

Abstract

A total synthesis of (−)-callipeltoside A (1) has been achieved. The core macrocycle was made via a dual macrolactonization/pyran hemiketal formation reaction, which was developed to circumvent issues related to the reversible nature of acylketene formation from β-keto lactone substrates. Initial approaches to the core of the natural product that revolved around ring-closing metathesis (RCM) and relay ring-closing metathesis (RRCM) reactions are also described.
Introduction
Callipeltoside A (1) is a cytotoxic macrolide isolated in 1996 from the marine lithistida sponge (Callipelta sp.).1 The stereochemical features of callipeltoside A were deduced on the basis of extensive NMR studies. When we initiated the research described here, issues still remained with respect to i) the absolute configuration and ii) the relative configuration of C(13) and C(20)/C(21) vis-à-vis those in the remainder of the molecule. The relative configuration of C(13) was established by Paterson and coworkers.2 The first total synthesis, which established the remaining relative configurational issues of C(20)/C(21) as well as the overall absolute configuration of (−)-callipeltoside A (1) was reported by the Trost labs in 2002.3a Additional total syntheses have been reported,3b–g and numerous related studies have appeared.4
Results/Discussion
Our approach to 1 involved a convergent union of the three key intermediates: macrolactone 2, carbohydrate 3, and phosphonate 4 (Scheme 1). Our initial approach to the synthesis of 2 aimed to utilize a RCM reaction from a suitable precursor, such as 5.5 Unfortunately, neither 5 or its C(13) epimer (epi-5) could be ring-closed to give a macrocyclic product (Scheme 2). Using Grubbs' first generation metathesis initiator (7),6 no initiation was observed at room temperature, as evidenced by lack of styrene formation by analysis of the NMR spectrum and recovery of starting material. Attempts to alter the reactivity of the substrate by using a more constrained, and perhaps geometrically favorable, conformer (cf. 6) also failed.7 At elevated temperatures (50–70 °C, benzene solvent) the catalyst [either 7 or the second generation initiator (8)]8 decomposed and starting material (5–6) could be recovered.
Scheme 1.

Callipeltoside Retrosynthesis.
Scheme 2.
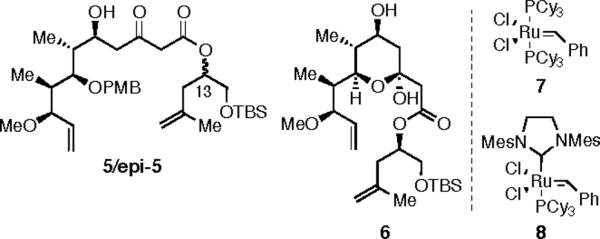
RCM Substrates.
Believing that the main problem with the lack of reactivity of substrates 5 and 6 was due to slow initiation, we hypothesized that the use of a relay strategy would be advantageous.5b,9 The concept, outlined in Scheme 3, was to use allyloxymethyl relays on substrates 9 and 10. Ruthenium loading onto the terminal alkene of the relay was envisioned to give intermediates 11 and 12. Intramolecular cyclization of each would give 13 or 14, respectively, with concomitant formation of 3,4-dihydrofuran. Finally, cyclization of 13 and 14 would give the desired β-keto lactone 15 and regenerate a ruthenium methylidene as the active catalyst. Since we had no way of knowing whether intermediate 13 or 14 would be more likely to macrocyclize, we chose to undertake the synthesis of both substrates 9 and 10.
Scheme 3.

Relay Ring-Closing Metathesis.
The synthesis of relay substrate 9 is outlined in Scheme 4. Known Weinreb amide 16 was protected as a PMB ether using PMB-trichloroacetimidate and catalytic CSA.10 Hydroboration/oxidation (9-BBN), followed by TBS protection and DIBAL-H reduction of the Weinreb amide gave aldehyde 17. Lithium-halogen exchange of alkenyl iodide 18 followed by addition to aldehyde 17 gave a 1:1.5 ratio of newly formed secondary alcohols. Subsequent methylation with methyl iodide gave the relay-armed substrate 19. Deprotection of the primary TBS ether using TBAF, oxidation with Dess-Martin periodinane, vinylogous Mukaiyama aldol reaction with diene 20,11 and TMS protection gave dioxinone substrate 21. Refluxing this substrate in benzene with secondary alcohol 22,12 followed by TMS deprotection with PPTS in acetonitrile gave 9.
Scheme 4. Synthesis of Relay Substrate 9.
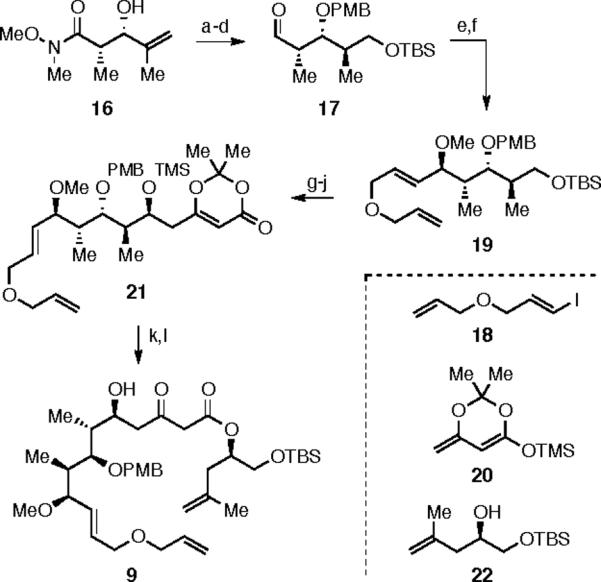
a) PMB acetimidate, CSA, 92%; b) 9-BBN, then NaOH/H2O2, 56%; c) TBSCl, TEA, 60%; d) DIBAL-H, 95%; e) tBuLi, then 18, dr = 1:1.5; SiO2 separation; 22%/33%; f) MeI, NaH, DMF, 64%; g) TBAF, THF; h) DMP; i) 20, BF3•OEt2; j) TMSCl, 65% over 4 steps; k) 22, benzene reflux; l) PPTS, ACN 69% over 2 steps.
The synthesis of the alternative relay substrate 10 is outlined in Scheme 5. The allyl ether of known bromo alkene 2313 was used, after lithium-halogen exchange (cat. CuI), to ring-open the TBS protected epoxide 2414 to give relay-armed alcohol 25. Aldehyde 17 was elaborated by addition of vinyl magnesium bromide, giving a separable mixture of diastereomeric (dr 1:1) alcohols. Methylation with methyl iodide, TBAF deprotection, and oxidation with Dess-Martin periodinane gave aldehyde 26. The vinylogous Mukaiyama aldol reaction of 26 with diene 20 and TMS protection of the C(5)-hydroxyl gave dioxinone 27. Refluxing a mixture of alcohol 25 and dioxinone 27 in benzene, followed by TMS deprotection, gave relay substrate 10.
Scheme 5. Synthesis of Relay Substrate 10.
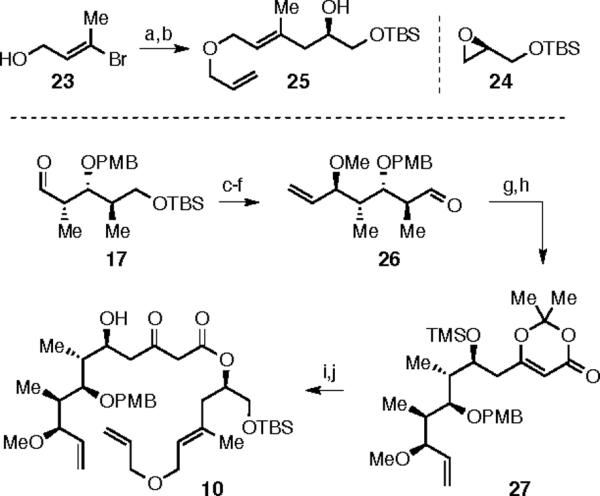
a) allyl bromide, NaH, 81%; b) tBuLi, CuI, 24; c) vinylMgBr, dr = 1:1; SiO2 separation; 34%/34%; d) MeI, NaH, DMF, 89%; e) TBAF, THF, 87%; f) DMP, 92%; g) 20, BF3·OEt2, 90%; h) TMSCl, TEA, 65%; i) 25, benzene reflux; j) PPTS, ACN, 77% over two steps.
Unfortunately for our cause, subjection of either 9 or 10 to the metathesis initiator 8 did not lead to the formation of desired product 15, but instead gave 5, the substrate for the initial RCM study. The process leading to 5 is outlined in Scheme 6; an event we refer to as truncation. When metathesis initiator 8 reacts with 10, the relay to produce 28 presumably proceeds as planned, as evidenced by formation of 3,4-dihydrofuran (1H NMR). In order for macrocyclic ring closure to occur within 28, the rate of its intramolecular cyclization (kintra) must be faster than the rate of bimolecular attack on the more reactive alkene in another molecule of 10 (kinter). Since truncation is bimolecular, we recognized that running the reaction at higher dilution would result in a more favorable kintra/kinter ratio, but even at 10−5 M, only the truncation product 5 was observed.7
Scheme 6.
