

Abstract
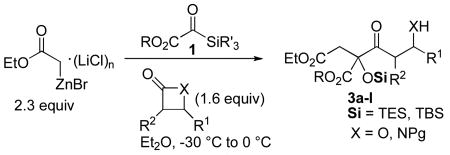
Reformatsky reagents react sequentially with silyl glyoxylates and β-lactones to give highly functionalized Claisen condensation products. A heretofore undocumented instance of stereochemical 1,4-induction results in efficient transmission of β-lactone stereochemistry to the emerging fully-substituted stereocenter. Second-stage transformations reveal that the five heteroatom-containing functionalities embedded within the products are entirely chemo-differentiated, a circumstance that permits rapid assembly of the leustroducsin B core substructure.
The Claisen condensation is a classic transformation in organic synthesis and remains the prototypical β-ketoester synthesis. In spite of its ubiquity, use of the Claisen condensation as an asymmetric C-acylation method is largely undeveloped because the thermodynamically-driven product deprotonation in most cases precludes maintenance of an intact α-stereocenter. The purpose of this communication is to report a three-component coupling reaction for the assembly of complex acyclic functional arrays; the key step of the sequence is a new strain release-driven stereoselective quaternary Claisen condensation.
Extant strategies directed toward stereoselective C-acylation include Evans's β-ketoimide synthesis,1 which forms products that are insulated from enolization by A1,3 strain, and several examples of silyl ketene acetal acylations that generate nonenolizable quaternary stereocenters.2a-d The formation of quaternary centers via the Claisen condensation under basic conditions is challenging due to an equilibrium that commonly favors the disubstituted ester enolate when deprotonation of the β-ketoester product cannot occur.2d-f In an effort to address this problematic transformation, we investigated reactions of Reformatsky reagents, silyl glyoxylates,3a and β-lactones, guided by the mechanistic considerations that follow.
Silyl glyoxylates (1)3 have emerged as useful conjunctive reagents for the geminal difunctionalization of glycolate units. We previously reported a double Reformatsky reaction cascade of silyl glyoxylates and ketones that afforded highly substituted γ-butyrolactone products bearing vicinal quaternary centers with surprisingly high diastereoselection (eq 1).3a The observed stereocontrol in that system was dependent on substitution on the Reformatsky reagent (R1 in eq 1). Equilibration of the kinetically formed (Z)-enolate3a,f to the more stable (E)-isomer, presumably driven by formation a stronger chelate with the pendant ethyl ester, allowed the β-stereocenter to influence electrophile approach to the unhindered diastereotopic face of (E)-2.
We envisioned a modification of this chemistry wherein acylation of the intermediate glycolate enolate, exposed after [1,2]-Brook rearrangement4 of the initial zinc aldolate, would be accomplished by nucleophilic attack on substituted β-lactones (eq 2). This transformation could in principle address two challenges associated with the quaternary Claisen condensation: first, the release of ring strain in the nucleophilic acyl substitution should disfavor the normally dominant retro-Claisen pathway; and second, equilibration of (Z)-2 and (E)-2 provides a mechanism for control of the enolate geometry through coordination with the ester carbonyl. β-Lactones are building blocks that are readily available in enantioenriched form,5 but the degree to which preexisting substrate chirality would impinge upon the incipient quaternary center lacked precedent.
