropoulos S, Ellul J, Paschalis C, Athanassiadou A, Papadimitriou A, Papapetropoulos T. Clinical characteristics of the alpha-synuclein mutation (G209A)-associated Parkinson's disease in comparison with other forms of familial Parkinson's disease in Greece. Eur J Neurol. 2003;10:281–286. doi: 10.1046/j.1468-1331.2003.00576.x. [DOI] [PubMed] [Google Scholar]
- Papapetropoulos S, Paschalis C, Athanassiadou A, Papadimitriou A, Ellul J, Polymeropoulos MH, Papapetropoulos T. Clinical phenotype in patients with alpha-synuclein Parkinson's disease living in Greece in comparison with patients with sporadic Parkinson's disease. J Neurol Neurosurg Psychiatry. 2001;70:662–665. doi: 10.1136/jnnp.70.5.662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papapetropoulos S, Singer C, Ross OA, Toft M, Johnson JL, Farrer MJ, Mash DC. Clinical heterogeneity of the LRRK2 G2019S mutation. Arch Neurol. 2006;63:1242–1246. doi: 10.1001/archneur.63.9.1242. [DOI] [PubMed] [Google Scholar]
- Park J, Lee G, Chung J. The PINK1-Parkin pathway is involved in the regulation of mitochondrial remodeling process. Biochem Biophys Res Commun. 2009;378:518–523. doi: 10.1016/j.bbrc.2008.11.086. [DOI] [PubMed] [Google Scholar]
- Parsian AJ, Racette BA, Zhao JH, Sinha R, Patra B, Perlmutter JS, Parsian A. Association of alpha-synuclein gene haplotypes with Parkinson's disease. Parkinsonism Relat Disord. 2007;13:343–347. doi: 10.1016/j.parkreldis.2006.12.006. [DOI] [PubMed] [Google Scholar]
- Pchelina SN, Yakimovskii AF, Emelyanov AK, Ivanova ON, Schwarzman AL, Singleton AB. Screening for LRRK2 mutations in patients with Parkinson's disease in Russia: identification of a novel LRRK2 variant. Eur J Neurol. 2008;15:692–696. doi: 10.1111/j.1468-1331.2008.02149.x. [DOI] [PubMed] [Google Scholar]
- Pellecchia MT, Varrone A, Annesi G, Amboni M, Cicarelli G, Sansone V, Annesi F, Rocca FE, Vitale C, Pappata S, Quattrone A, Barone P. Parkinsonism and essential tremor in a family with pseudo-dominant inheritance of PARK2: an FP-CIT SPECT study. Mov Disord. 2007;22:559–563. doi: 10.1002/mds.21262. [DOI] [PubMed] [Google Scholar]
- Periquet M, Latouche M, Lohmann E, Rawal N, De MG, Ricard S, Teive H, Fraix V, Vidailhet M, Nicholl D, Barone P, Wood NW, Raskin S, Deleuze JF, Agid Y, Durr A, Brice A. Parkin mutations are frequent in patients with isolated early-onset parkinsonism. Brain. 2003;126:1271–1278. doi: 10.1093/brain/awg136. [DOI] [PubMed] [Google Scholar]
- Periquet M, Lucking C, Vaughan J, Bonifati V, Durr A, De MG, Horstink M, Farrer M, Illarioshkin SN, Pollak P, Borg M, Brefel-Courbon C, Denefle P, Meco G, Gasser T, Breteler MM, Wood N, Agid Y, Brice A. Origin of the mutations in the parkin gene in Europe: exon rearrangements are independent recurrent events, whereas point mutations may result from Founder effects. Am J Hum Genet. 2001;68:617–626. doi: 10.1086/318791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry G, Zhu X, Babar AK, Siedlak SL, Yang Q, Ito G, Iwatsubo T, Smith MA, Chen SG. Leucine-rich repeat kinase 2 colocalizes with alpha-synuclein in Parkinson's disease, but not tau-containing deposits in tauopathies. Neurodegener Dis. 2008;5:222–224. doi: 10.1159/000113708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesah Y, Pham T, Burgess H, Middlebrooks B, Verstreken P, Zhou Y, Harding M, Bellen H, Mardon G. Drosophila parkin mutants have decreased mass and cell size and increased sensitivity to oxygen radical stress. Development. 2004;131:2183–2194. doi: 10.1242/dev.01095. [DOI] [PubMed] [Google Scholar]
