

Abstract
The key cytopathologies in the brains of Alzheimer's disease (AD) patients include mitochondrial dysfunction and energy hypometabolism, which are likely caused by the accumulation of small aggregates of amyloid-β (Aβ) peptides. Thus, targeting these two abnormalities of the AD brain may hold promising therapeutic value for delaying the onset of AD. In his paper, we discuss two potential approaches to delay the onset of AD. The first is the use of low dose of diaminophenothiazins (redox active agents) to prevent mitochondrial dysfunction and to attenuate energy hypometabolism. Diaminophenothiazines enhance mitochondrial metabolic activity and heme synthesis, both key factors in intermediary metabolism of the AD brain.The second is to use the naturally occurring osmolytes to prevent the formation of toxic forms of Aβ and prevent oxidative stress. Scientific evidence suggests that both approaches may change course of the basic mechanism of neurodegeneration in AD. Osmolytes are brain metabolites which accumulate in tissues at relatively high concentrations following stress conditions. Osmolytes enhance thermodynamic stability of proteins by stabilizing natively-folded protein conformation, thus preventing aggregation without perturbing other cellular processes. Osmolytes may inhibit the formation of Aβ oligomers in vivo, thus preventing the formation of soluble oligomers. The potential significance of combining diaminophenothiazins and osmolytes to treat AD is discussed.
1. Introduction
1.1. An Overview of Alzheimer's Disease
Alzheimer's disease (AD) is an irreversible brain disorder that slowly destroys memory and eventually a person's ability to perform the daily life tasks and activities. Memory problems are one of the first signs of AD, and as it progresses, decline in other cognitive abilities such as poor judgment and mood changes starts to surface. Eventually people with severe AD cannot communicate and become completely dependent on others for their care. Most people with AD have lateonset of disease, which usually develops after age 60. However, a silent preclinical phase which precedes the development of AD clinical symptoms may span 2–8 years.
AD is a disease with a complex etiology and no single therapeutic approach is likely to prevent or lead to a cure. All current treatments focus on several different aspects, including management of behavioral symptoms or temporarily slowing the progress of the disease. However, none of these treatments changes or alter the inevitable course of the disease. Thus, even after almost three decades of research, AD is still dementia with a progressive failure to form new memories, and thereby interfering with the basic mechanism of the disease.
In recent years, it has been well-accepted that one of the pathological mechanisms of AD relates to the accumulation amyloid-β (Aβ) peptide in certain brain regions [1]. Aβ is a product of Aβ precursor protein (AβPP) via natural proteolytic processing. AβPP is processed by three different proteases known as α-, β-, and γ-secretase. Each of these three proteases cleaves AβPP at different sites resulting in various Aβ species ranging from 39–43 amino acid residues [2]. It has been shown that long species of Aβ (Aβ42,43) are strongly amyloidogenic and form aggregates readily compared to the short forms (Aβ39,40). Aβ are the building blocks of insoluble extracellular Aβ deposits or senile plaques formation [3], a neuropathological hallmark of AD [3]. AD is also marked by neurofibrillary tangles, an intracellular filaments of highly phosphorylated tau protein. Impairment of cellular function in AD is demonstrated by a set of cytopathologies (reviewed in [4]) such as decline in cytochrome c oxidase (complex IV), mitochondrial dysfunction, abnormal iron homeostasis, oxidative stress, dimerization of AβPP and synaptic dysfunction [5–9], and energy hypometabolism. Several lines of evidence point towrads strong connection between small aggregates of Aβ and mitochondrial dysfunction.
1.2. Role of Amyloid-β and Oligomers in Alzheimer's Disease
According to the oldest so-called “cholinergic hypothesis”, AD is caused by reduced levels of the neurotransmitter acetylcholine, which is important for memory. However, medications intended to treat acetylcholine deficiency have not been very effective in modifying the course of the disease. In the early 90s, the “amyloid hypothesis” was postulated according to which Aβ deposits arising from AβPP cause AD. As a result, specific proteases that process AβPP were the focus of drug development as a possible mean to lower Aβ production. However, several complications stem from the significance of these proteases for other biological functions creating serious obstacles in this front.
It is now well-accepted fact that AD is a neurodegenerative disorder associated with protein aggregation and misfolding of AβPP, which may be triggered by genetic polymorphism, age-dependent alteration to Aβ metabolism, or environmental factors that may promote accumulation and aggregation of Aβ peptide [10–13]. Thus, therapies directed at reversing Aβ aggregation appeared promising. However, several limitations reduced the excitement in this approach as was the case for immunotherapy targeted using specific anti-Aβ antibodies. Additionally, the lack of chemical safety of the available drugs to prevent Aβ aggregation was also a major concern.
There are reports showing a correlation between an early cognitive impairment in AD and increased oligomerization of Aβ, which precede the appearance of senile plaques [14–16]. Oligomers of Aβ correlate with early cognitive impairment in AD [17, 18]. In recent years, studies using cells, mouse models, and human brain tissues strongly suggest that soluble Aβ oligomers could be a toxic forms of Aβ [19, 20]; however, experimental data for their direct in vivo toxicity is lacking [21]. Thus, preventive approaches may include also preventing the formation of Aβ aggregates. However, it is not entirely clear which cellular compartment is the primary target of Aβ toxicity. Interestingly, in addition to other extra- and intracellular compartments, Aβ is also found in the mitochondria [22, 23], suggesting that neurotoxic effects of Aβ may be widespread [24], unlike earlier views that the extracellular senile plaques are the only main neurotoxic factor in AD. Energy hypometabolism and synaptic dysfunctions are proposed to be the primary target of Aβ neurotoxicity [25, 26]. Thus, preventing mitochondrial dysfunction by identifying the primary metabolic pathway, specifically targeted by Aβ is a plausible approach to delay AD [27]. We propose that preventative approaches are more promising for lowering the prevalence of AD. These approaches could target the formation of Aβ oligomers and enhance mitochondrial activity to counter energy deficiency in AD.
2. Role of Mitochondrial Dysfunction in Alzheimer's Disease
Several lines of evidence suggest that impairment of mitochondrial function plays important role in the development of neurodegenerative diseases including AD. Mitochondrial dysfunction and impaired cellular energy is an early decline in metabolism seen in AD patients. In addition to the biochemical physiological changes, the brain mitochondria of AD patients exhibit substantial structural changes that included abnormal cristae, accumulation of osmophilic material, and smaller size compared to normal controls [23, 28]. It has been reported that mitochondrial fragmentation damages regions of nerve cell synapses. Excessive fragmentation of mitochondria causes synaptic injury leading to eventual nerve cell death. Since synapses are critical for learning and memory, their impairment leads to the dementia in AD patients. We have recently proposed that strong binding of Aβ with heme is a key factor associated with Aβ-mediated neurotoxicity [29, 30], which could be the primary metabolic pathway targeted by excess Aβ production [31–33], and thus interfering with mitochondrial structures and functions by increasing the production of nitric oxide (NO) leading to mitochondrial damage and impaired energy metabolism [34, 35]. One of the key cytopathologies of AD include decline in cytochrome c oxidase (complex IV) and αKGDH, which seems to contribute to mitochondrial dysfunction [36–38]. This, abnormal biology of the mitochondria may contribute to energy deficiency in AD [35, 39]. There are reports showing an abnormal interaction of Aβ with key brain metabolites such as zinc, copper, cholesterol, mitochondrial protein import machinery, HrtA2 protease, ABAD, and heme [22, 31, 40–43]; however, their relation to cytopathologies of AD is not clear.
The decline in complex IV, which occurs in heme deficient cells, leads to similar structural consequences on mitochondria (unpublished observations). Several lines of experimental evidence provided support that heme metabolism may be a specific metabolic pathway that is targeted by Aβ peptides [43–46]. We showed recently a specific heme-binding motif in human Aβ peptides [27]. Based on our recent work and other laboratories, we propose that depletion of regulatory heme and the formation of Aβ-heme peroxidase are key factors of mitochondrial dysfunction in the brains of AD patients. Due to phylogenic variation in the amino acid sequences of Aβ, differential heme-binding of Aβ could also explain why humans, but not rodents, develop AD-like neuropathology (Reviewed in [4]). Heme is responsible for the metabolic integrity of complex IV, which is a key factor in mitochondrial gene regulation systems; therefore, pathways that depend on heme may be properly regulated by lowering Aβ oligomeric forms, and enhanced heme synthesis may improve neuronal energy metabolism.