Preliminary studies focused on defining the reaction parameters necessary for achieving the desired Reformatsky/Claisen cascade leading to 3a (terminating electrophile: 4-((trimethylsilyl)-ethynyl)oxetan-2-one). In our prior work with Reformatsky reagents, a stepwise addition of the silyl glyoxylate and ketone was necessary to avoid competitive Reformatsky reactions with the secondary electrophile. Those chemoselectivity issues are significantly attenuated here: the synchronous addition of 1 and the β-lactone led to efficient formation of 3a without appreciable reaction between the excess Reformatsky reagent and β-lactone. These results are particularly gratifying given the known capacity of the latter to undergo Claisen condensation with magnesium and lithium acetate enolates.7 Moreover, the excellent diastereomeric ratio observed for the formation of 3a (>20:1) was a fortunate and unexpected divergence from what had been previously observed in reactions of an unsubstituted (acetate-derived) Reformatsky reagent and 1.3a
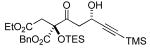
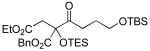
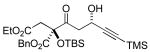
A variety of strained electrophiles exhibited similar reactivity. Asymmetric syntheses of β-lactones are prevalent in the literature, giving direct access to diverse functionality in the coupling products.5 β-lactones derived from α,β-unsaturated, alkyl, and aryl aldehydes were all viable substrates (3a, 3d, 3e, Table 1), as were lactones bearing potentially chelating functionalities (3f). The reaction was amenable to the use of α,β-disubstituted lactones, although lower diastereoselection was observed for certain substrates (dr3k > dr3i,j). γ-Butyrolactone provided analogous reactivity, but protection of the crude product as the corresponding silyl ether was necessary to avoid a potential retro-Claisen pathway during purification (3h).8 Finally, β-lactams afforded the corresponding β-amino ketone products 9 (3l,m), albeit as a 1:1 mixture of separable diastereomers.
Table 1. Substrate Scopea,b,d.
 | |||
|---|---|---|---|
 |
3a 61% yield >20:1 dr |
 |
3h 28% yieldc |
 |
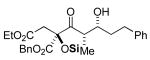
3b 67% yield >20:1 dr |
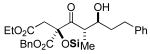 |
3i 49% yield 5:1 dr Si = TES |
 |
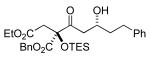
3c 35% yield >20:1 dr |
 |
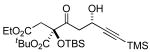
3j 72% yield 5:1 dr Si = TES |
 |
3d 64% yield >20:1 dr |
3k 63% yield >20:1 dr Si = TES |
|
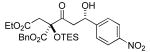 |
3e 33% yield >20:1 dr |
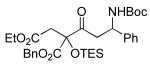 |
3l 65% yield 1:1 dr |
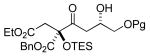 |
3f (Pg = Bn) 48% yield >20:1 dr |
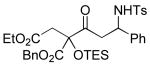 |
3m 55% yield 1:1 dr |
|
3g (Pg = TBDPS) 59% yield >20:1 dr |
|||
All reactions: [1]0 = 0.05 M.
Yields of isolated products; d.r. determined by 1H NMR spectroscopy.
Yield over two steps.
See Supporting Information for more details.6
The relevant stereochemical issues associated with these diastereoselective C-acylations are summarized in Figure 2. A closed transition state involving approach of the enolate on the diastereotopic face opposite the β-substituent of the lactone appears most plausible, but subtle stereocontrol elements must be at play to account for the dominance of 4 over 4′. The cyclic transition state depicted in Figure 2 provides a plausible means for differentiating the facial approach of the glycolate enolate, and dipole minimization of the coordinated lactone electrophile or lone pair repulsion with the benzyloxy substituent potentially suppresses formation of the minor diastereomer and favors reaction through 4. Circumstantial evidence for the proposed model was provided by the observation of reversed diastereoselection for β-keto ester 3i (derived from an anti-β-lactone); steric effects would be predicted to destabilize structure 4 with such an electrophile. Additional experimentation will be necessary to discount alternative models, but the poor diastereoselection observed with β-lactams suggests that they react through topographically distinct transition states.
Figure 2. Potential Competing Transition States.

The title reaction provides products immediately amenable to reliable diastereoselective reductions for the synthesis of functionalized diols. Reduction of 3b via the Evans10a and Prasad10b protocols afforded diols 5 and 7, respectively (Scheme 1). Cyclization of 5 (TsOH, PhMe, 80 °C) gave the mono(lactone) 6, while 7 afforded bis(lactone) 8 under the same conditions. This divergence in reactivity may be rationalized by the presumably higher energy barrier for lactonization of anti-diol 5 to a trans-fused bis(lactone).11 NOESY experiments on 6 and 8 were internally consistent and confirmed the configuration at the quaternary carbon.