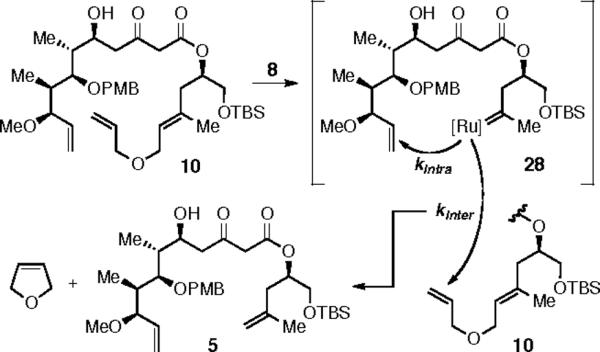
Truncation of Relay Substrate 10.
We examined other ways to favorably bias the ratio of kintra to kinter during this RRCM reaction. Since disubstituted alkenes are slower to engage in cross-metathesis (CM), it was hoped that using a 1,1-disubstituted alkene within the relay arm would slow the bimolecular truncation rate sufficiently so that macrocyclization could prevail. To this end, substrates 29 and 30 (Scheme 7) were synthesized in an analogous manner. While our reasoning was apparently sound, as evidenced by a significantly slower overall rate of reaction, no desired product was formed using these substrates having the modified relay subunit. Instead the truncation products 5 and 6 were observed, respectively. The clear implication is that the initial relay event occurred to deliver ruthenium to the more hindered carbon atom of the otherwise inactive alkene. As these results illustrate, the use of a RRCM strategy by no means guarantees a successful outcome. If the inherent rate of the crucial metathesis cyclization event is slower than competing events (e.g, CM or [Ru] decomposition), as is the case here, then the cyclization will not be efficient regardless of the process by which the key ruthenium alkylidene intermediate is formed.9c
Scheme 7.
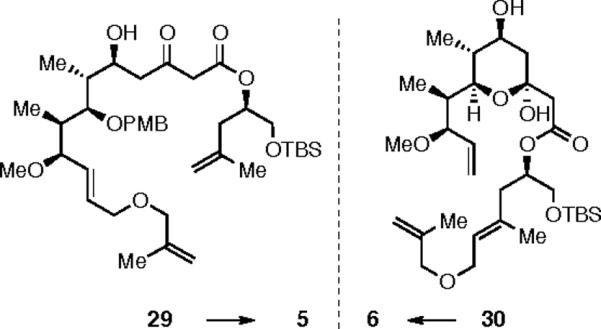
Additional RRCM Substrates.
We then turned to a strategy in which the Boeckman lactonization15 of an acylketene intermediate would be used to close the core macrocycle.5b This strategy formed the bases of both the Trost synthesis of 13a,c and the Marshall synthesis16 of the aglycone. Reaction of the TBS protected epoxide 31 with lithium TMS-acetylide, followed by a two-step deprotection gave diol 32.17,18 Negishi carboalumination/iodination19 and sequential protection of the 1,2-diol as TBS and PMB ethers gave 33. Lithium-halogen exchange in 33 and addition to aldehyde 34 gave a 1:1 mixture of separable secondary alcohols.20 Recycling of the undesired epimer by oxidation with Dess-Martin periodinane, followed by reduction with lithium triethylhydridoborate gave the desired epimer in a 10:1 dr. Subsequent methylation gave alkene 35, which was subjected to hydroboration/oxidation (9-BBN). The resulting primary alcohol was subjected to Dess-Martin periodinane oxidation and vinylogous Mukaiyama aldol reaction with diene 20. Each of the two new stereocenters at C(5) and C(6) were formed with essentially full diastereocontrol. Protection of the C(5)-OH with MOMCl and PMB removal at C(13) with DDQ gave the lactonization precursor 36.
The dioxinone-containing substrate 36 was heated in refluxing toluene to effect thermal extrusion of acetone and concomitant generation of the acylketene 37. Macrolactonization involves cylization of the C(13) hydroxyl group to give enol 38. Two products were isolated: the keto lactone 39 and the methyl ketone 40 (64% and 15%, respectively) (Scheme 9). The latter arises from competitive addition of water to 37 and subsequent decarboxylation of the resulting β-ketoacid, a phenomenon that has been observed by others.21 The relative amount of 40 increased over time. This suggests that the unimolecular lactonization of the acylketene 37 outcompetes its trapping by water and that the ketolactone 39 is able to revert via 38 to acylketene 37.22 While concentration effects are undoubtedly also important, it is relevant that i) addition of an O–H bond to an acylketene is a concerted event (cf., 37 to 38)23 and ii) the O–H bond dissociation energy of water is substantially higher than those of alcohols (119 vs. 104–107 kcal mol−1).24 We hypothesized that the detrimental outcome of competitive water addition could be mitigated if the β-ketone in a macrolactone like 39 could be sequestered. An attractive possibility was that it be trapped as its corresponding pyran-hemiketal by a free hydroxyl group at C(7).22
Scheme 9.
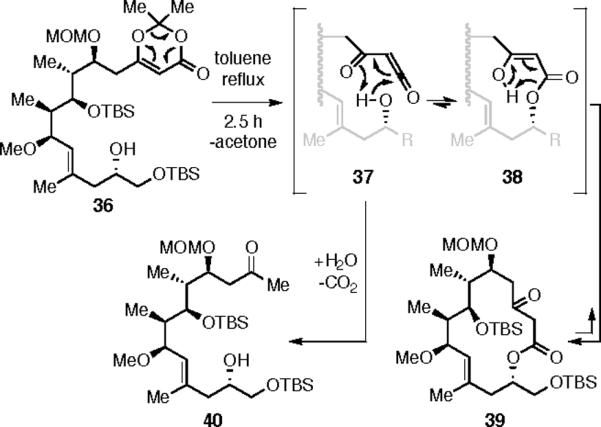
Boeckman Cyclization and Competitive Hydrolysis of 36.
To test this idea, we elected to prepare diol 43 (Scheme 10).25 Thus, the vinyllithium species derived from ent-33 was added to aldehyde 41 to give a 1:1 ratio of diols. Separation and methylation with methyl iodide gave alkene 42. Hydroboration/oxidation, Dess-Martin oxidation, and vinylogous Mukaiyama aldol reaction with diene 20 was performed. Protection of the resulting secondary alcohol, followed by DDQ removal of both PMB ethers afforded the dioxinone diol 43.
Scheme 10. Synthesis of Dual Macrolactonizion/Pyran-Hemiketal Precursor 43.
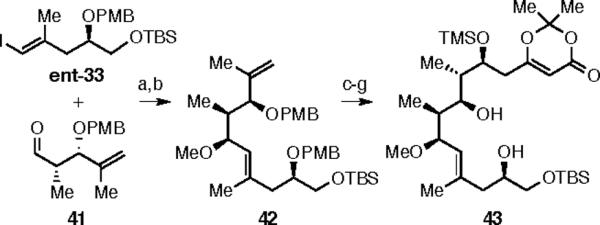
Reagents and conditions: (a) nBuLi, ent-33; then 41, dr = 1:1, SiO2 separation, 34%/34%; (b) MeI, NaH, 80%; (c) 9-BBN, then NaOH/H2O2, 70%; (d) DMP, 76%; (e) 20, BF3•OEt2, 90%; (f) TMSCl; g) DDQ, 78% over 2 steps.
To our delight, when refluxed in benzene for 12 hours, dioxinone 43 produced the desired dual macrocyclic pyran-hemiketal 44 in 76% yield (Scheme 11). We suspect that pyran-hemiketal formation within either enol 46 or ketone 47 follows macrolactonization (45 to 46). It is noteworthy that no products indicative of competitive trapping of the acylketene 45 by water could be detected (and no 8-membered lactone from reaction of the C(7)-OH with the ketene carbon was observed). It is clear that in situ protection of the C(3)-ketone as its hemiketal is advantageous.22
Scheme 11.

Formation of Pyran-Hemiketal 44.
The completion of the synthesis, following established lines, is outlined in Scheme 12. A three-step protecting group manipulation was used to convert 44 into 48. Parikh-Doerin'g oxidation afforded the corresponding aldehyde, from which aglycone 49 was obtained by Horner-Emmons olefination with the known phosphonate 4,26 followed by deprotection of the TMS ether at C(5). Glycosidation3a with known acetimidate 327 provided (−)-callipeltoside A (1).
Scheme 12. Completion of the Synthesis.
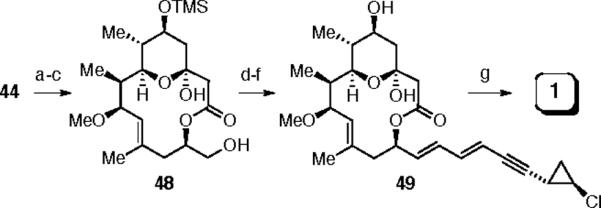
Reagents and conditions: (a) TBAF, THF; (b) TMSCl, TEA, DCM; (c) NaHCO3, MeOH, 70% over 3 steps; (d) SO3•pyr, DMSO, TEA; (e) 4, LiHMDS, THF; (f) TBAF, THF, 54% over 3 steps; (g) 5, TMSOTf, DCM, 70%.