3. Therapeutic Approaches for Alzheimer's Disease and Challenges
AD has a devastating impact on both personal and community levels. AD is the most common age-related dementia manifested by widespread progressive cognitive deterioration and impaired behavioral skills. With the aging US populations, and widespread prevalence of AD in these populations warrants immediate need for the management of this deleterious disease. In spite of efforts from scientific community for several decades, the available drug therapies for AD are only remedial without proper understanding of the underlying mechanisms involved therein. Unless new treatments are developed to decrease the likelihood of developing AD, the number of individuals with this disease in the United States is expected to be more than 10 million in next three decades.
There is currently no specific cure for AD patients, but scientific research is unraveling the mysteries of AD, including the causes and the mechanisms of the disease progression, which might one day effectively solve the Alzheimer's puzzle. In recent years, some understandings have already provided critical information about how to prevent, delay, or slow the nerve cell damage that leads to AD, which may help maximize quality of life of these patients. However, drug treatments currently available are used to only manage the cognitive symptoms of AD by slowing the progression of symptoms for a while.
Despite the intensive research on AD, a therapeutic or preventive strategy for AD remains elusive or limited at the most [47], which has been a key obstacle for the development of effective therapies for AD. The efficacy of currently available drugs has further been hampered by the fact that the effectiveness of these drugs progressively declines with the progression of disease. Thus, there is an urgent need for a new generation of drugs to prevent or delay the onset of AD.
Except for imaging techniques, a precise and accurate diagnostic biomarker for AD is also lacking [48]. However, images results are excellent in following disease progress, but limited in revealing preclinical before protein deposits occur AD. The ultimate biomarker would preferably be a blood- or CSF-borne metabolite that indicates the risk for AD in advance of the clinical signs or protein deposits. Aβ-heme peroxidase could serve a unique biomarker if found in blood or CSF of AD patients. Aβ-heme has the advantage of being dependant on Aβ and it is tightly linked to a key brain metabolite (heme as mitochondrial metabolite), thus could indicate impairment in brain metabolism that depends on Aβ accumulation.
4. Mitochondria As Targets for Delaying the Onset of Alzheimer's Disease
Mitochondrial dysfunction in AD could serve as a therapeutic target an interest in developing mitochondrial drugs is emerging. Enhancing mitochondrial function and maintaining structural integrity of mitochondria could delay the onset of AD. Below, we discuss pharmacological approaches to enhance mitochondrial function and prevent the formation of Aβ toxic oligomers.
Mitochondria are a major energy source, and it has been known that energy deficiency can result in synaptic dysfunction and neurodegeneration of the hippocampus and cortical regions of the brain [49]. The brain is particularly sensitive to mitochondrial dysfunction, the resulting oxidative stress, and impaired energy metabolism [50–52]. Thus, improved energy metabolism through enhanced mitochondrial activity in the brain might be an effective approach to delay the onset of AD.
Mitochondrial dysfunction in AD is associated with a decline in mitochondrial complex IV and energy deficiency. Due to involvement of mitochondria in cellular senescence and aging, it may contribute to neural dysfunction with age. Therefore, targeting mitochondria is an emerging field of research in finding therapeutic strategies to combat aging and neurodegenerative disorders. In fact, recent developments support this idea (reviewed in [53]), and potency of pharmacological agents to prevent or delay age-related neurodegeneration is under investigation [54]. Our recent results with methylene blue (3,7 Bis-dimethylamino-phenazathionium; MB) in countering some mitochondrial dysfunctions including mitochondrial complex IV formation, enhanced cellular oxygen consumption and heme synthesis, and reversed premature senescence are aimed at enhancing mitochondrial function, which could contribute to the antisenescence activity of MB [55–57]. Due to the ability of MB to cross the Blood Brain Barrier [58], our studies may provide a potential future therapeutic tool for AD and other related diseases using MB.
MB is known as a redox indicator with a low redox potential, which allows it to cycle readily in mitochondria (Figure 1), and is easily soluble in both water and organic solvents, thus, MB and its derivative MBH2 can enter the mitochondria and other intracellular compartments such as lysosomes [59]. MB is the first chemical to induce mitochondrial respiratory complex, and we propose a new medical use for MB by increasing brain's reserve of both complex IV and the capacity to synthesize heme [56]. Increasing the activity of complex IV is intriguing as a decrease in complex IV activity causes cytotoxicity leading to increased oxidants production and decreased energy charge of the mitochondria [60–62]. MB in turn may elevate the levels of complex IV and improve mitochondrial function. Complex IV consumes more than 95% of the O2 that reaches cells, and thus, excess complex IV may play a key role in lowering the production of oxidants by decreasing the steady-state concentration of intracellular O2 in the mitochondria. Complex IV activity correlates well with the metabolic activity of cells and thus could improve cognitive performance. On the other hand, enhancing heme synthesis should help neural cells in delaying the onset of the consequences of sequestration of heme by huAβ. Together, these findings suggest that MB may delay the onset of Alzheimer's dementia. Further, MB at higher concentrations (μM range) are neurotoxic, and our findings show that MB is effective in improving mitochondrial function at nM range of concentrations, which is consistent with the intrabrain concentration that can be achieved upon chronic treatment with MB [56]. MB has a long-standing, extensive history of medical uses [63] with an extended medical and safety record in humans, and its FDA approval for clinical trials in connotation to aging and age-related disorders may not be difficult to obtain on safety grounds.
Figure 1.
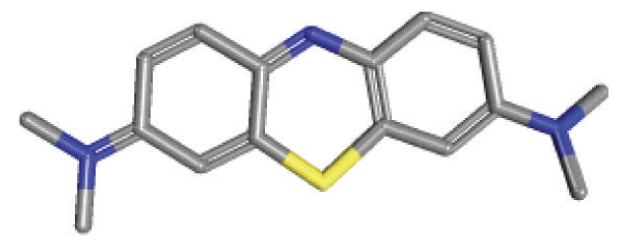
The chemical structure of methylene blue; a diaminophenothiazin.
5. Mechanisms of Action of Methylene Blue and Its Clinical Applications
In spite of widespread clinical uses of MB for decades, the mechanism(s) of its diverse biological actions are not clear. MB is readily absorbed by various organs including brain [64], and has a long history of clinical uses including chronic treatments of congenital methemoglobinemia, methemoglobinemia, psychiatric disorders, and more recently in the prevention of the side effects of ifosfamide-induced encepholopathy chemotherapy [65], and hypotension in septic shock [66–68]. MB has also been shown to protect against cyclosporine injury to kidney [69], streptozotocin injury to pancreas [70], ischemic-reperfusion injury [71], radiation [72], and enhances β-oxidation of long chain fatty acids [73]. Clinical doses of MB ranges between 1-2 mg/kg/day for up to 6 times over 24 hrs [74]. Higher doses (>7.5 mg/Kg) of MB cause the formation of Heinz bodies in erythrocytes [75]. In addition, MB administration has been reported to improve the cognitive function in rats, and increase the activity of cytochrome c oxidase (complex IV) [76, 77] and decrease of monoamine oxidase activity in the brain, which may result in an increased dopamine concentration [78]. Some reports have proposed that MB may be acting by inhibiting the NO-activating soluble guanylate cyclase [79], nitric oxide synthase [80], and MAO activity [78]. There are also reports suggesting that MB may be acting as an antioxidant precursor [81, 82]. However, in recent years, it has become quite clear that the biological effects of MB are not consistent with these mechanisms [55, 56, 83]. This discrepancy may in part be due to different doses of MB used in these experimental conditions, which have ranged from >10 μM to <1 μM, suggesting that uses of MB at doses in the nM concentrations may follow a different mechanism [55, 56]. For example the effect of MB on complex IV depends on the dose used [55]. Thus, interpretation of the experimental findings should take into consideration MB's dose especially when high (non-therapeutic) dose is used [84]. Based on our data and others, for the first time, we proposed a molecular mechanism explaining how MB might be affecting mitochondrial function [55–57]. We propose that diaminophenothiazins cycling between the reduced and the oxidized forms may explain, in part, their mitochondria-protecting activity (Figure 1). This mechanism is proposed in Figure 3 and in [55]. It is now an open secret that adequate assembly and activity of complex IV depends upon heme-a [85]; thus, the increased rate of heme synthesis with MB treatment could provide cells with heme to support the assembly of complex IV that could result in delaying mitochondrial dysfunction, cellular senescence, aging, and AD.