- Pigullo S, De LA, Barone P, Marchese R, Bellone E, Colosimo A, Scaglione C, Martinelli P, Di ME, Pizzuti A, Abbruzzese G, Dallapiccola B, Ajmar F, Mandich P. Mutational analysis of parkin gene by denaturing high-performance liquid chromatography (DHPLC) in essential tremor. Parkinsonism Relat Disord. 2004;10:357–362. doi: 10.1016/j.parkreldis.2004.04.012. [DOI] [PubMed] [Google Scholar]
- Pineda-Trujillo N, Carvajal-Carmona LG, Buritica O, Moreno S, Uribe C, Pineda D, Toro M, Garcia F, Arias W, Bedoya G, Lopera F, Ruiz-Linares A. A novel Cys212Tyr founder mutation in parkin and allelic heterogeneity of juvenile Parkinsonism in a population from North West Colombia. Neurosci Lett. 2001;298:87–90. doi: 10.1016/s0304-3940(00)01733-x. [DOI] [PubMed] [Google Scholar]
- Plun-Favreau H, Klupsch K, Moisoi N, Gandhi S, Kjaer S, Frith D, Harvey K, Deas E, Harvey RJ, McDonald N, Wood NW, Martins LM, Downward J. The mitochondrial protease HtrA2 is regulated by Parkinson's disease-associated kinase PINK1. Nat Cell Biol. 2007;9:1243–1252. doi: 10.1038/ncb1644. [DOI] [PubMed] [Google Scholar]
- Polymeropoulos MH, Higgins JJ, Golbe LI, Johnson WG, Ide SE, Di IG, Sanges G, Stenroos ES, Pho LT, Schaffer AA, Lazzarini AM, Nussbaum RL, Duvoisin RC. Mapping of a gene for Parkinson's disease to chromosome 4q21–q23. Science. 1996;274:1197–1199. doi: 10.1126/science.274.5290.1197. [DOI] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di IG, Golbe LI, Nussbaum RL. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- Poole AC, Thomas RE, Andrews LA, McBride HM, Whitworth AJ, Pallanck LJ. The PINK1/Parkin pathway regulates mitochondrial morphology. Proc Natl Acad Sci USA. 2008;105:1638–1643. doi: 10.1073/pnas.0709336105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poorkaj P, Nutt JG, James D, Gancher S, Bird TD, Steinbart E, Schellenberg GD, Payami H. parkin mutation analysis in clinic patients with early-onset Parkinson [corrected] disease. Am J Med Genet A. 2004;129A:44–50. doi: 10.1002/ajmg.a.30157. [DOI] [PubMed] [Google Scholar]
- Pramstaller PP, Schlossmacher MG, Jacques TS, Scaravilli F, Eskelson C, Pepivani I, Hedrich K, Adel S, Gonzales-McNeal M, Hilker R, Kramer PL, Klein C. Lewy body Parkinson's disease in a large pedigree with 77 Parkin mutation carriers. Ann Neurol. 2005;58:411–422. doi: 10.1002/ana.20587. [DOI] [PubMed] [Google Scholar]
- Prestel J, Gempel K, Hauser TK, Schweitzer K, Prokisch H, Ahting U, Freudenstein D, Bueltmann E, Naegele T, Berg D, Klopstock T, Gasser T. Clinical and molecular characterisation of a Parkinson family with a novel PINK1 mutation. J Neurol. 2008;255:643–648. doi: 10.1007/s00415-008-0763-4. [DOI] [PubMed] [Google Scholar]
- Pridgeon JW, Olzmann JA, Chin LS, Li L. PINK1 protects against oxidative stress by phosphorylating mitochondrial chaperone TRAP1. PLoS Biol. 2007;5:e172. doi: 10.1371/journal.pbio.0050172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puschmann A, Ross OA, Vilarino-Guell C, Lincoln SJ, Kachergus JM, Cobb SA, Lindquist SG, Nielsen JE, Wszolek ZK, Farrer M, Widner H, van WD, Hagerstrom D, Markopoulou K, Chase BA, Nilsson K, Reimer J, Nilsson C. A Swedish family with de novo alpha-synuclein A53T mutation: evidence for early cortical dysfunction. Parkinsonism Relat Disord. 2009;15:627–632. doi: 10.1016/j.parkreldis.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajput A, Dickson DW, Robinson CA, Ross OA, Dachsel JC, Lincoln SJ, Cobb SA, Rajput ML, Farrer MJ. Parkinsonism, Lrrk2 G2019S, and tau neuropathology. Neurology. 2006;67:1506–1508. doi: 10.1212/01.wnl.0000240220.33950.0c. [DOI] [PubMed] [Google Scholar]