In conclusion, we have completed a total synthesis of (−)-callipeltoside A (1). Initial attempts using an RCM reaction on substrates 5 and 6 were not successful; presumably these compounds are simply too hindered to load ruthenium to the requisite (and more hindered) alkene carbon atom. This led to the evolution of the RRCM strategy, which was explored via substrates 9, 10, 29, and 30. In each case, relay activation allowed ruthenium to be transiently installed to the necessary site (cf. 13, 14, and 28), but bimolecular truncation of these species thwarted the (inherently slow) final ring-closing event. Ultimately, the synthesis of 1 (20 linear steps from TBS-glycidol 24, 0.7% yield) was enabled by a highly efficient acylketene mediated dual macrolactonization/pyran-hemiketal formation reaction, converting the linear precursor 43 directly to the bridged bicyclic macrolactone 44.
Experimental Section
General. 1H NMR chemical shifts in CDCl3 are referenced to TMS (0.00 ppm) and in PhH-d6 to 7.16. 13C NMR chemical shifts in CDCl3 are referenced to chloroform (77.2 ppm). The following format has been used to report peaks: chemical shift in ppm [multiplicity, coupling constant(s) in Hz, integral]. MPLC refers to medium pressure liquid chromatography (25–60 psi) using hand-packed columns of E. Merck silica gel (230–400 mesh), a pump, and a differential refractive index detector. Reactions requiring anhydrous conditions were performed under an atmosphere of nitrogen or argon in oven-dried glassware. Dry diethyl ether and benzene were distilled from sodium benzophenone ketyl. Toluene was distilled from Na/K. Anhydrous THF and methylene chloride were passed through a column of activated alumina. Triethylamine and pyridine were distilled from KOH just prior to use. DMF was stored over 4 Angstrom molecular sieves. The structure of each intermediate numbered here as S## can be found in the Supporting Information.
(+)-(2S)-4-Pentyne-1,2-diol (32)28
To epoxide 31 (13.3 g, 71.0 mmol), TMS-acetylene (15 mL, 106.0 mmol), and THF (141 mL) was added n-BuLi (3.2 M in hexanes, 40.0 mL, 92.0 mmol) dropwise at −78 °C, followed by BF3•OEt2 (13.4 mL, 106.0 mmol) dropwise at the same temperature. The solution was stirred for 45 min at −78 °C before being quenched with aqueous NH4Cl (20 mL). The mixture was allowed to warm to rt and diluted with H2O (150 mL). The aqueous layer was extracted with DCM (3 × 100 mL) and the combined organic portions were dried over Na2SO4 and concentrated to give crude TMS alkyne as a colorless oil. This alkyne was immediately subjected to desilylation. To the crude oil was added K2CO3 (29.3 g, 212 mol) and MeOH (100 mL), and this mixture was stirred for 24 h at rt. The suspended K2CO3 was filtered through celite on a sintered glass funnel with MeOH. The filtrate was concentrated to afford the crude alkyne as a mixture of primary and secondary TBS ethers. The crude products were carried into the next step without purification. To the mixture of alkynes was added MeOH (40 mL), THF (40 mL), and KF (24 g, 420 mmol); and the resulting slurry was refluxed at 65 °C for 48 h. The mixture was cooled to rt, concentrated, placed on a flash column, and purified via flash chromatography (hexanes:EtOAc = 1:1 followed by 100% EtOAc) to provide a pale yellow oil that crystallized in a freezer overnight to afford diol 32 as white needles (3.2 g, 77%). 1H NMR (300 MHz, CDCl3) δ 3.91 (dddd, J = 3.7, 6.2, 6.2, and 6.2 Hz, 1H), 3.77 (dd, J = 3.7 and 11.1 Hz, 1H), 3.62 (dd, J = 6.2 and 10.0 Hz, 1H), 2.45 (dd, J = 2.7 and 6.2 Hz, 2H), 2.35 (bs, 1H), 1.90 (bs, 1H), and 2.08 (t, J = 2.7 Hz, 1H). 13C NMR (125 MHz, CDCl3) δ 80.3, 70.7, 70.2, 65.3, and 23.2. GC-MS TR = 2.66 min.; m/z: 100, 69, and 61. IR (neat) 3300, 3292, 2113, 1719, 1416, 1085, and 1044 cm−1. mp 44–51 °C. TLC Rf = 0.35, 100% EtOAc. [α]RT +8.22° (c = 1.1, CHCl3).
(−)-(2S,4E)-1-[[(1,1-Dimethylethyl)dimethylsilyl]oxy]-5-iodo-2-[(4-methoxyphenyl)methoxy]-4-methyl-4-pentene (33)
To zirconocene dichloride (2.5 g, 9.6 mmol) and 1,2-dichloroethane (90 mL) was added AlMe3 (2.0 M in toluene, 36 mL, 72 mmol) dropwise at rt followed by diol 32 (2.4 g, 24 mmol) in 1,2-dichloroethane (20 mL). The solution was stirred at 50 °C for 48 h and then cooled to −40 °C. Iodine (9.1 g, 36 mmol) in THF (26 mL) was added dropwise by cannula. The resulting dark brown solution was allowed to warm and was stirred for 30 min at rt before being poured into a mixture of hexanes and sodium potassium tartrate. The aqueous layer was extracted with EtOAc (1 × 50 mL), saturated with NaCl, and extracted again with EtOAc (3 × 50 mL). The combined organic layers were dried over Na2SO4, concentrated, and chromatographed (100% EtOAc) to provide iodo diol (Rf = 0.13 in 1:1 hexanes:EtOAc) as a pale yellow oil (3.4 g, 58%). The diol (3.0 g, 12.4 mmol) was dissolved in DCM (120 mL) and treated with TEA (7.0 mL, 50.0 mmol), DMAP (230 mg 1.86 mmol), and TBSCl (3.0 g 20.0 mmol). The resulting solution was stirred 24 h at rt before being quenched with aqueous NH4Cl (50 mL), diluted with H2O (100 mL) and extracted with DCM. The combined organic layers were dried over Na2SO4, concentrated, and chromatographed (hexanes:EtOAc = 19:1) to provide a primary TBS ether (Rf = 0.25 in 9:1 hexanes:EtOAc) as a colorless oil (3.2 g, 72%). To this ether (310 mg, 0.87 mmol) in DCM (5 mL) was added para-methoxybenzyl trichloroacetimidate (762 mg, 2.7 mmol) and CSA (46 mg, 0.2 mmol). The reaction mixture was stirred for 48 h before being quenched with aqueous NaOH (10%, 20 mL). The mixture was extracted with DCM, washed with aqueous NaOH (10%, 20 mL), dried over MgSO4, and concentrated. Purification by MPLC (hexanes:EtOAc = 12:1) gave 33 (330 mg, 82%) as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 7.23 (d, J = 8.5 Hz, 2H), 6.89 (d, J = 8.5 Hz, 2H), 5.97 (broad s, 1H), 4.60 (d, J = 11.5 Hz, 1H); 4.46 (d, J = 11.0 Hz, 1H), 3.81 (s, 3H), 3.67 (m, 1H), 3.55 (dd, J = 5.0 and 8.5 Hz, 1H), 3.52 (dd, J = 5.5 and 8.5 Hz, 1H), 2.44 (dd, J = 3.5 and 14.0 Hz, 1H), 2.36 (dd, J = 8.0 and 13.0 Hz, 1H), 1.78 (broad s, 3H), 0.90 (s, 9H), and 0.06 (s, 6H). 13C NMR (125 MHz, CDCl3) δ 159.2, 144.9, 130.6, 129.5, 113.8, 77.2, 77.1, 72.0, 65.0, 55.3, 41.8, 25.9, 24.3, 18.3, −5.32, and −5.37. IR (neat) 3063, 3032, 2996, 2958, 2931, 2857, 1612, 1512, and 1250 cm−1. GC-MS TR = 12.6 min.; m/z: 419, 349, and 121. HRMS (CI, 4% NH3 in CH4) Calcd for (C20H33IO3Si + NH4)+: 494.1582. Found: 494.1579. TLC Rf = 0.3, hexanes:EtOAc = 15:1. [α]RT −15.3° (c = 1.0, CHCl3).
