

Abstract
MicroRNAs (miRNAs) are small non-coding RNAs that inhibit expression of specific target genes at the post-transcriptional level. Sequence variations in miRNA genes, including pri-miRNAs, pre-miRNAs and mature miRNAs, could potentially influence the processing and/or target selection of miRNAs. In this study, we have found the single nucleotide polymorphism (SNP) at the twenty-fourth nucleotide (+24) of the mature miR-126 in the genome of RS4;11 cells, derived from a MLL-AF4 ALL patient. Through a series of in vivo analyzes, we found that this miR-126 SNP significantly blocks the processing of pri-miRNA to mature miRNA, as well as reduces miRNA-mediated translational suppression. Moreover, its frequency is different among races. Thus, our study emphasizes the importance of identifying new miRNA SNP and its contribution to miRNA biogenesis which is possible link to human genetic disease.
Keywords: miRNA, SNP, Drosha, Processing
1. Introduction
MicroRNAs (miRNAs) are 18–25 nt, single-stranded non-coding RNAs that are generated from primary mature through pre-miRNAs. miRNAs can suppress post-transcriptional gene expression by base pairing with their target messenger RNAs (mRNAs) and inducing either translational repression or mRNA degradation [1-4]. miRNAs regulate a wide range of biological processes in animal development and human disease. They have been implicated in the regulation development, growth control, apoptosis and stem cell maintenance [5]. A strong linkage between miRNAs and cancer has been demonstrated. miRNA expression profiling has also been shown to both faithfully reflect developmental lineages and classify disease states [6]. Thereafter, regulation of miRNA expression is studied extensively. One of these regulations is that of the miR-NA biogenesis. miRNAs are initially transcribed as long primary transcripts (pri-miRNAs), which are then processed into ~65 nt hairpin-shaped precursor miRNAs (pre-miRNA) by ‘Microprocessor’ complex. This nuclear complex consists of a member of the ribonuclease III family (RNaseIII), Drosha, and its cofactor, DGCR8[7,8]. The pri-miRNA processing is a critical step in miRNA biogenesis, because it defines the miRNA sequences embedded in long pri-miRNAs by generating one end of the molecule [7]. A typical pri-miRNA consists of a stem of ~33 bp, with a terminal loop and flanking segments. The terminal loop is inessential, whereas the flanking ssRNA segments are critical for processing. The cleavage site is determined chiefly by the distance (~11 bp) from the stem-ssRNA junction [9]. After processing by ‘Microprocessor’, pre-miRNAs are subsequently exported to cytoplasm by exportin-5 and cleaved by another RNaseIII, Dicer, to generate 18–22 nt mature miRNAs .
Single nucleotide polymorphism (SNP) are the most abundant form of DNA variation in the human genome and contribute to human phenotypic differences. Polymorphisms in miRNA genes could potentially alter various biological processes by influencing the processing and/or target selection of miRNAs [10-12]. In this study, we have identified SNP in pri-miR-126, which drastically alters its processing from pri-miRNA to pre-miRNA. Finally, we searched the frequency of the SNP in two population groups. Even though the sample size is small, the result implies that the frequency of the SNP is different among races. Our study revealed the SNP on pri-miR-126 is involved in pri-miRNA processing and underscores the need for continued exploration of other new miR-NA SNPs that may contribute to miRNA biogenesis and human disease.
2. Materials and methods
2.1. Cell culture and retroviral transfer
RS4;11 and SEM cell lines carrying the chromosomal translocation t(4;11)(q21;q23) were obtained from ATCC and DSMZ, respectively. The cells were maintained in RPMI 1640 containing 10% FCS. High-titer retroviral supernatant was created after the co-transfection of a miRNA expression vector and the pCLamph viral packaging construct into 293T cells using FuGENE 6 (Roche). SEM cells were plated at 4 × 105/mL to 5 × 105/mL in a 24-well plate and spin-infected with the desired retroviral supernatant for 2 h.