- Ramirez A, Heimbach A, Grundemann J, Stiller B, Hampshire D, Cid LP, Goebel I, Mubaidin AF, Wriekat AL, Roeper J, Al-Din A, Hillmer AM, Karsak M, Liss B, Woods CG, Behrens MI, Kubisch C. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat Genet. 2006;38:1184–1191. doi: 10.1038/ng1884. [DOI] [PubMed] [Google Scholar]
- Ramsey CP, Giasson BI. The E163K DJ-1 mutant shows specific antioxidant deficiency. Brain Res. 2008;1239:1–11. doi: 10.1016/j.brainres.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawal N, Periquet M, Lohmann E, Lucking CB, Teive HA, Ambrosio G, Raskin S, Lincoln S, Hattori N, Guimaraes J, Horstink MW, Dos Santos BW, Brousolle E, Destee A, Mizuno Y, Farrer M, Deleuze JF, De MG, Agid Y, Durr A, Brice A. New parkin mutations and atypical phenotypes in families with autosomal recessive parkinsonism. Neurology. 2003;60:1378–1381. doi: 10.1212/01.wnl.0000056167.89221.be. [DOI] [PubMed] [Google Scholar]
- Reetz K, Lencer R, Steinlechner S, Gaser C, Hagenah J, Buchel C, Petersen D, Kock N, Djarmati A, Siebner HR, Klein C, Binkofski F. Limbic and frontal cortical degeneration is associated with psychiatric symptoms in PINK1 mutation carriers. Biol Psychiatry. 2008;64:241–247. doi: 10.1016/j.biopsych.2007.12.010. [DOI] [PubMed] [Google Scholar]
- Rocca FE, Annesi F, Ciro CI, Carrideo S, Tarantino P, Provenzano G, Civitelli D, De Marco EV, Quattrone A, Annesi G. Novel human pathological mutations. Gene symbol: PARK2. Disease: Parkinson's disease. Hum Genet. 2007;122:415. [PubMed] [Google Scholar]
- Rogaeva E, Johnson J, Lang AE, Gulick C, Gwinn-Hardy K, Kawarai T, Sato C, Morgan A, Werner J, Nussbaum R, Petit A, Okun MS, McInerney A, Mandel R, Groen JL, Fernandez HH, Postuma R, Foote KD, Salehi-Rad S, Liang Y, Reimsnider S, Tandon A, Hardy J, St George-Hyslop P, Singleton AB. Analysis of the PINK1 gene in a large cohort of cases with Parkinson disease. Arch Neurol. 2004;61:1898–1904. doi: 10.1001/archneur.61.12.1898. [DOI] [PubMed] [Google Scholar]
- Rohe CF, Montagna P, Breedveld G, Cortelli P, Oostra BA, Bonifati V. Homozygous PINK1 C-terminus mutation causing early-onset parkinsonism. Ann Neurol. 2004;56:427–431. doi: 10.1002/ana.20247. [DOI] [PubMed] [Google Scholar]
- Romito LM, Contarino MF, Ghezzi D, Franzini A, Garavaglia B, Albanese A. High frequency stimulation of the subthalamic nucleus is efficacious in Parkin disease. J Neurol. 2005;252:208–211. doi: 10.1007/s00415-005-0638-x. [DOI] [PubMed] [Google Scholar]
- Ross OA, Gosal D, Stone JT, Lincoln SJ, Heckman MG, Irvine GB, Johnston JA, Gibson JM, Farrer MJ, Lynch T. Familial genes in sporadic disease: common variants of alpha-synuclein gene associate with Parkinson's disease. Mech Ageing Dev. 2007;128:378–382. doi: 10.1016/j.mad.2007.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross OA, Wu YR, Lee MC, Funayama M, Chen ML, Soto AI, Mata IF, Lee-Chen GJ, Chen CM, Tang M, Zhao Y, Hattori N, Farrer MJ, Tan EK, Wu RM. Analysis of Lrrk2 R1628P as a risk factor for Parkinson's disease. Ann Neurol. 2008;64:88–92. doi: 10.1002/ana.21405. [DOI] [PubMed] [Google Scholar]
- Samaranch L, Lorenzo-Betancor O, Arbelo JM, Ferrer I, Lorenzo E, Irigoyen J, Pastor MA, Marrero C, Isla C, Herrera-Henriquez J, Pastor P. PINK1-linked parkinsonism is associated with Lewy body pathology. Brain. 2010;133:1128–1142. doi: 10.1093/brain/awq051. [DOI] [PubMed] [Google Scholar]
- Sanchez MP, Gonzalo I, Avila J, de Yebenes JG. Progressive supranuclear palsy and tau hyperphosphorylation in a patient with a C212Y parkin mutation. J Alzheimers Dis. 2002;4:399–404. doi: 10.3233/jad-2002-4506. [DOI] [PubMed] [Google Scholar]