(−)-[3S-(3R*,4S*,5S*,9S*),6E]-5-Methoxy-3,9-Bis-[(4-methoxyphenyl)methoxy]-2,4,7-trimethyl-10-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-1,6-decadiene (42)
To vinyl iodide ent-33 (600 mg, 1.26 mmol, prepared by the same sequence described above) and diethyl ether (10 mL, stirred over CaH2 for 1 h) in a round bottom flask was added solid CaH2 (5 mg), and the mixture was stirred for 10 min at rt. The cloudy solution was cooled to −78 °C and n-BuLi (2.1 M in hexane, 0.66 mL, 1.38 mmol) was added dropwise and stirred for 10 min. At the same temperature aldehyde 4129 (342 mg, 1.38 mmol) in diethyl ether (2 mL) was added to the flask by cannula, and the solution was stirred for 15 min before warming to 0 °C. The reaction mixture was poured into a separatory funnel containing aqueous NH4Cl, diluted with water, and extracted with DCM. The combined organic layers were dried over Na2SO4, concentrated, and purified by MPLC (hexanes:EtOAc = 7:3) to afford (−)-[3S-(3R*,4S*,5S*,9S*),6E]-10-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-3,9-bis-[(4-methoxyphenyl)methoxy]-2,4,7-trimethyldeca-1,6-dien-5-ol (S30, 255 mg, 34%) and its C5-epimer (S30b, 265 mg, 34%), each as a colorless oil. S30: 1H NMR (500 MHz, CDCl3) δ 7.27 (d, J = 8.8 Hz, 2H), 7.24 (d, J = 8.7 Hz, 2H), 6.88 (d, J = 8.8 Hz, 2H), 6.84 (d, J = 8.8 Hz, 2H), 5.23 (broad d, J = 8.4 Hz, 1H), 5.03 (s, 1H), 5.00 (s, 1H), 4.59 (d, J = 11.4 Hz, 1H), 4.52 (d, J = 11.2 Hz, 1H), 4.49 (d, J = 11.2 Hz, 1H), 4.29 (ddd, J = 4.6, 7.3, and 9.0 Hz, 1H), 4.21 (d, J = 11.4 Hz, 1H), 4.00 (d, J = 4.0 Hz, 1H), 3.79 (s, 3H), 3.77 (s, 3H), 3.54 – 3.69 (m, 3H), 2.25 (dd, J = 4.3 and 13.9 Hz, 1H), 2.18 (dd, J = 7.7 and 13.9 Hz, 1H), 2.18 (d, J = 4.6 Hz, 1H), 1.73 (ddq, J = 4.3, 7.2 and 7.2 Hz, 1H), 1.70 (s, 3H), 1.66 (s, 3H), 0.90 (s, 9H), 0.82 (d, J = 7.1 Hz, 3H), and 0.05 (s, 6H). 13C NMR (125 MHz, CDCl3) δ 159.2, 159.0, 142.8, 135.7, 131.0, 130.6, 129.5, 129.2, 113.8, 113.6, 112.5, 82.0, 78.1, 71.7, 70.4, 70.3, 65.6, 55.2, 42.1, 41.3, 25.9, 19.5, 18.3, 17.2, 10.7, −5.31, and −5.38. IR (neat) 3476, 2952, 2928, 2856, 1613, 1514, 1248, 1090, 1037, and 836 cm−1. HRMS (FAB) Calcd for (C35H54O6Si + Na)+: 621.3582, found: 621.3593. TLC Rf = 0.3 in hexanes:EtOAc 4:1 [α]RT −10.31° (c = 0.38, DCM). S30b: 1H NMR (500 MHz, CDCl3) δ 7.26 (d, J = 8.2 Hz, 2H), 7.25 (d, J = 9.0 Hz, 2H), 6.87 (d, J = 8.2 Hz, 2H), 6.86 (d, J = 9.0 Hz, 2H), 5.37 (broad d, J = 8.2 Hz, 1H), 5.07 (s, 1H), 5.04 (s, 1H), 4.61 (d, J = 11.2 Hz, 1H), 4.50 (d, J = 11.2 Hz, 1H), 4.49 (d, J = 11.2 Hz, 1H), 4.44 (ddd, J = 2.9, 2.9, and 8.2, 1H), 4.19 (d, J = 11.2 Hz, 1H), 3.83 (d, J = 5.6 Hz, 1H), 3.81 (s, 3H), 3.79 (s, 3H), 3.53–3.66 (m, 3H), 2.25 (dd, J = 5.1 and 13.9 Hz, 1H), 2.17 (d, J = 2.9, 1H), 2.16 (dd, J = 7.2 and 13.9 Hz, 1H), 1.73 (ddq, J = 3.0, 5.6, and 6.9 Hz, 1H), 1.70 (s, 3H), 1.57 (s, 3H), 0.97 (d, J = 7.3 Hz, 3H), 0.90 (s, 9H), and 0.05 (s, 6H). 13C NMR (125 MHz, CDCl3) δ 159.1, 159.0, 142.6, 134.8, 131.0, 130.5, 129.4, 129.3, 129.2, 113.8, 113.7, 113.4, 85.6, 78.4, 71.8, 70.7, 70.3, 65.6, 55.2, 41.9, 41.0, 25.9, 18.5, 18.3, 17.3, 8.0, −5.31, and −5.38. IR (neat) 3476, 2952, 2928, 2855, 1612, 1513, 1464, 1247, 1081, 1037, and 836 cm−1. TLC Rf = 0.3 in hexanes:EtOAc 4:1. [α]RT −34.06° (c = 0.31, DCM).
To alcohol S30 (300 mg, 0.502 mmol) in DMF (3 mL) was added MeI (0.37 mL, 6.00 mmol) and NaH (25% dispersion in mineral oil, 240 mg, 2.51 mmol) at rt. The mixture was stirred for 24 h, quenched with aqueous NH4Cl (10 mL), diluted with H2O, extracted with Et2O (3 × 10 mL), washed with H2O, and dried over Na2SO4. MPLC (hexanes:EtOAc = 19:1) separation gave 42 (245 mg, 80%) as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 7.27 (d, J = 8.6 Hz, 2H), 7.24 (d, J = 8.6 Hz, 2H), 6.88 (d, J = 8.6 Hz, 2H), 6.84 (d, J = 8.6 Hz, 2H), 5.06 (broad d, J = 9.7 Hz, 1H), 4.98 (app s, 2H), 4.60 (d, J = 11.8 Hz, 1H), 4.50 (d, J = 11.8 Hz, 1H), 4.50 (d, J = 11.3 Hz, 1H), 4.19 (d, J = 11.3 Hz, 1H), 4.00 (d, J = 3.9 Hz, 1H), 3.80 (s, 3H), 3.79 (dd, J = 8.4 and 9.4 Hz, 1H), 3.78 (s, 3H), 3.57–3.69 (m, 3H), 3.07 (s, 3H), 2.30 (dd, J = 4.6 and 14.0 Hz, 1H), 2.23 (dd, J = 6.7 and 14.0 Hz, 1H), 1.73 (ddq, J = 3.9, 8.4, and 7.0 Hz, 1H), 1.68 (app s, 6H), 0.90 (s, 9H), 0.75 (d, J = 7.1 Hz, 3H), and 0.05 (s, 6H). 13C NMR (125 MHz, CDCl3) δ 159.0, 158.9, 143.5, 137.8, 131.5, 131.0, 129.3, 129.2, 126.8, 113.65, 113.62, 111.6, 81.2, 78.3, 77.8, 71.8, 70.7, 65.6, 55.5, 55.3, 55.2, 42.2, 40.4, 25.9, 19.3, 18.3, 17.5, 9.6, −5.32, and −5.39. IR (neat) 2952, 2928, 2856, 1613, 1513, 1463, 1247, 1082, and 837 cm−1. HRMS (FAB) Calcd for (C36H56O6Si + Na)+: 635.3738, found: 635.3772. TLC Rf = 0.5, hexanes:EtOAc = 4:1. [α]RT −3.00° (c = 0.95, DCM).