Scheme 1. Secondary Transformations of Ketone Products.

The two ester groups occupy distinct chemical environments that permit differentiation based on steric effects: the syn-acetonide 9 undergoes selective reduction to the mono(alcohol) with DIBAL-H (Scheme 2, eq 3) or to the corresponding diol with LiBHEt3 (eq 4).
Scheme 2. Diester Functionalization.

The potential application of this chemistry toward the assembly of stereochemically and functionally complex linear natural products is illustrated in Scheme 3. Mitsunobu reaction of diol 7 was highly selective for reaction at the more activated and less hindered propargylic site and proceeded with complete inversion of stereochemistry and concomitant protection of the hydroxyl group as the chloroacetate.12 Phosphorylation of the remaining hydroxyl functionality afforded 11, which constitutes the bulk of the carbon skeleton of leustroducsin B, a potent colony-stimulating factor inducer isolated from Streptomyces platensis.13 Notably, the preparation of 3b was achieved on multigram scale with no deterioration in yield.
Scheme 3. Synthesis of the Leustroducsin B Core.

In conclusion, a new reactivity pattern has been observed for Reformatsky reagents and silyl glyoxylates with lactone and lactam electrophiles. The resulting Reformatsky/Claisen cascade affords highly substituted ketone products bearing an α-quaternary center through an unprecedented acylation of fully-substituted glycolate enolate intermediates. Remarkable diastereoselection is observed for these transformations through the transfer of stereochemical information from the β-lactone terminating electrophiles to the emerging quaternary center. The products contain useful orthogonal functionality that can be selectively manipulated in downstream transformations.
Supplementary Material
Figure 1. Reformatsky Reactions of Silyl Glyoxylates.

Acknowledgments
The project described was supported by Award R01 GM084927 from the National Institute of General Medical Sciences. Additional support from Novartis and Amgen is gratefully acknowledged.
Footnotes
Supporting Information Available: Experimental details and characterization data for new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Evans DA, Ennis MD, Le T, Mandel N, Mandel G. J Am Chem Soc. 1984;106:1154. [Google Scholar]
- 2.See: Tanabe Y, Hamasaki R, Funakoshi S. Chem Commun. 2001:1674.Iida A, Nakazawa S, Okabayshi T, Horii A, Misaki T, Tanabe Y. Org Lett. 2006;8:5215. doi: 10.1021/ol0619361.Mermerian A, Fu GC. J Am Chem Soc. 2005;127:5604. doi: 10.1021/ja043832w.Iida A, Takai K, Okabayashi T, Misaki T, Tanabe Y. Chem Commun. 2005:3171. doi: 10.1039/b504750a.Hauser CR, Renfrow WB. J Am Chem Soc. 1937;59:1823.Yatluk YG, Chernyak SV, Suvorov AL, Khrustaleva EA, Abramova VI. Russ J Gen Chem. 2001;71:965.