Figure 3.

The proposed model for the interaction of methylene blue with specific mitochondrial and cellular components. ETC followed by roman number refers to the specific components of electron transport chain (ETC) of the mitochondria. MIM, MOM, and IMS refers to the mitochondrial inner membrane, mitochondrial outer membrane, and intermembrane space, respectively. MB and MBH2 refer to oxidized and reduced forms of methylene blue, respectively. The four complexes are: complex I (ETC I), complex II (ETC II), complex III (ETC III), and complex IV (ETC IV) in addition to ATP synthase (i.e., complex V). The electron transfer through each one of the ETC starts at ETC I, which catalyzes two electrons oxidation of NADH and continues until water is formed on ETC IV. Coenzyme Q serves as low-molecular weight electron carrier from ETCs I and II to III. Cytochrome c (cyt c) serves as electron carrier from ETC III to ETC IV. Production of superoxide radical from complex I is proposed to be prevented by MB, which serves as electron carrier that competes with molecular oxygen on the electrons “leaking” from complex I. During this process MB is converted to MBH2. Then MBH2, a reduce MB, carries the electrons to cytochrome c, which is then oxidized by ETC IV [55].
6. Preventing Aggregation and Oligmerization of Amyloid-β with Osmolytes
Protein misfolding and/or instability leads to aggregate formation. Many neurodegenerative diseases exhibit deposits of aggregated proteins such as Aβ oligomers, senile plaques, phosphorylated tau (p-tau), α-synuclein, and polyglutamine; and all are key neuropathologies in many CNS disorders. Aβ oligomers in AD may progress to form large insoluble fibrils that form the plaques, a key hall mark of AD. Accumulation of these protein deposits exacerbate neurodegeneration due to the fact that the human brain has a limited capacity to prevent the formation or removal of these protein aggregates. It is known that limited protein degradation may contribute to the accumulation of these peptide deposits with age [86]. The aging brain also exhibits limited antioxidant activity and self-repair capacities (e.g., limited neurogenesis).
Our knowledge of the kinetics of the formation of protein aggregates in vivo in brain disorders as well as the limited understanding for the mechanism by which these aggregates interfere with neuronal metabolism function has impaired our capabilities for the development of preventative therapeutic strategies. In general, the intermediate species in the cascade of protein misfolding appear to be highly toxic and may interfere with basic metabolic activity of the brain. As for Aβ oligomers it is not clear how toxic they would be in vivo [21]. However, removal or preventing the formation of such intermediate species may prove to be of great clinical value. We demonstrated that the binding of Aβ monomers and oligomers with heme results in sequestration of regulatory heme leading to impaired cellular metabolism. Therefore, preventative approaches to treating AD could be targeted at blocking the formation of Aβ oligomers, enhancing the synthesis of heme and complex IV, in addition to the use of antioxidants.
6.1. Oligmers as Targets for Delaying Alzheimer's Disease
Protein functions depends on maintaining and stabilizing their active conformation(s) under physiological and stress conditions [87]. Thus, under severe physical and chemical stress conditions, biological systems created mechanisms to maintain their functional conformations. These mechanisms are directed at preventing structural perturbations in proteins due to thermodynamic or chemical stressful conditions [88–91]. Biological systems that fail to provide proteins structure stabilizing conditions also fail to adapt to such conditions. As a result, often protein misfolding and aggregation occurs leading to a partial or complete loss of function, in addition to the formation of protein aggregates [92, 93]. Both conditions have serious consequences on cell function and metabolism.
In order to adapt to stressful conditions, tissues created certain mechanisms such as degradation of misfolded proteins or accumulation of small organic solutes at high concentrations. These solutes can serve as antioxidants or play stabilizing role for intracellular structures of macromolecules. Osmolytes [88, 89, 94] are group of endogenous chemicals produced by cells and accumulate to concentration as high as millimolars (Figure 2). The exact function of these metabolites and significance for intermediary metabolism and organ function is still under investigation. We are interested in finding scientific reasoning that links to identify the role of osmolytes and the high tissue connections. Experimental evidence points that osmolytes maintain stability and folding of proteins without perturbing other cellular processes, an action that requires relatively high intracellular concentrations [90, 91, 93, 95–97]. It is well known that following protein synthesis, a highly disordered unfolded state of the polypeptide chain passes through well-defined partially structured transition states before the fully folded protein forms. Molecular chaperones that deter aggregation of incompletely folded species also play role in correctly folding newly synthesized proteins [98]. If under certain conditions, the cellular environment becomes less than optimal for proper protein folding, then newly synthesized proteins become prone to aggregation [99–101]. Similarly, under these conditions, intermediates in protein processing pathways may also be subject to accumulation, misfolding, and aggregation (e.g., Aβ). Protein aggregates, if not quickly removed, may transform to fibrils and other possible aggregates that accumulate in tissue and interfere with cell metabolism [102–107]. It is likely that small aggregates, as well as the highly organized fibrils and plaques, can give rise to pathological conditions, a common feature among many neurodegenerative diseases, including AD [97, 108].
Figure 2.

Chemical structure of selected osmolytes. Upper panel: structure in 2D; lower panel, structure in 3D configurations, respectively.
The biological significance of naturally occurring osmolytes has intrigued scientists for many years. There are a number of well-known naturally occurring osmolytes, which fall into three chemical classes: methylamines (trimethylamine-N-oxide, Choline-O-sulphate, and sarcosine), polyols (sorbitol, glycerol, sucrose and trehalose), and certain amino acids and their derivatives (glycine, taurine, proline and betaine) (Table 1). The role of osmolytes in protein folding, cell senescence, cell homeostasis, and mitochondrial structure has been described in various studies. However, more investigations are still needed to evaluate the role of osmolytes in health and diseases and their therapeutic potential.
Table 1.
List of some known naturally occurring osmolytes in each class with their major presence in protecting the stability of specific proteins under harsh conditions.
| Type | Name(s) | Used by |
|---|---|---|
| Polyols | mannitol, glycerol, sorbitol, inositol, pinitol | plants, algae, mammalian kidneys, insects, reptiles, fish |
| Amino Acid | glycine, alanine, and proline | mammalian cells |
| Amino Acids Derivatives | taurine, octopine, alanine | marine invertebrates, prokaryotes |
| Methylamines | trimethylamine-N-oxide, sarcosine, phosphorylcholine, glycine betaine | marine invertebrates, plants, mammalian kidneys |
Osmolytes interact with the peptide backbone and amino acid side-chains [109]. The potency of an osmolyte to promote protein folding and solubility is determined by the balance of these interactions and the solvophobic effects of the osmolyte. There are several studies to support the view that the powerful solvophobic effects of osmolytes on the peptide backbone dominate, such that the relative Gibbs free energy (ΔG) of the unfolded state is less favorable than that of the folded state (ΔG of the peptide folding is more negative).
The presence of several osmolytes inside cells raises questions about their role in protecting intracellular macromolecules under stressful conditions. The antioxidative activity of the osmolytes has been also proposed. Since the protection provided by an osmolyte does not depend on specific chemical interactions with the macromolecules, in principle, any of the osmolytes should be capable of replacing each other, depending upon either endogenous or exogenous availability of particular osmolyte(s) [110]. Since the role of protein backbone is critical in determining thermodynamic stability and folding of proteins in osmolyte solutions [111–115], designing these small molecules (osmolytes) appears to be an excellent strategy and could be a critical step in preventing various critical proteins from misfolding or aggregation (6). This may have far-reaching consequences in understanding and preventing several deleterious diseases that are caused by protein misfolding/aggregation [116, 117]. Since organic osmolytes are naturally occurring molecules, they may have potential therapeutic applications without concerns of major toxic side effects [118].