- Santos-Reboucas CB, Abdalla CB, Baldi FJ, Martins PA, Correa JC, Goncalves AP, Cunha MS, Borges MB, Pereira JS, Laks J, Pimentel MM, et al. Co-occurrence of sporadic parkinsonism and late-onset Alzheimer's disease in a Brazilian male with the LRRK2 p.G2019S mutation. Genet Test. 2008;12:471–473. doi: 10.1089/gte.2008.0042. [DOI] [PubMed] [Google Scholar]
- Sasaki S, Shirata A, Yamane K, Iwata M. Parkin-positive autosomal recessive juvenile Parkinsonism with alpha-synuclein-positive inclusions. Neurology. 2004;63:678–682. doi: 10.1212/01.wnl.0000134657.25904.0b. [DOI] [PubMed] [Google Scholar]
- Satake W, Nakabayashi Y, Mizuta I, Hirota Y, Ito C, Kubo M, Kawaguchi T, Tsunoda T. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson's disease. Nat Genet. 2009;41:1303–1307. doi: 10.1038/ng.485. [DOI] [PubMed] [Google Scholar]
- Savettieri G, Annesi G, Civitelli D, Ciro CI, Salemi G, Ragonese P, Annesi F, Tarantino P, Terruso V, D'Amelio M, Quattrone A. Identification of the novel D297fsX318 PINK1 mutation and phenotype variation in a family with early-onset Parkinson's disease. Parkinsonism Relat Disord. 2008;14:509–512. doi: 10.1016/j.parkreldis.2007.10.014. [DOI] [PubMed] [Google Scholar]
- Scherfler C, Khan NL, Pavese N, Eunson L, Graham E, Lees AJ, Quinn NP, Wood NW, Brooks DJ, Piccini PP. Striatal and cortical pre- and postsynaptic dopaminergic dysfunction in sporadic parkin-linked parkinsonism. Brain. 2004;127:1332–1342. doi: 10.1093/brain/awh150. [DOI] [PubMed] [Google Scholar]
- Schlitter AM, Woitalla D, Mueller T, Epplen JT, Dekomien G. The LRRK2 gene in Parkinson's disease: mutation screening in patients from Germany. J Neurol Neurosurg Psychiatry. 2006;77:891–892. doi: 10.1136/jnnp.2005.083022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimura H, Hattori N, Kubo S, Mizuno Y, Asakawa S, Minoshima S, Shimizu N, Iwai K, Chiba T, Tanaka K, Suzuki T. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet. 2000;25:302–305. doi: 10.1038/77060. [DOI] [PubMed] [Google Scholar]
- Shyu WC, Lin SZ, Chiang MF, Pang CY, Chen SY, Hsin YL, Thajeb P, Lee YJ, Li H. Early-onset Parkinson's disease in a Chinese population: 99mTc-TRODAT-1 SPECT, Parkin gene analysis and clinical study. Parkinsonism Relat Disord. 2005;11:173–180. doi: 10.1016/j.parkreldis.2004.12.004. [DOI] [PubMed] [Google Scholar]
- Sim CH, Lio DS, Mok SS, Masters CL, Hill AF, Culvenor JG, Cheng HC. C-terminal truncation and Parkinson's disease-associated mutations down-regulate the protein serine/threonine kinase activity of PTEN-induced kinase-1. Hum Mol Genet. 2006;15:3251–3262. doi: 10.1093/hmg/ddl398. [DOI] [PubMed] [Google Scholar]
- Simon-Sanchez J, Marti-Masso JF, Sanchez-Mut JV, Paisan-Ruiz C, Martinez-Gil A, Ruiz-Martinez J, Saenz A, Singleton AB, Lopez de MA, Perez-Tur J. Parkinson's disease due to the R1441G mutation in Dardara founder effect in the Basques. Mov Disord. 2006;21:1954–1959. doi: 10.1002/mds.21114. [DOI] [PubMed] [Google Scholar]
- Simon-Sanchez J, Scholz S, Matarin MM, Fung HC, Hernandez D, Gibbs JR, Britton A, Hardy J, Singleton A. Genomewide SNP assay reveals mutations underlying Parkinson disease. Hum Mutat. 2008;29:315–322. doi: 10.1002/humu.20626. [DOI] [PubMed] [Google Scholar]
- Simon-Sanchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, Paisan-Ruiz C, Lichtner P. Genome-wide association study reveals genetic risk underlying Parkinson's disease. Nat Genet. 2009;41:1308–1312. doi: 10.1038/ng.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J. alpha-Synuclein locus triplication causes Parkinson's disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- Sironi F, Primignani P, Zini M, Tunesi S, Ruffmann C, Ricca S, Brambilla T. Parkin analysis in early onset Parkinson's disease. Parkinsonism Relat Disord. 2008;14:326–333. doi: 10.1016/j.parkreldis.2007.10.003. [DOI] [PubMed] [Google Scholar]