(−)-6-{[2S-(2R*,3R*,4R*,5R*,6S*,10S*),7E]-4,10-Dihydroxy-6-methoxy-3,5,8-trimethyl-11-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-2-[(trimethylsilyl)oxy]-undec-7-enyl}-2,2-dimethyl-4H-1,3-dioxin-4-one (43)
A flask containing 42 (235 mg, 0.384 mmol) was brought into a dry box where solid 9-BBN dimer (235 mg, 1.91 mmol) was added. The flask was taken out of the dry box, THF (4 mL) was added at −78 °C, and the solution was warmed to rt. The system was stirred for 18 h before H2O2 (30%, 0.6 mL) and aqueous NaOH (10%, 3.8 mL) were added at 0 °C. After 45 min the reaction mixture was extracted with DCM and dried over MgSO4. Separation by flash chromatography (hexanes:EtOAc = 7:3) gave a mixture of both diastereomers (7:1 ratio). MPLC (hexanes:EtOAc = 7:3) separation afforded the desired alcohol (+)-[2R-(2R*,3R*,4R*,5R*,9R*),6E]-5-methoxy-3,9-bis-[(4-methoxyphenyl)-methoxy]-2,4,7-trimethyl-10-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-deca-6-en-1-ol (S31, 168 mg, 70%) and a minor amount of the C2-epimer (S32), each as a colorless oil. S31: 1H NMR (500 MHz, CDCl3) δ 7.31 (d, J = 8.8 Hz, 2H), 7.25 (d, J = 8.7 Hz, 2H), 6.90 (d, J = 8.8 Hz, 2H), 6.85 (d, J = 8.7 Hz, 2H), 5.01 (dq, J = 9.1 and 1.2 Hz, 1H), 4.62 (d, J = 11.3 Hz, 1H), 4.59 (d, J = 11.0 Hz, 1H), 4.56 (d, J = 11.0 Hz, 1H), 4.51 (d, J = 11.3 Hz, 1H), 3.85 (dd, J = 1.8 and 9.0 Hz, 1H), 3.81 (s, 3H), 3.80 (s, 3H), 3.75 (dd, J = 9.7 and 9.7 Hz, 1H) 3.50–3.71 (m, 5H), 3.17 (s, 3H), 2.73 (dd, J = 3.3 and 8.7 Hz, 1H), 2.33 (dd, J = 5.1 and 14.0 Hz, 1H), 2.25 (dd, J = 7.5 and 14.0 Hz, 1H), 1.92 (ddq, J = 4.0, 9.0, and 7.0 Hz, 1H), 1.65 (ddq, J = 1.4, 8.3, and 6.8 Hz, 1H), 1.68 (d, J = 1.5 Hz, 3H), 0.90 (s, 9H), 0.83 (d, J = 7.1 Hz, 3H), 0.80 (d, J = 7.1 Hz, 3H), 0.061 (s, 3H), and 0.059 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 159.2, 159.1, 138.2, 131.0, 130.9, 129.3, 129.3, 127.6, 113.9, 113.6, 81.8, 78.4, 78.2, 74.2, 71.8, 67.2, 65.5, 55.3, 55.3, 55.1, 42.2, 40.4, 38.3, 25.9, 18.3, 17.4, 14.1, 9.8, −5.33, and −5.40. IR (neat) 3470, 2953, 2928, 2857, 1613, 1513, 1248, 1090, 1036, and 837 cm−1. HRMS (FAB) Calcd for (C36H58O7Si + Na)+: 653.3844, found: 653.3851. TLC Rf = 0.2, hexanes:EtOAc = 7:3. [α]RT +23.2° (c = 0.45, DCM). S32: 1H NMR (500 MHz, CDCl3) δ 7.29 (d, J = 8.6 Hz, 2H), 7.25 (d, J = 8.6 Hz, 2H), 6.89 (d, J = 8.6 Hz, 2H), 6.85 (d, J = 8.6 Hz, 2H), 5.01 (d, J = 9.8 Hz, 1H), 4.62 (d, J = 10.9 Hz, 1H), 4.58 (d, J = 10.4 Hz, 1H), 4.48 (d, J = 10.9 Hz, 1H), 4.51 (d, J = 10.3 Hz, 1H), 3.80 (s, 3H), 3.79 (s, 3H), 3.78 (dd, J = 2.3 and 7.0 Hz, 1H), 3.50 – 3.70 (m, 6H), 3.12 (s, 3H), 2.31 (dd, J = 5.1 and 14.2 Hz, 1H), 2.24 (dd J = 7.2 and 14.2 Hz, 1H), 1.96 (ddq, J = 6.0, 7.0, and 7.0 Hz, 1H), 1.72 (ddq, J = 2.1, 6.6, and 9.0 Hz, 1H), 1.67 (d, J = 1.5 Hz, 3H), 1.01 (d, J = 7.3 Hz, 3H), 0.90 (s, 9H), 0.85 (d, J = 6.9 Hz, 3H), and 0.06 (s, 6H). IR (neat) 3434, 2953, 2928, 2856, 1613, 1513, 1247, 1096, and 837 cm−1. HRMS (FAB) Calcd for (C36H58O7Si + Na)+: 653.3844, Found: 653.3857. TLC Rf = 0.2, hexanes:EtOAc = 7:3. [α]RT +6.62° (c = 0.64, DCM).
Dess-Martin periodinane (158 mg, 0.37 mmol) was added to alcohol S31 (158 mg, 0.25 mmol) and DCM (12 mL) at rt. The resulting cloudy mixture was stirred 1 h before being placed directly onto a flash column. Flash chromatography (hexanes:EtOAc = 9:1 to 4:1) afforded (+)–[2S-(2R*,3R*,4S*,5S*,9S*),6E]-5-methoxy-3,9-bis-[(4-methoxyphenyl)-methoxy]-2,4,7-trimethyl-10-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-6-decenal (S33, 119 mg, 76%) as a colorless oil. S33 1H NMR (500 MHz, CDCl3) δ 9.73 (d, J = 3.0 Hz, 1H), 7.25 (d, J = 8.7 Hz, 2H), 7.24 (d, J = 8.7 Hz, 2H), 6.88 (d, J = 8.7 Hz, 2H), 6.85 (d, J = 8.7 Hz, 2H), 5.02 (d, J = 9.6 Hz, 1H), 4.62 (d, J = 11.4 Hz, 1H), 4.51 (s, 2H), 4.50 (d, J = 11.4 Hz, 1H), 4.15 (dd, J = 2.2 and 9.3 Hz, 1H), 3.80 (s, 3H), 3.79 (s, 3H), 3.82 (m, 1H), 3.57–3.70 (m, 3H), 3.17 (s, 3H), 2.67 (ddq, J = 3.0, 9.3, and 7.0 Hz, 1H), 2.33 (dd, J = 5.0 and 14.0 Hz, 1H), 2.25 (dd, J = 7.4 and 14.0 Hz, 1H), 1.68 (d, J = 1.4 Hz, 3H), 1.62 (ddq, J = 2.2, 9.1, and 7.0 Hz, 1H), 0.97 (d, J = 7.5 Hz, 3H), 0.91 (s, 9H), 0.80 (d, J = 7.5 Hz, 3H), 0.061 (s, 3H), and 0.059 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 205.1, 159.1, 159.2, 138.5, 130.9, 130.9, 129.3, 129.2, 127.4, 113.8, 113.7, 78.4, 78.1, 77.7, 73.9, 71.8, 65.5, 55.28, 55.27, 55.0, 49.8, 42.2, 40.5, 25.9, 18.3, 17.4, 11.1, 9.5, −5.32, and −5.39. IR (neat) 2952, 2932, 2856, 1724, 1613, 1513, 1248,1036, and 837 cm−1. Anal. Calcd for C36H56O7Si: C, 68.75; H: 8.97. found: C, 69.14; H, 8.65. TLC Rf = 0.4, hexanes:EtOAc = 4:1. [α]RT +2.0° (c = 0.84, DCM).
BF3•Et2O (45 μL, 0.36 mmol) was added over 2 min to aldehyde S33 (113 mg, 0.18 mmol) and ketene acetal 20 (385 mg, 1.8 mmol) in DCM (9 mL) at −78 °C. After 45 min the reaction mixture was allowed to warm to room temperature before being quenched with saturated aqueous NaHCO3. The resulting mixture was extracted with DCM, dried over MgSO4, and concentrated. Flash chromatography (hexanes:EtOAc = 7:3 to 1:1) gave (+)-6-{[2S-(2R*,3S*,4S*,5S*,6S*,10S*),7E]-2-hydroxy-6-methoxy-5,10-bis-[(4-methoxyphenyl)methoxy]-3,5,8-trimethyl-11-[[(1,1-dimethylethyl)dimethylsilyl]-oxy]-undec-7-enyl}-2,2-dimethyl-4H-1,3-dioxin-4-one (S34, 125 mg, 90%). S34 1H NMR (500 MHz, CDCl3) δ 7.29 (d, J = 8.4 Hz, 2H), 7.25 (d, J = 8.4 Hz, 2H), 6.90 (d, J = 8.4 Hz, 2H), 6.85 (d, J = 8.4 Hz, 2H), 5.27 (s, 1H), 5.02 (d, J = 9.6 Hz, 1H), 4.63 (d, J = 10.6 Hz, 1H), 4.61 (d, J = 10.6 Hz, 1H), 4.51 (d, J = 10.6 Hz, 1H), 4.50 (d, J = 10.6 Hz, 1H), 4.16 (m, 1H), 3.87 (dd, J = 2.4 and 8.2 Hz, 1H), 3.81 (s, 3H), 3.79 (s, 3H), 3.78 (dd, J = 9.6 and 9.6 Hz, 1H), 3.58–3.72 (m, 3H), 3.20 (s, 3H), 2.38 (dd, J = 9.45 and 14.4 Hz, 2H), 2.35 (dd, J = 4.53 and 12.8 Hz, 1H), 2.33 (d, J = 5.8 Hz, 1H), 2.26 (dd, J = 6.8 and 12.8 Hz, 1H), 2.23 (dd, J = 4.15 and 14.4 Hz, 1H), 1.70 (m, 2H), 1.69 (s, 3H), 0.91 (s, 9H), 0.85 (d, J = 7.0 Hz, 3H), 0.81 (d, J = 7.0 Hz, 3H), and 0.06 (s, 6H). 13C NMR (125 MHz, CDCl3) δ 169.9, 161.1, 159.3, 159.0, 138.3, 130.8, 130.7, 129.4, 129.3, 127.3, 113.8, 113.6, 106.4, 94.7, 79.9, 78.4, 78.1, 74.2, 71.7, 68.7, 65.4, 55.2, 55.2, 55.0, 42.1, 40.8, 40.7, 39.3, 25.9, 25.2, 24.7, 18.2, 17.4, 10.6, 9.8, −5.37, and −5.43. IR (neat) 3457, 2928, 2856, 1726, 1634, 1613, 1512, 1389, and 1375 cm−1. HRMS (FAB) Calcd for (C43H66O10Si + Na)+: 793.4318, found: 793.4336. TLC Rf = 0.35, hexanes:EtOAc = 1:1. [α]RT +7.2° (c = 0.31, DCM).