2.2. Constructs
Genomic DNA from RS4;11 was extracted using DNeasy@ Tissue extraction kit (QIAGEN). The genomic sequences of the segments in pri-miR-126 were amplified by genomic PCR using Pfx polymerase (Invitrogen). The oligonucleotide sequences used for the PCR were F;AATTATATCTCGAGGAGGGAGGATAGGTGGGTTC, R;GCTCGAATTCCAGAGGTCTCAGGGCTATGC. The reaction parameters were 35 cycles of 94 °C for 30 s, 55 °C for 30 s, and 72 °C for 1 min and 30 s. PCR products were purified using a PCR purification kit (Qiagen) and sequenced. The purified genomic PCR product was cloned into the Xho1-EcoR1 site of a mammalian retroviral expression vector, pXZ201 [25], and the constructed vectors were sequenced to identify the insertion of either the major or the minor allele of miR-126.
Segments in 3′UTR of the human IRS1 were amplified by genomic PCR and cloned in between the Xho1-Not1 sites of psicheck-2 (Promega). IRS1-UTR contains one potential miR-126 binding site. The site is located at nucleotides 117–137, of the 3′UTR of IRS1mRNA (NM_005544). The oligonucleotide sequences used for the PCR are F; TAAATAGCGGCCGCTGACCTCAGCAAATCCTCTTC, R; TTATATATCTCGAGCTCTCTCCACCCAACGTGA. The PCR condition was 35 cycles of 94 °C for 30 s, 55 °C for 30 s, and 72 °C for 1 min and 30 s.
2.3. Quantitative PCR
Total RNA was isolated with Trizol (Invitrogen) and used to synthesize cDNA with the superscript II cDNA synthesis kit (Invitrogen)by use of random primers or the pri-miR-126 specific primer 5′-GCCAGCGGAG-3′. The expression levels of pri-miR-126 by quantitative PCR by using SYBR green master mix (ABI). The oligonucleotide sequences used for the PCR were F; TATCAGCCAAGAAGGCAGAA, R; CGTGGCGTCTTCCAGAAT. The expressions of mature miR-126 were measured using a TaqMan® MicroRNA Assay (ABI). The PCR condition was as follows: 40 cycles of 95 °C for 15 s, and 60 °C for 1 min. All reactions were performed in triplicate.
2.4. Dual luciferase assay
In each well of a 96-well plate, 293T cells were cotransfected with 20 ng psiCHECK-2s (Promega) and 100 ng miRNA expression vectors. After 48 h of transfection, the relative amounts of Renilla and firefly luciferase were analyzed by a dual luciferase assay (Promega). The Renilla/firefly luciferase ratio was calculated and normalized against the control.
2.5. Statistical analysis
We used the Student t-tests to analyze the effect of the micro RNA overexpression by comparing the miRNA induced change to that of the respective controls. The test was used in dual luciferase assays, and quantitative PCR, and values of P < 0.05 were considered statistically significant. We used the Chi-square tests to analyze the difference in frequency of the alleles between two populations. All analyzes were performed with Microsoft Excel.
3. Results
3.1. Heterozygous polymorphism was identified in the pri-miR-126 in RS4;11 MLL-AF4 cells
Previously Lu et al. [6] reported that many miRNAs are down-regulated in MLL-AF4 ALL samples, compared with other types of ALLs, in which miR-126 is among them. In trying to understand why the level of miR-126 might be reduced in MLL-AF4 ALL patients, we cloned and sequenced the miR-126 genes from RS4;11, a cell line derived from an MLL-AF4 ALL patient. Fig. 1A shows that the cells have a single nucleotide change, A24G, located 24 bp from the 3′end of mature miR-126. The base substitution is located on the ssRNA regions adjacent to the stem loop structure (Fig. 1B). In the Ensemble genome server, the human reference base is “A”. This base substitution “A to G” has been reported as a polymorphism. (Entrez SNP, rs4636297).
Fig. 1.