- Skipper L, Shen H, Chua E, Bonnard C, Kolatkar P, Tan LC, Jamora RD, Puvan K, Puong KY, Zhao Y, Pavanni R, Wong MC, Yuen Y, Farrer M, Liu JJ, Tan EK. Analysis of LRRK2 functional domains in nondominant Parkinson disease. Neurology. 2005;65:1319–1321. doi: 10.1212/01.wnl.0000180517.70572.37. [DOI] [PubMed] [Google Scholar]
- Spanaki C, Latsoudis H, Plaitakis A. LRRK2 mutations on Crete: R1441H associated with PD evolving to PSP. Neurology. 2006;67:1518–1519. doi: 10.1212/01.wnl.0000239829.33936.73. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M. alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson's disease and dementia with lewy bodies. Proc Natl Acad Sci USA. 1998;95:6469–6473. doi: 10.1073/pnas.95.11.6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spira PJ, Sharpe DM, Halliday G, Cavanagh J, Nicholson GA. Clinical and pathological features of a Parkinsonian syndrome in a family with an Ala53Thr alpha-synuclein mutation. Ann Neurol. 2001;49:313–319. [PubMed] [Google Scholar]
- Strauss KM, Martins LM, Plun-Favreau H, Marx FP, Kautzmann S, Berg D, Gasser T, Wszolek Z, Muller T, Bornemann A, Wolburg H, Downward J, Riess O, Schulz JB, Kruger R. Loss of function mutations in the gene encoding Omi/HtrA2 in Parkinson's disease. Hum Mol Genet. 2005;14:2099–2111. doi: 10.1093/hmg/ddi215. [DOI] [PubMed] [Google Scholar]
- Sun M, Latourelle JC, Wooten GF, Lew MF, Klein C, Shill HA, Golbe LI, Mark MH. Influence of heterozygosity for parkin mutation on onset age in familial Parkinson disease: the GenePD study. Arch Neurol. 2006;63:826–832. doi: 10.1001/archneur.63.6.826. [DOI] [PubMed] [Google Scholar]
- Taira T, Saito Y, Niki T, Iguchi-Ariga SM, Takahashi K, Ariga H. DJ-1 has a role in antioxidative stress to prevent cell death. EMBO Rep. 2004;5:213–218. doi: 10.1038/sj.embor.7400074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan EK, Chai A, Teo YY, Zhao Y, Tan C, Shen H, Chandran VR, Teoh ML, Yih Y, Pavanni R, Wong MC, Puvan K, Lo YL, Yap E. Alpha-synuclein haplotypes implicated in risk of Parkinson's disease. Neurology. 2004;62:128–131. doi: 10.1212/01.wnl.0000101721.25345.dc. [DOI] [PubMed] [Google Scholar]
- Tan EK, Peng R, Wu YR, Wu RM, Wu-Chou YH, Tan LC, An XK, Chen CM, Fook-Chong S, Lu CS. LRRK2 G2385R modulates age at onset in Parkinson's disease: a multi-center pooled analysis. Am J Med Genet B Neuropsychiatr Genet. 2009;150B:1022–1023. doi: 10.1002/ajmg.b.30923. [DOI] [PubMed] [Google Scholar]
- Tan EK, Skipper L, Chua E, Wong MC, Pavanni R, Bonnard C, Kolatkar P, Liu JJ. Analysis of 14 LRRK2 mutations in Parkinson's plus syndromes and late-onset Parkinson's disease. Mov Disord. 2006a;21:997–1001. doi: 10.1002/mds.20875. [DOI] [PubMed] [Google Scholar]
- Tan EK, Tan LC, Lim HQ, Li R, Tang M, Yih Y, Pavanni R, Prakash KM, Fook-Chong S, Zhao Y. LRRK2 R1628P increases risk of Parkinson's disease: replication evidence. Hum Genet. 2008;124:287–288. doi: 10.1007/s00439-008-0544-2. [DOI] [PubMed] [Google Scholar]
- Tan EK, Yew K, Chua E, Puvan K, Shen H, Lee E, Puong KY, Zhao Y, Pavanni R, Wong MC, Jamora D, de SD, Moe KT, Woon FP, Yuen Y, Tan L. PINK1 mutations in sporadic early-onset Parkinson's disease. Mov Disord. 2006b;21:789–793. doi: 10.1002/mds.20810. [DOI] [PubMed] [Google Scholar]
- Tan EK, Yew K, Chua E, Shen H, Jamora RD, Lee E, Puong KY, Zhao Y, Pavanni R, Wong MC, Puvan K, Yih Y, Tan LC. Analysis of PINK1 in Asian patients with familial parkinsonism. Clin Genet. 2005;68:468–470. doi: 10.1111/j.1399-0004.2005.00500.x. [DOI] [PubMed] [Google Scholar]