TMSCl (0.046 mL, 0.36 mmol) was added to a solution of S34 (28 mg, 0.036 mmol), Et3N (0.15 mL, 1.08 mmol), and DCM (0.3 mL) at rt. The resulting mixture was stirred until the reaction was deemed complete by TLC, ca. 4 h, at which time the solution had taken on a pink color. The mixture was quenched with aqueous NaHCO3, extracted with DCM, dried over Na2SO4, and concentrated to furnish crude silyl ether, which was carried forward without purification. This oil was first dissolved in wet DCM (1.2 mL) and H2O (100 μL). DDQ (65 mg, 0.29 mmol) was added at rt, and the mixture was stirred for 0.5 h. Aqueous NaHCO3 was added, the mixture was stirred for 5 min, diluted with water, and extracted with DCM. The organic layers were dried over Na2SO4 and concentrated. Flash chromatography (hexanes:EtOAc = 7:3 to 100% EtOAc) furnished diol 43 (17 mg, 78%) as a colorless oil. 43 1H NMR (500 MHz, CDCl3) δ 5.29 (s, 1H), 5.20 (d, J = 9.3 Hz, 1H), 4.36 (ddd, J = 2.2, 4.3, 8.6 Hz, 1H), 3.95 (dd, J = 6.7 and 9.4 Hz, 1H), 3.83 (ddd, J = 1.5, 1.5, and 9.7 Hz, 1H), 3.81 (m, 1H), 3.63 (dd, J = 3.6, 9.9 Hz, 1H), 3.46 (dd, J = 6.8 and 9.5 Hz, 1H), 3.44 (d, J = 1.8 Hz, 1H), 3.25 (s, 3H), 2.47 (dd, J = 8.8 and 13.8 Hz, 2H), 2.41 (d, J = 3.6, 1H), 2.31 (dd, J = 4.3 and 13.8 Hz, 1H), 2.21 (bs, 1H), 2.19 (bs, 1H), 1.75 (d, J = 1.4 Hz, 3H), 1.70 (s, 3H), 1.68 (s, 3H), 1.63 (m, 2H) 0.91 (s, 9H), 0.86 (d, J = 7.2 Hz, 3H), 0.74 (d, J = 6.8 Hz, 3H), 0.12 (s, 9H), and 0.07 (s, 6H). 13C NMR (125 MHz, CDCl3) δ 169.6, 161.1, 137.0, 127.6, 106.4, 95.2, 80.2, 70.9, 70.9, 69.7, 66.9, 56.1, 43.6, 40.6, 39.6, 39.2, 26.2, 25.8, 24.0, 18.3, 17.0, 10.6, 8.4, 0.19, −5.38, and −5.41. IR (neat) 3502, 2953, 2931, 2857, 1734, 1635, 1464, 1390, 1375, 1273, 1251, 1085 and 840 cm−1. HRMS (FAB) Calcd for (C30H58O8Si2 + Na)+: 625.3562, found: 625.3570. TLC Rf = 0.6, hexanes:EtOAc = 1:1. [α]RT −22.8° (c = 0.29, DCM).
(−)-[1S*(1R*,5S*,9S*,10S*,11R*,12R*,13R*)7E)-1-Hydroxy-9-methoxy-7,10,12-trimethyl-5-[[[(1,1-dimethylethyl)dimethylsilyl]oxy]methyl]-13-[(trimethylsilyl)oxy]-4,15-dioxabicyclo[9.3.1]pentadec-7-en-3-one (44)
Benzene (200 mL) was placed in a round bottom flask equipped with a Dean-Stark trap. The flask was heated and 40 mL of benzene was removed through the trap. The condenser was removed and diol 43 (16 mg, 0.027 mmol) in benzene (2 mL) was transferred into the flask. The condenser was replaced and the mixture was refluxed for 12 h. The reaction mixture was cooled to rt, concentrated, and purified by flash chromatography (hexanes:EtOAc = 9:1) to afford macrolactone 44 (11 mg, 78%). 1H NMR (500 MHz, CDCl3) δ 5.37 (dddd, J = 11.6, 6.8, 4.4, 2.5 Hz, 1H), 5.28 (dq, J = 9.5, 1.2 Hz, 1H), 5.07 (d, J = 2.5 Hz, 1H), 3.80 (dd, J = 9.3, 2.4 Hz, 1H), 3.77 (m, 1H), 3.70 (dd, J = 11.0, 6.7 Hz, 1H), 3.66 (dd, J = 11.1, 4.5 Hz, 1H), 3.61 (dd, J = 10.4, 2.7 Hz, 1H), 3.24 (s, 3H), 2.50 (d, J = 12.8 Hz, 1H), 2.40 (d, J = 12.8 Hz, 1H), 2.24 (dd, J = 13.4, 2.6 Hz, 1H), 2.20 (ddq, J = 2.4, 2.4, 7.0 Hz, 2H), 2.16 (dd, J =13.4, 11.7 Hz, 1H), 1.97 (dd, J = 12.0, 4.7 Hz, 1H), 1.72 (s, 3H), 1.42 (ddq, J = 10.2, 10.2, 6.1 Hz, 1H), 1.35 (ddd, J = 11.9, 10.7, 2.4 Hz, 1H), 0.97 (d, J = 7.1 Hz, 3H), 0.89 (d, J = 6.7 Hz, 3H), 0.88 (s, 9H), 0.13 (s, 9H), 0.05 (s, 3H), and 0.05 (s, 3H). 13C NMR (125
MHz, CDCl3) δ 172.4, 132.6, 127.3, 95.4, 79.9, 72.7, 70.1, 65.17, 65.12, 55.2, 44.6, 44.4, 42.6, 40.1, 37.1, 25.7, 18.2, 16.1, 12.3, 6.6, −0.02, −5.30, and −5.33. IR (neat) 3451, 2956, 2929, 2859, 1706, 1251, 1075, and 838 cm−1. HRMS (FAB) calcd for (C27H52O7Si2 + Na)+: 567.3144, found: 567.3159. TLC Rf = 0.4, hexanes:EtOAc = 9:1. [α]rt −19.8° (c = 0.93, DCM).
(−)-[1S*(1R*,5S*,9S*,10S*,11R*,12R*,13R*)7E]-1-Hydroxy-5-(hydroxymethyl)-9-methoxy-7,10,12-trimethyl-13-[(trimethylsilyl)oxy]-4,15-dioxabicyclo[9.3.1]-pentadec-7-en-3-one (48)
TBAF (0.28 mL, 1.0 M in THF, 0.28 mmol) was added to 44 (11 mg, 0.020 mmol) in THF (0.5 mL) at rt and stirred for 10 min. The solution was placed on a pipette column and purified by flash chromatography (100% EtOAc) to afford a diol that was immediately carried into the next reaction. TMSCl (0.05 mL, 0.40 mmol) was added to a mixture of the diol, NEt3 (0.17 mL, 1.21 mmol), and DCM (0.2 mL) at rt. The mixture was stirred for 2 h, quenched with aqueous NaHCO3, diluted with water, extracted with DCM, dried over Na2SO4, and concentrated to give a crude oil. This lactone was dissolved in MeOH (1 mL), NaHCO3 (5 mg) was added, and the resulting mixture was stirred for 2 h. The mixture was diluted with water, extracted with DCM, dried over Na2SO4, and concentrated. Purification by flash chromatography (hexanes:EtOAc = 19:1) afforded 48 (6 mg, 70% over three steps). 1H NMR (500 MHz, CDCl3) δ 5.42 (dddd, J = 2.4, 3.7, 6.2, and 8.7 Hz, 1H), 5.31 (d, J = 9.4 Hz, 1H), 5.00 (d, J = 2.4 Hz, 1H), 3.8 (dd, J = 2.2 and 9.6 Hz, 1H), 3.76 (m, 3H), 3.61 (dd, J = 2.5, 10.3 Hz, 1H), 3.24 (s, 3H), 2.54 (d, J = 13.0 Hz, 1H), 2.44 (d, J = 13.0 Hz, 1H), 2.30 (dd, J = 12.0 and 12.0 Hz, 1H), 2.21 (m, 2H), 1.98 (dd, J = 4.6 and 12.0 Hz, 1H), 1.74 (s, 3H), 1.61 (dd, J = 5.9 and 5.9 Hz, 1H), 1.43 (ddq, J = 10.3, 10.3, and 6.5 Hz, 1H), 1.36 (ddd, J = 2.4, 10.7, and 12.0 Hz, 1H), 0.97 (d, J = 7.3 Hz, 3H), 0.90 (d, J = 6.4 Hz, 3H), 0.14 (s, 9H). 13C NMR (125 MHz, CDCl3) δ 172.4, 132.2, 127.7, 99.4, 79.8, 75.1, 72.8, 70.0, 64.9, 55.2, 44.7, 44.3, 42.4, 40.1, 37.0, 16.2, 12.4, 6.6, and 0.30. IR (neat) 3434, 2962, 2930, 2874, 1704, 1251, 1227, 1182, 1075, 891, and 841 cm−1. HRMS (FAB) Calcd for (C7H38O7Si + Na)+: 453.2279, found: 453.2291. TLC Rf = 0.5, hexanes:EtOAc = 1:1. [α]RT −23.9° (c = 0.33, DCM).