Because of their capabilities to fold proteins into native-like functional species, osmolytes have been the focus of several studies related to neurodegenerative diseases in which the pathogenesis is associated with the misfolding of specific proteins [119, 120]. These diseases include AD, Huntington's disease (HD), and muscular dystrophy (MD). Trehalose, an osmoluyte, can significantly inhibit polyglutamine-mediated protein aggregation when orally administrated to the transgenic mouse model of Huntington's disease (a neurodegenerative disease) [114], and can increase the life span of Huntington's disease mouse model [114], indicating that trehalose is readily bioavailable. Osmolytes have similar effect of the folding of androgen receptor containing elongated polyglutamine chain length. Elongated polyglutamine chain in androgen receptor leads to formation of its aggregation and thus play role in causing neurodegeneration in Kennedy's disease [116].
Experimental finding suggests that AD toxicity could be linked to the formation of oligomeric forms of Aβ peptides and AD progression correlates with increasing aggregate formation of Aβ. At physiological concentration, Aβ40 peptide incubated in the presence of trehalose inhibits aggregation of this peptide in a dose-dependent manner [121], and this osmolyte-mediated inhibition of Aβ40 peptide aggregation correlates with its toxic effects in neuronal cell system [121].
In these pathological conditions, specific misfolded aggregate-prone proteins are resistant to the normal cellular processes of protein folding and turnover and we propose that osmolytes may interfere with the production and/or the removal of these toxic intermediate aggregates. Further, these osmolytes can be attractive molecules for delaying the onset of neurodegenerative diseases characterized by protein misfolding and toxic aggregation. In particular, osmolytes may have therapeutic potential for treating AD, because of their effect on Aβ oligomers. Osmolytes may also help stabilize the senile plaques, preventing the shedding of Aβ oligomers. Osmolytes are also known to function as antioxidants [122, 123], and their level seems to decline in AD patients [124].
The research on naturally occurring osmolytes suggests that they have a protective role in promoting brain health, including resistance to neurodegeneration. Thus, there is potential for both prevention and treatment of neurodegenerative diseases using osmolytes [125, 126]. The prospect of using natural osmolytes as a therapeutic tool for AD appears to be quite exciting and can have far reaching consequences in developing therapeutic tools for its prevention and/or management. However, while these proposals appear to be quite promising, more studies are needed to validate their effectiveness as a potential therapeutic target.
7. Summary
The mitochondrial role in health and disease has recently received immense scientific interest, particularly, because of the emerging field of mitochondria as a potential therapeutic target. Mitochondria play intricate role in energy, redox, and intermediary metabolism positioning them in cross road for health or disease. Genetic, environmental, and life style factors can lead to impairments in mitochondrial function. Impairment to mitochondrial function is found in numerous age-related degenerative diseases and disorders. Mitochondrial dysfunction leads to an increase in oxidative stress, energy hypometabolism, and impairs calcium homeostasis, ultimately leading to inadequate cellular function. Preventing mitochondrial dysfunction is presumed to have functional benefits, regardless of whether mitochondrial dysfunction is primary or secondary [57]. Thus, therapeutic strategies to improve mitochondrial function and delay the onset of age-related degenerative disorders are currently under investigation. We have shown that diaminophenothiazins (e.g., methylene blue, thionine, Figure 1) can delay cellular senescence by enhancing mitochondrial function, which are impaired in AD brains.
Mounting evidence suggests a role of small aggregates of amyloid-β (Aβ) in the etiology of AD. Aβ aggregates impair mitochondrial function, synaptic function, Ca++ homeostasis and ultimately leading to cellular hypometabolism and neurdegeneration. Aβ peptide can also be localized within the mitochondria of AD patients. Experimental evidence show that osmolytes can stabilize and enhance cellular proteins to adopt physiologically compatible conformation. Osmolytes are efficient antioxidants that may also increase neural resistance to oxidative stress caused by Aβ. Thus, osmolytes may interfere with the aggregation of Aβ, enhance their proteolytic clearance, and counter oxidative stress [57].
We propose two different approaches to prevent or delay the onset of AD. The first is directed at enhancing mitochondrial activity using MB to enhance mitochondrial function (Figure 3). Energy deficiency in AD may be contributed by impaired insulin (glucose) metabolism and mitochondrial function. Thus, concentrating on single impairment at the time would not be enough to resolve the energy hypometabolism in AD. Glucose metabolism depends on adequately functioning mitochondria and vice versa. Since both glucose and mitochondrial metabolism are interconnected, it might be more beneficial for AD patients to develop a therapeutic approach that resolves (or delay) both impairments. Successful merger of treatment with MB and intranasal delivery of insulin to the brain may prove valuable for AD patients. MB exerts its effect at very low (nM) concentration, which in conjunction with its safety record in humans further minimizes any risk of side effects of chronic exposure to MB. The second approach is directed at preventing the aggregation of Aβ by using osmolytes, natural metabolites synthesized in the brain. Preventing the aggregation of Aβ may enhance their proteolytic removal and decrease the risk of their interference with heme and mitochondrial metabolism. MB can also induce heme synthesis, thus, when combined with osmolytes, may assist in preventing heme deficiency. We propose MB and osmolytes could help delay the onset of AD by preventing Aβ oligomers formation, enhancing mitochondrial function, and attenuating heme deficiency.
We propose that preventative approaches for AD could be targeted at blocking the formation of Aβ oligomers, enhancing the synthesis of heme and complex IV, in addition to the use of antioxidants.
Conflict of Interests
Dr. Atamna has applied for patent on MB to treat mitochondrial dysfunction.
Acknowledgments
The study was supported in part by Ames Foundation, AFAR (H. Atamna), and NIH (R. Kumar).