A base substitution in pri-miR-126 in RS4;11 cells is identified. (A) DNA chromatograms illustrating the genomic sequence of miR-126 DNA from RS4;11 cells. Nucleotides are indicated by capital letters. The green, black, and blue wave indicates A, G, and C, respectively. The arrows indicate the base position of A24G base substitution. Note that G and A sequence are both present. The base substitution is located 24 bp from the 3′end of mature miR-126 miRNA. (B) The location of the base substitution in the pri-miR-126. The red capital indicates A24G. (C) The structure of the pXZ201-pri-miR-126. Pri-miR-126 genomic sequence was cloned into the pXZ201 retroviral expression vector. (D) The structure of the pri-miR-126 cloned from the genome of RS4;11 cells. 5′ and 3′ flanking regions was 134 and 117 base long, respectively, which are enough length for processed from pri-miRNA to pre-miRNA [27].
3.2. The A24G polymorphism reduces the processing efficiency of pri-miR-126
The prediction of the secondary structure of these pri-miR-126-24G and pri-miR-126-24A was done by mfold [13]. Following the ‘ssRNA–dsRNA junction–anchoring’ model proposed recently, the ssRNA flanking regions of the stem loop structure, which is processed by Drosha/DGCR8 complex, is critical structure for Drosha cleavage [8]. Significant difference was shown between the ssRNA regions of secondary structures of pri-miR-126-24G and 24A (Fig. 2D). That of pri-miR-126-24G showed more closed structure, compared with that of 24A, suggesting that the cleavage of pri-miR-126-24G by Drosha is blocked by the secondary structure. In order to identify the molecular effects of the A24G polymorphism, expression vectors were constructed, encoding a 333 bp segment of either the reference type (miR-126-24A) or the polymorphism type (miR-126-24G) of a pri-miR-126 gene (Fig. 1C and D). These are transduced into SEM cells by retroviral infection, which also derived from MLL-AF4 ALL patient expressing a low level of endogenous mature miR-126 (Fig. 2A).
Fig. 2.
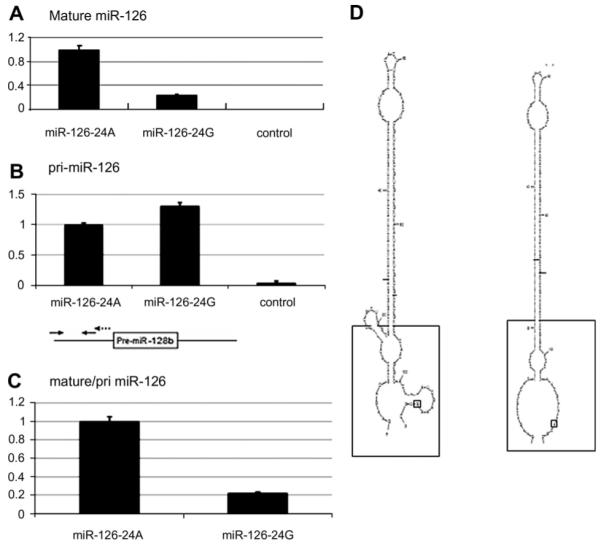
pri-miR-126-24G blocks the processing of pri-miR-126 to mature miR-126. (A) Quantitative PCR for mature miR-126 in SEM cells transduced by miR-126-24A, -24G, and control. (B) Quantitative PCR for the unprocessed and processed pri-miR-126 in SEM cells transduced by miR-126-24A, -24G, and control (upper panel). The position of primers that detect both unprocessed and processed pri-miR-126 is shown (lower panel). The arrows with solid lines and dotted lines indicate the position of the primers for PCR and RT, respectively. (C) The processing efficiency is calculated as the ratio of expression levels of mature and primary miR-126. (D) Sequence variation in pri-miR-126 can alter their secondary structure. RNA secondary structure prediction using Mfold revealed conformational changes in the secondary structure of the primary transcripts of miR-126 that could affect the ssRNA region (big black square). Depicted ones are only the most stable secondary structures with the lowest free energy. Left panel indicates pri-miR-126-24G; right, pri-miR-126-24A. Bold black lines indicate cleaved sites by Drosha; small black squares, the SNP.