- Tan EK, Zhao Y, Skipper L, Tan MG, Di FA, Sun L, Fook-Chong S, Tang S, Chua E, Yuen Y, Tan L, Pavanni R, Wong MC, Kolatkar P, Lu CS, Bonifati V, Liu JJ. The LRRK2 Gly2385Arg variant is associated with Parkinson's disease: genetic and functional evidence. Hum Genet. 2007;120:857–863. doi: 10.1007/s00439-006-0268-0. [DOI] [PubMed] [Google Scholar]
- Tan LC, Tanner CM, Chen R, Chan P, Farrer M, Hardy J, Langston JW. Marked variation in clinical presentation and age of onset in a family with a heterozygous parkin mutation. Mov Disord. 2003;18:758–763. doi: 10.1002/mds.10432. [DOI] [PubMed] [Google Scholar]
- Tang B, Xiong H, Sun P, Zhang Y, Wang D, Hu Z, Zhu Z, Ma H, Pan Q, Xia JH, Xia K, Zhang Z. Association of PINK1 and DJ-1 confers digenic inheritance of early-onset Parkinson's disease. Hum Mol Genet. 2006;15:1816–1825. doi: 10.1093/hmg/ddl104. [DOI] [PubMed] [Google Scholar]
- Tarantino P, Ciro CI, Annesi F, Rocca FE, Carrideo S, Provenzano G, Civitelli D, De Marco EV, Quattrone A, Annesi G. Novel human pathological mutations. Gene symbol: PARK2. Disease: Parkinson's disease. Hum Genet. 2007;122:415. [PubMed] [Google Scholar]
- Tarantino P, Civitelli D, Annesi F, De Marco EV, Rocca FE, Pugliese P, Nicoletti G, Carrideo S, Provenzano G, Annesi G, Quattrone A. Compound heterozygosity in DJ-1 gene non-coding portion related to parkinsonism. Parkinsonism Relat Disord. 2009;15:324–326. doi: 10.1016/j.parkreldis.2008.07.001. [DOI] [PubMed] [Google Scholar]
- Terreni L, Calabrese E, Calella AM, Forloni G, Mariani C. New mutation (R42P) of the parkin gene in the ubiquitinlike domain associated with parkinsonism. Neurology. 2001;56:463–466. doi: 10.1212/wnl.56.4.463. [DOI] [PubMed] [Google Scholar]
- Toft M, Myhre R, Pielsticker L, White LR, Aasly JO, Farrer MJ. PINK1 mutation heterozygosity and the risk of Parkinson's disease. J Neurol Neurosurg Psychiatry. 2007;78:82–84. doi: 10.1136/jnnp.2006.097840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomiyama H, Li Y, Funayama M, Hasegawa K, Yoshino H, Kubo S, Sato K, Hattori T. Clinicogenetic study of mutations in LRRK2 exon 41 in Parkinson's disease patients from 18 countries. Mov Disord. 2006;21:1102–1108. doi: 10.1002/mds.20886. [DOI] [PubMed] [Google Scholar]
- Tomiyama H, Mizuta I, Li Y, Funayama M, Yoshino H, Li L, Murata M, Yamamoto M, Kubo S, Mizuno Y, Toda T, Hattori N. LRRK2 P755L variant in sporadic Parkinson's disease. J Hum Genet. 2008;53:1012–1015. doi: 10.1007/s10038-008-0336-5. [DOI] [PubMed] [Google Scholar]
- Uversky VN, Lee HJ, Li J, Fink AL, Lee SJ. Stabilization of partially folded conformation during alpha-synuclein oligomerization in both purified and cytosolic preparations. J Biol Chem. 2001a;276:43495–43498. doi: 10.1074/jbc.C100551200. [DOI] [PubMed] [Google Scholar]
- Uversky VN, Li J, Fink AL. Evidence for a partially folded intermediate in alpha-synuclein fibril formation. J Biol Chem. 2001b;276:10737–10744. doi: 10.1074/jbc.M010907200. [DOI] [PubMed] [Google Scholar]
- Valente EM, Bentivoglio AR, Dixon PH, Ferraris A, Ialongo T, Frontali M, Albanese A, Wood NW. Localization of a novel locus for autosomal recessive early-onset parkinsonism, PARK6, on human chromosome 1p35-p36. Am J Hum Genet. 2001;68:895–900. doi: 10.1086/319522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente EM, bou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del TD, Bentivoglio AR, Healy DG, Albanese A, Nussbaum R, Gonzalez-Maldonado R, Deller T, Salvi S, Cortelli P, Gilks WP, Latchman DS, Harvey RJ, Dallapiccola B, Auburger G, Wood NW. Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science. 2004a;304:1158–1160. doi: 10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- Valente EM, Brancati F, Ferraris A, Graham EA, Davis MB, Breteler MM, Gasser T. PARK6-linked parkinsonism occurs in several European families. Ann Neurol. 2002;51:14–18. [PubMed] [Google Scholar]