References
- 1.Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid β-peptide. Nature Reviews Molecular Cell Biology. 2007;8(2):101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- 2.Nunan J, Small DH. Regulation of APP cleavage by α-, β- and γ-secretases. FEBS Letters. 2000;483(1):6–10. doi: 10.1016/s0014-5793(00)02076-7. [DOI] [PubMed] [Google Scholar]
- 3.Atwood CS, Martins RN, Smith MA, Perry G. Senile plaque composition and posttranslational modification of amyloid-β peptide and associated proteins. Peptides. 2002;23(7):1343–1350. doi: 10.1016/s0196-9781(02)00070-0. [DOI] [PubMed] [Google Scholar]
- 4.Atamna H. Heme binding to Amyloid-β peptide: mechanistic role in Alzheimer's disease. Journal of Alzheimer's Disease. 2006;10(2-3):255–266. doi: 10.3233/jad-2006-102-310. [DOI] [PubMed] [Google Scholar]
- 5.Parker WD, Filley CM, Parks JK. Cytochrome oxidase deficiency in Alzheimer's disease. Neurology. 1990;40(8):1302–1303. doi: 10.1212/wnl.40.8.1302. [DOI] [PubMed] [Google Scholar]
- 6.Valla J, Berndt JD, Gonzalez-Lima F. Energy hypometabolism in posterior cingulate cortex of Alzheimer's patients: superficial laminar cytochrome oxidase associated with disease duration. Journal of Neuroscience. 2001;21(13):4923–4930. doi: 10.1523/JNEUROSCI.21-13-04923.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Daly C, Sugimori M, Moreira JE, Ziff EB, Llinás R. Synaptophysin regulates clathrin-independent endocytosis of synaptic vesicles. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(11):6120–6125. doi: 10.1073/pnas.97.11.6120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Markesbery WR. Oxidative stress hypothesis in Alzheimer's disease. Free Radical Biology and Medicine. 1997;23(1):134–147. doi: 10.1016/s0891-5849(96)00629-6. [DOI] [PubMed] [Google Scholar]
- 9.Connor JR, Menzies SL, Martin SM, Mufson EJ. A histochemical study of iron, transferrin, and ferritin in Alzheimer's diseased brains. Journal of Neuroscience Research. 1992;31(1):75–83. doi: 10.1002/jnr.490310111. [DOI] [PubMed] [Google Scholar]
- 10.Blasko I, Beer R, Bigl M, et al. Experimental traumatic brain injury in rats stimulates the expression, production and activity of Alzheimer's disease β-secretase (BACE-1) Journal of Neural Transmission. 2004;111(4):523–536. doi: 10.1007/s00702-003-0095-6. [DOI] [PubMed] [Google Scholar]
- 11.Farrer LA, Cupples LA, Haines JL, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease: a meta-analysis. Journal of the American Medical Association. 1997;278(16):1349–1356. [PubMed] [Google Scholar]
- 12.Landrigan PJ, Sonawane B, Butler RN, Trasande L, Callan R, Droller D. Early environmental origins of neurodegenerative disease in later life. Environmental Health Perspectives. 2005;113(9):1230–1233. doi: 10.1289/ehp.7571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Prasad GS, Wahlberg M, Sridhar V, et al. Crystal structures of transhydrogenase domain I with and without bound NADH. Biochemistry. 2002;41(42):12745–12754. doi: 10.1021/bi020251f. [DOI] [PubMed] [Google Scholar]
- 14.Hartmann T, Bieger SC, Brühl B, et al. Distinct sites of intracellular production for Alzheimer's disease Aβ40/42 amyloid peptides. Nature Medicine. 1997;3(9):1016–1020. doi: 10.1038/nm0997-1016. [DOI] [PubMed] [Google Scholar]
- 15.Fezoui Y, Hartley DM, Harper JD, et al. An improved method of preparing the amyloid β-protein for fibrillogenesis and neurotoxicity experiments. Amyloid. 2000;7(3):166–178. doi: 10.3109/13506120009146831. [DOI] [PubMed] [Google Scholar]
- 16.Mucke L, Masliah E, Yu GQ, et al. High-level neuronal expression of Aβ(1-42) in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. Journal of Neuroscience. 2000;20(11):4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kern A, Behl C. The unsolved relationship of brain aging and late-onset Alzheimer disease. Biochimica et Biophysica Acta. 2009;1790(10):1124–1132. doi: 10.1016/j.bbagen.2009.07.016. [DOI] [PubMed] [Google Scholar]
- 18.Bierer LM, Hof PR, Purohit DP, et al. Neocortical neurofibrillary tangles correlate with dementia severity in Alzheimer's disease. Archives of Neurology. 1995;52(1):81–88. doi: 10.1001/archneur.1995.00540250089017. [DOI] [PubMed] [Google Scholar]
- 19.Aliev G, Seyidova D, Lamb BT, et al. Mitochondria and vascular lesions as a central target for the development of Alzheimer's disease and Alzheimer disease-like pathology in transgenic mice. Neurological Research. 2003;25(6):665–674. doi: 10.1179/016164103101201977. [DOI] [PubMed] [Google Scholar]
- 20.Gong Y, Chang L, Viola KL, et al. Alzheimer's disease-affected brain: presence of oligomeric Aβ ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(18):10417–10422. doi: 10.1073/pnas.1834302100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Castellani RJ, Lee HG, Zhu X, Perry G, Smith MA. Alzheimer disease pathology as a host response. Journal of Neuropathology and Experimental Neurology. 2008;67(6):523–531. doi: 10.1097/NEN.0b013e318177eaf4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Devi L, Prabhu BM, Galati DF, Avadhani NG, Anandatheerthavarada HK. Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer's disease brain is associated with mitochondrial dysfunction. Journal of Neuroscience. 2006;26(35):9057–9068. doi: 10.1523/JNEUROSCI.1469-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baloyannis SJ. Mitochondrial alterations in Alzheimer's disease. Journal of Alzheimer's Disease. 2006;9(2):119–126. doi: 10.3233/jad-2006-9204. [DOI] [PubMed] [Google Scholar]
- 24.Golde TE, Janus C. Homing in on intracellular Aβ? Neuron. 2005;45(5):639–642. doi: 10.1016/j.neuron.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 25.Walsh DM, Klyubin I, Fadeeva JV, et al. Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416(6880):535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 26.Lambert MP, Barlow AK, Chromy BA, et al. Diffusible, nonfibrillar ligands derived from Aβ are potent central nervous system neurotoxins. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(11):6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Atamna H. Amino acids variations in Amyloid-β peptides, mitochondrial dysfunction, and new therapies for Alzheimer's disease. Journal of Bioenergetics and Biomembranes. 2009;41(5):457–464. doi: 10.1007/s10863-009-9246-2. [DOI] [PubMed] [Google Scholar]
- 28.Su B, Wang X, Bonda D, Perry G, Smith M, Zhu X. Abnormal mitochondrial dynamics-a novel therapeutic target for Alzheimer's disease? Molecular Neurobiology. 2010;41(2-3):87–96. doi: 10.1007/s12035-009-8095-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Atamna H. Heme, iron, and the mitochondrial decay of ageing. Ageing Research Reviews. 2004;3(3):303–318. doi: 10.1016/j.arr.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 30.Atamna H, Killilea DW, Killilea AN, Ames BN. Heme deficiency may be a factor in the mitochondrial and neuronal decay of aging. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(23):14807–14812. doi: 10.1073/pnas.192585799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lustbader JW, Cirilli M, Lin C, et al. ABAD directly links Aβ to mitochondrial toxicity in Alzheimer's disease. Science. 2004;304(5669):448–452. doi: 10.1126/science.1091230. [DOI] [PubMed] [Google Scholar]
- 32.Yamaguchi H, Yamazaki T, Ishiguro K, Shoji M, Nakazato Y, Hirai S. Ultrastructural localization of Alzheimer amyloid β/A4 protein precursor in the cytoplasm of neurons and senile plaque-associated astrocytes. Acta Neuropathologica. 1992;85(1):15–22. doi: 10.1007/BF00304629. [DOI] [PubMed] [Google Scholar]