The results of quantitative PCR measurements of the cells transduced by miR-126-24A expressed primary miR-126 at a level 10 times that of control cells, while cells transduced by miR-126-24G expressed primary miR-126 at a level 12 times that of the endogenous primary miR-126 (Fig. 2B). In contrast, quantitative PCR measurements of mature miR-126 in the cells expressing miR-126-24G, showed only 20% of that in cells expressing miR-126-24A (Fig. 2A). The PCR products of both pri-miR-126 were sequenced to verify the correct sequence (data not shown). The processing efficiency was calculated by dividing the expression level of mature miR-126 by that of the pri-miR-126. The processing efficiency in cells expressing exogenous miR-126-24G was only 16% of cells expressing miR-126-24A (Fig. 2C).
3.3. The novel convenient and sensitive method for detecting the processing alteration indicates that the processing of primary miR-126-24G is dramatically reduced in vivo compared with primary miR-126-24A
The SNP is located on the flanking region of the stem loop structure, which is cleaved by Drosha. After the cleavage, the stem loop is transported to the cytosol from the nucleus. The prediction of the RNA secondary structure indicated that the first step of biogenesis of miRNA, the cleavage by Drosha, is affected by the SNP. To confirm that the SNP interferes with the processing step, we designed the primers to recognize the unprocessed pri-miR-126 (Fig. 3A). Using these primers, we performed both genomic PCR and RT-PCR using RS4;11 cells, then the PCR products were sequenced. Genomic PCR products revealed that RS4;11 contain both allele, 24A and 24G, while the chromograph of RT-PCR products showed that the unprocessed pri-miR-126 dominantly produced by 24G allele (Fig. 3B). These results demonstrate two possibilities. One is that in RS4;11, the only 24G allele is dominantly transcribed, but not 24A. Alternatively, the processing of the 24G by Drosha/DGCR8 is reduced, compared with that of 24A. The majority of heterozygous miR-126 deficient mouse (+/ −) are alive [14], suggesting that both alleles are transcribed in vivo. Together with the results shown in Fig. 2, we concluded that the SNP within the pri- miR-126 sequence alters the processing of pri-miRNA to pre-miRNA in RS4;11.
Fig. 3.

Endogenous expression of unprocessed pri-miR-126 in RS4;11. (A) pri-miR-126-24A is processed, while pri-miR-126-24G is the less processed than pri-miR-126-24A. (B) Expression of unprocessed pri-miR-126 derived from 24G was much higher than that from 24A. The DNA chromatograms illustrates the genomic sequence of pri-miR-126 DNA from RS4;11 cells (upper column). Nucleotides are indicated by capital letters. R indicates the A and G. The DNA chromatograms illustrates the cDNA sequence of pri-miR-126 from RS4;11 cells (lower column). The sequence of the unprocessed pri-miR-126 dominantly shows G, derived from miR-126-24G.
3.4. miR-126-24G attenuates miRNA-mediated translational suppression
To determine the functionality of this SNP, we examined whether it could modulate translational regulation mediated by miR-126 in a reporter gene system. In this previously established system [15], miR-126 could suppress the translation of a luciferase reporter containing the 3′UTR of the human IRS1 mRNA. The suppression depends on the presence of miR-126 targeting sequences in the 3′UTR. Briefly, one 9 nucleotide sequences in the 3′UTR of human IRS1 mRNA is complementary to the miR-126 “seed” region (Fig. 4A). The 3′UTR regions containing these miR-126 binding sites were cloned downstream of the reporter Renilla luciferase open reading frame (ORF). We performed co-transfection experiments using luciferase constructs containing either IRS1 or lacking miR-126 target sequences, in combination with the control, miR-126-24A, or miR-126-24G expression vectors. Overexpression of miR-126-24A significantly reduced reporter luciferase expression of the vectors containing IRS1-UTR segments which have a s the putative miR-126 binding site, (Fig. 4B left panel) but not the expression of control vectors containing no segments of the 3′UTRs of IRS1 (Fig. 4B right panel). On the other hand, miR-126-24G reduced lesser amount of luciferase activity in reporter luciferase expression of the vectors containing the segments of the IRS1-UTR. miR-126-24G is less processed into mature form, approximately 20% of mature miR-126 that being processed from miR-126-24A. It was also shown in the translational repression. The average of reporter luciferase expression in miR-126-24G was less than that of the control, though no statistical significance was detected between miR-126-24G and the control. The miR-126-mediated translational suppression was substantially more compromised in miR-126-24G overexpressing 293T cells, suggesting that this SNP could also alter the efficiency of miR-126-mediated translational suppression.