(−)-[1S(1R*,5S*,9S*,10S*,11S*,12S*,13R*)7E)-5-[(1E,3E)-6-((1R*,2S*)-2-Chlorocyclopropyl)-1,3-hexadien-5-yn-1-yl]-1,13-dihydroxy-9-methoxy-7,10,12-trimethyl-4-15-dioxabicyclo[9.3.1]pentadec-7-en-3-one (49)2,3a,3c,3d,3e
Sulfur trioxide•pyridine (19 mg, 0.12mmol) was added to a mixture of alcohol 48 (5 mg, 0.01 mmol), TEA (24.0 μL, 0.174 mmol), DCM (100 μL), and DMSO (300 μL) at 0 °C. The mixture was stirred 0.5 h at which time it was diluted with EtOAc (500 μL), quenched with aqueous NH4Cl, and diluted with H2O (5 mL). The aqueous layer was extracted with EtOAc (3 × 5 mL). The combined organic layers were washed with brine (5 mL), washed with water (5 mL), dried over Na2SO4, and concentrated. The crude product was passed through a plug of silica gel (hexanes:EtOAc = 1:1) to afford crude aldehyde, which was immediately carried on to the next reaction. A solution of n-BuLi (0.3 mL, 2.05 M, 0.67 mmol) was added to neat hexamethyldisilazane (0.14 mL, 0.67 mmol) in a round bottom flask at 0 °C. The solution was stirred for 0.5 h at which time a white precipitate had formed. THF (2.9 mL) was then added to make a 0.2 M solution of LiHMDS. Phosphonate 433c,3e (14 mg, 0.051 mmol) was placed in a vial and dissolved in THF (0.3 mL). To this solution was added LiHMDS (255 μL, 0.2 M in THF, 0.051 mmol) at −78 °C, resulting in a dark brown color. The mixture was stirred 3 min at −78 °C before the aldehyde from the previous step (in THF, 200 μL) was added. The vessel was immediately removed from the dry ice bath and allowed to warm to rt over 4 min before being quenched with aqueous NH4Cl, extracted with EtOAc, dried over Na2SO4, and concentrated. The crude product was passed through a pipette column of silica gel (hexanes:EtOAc = 4:1) to afford the TMS protected product. The silylated product was dissolved in THF (0.5 mL), TBAF (30 μL, 1.0 M in THF) was added, and the resulting mixture was stirred for 5 min at rt. The solvent was removed with a stream of N2 and the resulting oil was loaded onto a flash column with DCM. Flash chromatography (hexanes:EtOAc = 7:3 to 1:1) afforded 49 (3 mg, 54%). 1H NMR (500 MHz, CDCl3) δ 6.48 (dd, J = 10.9 and 15.4 Hz, 1H), 6.27 (dd, J = 10.9 and 15.1 Hz, 1H), 5.83 (m, 1H), 5.76 (dd, J = 6.2 and 15.1 Hz, 1H), 5.57 (dd, J = 2.2 and 15.5 Hz, 1H), 5.31 (d, J = 9.7 Hz, 1H), 5.03 (d, J = 2.4 Hz, 1H), 3.80 (dd, J = 2.4 and 9.5 Hz, 1H), 3.79 (m, 1H), 3.61 (dd, J = 2.6 and 10.2 Hz, 1H), 3.24 (s, 3H), 3.18 (ddd, J = 3.2, 5.6, and 6.2 Hz, 1H), 2.55 (d, J = 12.9 Hz, 1H), 2.44 (d, J = 12.9 Hz, 1H), 2.29 (m, 2H), 2.22 (ddq, J = 2.5, 2.5, and 6.9 Hz, 1H), 2.10 (dd, J = 4.6 and 11.8 Hz, 1H), 1.78 (m, 1H), 1.74 (s, 3H), 1.39 (m, 1H) 1.30 (m, 4H), and 0.98 (app d, J = 6.8 Hz, 6H). 13C NMR (125 MHz, CDCl3) δ 171.8, 140.1, 132.7, 132.3, 130.9, 127.8, 112.5, 95.3, 92.2, 79.8, 77.2, 75.0, 71.5, 69.5, 55.2, 46.9, 44.7, 43.7, 40.1, 36.8, 34.3, 19.3, 16.1, 12.06, 12.03, and 6.5. IR (neat) 3452, 3364, 2953, 2923, 2853, 1700, 1465, 1457, and 1225 cm−1. HRMS (FAB) Calcd for (C26H35ClO6 + Na)+: 501.2014, found: 501.2037. TLC Rf = 0.5, hexanes:EtOAc = 1:1. [α]RT −53.8° (c = 0.5, CHCl3).
(−)-{3aR(3aR*,4S*,6R,7R,7aR*)}-6-[[(1S*,5R*,7E,9R*,10R*,11R*,12R*,13S*)-5-[(1E,3E)-6-[(1S*,2R*)-2-Chlorocyclopropyl]-1,3-hexadien-5-yn-1-yl]-1-hydroxy-9-methoxy-7,10,12-trimethyl-3-oxo-4,15-dioxabicyclo[9.3.1]pentadec-7-en-13-yl]oxy]-7-methoxy-4,7a-dimethyl-4H-pyrano[3,4-d]oxazol-2(3H)-one (1)
To a mixture of the aglycon 49 (2 mg, 0.004 mmol), carbohydrate 343f (3 mg, 0.008), and powdered molecular sieves (20 mg) in DCM (300 μL) was added TMSOTf (0.055 M in DCM, 15 μL, 0.836 μmol) at 0 °C. TLC still showed starting material after stirring for 30 min and so additional carbohydrate 3 (1.5 mg, 0.004 mmol) was added. After the addition, TLC showed absence of starting material within 10 min. The mixture was quenched with saturated aqueous NaHCO3. The resulting solution was diluted with H2O, extracted with DCM, dried over Na2SO4, and purified by HPLC (100% EtOAc) to afford (−)-callipeltoside A (1) (2 mg, 70%). 1H NMR (500 MHz, CD3OD) δ 6.49 (dd, J = 10.8 and 15.4 Hz, 1H), 6.33 (dd, J = 10.8 and 14.1 Hz, 1H), 5.83 (m, 1H), 5.80 (m, 1H), 5.64 (dd, J = 1.9 and 15.4 Hz, 1H), 5.26 (d, J = 9.7 Hz, 1H), 4.71 (d, J = 6.2 Hz, 1H), 3.95 (dd, J = 1.9 and 6.5 Hz, 1H), 3.87 (dd, J = 2.4 and 9.5 Hz, 1H), 3.72 (dd, J = 4.7, 10.7, and 10.7 Hz, 1H), 3.64 (dd, J = 2.4 and 10.4 Hz, 1H), 3.59 (s, 3H), 3.44 (d, J = 1.9 Hz, 1H), 3.42 (d, J = 6.1 Hz, 1H), 3.24 (m), 3.21 (s, 3H), 2.52 (d, J = 12.8 Hz, 1H), 2.46 (d, J = 12.8 Hz), 2.33 (bd, J = 13.3 Hz, 1H), 2.26 (dd, J = 11.1 and 13.3 Hz, 1H), 2.21 (m, 1H), 2.21 (ddq, 2.3, 2.3, and 7.2 Hz, 1H), 1.80 (m, 1H), 1.74 (s, 3H), 1.50 (s, 3H), 1.50 (ddq, J = 3.9, 10.3, and 6.6 Hz, 1H), 1.39 (dd, J = 11.1 and 12.2 Hz, 1H), 1.27 (m, 2H), 1.08 (d, J = 6.5 Hz, 3H), 0.99 (d, J = 6.5 Hz, 3H), and 0.95 (d, J = 7.0 Hz, 3H). 13C NMR* (125 MHz, CD3OD) δ 172.9, 141.6, 134.4, 134.3, 132.0, 128.4, 113.5, 103.6, 96.6, 92.8, 83.9, 83.0, 81.4, 78.7, 78.4, 76.4, 72.7, 65.3, 62.7, 62.0, 55.4, 47.8, 46.1, 44.5, 36.8, 39.9, 38.2, 35.9, 35.1, 30.8, 23.0, 19.8, 16.3, 15.9, 12.7, and 6.8. *The weak carbamate carbonyl resonance at δ 161.1 ppm reported for the natural sample of callipeltoside A was not observed. IR (neat) 3452, 3266, 2921, 2852, 1750, 1699, 1464, 1358, and 988 cm−1. HRMS (FAB) Calcd for (C35H48ClNO10 + Na)+: 700.2864, Found: 700.2883. TLC Rf = 0.5, hexanes:EtOAc = 1:1. [α]RT −53.8° (c = 0.5, CHCl3).
Supplementary Material
Scheme 8. Synthesis of Lactonization Precursor 38.