- 33.Crouch PJ, Blake R, Duce JA, et al. Copper-dependent inhibition of human cytochrome c oxidase by a dimeric conformer of amyloid-β. Journal of Neuroscience. 2005;25(3):672–679. doi: 10.1523/JNEUROSCI.4276-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cho DH, Nakamura T, Fang J, et al. β-amyloid-related mitochondrial fission and neuronal injury. Science. 2009;324(5923):102–105. doi: 10.1126/science.1171091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang X, Su B, Siedlak SL, et al. Amyloid-β overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(49):19318–19323. doi: 10.1073/pnas.0804871105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fu M, Zhu X, Zhang J, et al. Egr-1 target genes in human endothelial cells identified by microarray analysis. Gene. 2003;315(1-2):33–41. doi: 10.1016/s0378-1119(03)00730-3. [DOI] [PubMed] [Google Scholar]
- 37.Sohal RS. Aging, cytochrome oxidase activity, and hydrogen peroxide release by mitochondria. Free Radical Biology and Medicine. 1993;14(6):583–588. doi: 10.1016/0891-5849(93)90139-l. [DOI] [PubMed] [Google Scholar]
- 38.Boveris A, Chance B. The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochemical Journal. 1973;134(3):707–716. doi: 10.1042/bj1340707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Du H, Guo L, Fang F, et al. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer's disease. Nature Medicine. 2008;14(10):1097–1105. doi: 10.1038/nm.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lovell MA, Robertson JD, Teesdale WJ, Campbell JL, Markesbery WR. Copper, iron and zinc in Alzheimer's disease senile plaques. Journal of the Neurological Sciences. 1998;158(1):47–52. doi: 10.1016/s0022-510x(98)00092-6. [DOI] [PubMed] [Google Scholar]
- 41.Avdulov NA, Chochina SV, Igbavboa U, Warden CS, Vassiliev AV, Wood WG. Lipid binding to amyloid β-peptide aggregates: preferential binding of cholesterol as compared with phosphatidylcholine and fatty acids. Journal of Neurochemistry. 1997;69(4):1746–1752. doi: 10.1046/j.1471-4159.1997.69041746.x. [DOI] [PubMed] [Google Scholar]
- 42.Bartzokis G, Tishler TA, Lu PH, et al. Brain ferritin iron may influence age- and gender-related risks of neurodegeneration. Neurobiology of Aging. 2007;28(3):414–423. doi: 10.1016/j.neurobiolaging.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 43.Atamna H, Frey WH, Ko N. Human and rodent amyloid-β peptides differentially bind heme: relevance to the human susceptibility to Alzheimer's disease. Archives of Biochemistry and Biophysics. 2009;487(1):59–65. doi: 10.1016/j.abb.2009.05.003. [DOI] [PubMed] [Google Scholar]
- 44.Cullen KM, Kócsi Z, Stone J. Microvascular pathology in the aging human brain: evidence that senile plaques are sites of microhaemorrhages. Neurobiology of Aging. 2006;27(12):1786–1796. doi: 10.1016/j.neurobiolaging.2005.10.016. [DOI] [PubMed] [Google Scholar]
- 45.Gatta LB, Vitali M, Verardi R, Arosio P, Finazzi D. Inhibition of heme synthesis alters Amyloid Precursor Protein processing. Journal of Neural Transmission. 2009;116(1):79–88. doi: 10.1007/s00702-008-0147-z. [DOI] [PubMed] [Google Scholar]
- 46.Perry RT, Gearhart DA, Wiener HW, et al. Hemoglobin binding to Aβ and HBG2 SNP association suggest a role in Alzheimer's disease. Neurobiology of Aging. 2008;29(2):185–193. doi: 10.1016/j.neurobiolaging.2006.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kelley BJ, Knopman DS. Alternative medicine and Alzheimer disease. Neurologist. 2008;14(5):299–306. doi: 10.1097/NRL.0b013e318172cf4d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bellinger DC, Trachtenberg F, Barregard L, et al. Neuropsychological and renal effects of dental amalgam in children: a randomized clinical trial. Journal of the American Medical Association. 2006;295(15):1775–1783. doi: 10.1001/jama.295.15.1775. [DOI] [PubMed] [Google Scholar]
- 49.Beal MF. Energetics in the pathogenesis of neurodegenerative diseases. Trends in Neurosciences. 2000;23(7):298–304. doi: 10.1016/s0166-2236(00)01584-8. [DOI] [PubMed] [Google Scholar]
- 50.Beal MF. Oxidative damage as an early marker of Alzheimer's disease and mild cognitive impairment. Neurobiology of Aging. 2005;26(5):585–586. doi: 10.1016/j.neurobiolaging.2004.09.022. [DOI] [PubMed] [Google Scholar]
- 51.Butterfield DA. Amyloid β-peptide (1-42)-induced oxidative stress and neurotoxicity: implications for neurodegeneration in Alzheimer's disease brain. A review. Free Radical Research. 2002;36(12):1307–1313. doi: 10.1080/1071576021000049890. [DOI] [PubMed] [Google Scholar]
- 52.Hirai K, Aliev G, Nunomura A, et al. Mitochondrial abnormalities in Alzheimer's disease. Journal of Neuroscience. 2001;21(9):3017–3023. doi: 10.1523/JNEUROSCI.21-09-03017.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wagner G. Towards a life prolonging pill? Small molecules with anti-ageing properties. Current Drug Targets. 2006;7(11):1531–1537. doi: 10.2174/1389450110607011531. [DOI] [PubMed] [Google Scholar]
- 54.Miller RA, Harrison DE, Astle CM, et al. An aging interventions testing program: study design and interim report. Aging Cell. 2007;6(4):565–575. doi: 10.1111/j.1474-9726.2007.00311.x. [DOI] [PubMed] [Google Scholar]
- 55.Atamna H, Nguyen A, Schultz C, et al. Methylene blue delays cellular senescence and enhances key mitochondrial biochemical pathways. FASEB Journal. 2008;22(3):703–712. doi: 10.1096/fj.07-9610com. [DOI] [PubMed] [Google Scholar]
- 56.Atamna H, Gharib A. Methylene Blue Induces Mitochondrial Complex IV and Improves Cognitive Function and Grip Strength in Old Mice. Huntington, NY, USA: Nova Science; 2010. [Google Scholar]
- 57.Atamna H, Kumar R. Protective role of methylene blue in Alzheimer's disease via mitochondria and cytochrome c oxidase. Journal of Alzheimer's Disease. 2010;20(supplement 2):S439–S452. doi: 10.3233/JAD-2010-100414. [DOI] [PubMed] [Google Scholar]
- 58.Peter C, Hongwan D, Küpfer A, Lauterburg BH. Pharmacokinetics and organ distribution of intravenous and oral methylene blue. European Journal of Clinical Pharmacology. 2000;56(3):247–250. doi: 10.1007/s002280000124. [DOI] [PubMed] [Google Scholar]
- 59.Mellish KJ, Cox RD, Vernon DI, Griffiths J, Brown SB. In vitro photodynamic activity of a series of methylene blue analogues. Photochemistry and Photobiology. 2002;75(4):392–397. doi: 10.1562/0031-8655(2002)075<0392:ivpaoa>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 60.Shoubridge EA. Cytochrome c oxidase deficiency. American Journal of Medical Genetics. 2001;106:46–52. doi: 10.1002/ajmg.1378. [DOI] [PubMed] [Google Scholar]
- 61.Kushnareva Y, Murphy AN, Andreyev A. Complex I-mediated reactive oxygen species generation: modulation by cytochrome c and NAD(P)+ oxidation-reduction state. Biochemical Journal. 2002;368(2):545–553. doi: 10.1042/BJ20021121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Barros MH, Bandy B, Tahara EB, Kowaltowski AJ. Higher respiratory activity decreases mitochondrial reactive oxygen release and increases life span in Saccharomyces cerevisiae. Journal of Biological Chemistry. 2004;279(48):49883–49888. doi: 10.1074/jbc.M408918200. [DOI] [PubMed] [Google Scholar]
- 63.Moura JCVP, Cordeiro N. 3,7-bis(dialkylamino)phenothiazin-5-ium derivatives: biomedical applications and biological activity. Current Drug Targets. 2003;4(2):133–141. doi: 10.2174/1389450033346902. [DOI] [PubMed] [Google Scholar]
- 64.Rengelshausen J, Burhenne J, Fröhlich M, et al. Pharmacokinetic interaction of chloroquine and methylene blue combination against malaria. European Journal of Clinical Pharmacology. 2004;60(10):709–715. doi: 10.1007/s00228-004-0818-0. [DOI] [PubMed] [Google Scholar]