Fig. 4.
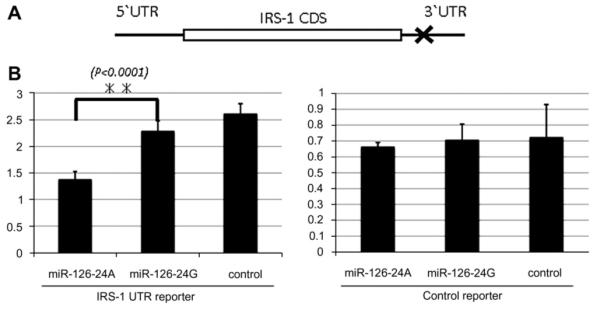
miR-126-24G induces much less translational repression than miR-126-24A. (A) Positions of the predicted (Pictar) miR-126 target sequences are marked (X) in the 3′UTRs of human IRS-1 mRNAs. (B) Expression of the Lenilla luciferase reporter is significantly reduced when IRS1-UTR reporter vectors, containing the miR-126 binding site is cotransfected together with miR-126-24A. This reduction is not observed when miR-126-24G or control expression vectors are used (left panel) (n =3; ** indicates p < 0.05.). While, That shows no significant difference between miR-126-24A, miR-126-24G and control, when psiceck-two control vectors is cotransfected (right panel). The vertical axis indicates the Renilla/firefly luciferase ratio.
3.5. The frequency of miR-126-24G and miR-126-24A
NCBI released an updated version of the human genome reference genome assembly and updated annotation for all available assemblies. The reference assembly update named GRCh37, was provided by the Genome Reference Consortium (GRC). The reference sequence is identical to miR-126-24A. To further confirm the frequency of this SNP in the general population, we searched for SNP on NCBI Reference Assembly, and found that the average frequency of the heterozygousity (A/G) is 0.375 ± 0.217.
(http://www.ncbi.nlm.nih.gov/projects/SNP/snp_Ref.c-gi?rs=4636297). As for population diversity, we searched the same database and found that in North America the allele frequency of A and G is almost the same (A:G = 0.417:0.583), while in East Asia, that of G is more than A (A:G = 0.25:0.75). Though the sample numbers in both populations were small (n = 36, n = 44), the chitest indicates that the frequency of A and G alleles was significantly different between two populations (p = 0.004). The result raised the possibility that the frequency of the SNP is different among the races.
4. Discussion
A series of studies about the mutation and polymorphism in primary miRNAs has been published. Calin reported that in 92 chronic lymphocytic leukemia (CLL) patient sample, five mutations were identified, and one of them is homozygous, and displayed complete abolishment of processing of both miR-15 and miR-16 [11]. Other group identified as many as 24 mutations in let-7 family, miR-15, −16, −124, etc. of the genome in all kinds of cancer cell lines, but concluded that none of them affected the processing of micro RNAs [16]. However, they did not analyze it quantitatively. If they use the methods successfully detecting the different processing efficiency in our study, they might detect some difference between wild type and mutant. In that case, the mutations they identified might bring an important meaning on tumorigenesis. Similarly, it has been reported many polymorphism in primary miRNAs from the 900 Japanese healthy individuals, which have not been exploited yet [10]. Our observations suggest that these polymorphisms might alter the processing of miRNAs. We identified the A24G SNP in pri-miR-126 gene in RS4;11, a cell line derived from an MLL-AF4 ALL patient. Through a series of in vivo analyzes, we demonstrate that this miR-126-24G SNP blocks the processing of pri-miRNA to pre-miRNA significantly, resulting in the dramatically reduced mature miR-126 expression. In the luciferase assay, the pri-miR-126-24G showed the reduced miRNA-mediated translational suppression, compared with miR-126-A. These data suggest that the SNPs that reside within the primary miR-126 gene could indeed regulate miRNA biogenesis, thereby potentially having profound biological effects.