- 3.(a) Greszler SN, Johnson JS. Angew Chem Int Ed. 2009;48:3689. doi: 10.1002/anie.200900215. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Nicewicz DA, Johnson JS. J Am Chem Soc. 2005;127:6170. doi: 10.1021/ja043884l. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Linghu X, Satterfield AD, Johnson JS. J Am Chem Soc. 2006;128:9302. doi: 10.1021/ja062637. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Nicewicz DA, Satterfield AD, Schmitt DC, Johnson JS. J Am Chem Soc. 2008;130:17281. doi: 10.1021/ja808347q. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Greszler SN, Johnson JS. Org Lett. 2009;11:827. doi: 10.1021/ol802828d. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Schmitt DC, Johnson JS. Org Lett. 2010;12:944. doi: 10.1021/ol9029353. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Nicewicz DA, Breteche G, Johnson JS. Org Synth. 2008;85:278. [Google Scholar]
- 4.Brook AG. Acc Chem Res. 1974;7:77. [Google Scholar]
- 5.(a) Nelson SG, Peelen TJ, Wan Z. J Am Chem Soc. 1999;121:9742. [Google Scholar]; (b) Nelson SG, Zhu C, Shen X. J Am Chem Soc. 2004;126:14. doi: 10.1021/ja0391208. [DOI] [PubMed] [Google Scholar]; (c) Nelson SG, Zhu C, Shen X. J Am Chem Soc. 2004;126:5352. doi: 10.1021/ja0492900. [DOI] [PubMed] [Google Scholar]; (d) Evans DA, Janey JM. Org Lett. 2001;3:2125. doi: 10.1021/ol016096z. [DOI] [PubMed] [Google Scholar]; (e) Oh SH, Cortez GS, Romo D. J Org Chem. 2005;70:2835. doi: 10.1021/jo050024u. [DOI] [PubMed] [Google Scholar]; (f) Purohit VC, Richardson RD, Smith JW, Romo D. J Org Chem. 2006;71:4549. doi: 10.1021/jo060392d. [DOI] [PubMed] [Google Scholar]
- 6.Lithium chloride has previously been shown to increase the reactivity of other organozinc species: Krasovskiy A, Malakhov V, Gavryushin A, Knochel P. Angew Chem. 2006;118:6186. doi: 10.1002/anie.200601450.Angew Chem Int Ed Engl. 2006;45:6040.Metzger A, Schade MA, Knochel P. Org Lett. 2008;10:1107. doi: 10.1021/ol7030697.
- 7.For reactions of β-lactones with unsubstituted enolates, see: Shen X, Wasmuth AS, Zhao J, Zhu C, Nelson SG. J Am Chem Soc. 2006;128:7438. doi: 10.1021/ja061938g.Green ME, Rech JC, Floreancig PE. Angew Chem Int Ed. 2008;47:7317. doi: 10.1002/anie.200802548.Shimizu M, Ishii K, Fujisawa T. Chem Lett. 1997:765.Wang Y, Tennyson RL, Romo D. Heterocycles. 2004;64:605. For a review of reactivity, see:
- 8.(a) Wang MX, Liu Y, Gao HY, Zhang Y, Yu CY, Huang ZT, Fleet GWJ. J Org Chem. 2003;68:3281. doi: 10.1021/jo026560t. [DOI] [PubMed] [Google Scholar]; (b) Rao D, Best D, Yoshihara A, Gullipalli P, Morimoto K, Wormald MR, Wilson FX, Izumori K, Fleet GWJ. Tetrahedron Lett. 2009;50:3559. [Google Scholar]
- 9.Ojima I, Ng EW, Sun CM. Tetrahedron Lett. 1995;36:4547. [Google Scholar]
- 10.(a) Evans DA, Chapman KT, Carreira EM. J Am Chem Soc. 1988;110:3560. [Google Scholar]; (b) Chen KM, Gunderson KG, Hardtmann GE, Prasad K, Repic O, Shapiro MJ. Chem Lett. 1987:1923. [Google Scholar]
- 11.Jahn U, Hartmann P, Kaasalainen E. Org Lett. 2004;6:257. doi: 10.1021/ol036233n. [DOI] [PubMed] [Google Scholar]
- 12.Saiah M, Bessodes M, Antonakis K. Tetrahedron Lett. 1992;33:4317. [Google Scholar]
- 13.(a) Kohama T, Enokita R, Okazaki T, Miyaoka H, Torikata A, Inukai M, Kaneko I, Kagasaki T, Sakaida Y, Satoh A, Shirashi A. J Antibiot. 1993;46:1503. doi: 10.7164/antibiotics.46.1503. [DOI] [PubMed] [Google Scholar]; (b) Kohama T, Nakamura T, Kinoshita T, Kaneko I, Shiraishi A. J Antibiot. 1993;46:1512. doi: 10.7164/antibiotics.46.1512. [DOI] [PubMed] [Google Scholar]; (c) Matsuhashi H, Shimada K. Tetrahedron. 2002;58:5619. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.