- Valente EM, Salvi S, Ialongo T, Marongiu R, Elia AE, Caputo V, Romito L, Albanese A, Dallapiccola B, Bentivoglio AR. PINK1 mutations are associated with sporadic early-onset parkinsonism. Ann Neurol. 2004b;56:336–341. doi: 10.1002/ana.20256. [DOI] [PubMed] [Google Scholar]
- van Duijn CM, Dekker MC, Bonifati V, Galjaard RJ, Houwing-Duistermaat JJ, Snijders PJ, Testers L, Breedveld GJ, Horstink M, Sandkuijl LA, van Swieten JC, Oostra BA, Heutink P. Park7, a novel locus for autosomal recessive early-onset parkinsonism, on chromosome 1p36. Am J Hum Genet. 2001;69:629–634. doi: 10.1086/322996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varrone A, Pellecchia MT, Amboni M, Sansone V, Salvatore E, Ghezzi D, Garavaglia B, Brice A, Brunetti A, Bonavita V, De MG, Salvatore M, Pappata S, Barone P. Imaging of dopaminergic dysfunction with [123I]FP-CIT SPECT in early-onset parkin disease. Neurology. 2004;63:2097–2103. doi: 10.1212/01.wnl.0000145765.19094.94. [DOI] [PubMed] [Google Scholar]
- Wakabayashi K, Hayashi S, Ishikawa A, Hayashi T, Okuizumi K, Tanaka H, Tsuji S, Takahashi H. Autosomal dominant diffuse Lewy body disease. Acta Neuropathol. 1998;96:207–210. doi: 10.1007/s004010050883. [DOI] [PubMed] [Google Scholar]
- Wang HL, Chou AH, Yeh TH, Li AH, Chen YL, Kuo YL, Tsai SR, Yu ST. PINK1 mutants associated with recessive Parkinson's disease are defective in inhibiting mitochondrial release of cytochrome c. Neurobiol Dis. 2007;28:216–226. doi: 10.1016/j.nbd.2007.07.010. [DOI] [PubMed] [Google Scholar]
- Wang M, Hattori N, Matsumine H, Kobayashi T, Yoshino H, Morioka A, Kitada T, Asakawa S, Minoshima S, Shimizu N, Mizuno Y. Polymorphism in the parkin gene in sporadic Parkinson's disease. Ann Neurol. 1999;45:655–658. doi: 10.1002/1531-8249(199905)45:5<655::aid-ana15>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- Wang X, Schwarz TL. The mechanism of Ca2+-dependent regulation of kinesin-mediated mitochondrial motility. Cell. 2009;136:163–174. doi: 10.1016/j.cell.2008.11.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Clark LN, Louis ED, Mejia-Santana H, Harris J, Cote LJ, Waters C, Andrews H, Ford B, Frucht S, Fahn S, Ottman R, Rabinowitz D, Marder K. Risk of Parkinson disease in carriers of parkin mutations: estimation using the kin-cohort method. Arch Neurol. 2008;65:467–474. doi: 10.1001/archneur.65.4.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters CH, Miller CA. Autosomal dominant Lewy body parkinsonism in a four-generation family. Ann Neurol. 1994;35:59–64. doi: 10.1002/ana.410350110. [DOI] [PubMed] [Google Scholar]
- Weihofen A, Thomas KJ, Ostaszewski BL, Cookson MR, Selkoe DJ. Pink1 forms a multiprotein complex with Miro and Milton, linking Pink1 function to mitochondrial trafficking. Biochemistry. 2009;48:2045–2052. doi: 10.1021/bi8019178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng YH, Chou YH, Wu WS, Lin KJ, Chang HC, Yen TC, Chen RS, Wey SP, Lu CS. PINK1 mutation in Taiwanese early-onset parkinsonism: clinical, genetic, and dopamine transporter studies. J Neurol. 2007;254:1347–1355. doi: 10.1007/s00415-007-0534-7. [DOI] [PubMed] [Google Scholar]