As shown in Fig. 3, our methods combining RT-PCR and chromatograph of the sequence for detecting the unprocessed pri-miRNAs can be applied for screening the difference in processing efficiency by Drosha/DGCR8 complex between two alleles. This method is low cost and requires only a few days. The large scale screening of the heterozygous SNPs or mutations whether their processing efficiency is affected by the base alteration or not, is under investigation.miR-126 is one of the most extensively studied miRNAs. Its expression is known to be highest in vascularized tissues such as the lung, heart, and kidney [17-19]. The miR-126 deficient mice are embryonic lethal due to its vascular malformation [22]. In hematopoietic system, though Landgraf et al. reported qualitative detection of miR-126 in the CD34+ pool that contains HSCs [23]. We investigated the function of miR-126 in hematopoietic system and found that it facilitates B cell differentiation (preparation for manuscript). In this study, we found that miR-126-24G induces much less effect on B cells differentiation than miR-126-24A, which is compatible to our findings in this study. miR-126 is downregulated in a number of malignancies [20,25] and to act as a tumor suppressor in breast cancer [26]. These observations raise the possibility that miR-126 could play important roles in the pathogenesis of breast cancer or other malignancy. Given that the SNP associated with miR-126 identified here could block the processing of pri-miR-126 and reduce the production of mature miR-126, further exploration of the relationship between this SNP and development of human breast cancer and other malignancy could be promising.
5. Conclusions
In summary, we identified a particular SNP in miR-126 that revealed the alteration of processing of pri-miR-126 which leads to its inability to suppress its target genes at translational level. Further characterization of these miRNA-associated SNPs would likely improve our understanding of miRNA biogenesis and the potential contribution of these SNPs to normal human variation and disease pathogenesis.
Acknowledgments
We thank Dr. Mikita Suyama for helpful discussion. This work was supported by the Japan Society for the Promotion of Science, Mitsubishi Pharma Research Foundation, Japan Leukaemia Research Fund (to A.K.), and US National Institutes of Health Grant R01 DK068348 (to H.F.L.).
Footnotes
Conflict-of-interest disclosure: The authors declare no competing financial interests.
References
- [1].Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- [2].Bagga S, Bracht J, Hunter S, Massirer K, Holtz J, Eachus R, Pasquinelli AE. Regulation by let-7 and lin-4 miRNAs results in target mRNA degradation. Cell. 2005;122:553–563. doi: 10.1016/j.cell.2005.07.031. [DOI] [PubMed] [Google Scholar]
- [3].Yekta S, Shih IH, Bartel DP. MicroRNA-directed cleavage of HOXB8 mRNA. Science. 2004;304:594–596. doi: 10.1126/science.1097434. [DOI] [PubMed] [Google Scholar]
- [4].Pillai R, Bhattacharyya S, Artus C, Zoller T, Cougot N, Basyuk E, Bertrand E, Filipowicz W. Inhibition of translational initiation by let-7 microRNA in human cells. Science. 2005;309:1573–1576. doi: 10.1126/science.1115079. [DOI] [PubMed] [Google Scholar]
- [5].Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–355. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- [6].Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA, Downing JR, Jacks T, Horvitz HR, Golub TR. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–838. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- [7].Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J, Lee J, Provost P, Rådmark O, Kim S, Kim V. The nuclear RNase III Drosha initiates microRNA processing. Nature. 2003;425:415–419. doi: 10.1038/nature01957. [DOI] [PubMed] [Google Scholar]