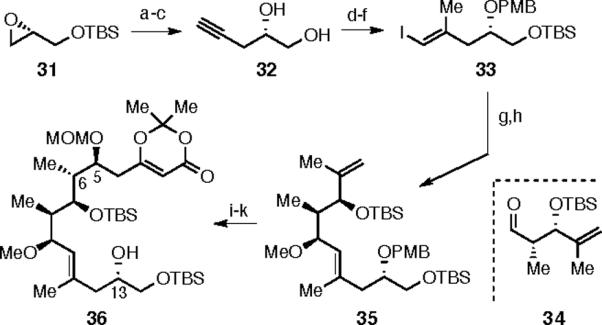
a) Li-TMS-acetylide, BF3•OEt2; b) K2CO3, MeOH; c) KF, MeOH reflux, 77% over 3 steps; d) AlMe3, ZrCp2Cl2; I2, 65%; e) TBSCl, imidazole, 72%; f) PMB acetimidate, CSA, 82%; g) n-BuLi, 34, dr = 1:1; h) MeI, NaH, 80%; i) 9-BBN, then NaOH/H2O2, 99%; j) DMP, 79%; k) 20, BF3·OEt2, 70%; l) MOMCl, 95%; m) DDQ, 100%.
Acknowledgement
These studies were supported by the National Cancer Institute (CA-76497) of the United States National Institutes of Health. We thank Mr. Aaron M. Johnson for his preparation of intermediates enroute to fragments 3 and 4.
Footnotes
Supporting Information Available: Experimental procedures, characterization data, and copies of 1H and 13C NMR spectra for new compounds (182 pages). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).a) Zampella A, D'Auria MV, Minale L, Debitus C, Roussakis C. J. Am. Chem. Soc. 1996;118:11085–11088. [Google Scholar]; b) Zampella A, D'Auria V, Minale L. Tetrahedron. 1997;53:3243–3248. [Google Scholar]
- (2).Paterson I, Davies RDM, Marquez R. Angew. Chem. Int. Ed. 2001;40:603–607. doi: 10.1002/1521-3773(20010202)40:3<603::AID-ANIE603>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- (3).Total syntheses: Trost BM, Dirat O, Gunzner JL. Angew. Chem. Int. Ed. 2002;41:841–843. doi: 10.1002/1521-3773(20020301)41:5<841::aid-anie841>3.0.co;2-2. Evans DA, Hu J, Burch JD, Jaeschke G. J. Am. Chem. Soc. 2002;124:5654–5655. doi: 10.1021/ja026235n. Trost BM, Gunzner JL, Dirat O, Rhee YH. J. Am. Chem. Soc. 2002;124:10396–10415. doi: 10.1021/ja0205232. Paterson I, Davies RDM, Heimann AC, Marquez R, Meyer A. Org. Lett. 2003;5:4477–4480. doi: 10.1021/ol0357853. Huang H, Panek JS. Org. Lett. 2004;6:4383–4385. doi: 10.1021/ol0480325. Evans DA, Burch JD, Hu E, Jaeschke G. Tetrahedron. 2008;64:4671–4699. doi: 10.1016/j.tet.2008.02.001. Carpenter J, Northrup AB, Chung D, Wiener JJM, Kim S-G, MacMillan DWC. Angew. Chem. Int. Ed. 2008;47:3568–3572. doi: 10.1002/anie.200800086.
- (4).For a recent compilation of approaches to the synthesis of the macrolactone fragment, see ref 3f.
- (5).(a) Hoye TR, Zhao H. Org. Lett. 1999;1:169–171. doi: 10.1021/ol9906514. [DOI] [PubMed] [Google Scholar]; (b) Zhao H. PhD Thesis. University of Minnesota; 2000. [Google Scholar]
- (6).Schwab P, France MB, Ziller JW, Grubbs RH. Angew. Chem. Int. Ed. 1995;34:2039–2041. [Google Scholar]
- (7).Danielson ME. PhD Thesis. University of Minnesota; 2003. [Google Scholar]
- (8).Scholl M, Ding S, Lee CW, Grubbs RH. Org. Lett. 1999;1:953–956. doi: 10.1021/ol990909q. [DOI] [PubMed] [Google Scholar]
- (9).(a) Hoye TR, Jeffrey CS, Tennakoon MA, Wang J, Zhao H. J. Am. Chem. Soc. 2004;126:10210–10211. doi: 10.1021/ja046385t. [DOI] [PubMed] [Google Scholar]; (b) Wang X, Bowman EJ, Bowman BJ, Porco JA. Angew. Chem. Int. Ed. 2004;43:3601–3605. doi: 10.1002/anie.200460042. [DOI] [PubMed] [Google Scholar]; (c) Hoye TR, Jeon J. Methathesis Involving a Relay and Applications in Natural Product Synthesis. In: Cossy J, Arseniyadis S, Meyer C, editors. Metathesis in Natural Product Synthesis. Wiley-VCH; Weinheim: 2010. pp. 261–285. [Google Scholar]
- (10).Schoening KU, Wittenberg R, Kirschning A. Synlett. 1999;10:1624–1626. [Google Scholar]
- (11).Sato M, Sugita Y, Abiko Y, Kaneko C. Tetrahedron: Asymmetry. 1992;3:1157–1160. [Google Scholar]
- (12).Guo J, Duffy KJ, Stevens KL, Dalko PI, Roth RM, Hayward MM, Kishi Y. Angew. Chem. Int. Ed. 1998;37:187–192. [Google Scholar]
- (13).Corey EJ, Bock MG, Kozikowski AP, Rama Rao AV, Floyd D, Lipshutz B. Tetrahedron Lett. 1978;19:1051–1054. [Google Scholar]
- (14).Schaus SE, Brandes BD, Larrow JF, Tokunaga M, Hansen KB, Gould AE, Furrow ME, Jacobsen EN. J. Am. Chem. Soc. 2002;124:1307–1315. doi: 10.1021/ja016737l. [DOI] [PubMed] [Google Scholar]
- (15).Boeckman RK, Jr., Pruitt JR. J. Am. Chem. Soc. 1989;111:8286–8288. [Google Scholar]
- (16).Marshall JA, Eidam PM. Org. Lett. 2008;10:93–96. doi: 10.1021/ol702024b. [DOI] [PubMed] [Google Scholar]
- (17).Smith AB, III, Verhoest PR, Minbiole KP, Schelhaas M. J. Am. Chem. Soc. 2001;123:4834–4836. doi: 10.1021/ja0105055. [DOI] [PubMed] [Google Scholar]
- (18).Jones ERH, Stephenson JS, Turner WB, Whiting MC. J. Chem. Soc. 1963:2048–2055. [Google Scholar]
- (19).Rand CL, Van Horn DE, Moore MW, Negishi E. J. Org. Chem. 1981;46:4096–4097. [Google Scholar]
- (20).Smith AB, Barbosa J, Wong W, Wood JL. J. Am. Chem. Soc. 1996;118:8316–8328. [Google Scholar]
- (21).Boeckman RK, Jr., Shao P, Wrobleski ST, Boehmler DJ, Heintzelman GR, Barbosa AJ. J. Am. Chem. Soc. 2006;128:10572–10588. doi: 10.1021/ja0581346. [DOI] [PubMed] [Google Scholar]
- (22).Hoye TR, Danielson ME, May AE, Zhao H. Angew. Chem. Int. Ed. 2008;47:9743–9746. doi: 10.1002/anie.200804049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).a) Freiermuth B, Wentrup C. J. Org. Chem. 1991;56:2286–2289. [Google Scholar]; b) Birney DM, Wagenseller PE. J. Am. Chem. Soc. 1994;116:6262–6270. [Google Scholar]
- (24).Blanksby SJ, Ellison GB. Acc. Chem. Res. 2003;36:255–263. doi: 10.1021/ar020230d. [DOI] [PubMed] [Google Scholar]
- (25).By this juncture, the configuration at C(13) in 1 had been established,2 so our subsequent studies described here all involved intermediates directed toward the 13R configuration.
- (26).Chlorocyclopropane sidechain syntheses: Olivo HF, Velazquez F, Trevisan HC. Org. Lett. 2000;2:4055–4058. doi: 10.1021/ol006696i. Evans DA, Burch JD. Org. Lett. 2001;3:503–505. doi: 10.1021/ol0155182.
- (27).Evans DA, Hu E, Tedrow JS. Org. Lett. 2001;3:3133–3136. doi: 10.1021/ol016416e. [DOI] [PubMed] [Google Scholar]
- (28).a) Heathcock CH, McLaughlin M, Medina J, Hubbs JL, Wallace GA, Scott R, Claffey MM, Hayes CJ, Ott GR. J. Am. Chem. Soc. 2003;125:12844–12849. doi: 10.1021/ja030317+. [DOI] [PubMed] [Google Scholar]; b) Tang B, Bray CD, Pattenden G. Tetrahedron Lett. 2006;47:6401–6404. [Google Scholar]; c) Ding F, Jennings MP. J. Org. Chem. 2008;73:5965–5976. doi: 10.1021/jo8009853. [DOI] [PubMed] [Google Scholar]
- (29).Hoffman RW, Stuermer R. Chem. Ber. 1994;127:2511–2518. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.