- 65.Patel PN. Methylene blue for management of ifosfamide-induced encephalopathy. Annals of Pharmacotherapy. 2006;40(2):299–303. doi: 10.1345/aph.1G114. [DOI] [PubMed] [Google Scholar]
- 66.Betten DP, Vohra RB, Cook MD, Matteucci MJ, Clark RF. Antidote use in the critically ill poisoned patient. Journal of Intensive Care Medicine. 2006;21(5):255–277. doi: 10.1177/0885066606290386. [DOI] [PubMed] [Google Scholar]
- 67.Naylor GJ, Martin B, Hopwood SE, Watson Y. A two-year double-blind crossover trial of the prophylactic effect of methylene blue in manic-depressive psychosis. Biological Psychiatry. 1986;21(10):915–920. doi: 10.1016/0006-3223(86)90265-9. [DOI] [PubMed] [Google Scholar]
- 68.De-Oliveira RW, Guimarães FS. Anxiolytic effect of methylene blue microinjected into the dorsal periaqueductal gray matter. Brazilian Journal of Medical and Biological Research. 1999;32(12):1529–1532. doi: 10.1590/s0100-879x1999001200012. [DOI] [PubMed] [Google Scholar]
- 69.Rezzani R, Rodella L, Corsetti G, Bianchi R. Does methylene blue protect the kidney tissues from damage induced by ciclosporin A treatment? Nephron. 2001;89(3):329–336. doi: 10.1159/000046094. [DOI] [PubMed] [Google Scholar]
- 70.Haluzik M, Nedvídková J, Škrha J. Treatment with the NO-synthase inhibitor, methylene blue, moderates the decrease in serum leptin concentration in streptozotocin-induced diabetes. Endocrine Research. 1999;25(2):163–171. doi: 10.1080/07435809909066138. [DOI] [PubMed] [Google Scholar]
- 71.Salaris SC, Babbs CF, Voorhees WD. Methylene blue as an inhibitor of superoxide generation by xanthine oxidase. A potential new drug for the attenuation of ischemia/reperfusion injury. Biochemical Pharmacology. 1991;42(3):499–506. doi: 10.1016/0006-2952(91)90311-r. [DOI] [PubMed] [Google Scholar]
- 72.Teicher BA, Herman TS, Kaufmann ME. Cytotoxicity, radiosensitization, and DNA interaction of platinum complexes of thiazin and xanthene dyes. Radiation Research. 1990;121(2):187–195. [PubMed] [Google Scholar]
- 73.Visarius TM, Stucki JW, Lauterburg BH. Inhibition and stimulation of long-chain fatty acid oxidation by chloroacetaldehyde and methylene blue in rats. Journal of Pharmacology and Experimental Therapeutics. 1999;289(2):820–824. [PubMed] [Google Scholar]
- 74.Clifton J, II, Leikin JB. Methylene blue. American Journal of Therapeutics. 2003;10(4):289–291. doi: 10.1097/00045391-200307000-00009. [DOI] [PubMed] [Google Scholar]
- 75.Sills MR, Zinkham WH. Methylene blue-induced Heinz body hemolytic anemia. Archives of Pediatrics and Adolescent Medicine. 1994;148(3):306–310. doi: 10.1001/archpedi.1994.02170030076017. [DOI] [PubMed] [Google Scholar]
- 76.Martinez JL, Jr., Jensen RA, Vasquez BJ, McGuinness T, McGaugh JL. Methylene blue alters retention of inhibitory avoidance responses. Physiological Psychology. 1978;6(3):387–390. [Google Scholar]
- 77.Callaway NL, Riha PD, Bruchey AK, Munshi Z, Gonzalez-Lima F. Methylene blue improves brain oxidative metabolism and memory retention in rats. Pharmacology Biochemistry and Behavior. 2004;77(1):175–181. doi: 10.1016/j.pbb.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 78.Ramsay RR, Dunford C, Gillman PK. Methylene blue and serotonin toxicity: inhibition of monoamine oxidase A (MAO A) confirms a theoretical prediction. British Journal of Pharmacology. 2007;152(6):946–951. doi: 10.1038/sj.bjp.0707430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dierks EA, Burstyn JN. The deactivation of soluble guanylyl cyclase by redox-active agents. Archives of Biochemistry and Biophysics. 1998;351(1):1–7. doi: 10.1006/abbi.1997.0408. [DOI] [PubMed] [Google Scholar]
- 80.Mayer B, Brunner F, Schmidt K. Inhibition of nitric oxide synthesis by methylene blue. Biochemical Pharmacology. 1993;45(2):367–374. doi: 10.1016/0006-2952(93)90072-5. [DOI] [PubMed] [Google Scholar]
- 81.Meissner PE, Mandi G, Witte S, et al. Safety of the methylene blue plus chloroquine combination in the treatment of uncomplicated falciparum malaria in young children of Burkina Faso [ISRCTN27290841] Malaria Journal. 2005;4 doi: 10.1186/1475-2875-4-45. Article ID 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kelner MJ, Bagnell R, Hale B, Alexander NM. Methylene blue competes with paraquat for reduction by flavo-enzymes resulting in decreased superoxide production in the presence of heme proteins. Archives of Biochemistry and Biophysics. 1988;262(2):422–426. doi: 10.1016/0003-9861(88)90393-1. [DOI] [PubMed] [Google Scholar]
- 83.Zacharakis N, Tone P, Flordellis CS, Maragoudakis ME, Tsopanoglou NE. Methylene blue inhibits angiogenesis in chick chorioallontoic membrane through a nitric oxide-independent mechanism. Journal of Cellular and Molecular Medicine. 2006;10(2):493–498. doi: 10.1111/j.1582-4934.2006.tb00414.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Medina DX, Caccamo A, Oddo S. Methylene blue reduces Aβ levels and rescues early cognitive deficit by increasing proteasome activity. Brain Pathology. 2011;21(2):140–149. doi: 10.1111/j.1750-3639.2010.00430.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Carr HS, Winge DR. Assembly of cytochrome c oxidase within the mitochondrion. Accounts of Chemical Research. 2003;36(5):309–316. doi: 10.1021/ar0200807. [DOI] [PubMed] [Google Scholar]
- 86.Luo GR, Le WD. Collective roles of molecular chaperones in protein degradation pathways associated with neurodegenerative diseases. Current Pharmaceutical Biotechnology. 2010;11(2):180–187. doi: 10.2174/138920110790909740. [DOI] [PubMed] [Google Scholar]
- 87.Hochachka PW, Somero GN. Biochemical Adaptation. Mechanism and Process in Physiological Evolution. Oxford, UK: Oxford University Press; 2002. [Google Scholar]
- 88.Bolen DW, Baskakov IV. The osmophobic effect: natural selection of a thermodynamic force in protein folding. Journal of Molecular Biology. 2001;310(5):955–963. doi: 10.1006/jmbi.2001.4819. [DOI] [PubMed] [Google Scholar]
- 89.Baldwin RL, Rose GD. Is protein folding hierarchic? I. Local structure and peptide folding. Trends in Biochemical Sciences. 1999;24(1):26–33. doi: 10.1016/s0968-0004(98)01346-2. [DOI] [PubMed] [Google Scholar]
- 90.Burg MB. Molecular basis of osmotic regulation. American Journal of Physiology. 1995;268(6):F983–F996. doi: 10.1152/ajprenal.1995.268.6.F983. [DOI] [PubMed] [Google Scholar]
- 91.Bai C, Biwersi J, Verkman AS, Matthay MA. A mouse model to test the in vivo efficacy of chemical chaperones. Journal of Pharmacological and Toxicological Methods. 1998;40(1):39–45. doi: 10.1016/s1056-8719(98)00034-3. [DOI] [PubMed] [Google Scholar]
- 92.Pace CN. The stability of globular proteins. CRC Critical Reviews in Biochemistry. 1975;3(1):1–43. doi: 10.3109/10409237509102551. [DOI] [PubMed] [Google Scholar]
- 93.Yancey PH, Clark ME, Hand SC, Bowlus RD, Somero GN. Living with water stress: evolution of osmolyte systems. Science. 1982;217(4566):1214–1222. doi: 10.1126/science.7112124. [DOI] [PubMed] [Google Scholar]
- 94.Atamna H, Ginsburg H. Heme degradation in the presence of glutathione. A proposed mechanism to account for the high levels of non-heme iron found in the membranes of hemoglobinopathic red blood cells. Journal of Biological Chemistry. 1995;270(42):24876–24883. doi: 10.1074/jbc.270.42.24876. [DOI] [PubMed] [Google Scholar]
