

Abstract
Pathological as well as physiological angiogenesis is known to be regulated by such factors as nucleotides and Vascular Endothelial Growth Factor (VEGF). Activated P2Y nucleotide receptors have been observed to associate and transactivate VEGF Receptor 2 (VEGFR2), suggesting a cooperation between nucleotide and VEGF signaling in angiogenesis. P2YR mediated VEGFR2 signaling therefore may be important in describing the angiogenic signaling of nucleotides such as ATP. Here, we provide evidence that supports the notion of P2YR-VEGFR2 signaling. The significant angiogenic effect of P2Y1/2 receptor agonists (100 μM ATP and 10 μM 2MS-ATP) on endothelial cell tubulogenesis was suppressed back to near control levels upon addition of 1 μM SU1498 (specific VEGFR2 tyrosine kinase inhibitor). We believe that this P2YR-VEFGR2 signaling is an important component of pathological, as well as physiological angiogenesis.
INTRODUCTION
The secretion of nucleoside diphosphate kinase (NDPK) orthologues by intracellular parasites [1;2], NDPK secretion by various carcinomas [3;4], and NDPK’s role in blood flow regulation [5] lead us to first propose a pathological role for secreted NDPK in cancer and tumor angiogenesis. We recently provided evidence for a purinergic regulation of angiogenesis by cancer secreted NDPK [6] which supports this hypothesis. Activated P2Y receptors have been observed to transactivate Vascular Endothelial Growth Factor Receptor 2 (VEGFR2), directly linking extracellular nucleotide regulation to established tumor angiogenesis signaling [7]. P2YR activation and subsequent VEGFR2 signaling therefore may be important in describing and delineating the angiogenic signaling of nucleotides such as ATP. Given this evidence, we hypothesize that P2YR activation stimulates angiogenesis via VEGFR2 signaling and human breast cancer NDPK exploits this to induce pathological angiogenesis.
Anti-vascular growth factor (VEGF) antibody bevacizumab (Avastin®) is currently approved for first-line treatment of both metastatic colorectal and non-squamous, non-small cell lung carcinomas. Its participation in more than 300 current clinical trials for the treatment of diverse cancers such as breast, prostate, ovarian, renal, and pancreatic further emphasizes the importance of VEGF signaling in tumor angiogenesis [8]. VEGFR2, the major mediator of angiogenic and permeability enhancing effects of VEGF [9], has been shown to compartmentalized to caveolar domains on the surface of endothelial cells [10;11] along with P2Y receptors [12]. This close proximity along with the observed interaction between P2Y receptors and VEGFR2 further supports the notion of cooperation in angiogenic signaling. Here, we provide evidence that P2YR signaling utilizes VEGFR2 intracellular signaling to induce endothelial tubulogenesis in vitro. This following data suggests that P2YR activation may be important in describing VEFGR2 signaling in pathological, as well as physiological angiogenesis.
MATERIALS AND METHODS
Cell Culture
Human cardiac ECs were previously isolated by Fluorescence Activated Cell Sorting (FACS) for CD31 (PECAM) and immortalized by human telomerase reverse transcriptase (hTRT) – referred to as CD31+ cells. Unless specifically stated, cells were maintained and incubated in Dulbecco’s Modified Eagle Medium (DMEM) (HyClone, Logan, UT) supplemented with 10% Fetal Bovine Serum (FBS) (Atlanta Biological, Lawrenceville, GA), Penicillin-Streptomycin, (1500 U/L-100 mg/L) and 0.5 mg/L Fungizone (Invitrogen, Carlsbad, CA) at 37°C in a humidified 5% CO2/95% air atmosphere.
In Vitro Angiogenesis Scoring Technique
CD31+ cell tubulogenesis and subsequent angiogenesis scores were determined from these images as previously described (6). Briefly, the number of branch points (bp), length (l), and cell surface area (a) were multiplied to produce a relative angiogenesis score (s) for comparison between groups.
Effect of Disrupting VEGFR2 Signaling on P2YR Mediated Angiogenesis
To determine if inhibiting VEGFR2 intracellular signaling suppresses P2Y1/2 receptor mediated in vitro angiogenesis, 3 × 104 CD31+ cells per well were first seeded onto 24-well tissue culture plates coated with 1 mg/mL collagen (Rat type I; BD Biosciences) and allowed to attach for 40 min. The P2Y receptor agonists 2MS-ATP (P2Y1R; 10 μM; Sigma, St. Louis, MO) and ATP (P2Y1/2R; 100 μM; Sigma) were added to their respective wells and incubated with CD31+ cells for 24 hr. CD31+ cell tubulogenesis was also observed in the presence of VEGFR2 tyrosine kinase inhibitor SU1498 (1 μM; Sigma) with either 10 μM 2MS-ATP or 100 μM ATP. Endothelial growth medium-2 (EGM-2™; Clonetics, East Rutherford, NJ) was used as a positive control to confirm that this modified assay could successfully detect angiogenic stimulation. Non-treatment controls were performed for normalization and comparison. The following experiments were performed with 2% FBS supplementation. SU1498 was better soluble in DMSO (Sigma), thus all experimental groups were matched with 0.01% DMSO.