- West AB, Moore DJ, Biskup S, Bugayenko A, Smith WW, Ross CA, Dawson VL, Dawson TM. Parkinson's disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci USA. 2005;102:16842–16847. doi: 10.1073/pnas.0507360102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AB, Moore DJ, Choi C, Andrabi SA, Li X, Dikeman D, Biskup S, Zhang Z, Lim KL, Dawson VL, Dawson TM. Parkinson's disease-associated mutations in LRRK2 link enhanced GTP-binding and kinase activities to neuronal toxicity. Hum Mol Genet. 2007;16:223–232. doi: 10.1093/hmg/ddl471. [DOI] [PubMed] [Google Scholar]
- Winkler S, Hagenah J, Lincoln S, Heckman M, Haugarvoll K, Lohmann-Hedrich K, Kostic V, Farrer M, Klein C. alpha-Synuclein and Parkinson disease susceptibility. Neurology. 2007;69:1745–1750. doi: 10.1212/01.wnl.0000275524.15125.f4. [DOI] [PubMed] [Google Scholar]
- Wu RM, Bounds R, Lincoln S, Hulihan M, Lin CH, Hwu WL, Chen J, Gwinn-Hardy K, Farrer M. Parkin mutations and early-onset parkinsonism in a Taiwanese cohort. Arch Neurol. 2005;62:82–87. doi: 10.1001/archneur.62.1.82. [DOI] [PubMed] [Google Scholar]
- Xiong H, Wang D, Chen L, Choo YS, Ma H, Tang C, Xia K, Jiang W, Ronai Z, Zhuang X, Zhang Z. Parkin, PINK1, and DJ-1 form a ubiquitin E3 ligase complex promoting unfolded protein degradation. J Clin Invest. 2009;119:650–660. doi: 10.1172/JCI37617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiromerisiou G, Hadjigeorgiou GM, Gourbali V, Johnson J, Papakonstantinou I, Papadimitriou A, Singleton AB. Screening for SNCA and LRRK2 mutations in Greek sporadic and autosomal dominant Parkinson's disease: identification of two novel LRRK2 variants. Eur J Neurol. 2007;14:7–11. doi: 10.1111/j.1468-1331.2006.01551.x. [DOI] [PubMed] [Google Scholar]
- Xu Y, Liu Z, Wang Y, Tao E, Chen G, Chen B. [A new point mutation on exon 2 of parkin gene in Parkinson's disease] Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2002;19:409–411. [PubMed] [Google Scholar]
- Zabetian CP, Samii A, Mosley AD, Roberts JW, Leis BC, Yearout D, Raskind WH, Griffith A. A clinic-based study of the LRRK2 gene in Parkinson disease yields new mutations. Neurology. 2005;65:741–744. doi: 10.1212/01.wnl.0000172630.22804.73. [DOI] [PubMed] [Google Scholar]
- Zabetian CP, Yamamoto M, Lopez AN, Ujike H, Mata IF, Izumi Y, Kaji R, Maruyama H, Morino H, Oda M, Hutter CM, Edwards KL, Schellenberg GD, Tsuang DW, Yearout D, Larson EB, Kawakami H. LRRK2 mutations and risk variants in Japanese patients with Parkinson's disease. Mov Disord. 2009;24:1034–1041. doi: 10.1002/mds.22514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atares B, Llorens V, Gomez TE, del ST, Munoz DG, de Yebenes JG. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. 2004;55:164–173. doi: 10.1002/ana.10795. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Gao J, Chung KK, Huang H, Dawson VL, Dawson TM. Parkin functions as an E2-dependent ubiquitin–protein ligase and promotes the degradation of the synaptic vesicle-associated protein, CDCrel-1. Proc Natl Acad Sci USA. 2000;97:13354–13359. doi: 10.1073/pnas.240347797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB, Stoessl AJ, Pfeiffer RF, Patenge N, Carbajal IC, Vieregge P, Asmus F, Muller-Myhsok B, Dickson DW, Meitinger T, Strom TM, Wszolek ZK, Gasser T. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004a;44:601–607. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Zimprich A, Muller-Myhsok B, Farrer M, Leitner P, Sharma M, Hulihan M, Lockhart P. The PARK8 locus in autosomal dominant parkinsonism: confirmation of linkage and further delineation of the disease-containing interval. Am J Hum Genet. 2004b;74:11–19. doi: 10.1086/380647. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.