- 95.Mello CC, Barrick D. Measuring the stability of partly folded proteins using TMAO. Protein Science. 2003;12(7):1522–1529. doi: 10.1110/ps.0372903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yancey PH. Organic osmolytes as compatible, metabolic and counteracting cytoprotectants in high osmolarity and other stresses. Journal of Experimental Biology. 2005;208(15):2819–2830. doi: 10.1242/jeb.01730. [DOI] [PubMed] [Google Scholar]
- 97.Baskakov I, Bolen DW. Forcing thermodynamically unfolded proteins to fold. Journal of Biological Chemistry. 1998;273(9):4831–4834. doi: 10.1074/jbc.273.9.4831. [DOI] [PubMed] [Google Scholar]
- 98.Quinones QJ, de Ridder GG, Pizzo SV. GRP78: a chaperone with diverse roles beyond the endoplasmic reticulum. Histology and Histopathology. 2008;23(11):1409–1416. doi: 10.14670/HH-23.1409. [DOI] [PubMed] [Google Scholar]
- 99.Kelly JW. The alternative conformations of amyloidogenic proteins and their multi-step assembly pathways. Current Opinion in Structural Biology. 1998;8(1):101–106. doi: 10.1016/s0959-440x(98)80016-x. [DOI] [PubMed] [Google Scholar]
- 100.Lansbury PT., Jr. Evolution of amyloid: what normal protein folding may tell us about fibrillogenesis and disease. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(7):3342–3344. doi: 10.1073/pnas.96.7.3342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Davies SW, Turmaine M, Cozens BA, et al. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;90(3):537–548. doi: 10.1016/s0092-8674(00)80513-9. [DOI] [PubMed] [Google Scholar]
- 102.Warrick JM, Paulson HL, Gray-Board GL, et al. Expanded polyglutamine protein forms nuclear inclusions and causes neural degeneration in Drosophila. Cell. 1998;93(6):939–949. doi: 10.1016/s0092-8674(00)81200-3. [DOI] [PubMed] [Google Scholar]
- 103.Klement IA, Skinner PJ, Kaytor MD, et al. Ataxin-1 nuclear localization and aggregation: role in polyglutarnine-induced disease in SCA1 transgenic mice. Cell. 1998;95(1):41–53. doi: 10.1016/s0092-8674(00)81781-x. [DOI] [PubMed] [Google Scholar]
- 104.Strong TV, Tagle DA, Valdes JM, et al. Widespread expression of the human and rat Huntington's disease gene in brain and nonneural tissues. Nature Genetics. 1993;5(3):259–265. doi: 10.1038/ng1193-259. [DOI] [PubMed] [Google Scholar]
- 105.Kennedy WR, Alter M, Sung JH. Progressive proximal spinal and bulbar muscular atrophy of late onset. A sex-linked recessive trait. Neurology. 1968;18(7):671–680. doi: 10.1212/wnl.18.7.671. [DOI] [PubMed] [Google Scholar]
- 106.La Spada AR, Wilson EM, Lubahn DB, Harding AE, Fischbeck KH. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature. 1991;352(6330):77–79. doi: 10.1038/352077a0. [DOI] [PubMed] [Google Scholar]
- 107.The Huntington's Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993;72(6):971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- 108.DiFiglia M, Sapp E, Chase KO, et al. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277(5334):1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- 109.Kumar R. Role of naturally occurring osmolytes in protein folding and stability. Archives of Biochemistry and Biophysics. 2009;491(1-2):1–6. doi: 10.1016/j.abb.2009.09.007. [DOI] [PubMed] [Google Scholar]
- 110.Kempf B, Bremer E. Uptake and synthesis of compatible solutes as microbial stress responses to high-osmolality environments. Archives of Microbiology. 1998;170(5):319–330. doi: 10.1007/s002030050649. [DOI] [PubMed] [Google Scholar]
- 111.Lin TY, Timasheff SN. Why do some organisms use a urea-methylamine mixture as osmolyte? Thermodynamic compensation of urea and trimethylamine N-oxide interactions with protein. Biochemistry. 1994;33(42):12695–12701. doi: 10.1021/bi00208a021. [DOI] [PubMed] [Google Scholar]
- 112.Singh R, Haque I, Ahmad F. Counteracting osmolyte trimethylamine N-oxide destabilizes proteins at pH below its pK: measurements of thermodynamic parameters of proteins in the presence and absence of trimethylamine N-oxide. Journal of Biological Chemistry. 2005;280(12):11035–11042. doi: 10.1074/jbc.M410716200. [DOI] [PubMed] [Google Scholar]
- 113.Anjum F, Rishi V, Ahmad F. Compatibility of osmolytes with Gibbs energy of stabilization of proteins. Biochimica et Biophysica Acta. 2000;1476(1):75–84. doi: 10.1016/s0167-4838(99)00215-0. [DOI] [PubMed] [Google Scholar]
- 114.Tanaka M, Machida Y, Niu S, et al. Trehalose alleviates polyglutamine-mediated pathology in a mouse model of Huntington disease. Nature Medicine. 2004;10(2):148–154. doi: 10.1038/nm985. [DOI] [PubMed] [Google Scholar]
- 115.Davies P, Watt K, Kelly SM, Clark C, Price NC, McEwan IJ. Consequences of poly-glutamine repeat length for the conformation and folding of the androgen receptor amino-terminal domain. Journal of Molecular Endocrinology. 2008;41(5-6):301–314. doi: 10.1677/JME-08-0042. [DOI] [PubMed] [Google Scholar]
- 116.Duff J, Davies P, Watt K, McEwan IJ. Structural dynamics of the human androgen receptor: implications for prostate cancer and neurodegenerative disease. Biochemical Society Transactions. 2006;34(6):1098–1102. doi: 10.1042/BST0341098. [DOI] [PubMed] [Google Scholar]
- 117.Irvine RA, Ma H, Yu MC, Ross RK, Stallcup MR, Coetzee GA. Inhibition of p160-mediated coactivation with increasing androgen receptor polyglutamine length. Human Molecular Genetics. 2000;9(2):267–274. doi: 10.1093/hmg/9.2.267. [DOI] [PubMed] [Google Scholar]
- 118.Becker EM, Greer JM, Ponka P, Richardson DR. Erythroid differentiation and protoporphyrin IX down-regulate frataxin expression in Friend cells: characterization of frataxin expression compared to molecules involved in iron metabolism and hemoglobinization. Blood. 2002;99(10):3813–3822. doi: 10.1182/blood.v99.10.3813. [DOI] [PubMed] [Google Scholar]
- 119.Walsh DM, Klyubin I, Shankar GM, et al. The role of cell-derived oligomers of Aβ in Alzheimer's disease and avenues for therapeutic intervention. Biochemical Society Transactions. 2005;33(5):1087–1090. doi: 10.1042/BST20051087. [DOI] [PubMed] [Google Scholar]
- 120.Singer MA, Lindquist S. Multiple effects of trehalose on protein folding in vitro and in vivo. Molecular Cell. 1998;1(5):639–648. doi: 10.1016/s1097-2765(00)80064-7. [DOI] [PubMed] [Google Scholar]
- 121.Liu R, Barkhordarian H, Emadi S, Chan BP, Sierks MR. Trehalose differentially inhibits aggregation and neurotoxicity of beta-amyloid 40 and 42. Neurobiology of Disease. 2005;20(1):74–81. doi: 10.1016/j.nbd.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 122.Reddy VP, Garrett MR, Perry G, Smith MA. Carnosine: a versatile antioxidant and antiglycating agent. Science of Aging Knowledge Environment. 2005;2005(18):p. pe12. doi: 10.1126/sageke.2005.18.pe12. [DOI] [PubMed] [Google Scholar]
- 123.Mozdzan M, Szemraj J, Rysz J, Nowak D. Antioxidant properties of carnosine re-evaluated with oxidizing systems involving iron and copper ions. Basic and Clinical Pharmacology and Toxicology. 2005;96(5):352–360. doi: 10.1111/j.1742-7843.2005.pto_03.x. [DOI] [PubMed] [Google Scholar]
- 124.Fonteh AN, Harrington RJ, Tsai A, Liao P, Harrington MG. Free amino acid and dipeptide changes in the body fluids from Alzheimer's disease subjects. Amino Acids. 2007;32(2):213–224. doi: 10.1007/s00726-006-0409-8. [DOI] [PubMed] [Google Scholar]
- 125.Qi W, Zhang A, Good TA, Fernandez EJ. Two disaccharides and trimethylamine N-oxide affect Aβ aggregation differently, but all attenuate oligomer-induced membrane permeability. Biochemistry. 2009;48(37):8908–8919. doi: 10.1021/bi9006397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Yang DS, Yip CM, Huang THJ, Chakrabartty A, Fraser PE. Manipulating the amyloid-β aggregation pathway with chemical chaperones. Journal of Biological Chemistry. 1999;274(46):32970–32974. doi: 10.1074/jbc.274.46.32970. [DOI] [PubMed] [Google Scholar]