Statistical Analyses
All graphs were prepared using Prism Graphing Software (V5.01; GraphPad Software, San Diego, CA) and statistical analyses were performed using InStat Statistical Software (V3.06; GraphPad Software), with P < 0.05 considered to be statistically significant. Angiogenesis scores were tested for statistical significance using ANOVA and Kruskal-Wallis multiple comparisons post-test. Data points and error bars represent means ± SEM.*, P < 0.05; ***, P < 0.001 (vs. negative control).
RESULTS AND DISCUSSION
Disrupting VEGFR2 Signaling Suppresses P2YR Mediated Angiogenesis
CD31+ cells incubated for 24 hr with 10 μM 2MS-ATP (P2Y1R agonist) or 100 μM ATP (P2Y1/2R agonist) indicate a similar and significant induction of in vitro angiogenesis, respectively ~1.9 and ~1.5 fold above control levels (P ≤ 0.05; Fig 1). The addition of 1 μM SU1498 (specific VEGFR2 kinase inhibitor) to either 10 μM 2MS-ATP or 100 μM ATP stimulations reduced tubulogenesis back to near control levels (Fig 1). The angiogenic stimulation control EGM-2™ (containing VEGF) produced significant angiogenesis over the 24 hr duration, ~2.4 fold above control levels (P ≤ 0.001; Fig 1), with no detectable inhibition upon addition of 1 μM SU1498. No effect on tubulogenesis was observed with SU1498 (1 μM) alone (data not shown).
Figure 1. P2Y Receptor Mediated Angiogenesis Utilizes VEGFR2 Signaling.

Inhibition of VEGFR2 intracellular signaling by SU1498 (1 μM) suppressed the pro-angiogenic potential of P2Y1/2 receptor agonists (ATP and 2MS-ATP) during a 24 h tubulogenesis assay. Control mean = 979.4 ± 403.6 angiogenesis units. Control consisted of CD31+ cells incubated in CDMEM supplemented with 2% FBS and 0.01% DMSO. The angiogenic stimulation control used was endothelial growth media-2 (EGM-2) containing VEGF.
Our results implicate VEGFR2 signaling in our previously observed purinergic regulation of angiogenesis by cancer secreted NDPK [6], providing a direct link to well established VEGF signaling. The observed P2Y1R mediated tubulogenesis is consistent with our previously reported data; less pronounced effects were due to the addition of DMSO for the solubility of SU1498. The inhibition of ATP and 2MS-ATP mediated in vitro angiogenesis by SU1498 at 1 μM (approximate VEGFR2kinase IC50) suggests that VEGFR2 intracellular signaling is a substantial component of P2YR mediated tubulogenesis. The unaffected angiogenic potential of EGM-2™ (includes many angiogenic factors) by SU1498 is consistent with the presence of multiple angiogenic pathways that can compensate for impaired VEGF signaling.
We now further expand our original hypothesis to include VEGFR2 signaling as a significant contributor to P2YR induced angiogenesis, which cancer cells exploit to their pathological advantage (Fig 2). Interestingly, this P2YR activation of VEGFR2 intracellular signaling may in part explain constitutive VEGFR2 signaling in the absence of exogenous VEGF [13]. We therefore also propose that P2YR-VEGFR2 signaling may be important in describing and understanding the VEGF signaling required for vascular homeostasis in both tumor as well as normal vasculature.
Figure 2. Putative Role of NDPK-B and P2Y Receptor Activation in Tumor Angiogenesis.
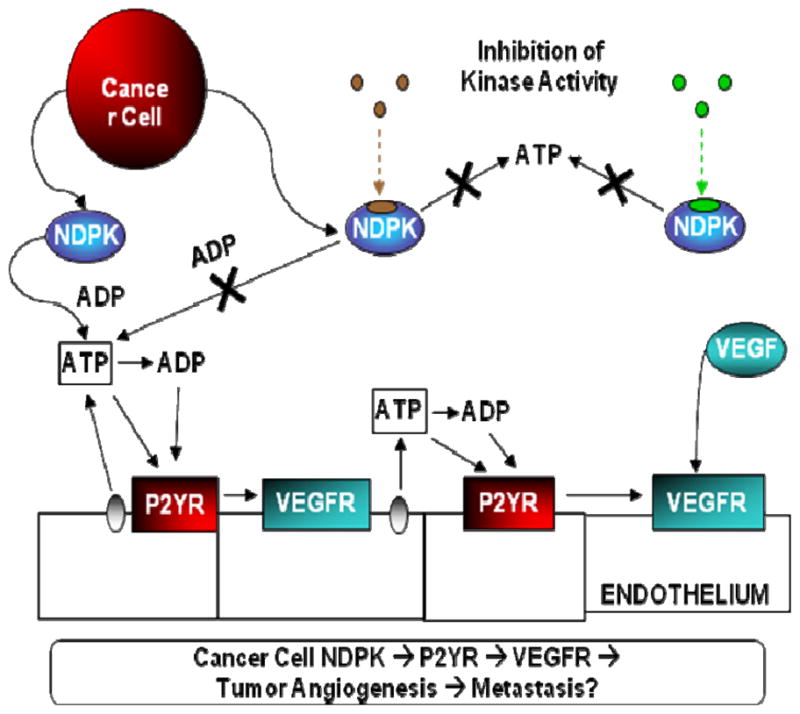
We have observed breast cancer-secreted NDPK-B to be a significant contributor to promoting angiogenesis (ref). Extracellular NDPK would modulate nucleotides such as elevating ATP levels. This scenario would subsequently lead to P2Y purinergic receptor activation above an unknown threshold to produce conditions favorable to pathological angiogenesis. Our current data suggests that this P2Y angiogenic signaling promotes tumor angiogenesis in concert with established VEGF angiogenic signal.
Acknowledgments
This work was supported by NIH T32 CA09563 to SR and a grant from the Clayton Foundation for Research to ILOB.
References
- 1.Chopra P, Singh A, Koul A, Ramachandran S, Drlica K, Tyagi AK, et al. Eur J Biochem. 2003 Feb;270(4):625–34. doi: 10.1046/j.1432-1033.2003.03402.x. [DOI] [PubMed] [Google Scholar]
- 2.Gounaris K, Thomas S, Najarro P, Selkirk ME. Infect Immun. 2001 Jun;69(6):3658–62. doi: 10.1128/IAI.69.6.3658-3662.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anzinger J, Malmquist NA, Gould J, Buxton IL. Proc West Pharmacol Soc. 2001;44:61–3. [PubMed] [Google Scholar]
- 4.Okabe-Kado J, Kasukabe T. J Bioenerg Biomembr. 2003 Feb;35(1):89–93. doi: 10.1023/a:1023402125186. [DOI] [PubMed] [Google Scholar]
- 5.Buxton IL, Kaiser RA, Oxhorn BC, Cheek DJ. Am J Physiol Heart Circ Physiol. 2001 Oct;281(4):H1657–H1666. doi: 10.1152/ajpheart.2001.281.4.H1657. [DOI] [PubMed] [Google Scholar]
- 6.Rumjahn SM, Javed MA, Wong N, Law WE, Buxton IL. Br J Cancer. 2007 Oct 16; doi: 10.1038/sj.bjc.6604019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seye CI, Yu N, Gonzalez FA, Erb L, Weisman GA. J Biol Chem. 2004 Aug 20;279(34):35679–86. doi: 10.1074/jbc.M401799200. [DOI] [PubMed] [Google Scholar]
- 8.Clinical Trials. Current Avastin Clinical Trials. 2007 http://clinicaltrials.gov/
- 9.Shibuya M. J Biochem Mol Biol. 2006 Sep 30;39(5):469–78. doi: 10.5483/bmbrep.2006.39.5.469. [DOI] [PubMed] [Google Scholar]
- 10.Cho CH, Lee CS, Chang M, Jang IH, Kim SJ, Hwang I, et al. Am J Physiol Heart Circ Physiol. 2004 May;286(5):H1881–H1888. doi: 10.1152/ajpheart.00786.2003. [DOI] [PubMed] [Google Scholar]
- 11.Labrecque L, Royal I, Surprenant DS, Patterson C, Gingras D, Beliveau R. Mol Biol Cell. 2003 Jan;14(1):334–47. doi: 10.1091/mbc.E02-07-0379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaiser RA, Oxhorn BC, Andrews G, Buxton IL. Circ Res. 2002 Aug 23;91(4):292–9. doi: 10.1161/01.res.0000030711.21521.ac. [DOI] [PubMed] [Google Scholar]
- 13.Lee S, Chen TT, Barber CL, Jordan MC, Murdock J, Desai S, et al. Cell. 2007 Aug 24;130(4):691–703. doi: 10.1016/j.cell.2007.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]