- [8].Han J, Lee Y, Yeom K, Kim Y, Jin H, Kim V. The Drosha-DGCR8 complex in primary microRNA processing. Genes Dev. 2004;18:3016–3027. doi: 10.1101/gad.1262504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Han J, Lee Y, Yeom KH, Nam JW, Heo I, Rhee JK, Sohn SY, Cho Y, Zhang BT, Kim VN. Molecular basis for the recognition of primary microRNAs by the Drosha-DGCR8 complex. Cell. 2006;125:887–901. doi: 10.1016/j.cell.2006.03.043. [DOI] [PubMed] [Google Scholar]
- [10].Iwai N, Naraba H. Polymorphisms in human pre-miRNAs. Biochem. Biophys. Res. Commun. 2005;331:1439–1444. doi: 10.1016/j.bbrc.2005.04.051. [DOI] [PubMed] [Google Scholar]
- [11].Calin GA, Ferracin M, Cimmino A, Di Leva G, Shimizu M, Wojcik SE, Iorio MV, Visone R, Sever NI, Fabbri M, Iuliano R, Palumbo T, Pichiorri F, Roldo C, Garzon R, Sevignani C, Rassenti L, Alder H, Volinia S, Liu CG, Kipps TJ, Negrini M, Croce CM. A microRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N. Engl. J. Med. 2005;353:1793–1801. doi: 10.1056/NEJMoa050995. [DOI] [PubMed] [Google Scholar]
- [12].Duan R, Pak C, Jin P. Single nucleotide polymorphism associated with mature miR-125a alters the processing of pri-miRNA. Hum. Mol. Genet. 2007;16:1124–1131. doi: 10.1093/hmg/ddm062. [DOI] [PubMed] [Google Scholar]
- [13].Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wang M, Tan L, Dijkstra M, van Lom K, Robertus J, Harms G, Blokzijl T, Kooistra K, van T’veer M, Rosati S, Visser L, Jongen-Lavrencic M, Kluin P, van den Berg A. miRNA analysis in B-cell chronic lymphocytic leukaemia: proliferation centres characterized by low miR-150 and high BIC/miR-155 expression. J. Pathol. 2008;215:13–20. doi: 10.1002/path.2333. [DOI] [PubMed] [Google Scholar]
- [15].Zhang J, Du Y, Lin Y, Chen Y, Yang L, Wang H, Ma D. The cell growth suppressor, mir-126, targets IRS-1. Biochem. Biophys. Res. Commun. 2008;377:136–140. doi: 10.1016/j.bbrc.2008.09.089. [DOI] [PubMed] [Google Scholar]
- [16].Diederichs S, Haber DA. Sequence variations of microRNAs in human cancer: alterations in predicted secondary structure do not affect processing. Cancer Res. 2006;66:6097–6104. doi: 10.1158/0008-5472.CAN-06-0537. [DOI] [PubMed] [Google Scholar]
- [17].Baskerville S, Bartel D. Microarray profiling of microRNAs reveals frequent coexpression with neighboring miRNAs and host genes. RNA. 2005;11:241–247. doi: 10.1261/rna.7240905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Harris T, Yamakuchi M, Ferlito M, Mendell J, Lowenstein C. MicroRNA-126 regulates endothelial expression of vascular cell adhesion molecule 1. Proc. Natl. Acad. Sci. USA. 2008;105:1516–1521. doi: 10.1073/pnas.0707493105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Wang Y, Weng T, Gou D, Chen Z, Chintagari N, Liu L. Identification of rat lung-specific microRNAs by micoRNA microarray: valuable discoveries for the facilitation of lung research. BMC Genomics. 2007;8:29. doi: 10.1186/1471-2164-8-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Crawford M, Brawner E, Batte K, Yu L, Hunter M, Otterson G, Nuovo G, Marsh C, Nana-Sinkam S. MicroRNA-126 inhibits invasion in non-small cell lung carcinoma cell lines. Biochem. Biophys. Res. Commun. 2008;373:607–612. doi: 10.1016/j.bbrc.2008.06.090. [DOI] [PubMed] [Google Scholar]
- [22].Wang S, Aurora A, Johnson B, Qi X, McAnally J, Hill J, Richardson J, Bassel-Duby R, Olson E. The endothelial-specific microRNA miR-126 governs vascular integrity and angiogenesis. Dev. Cell. 2008;15:261–271. doi: 10.1016/j.devcel.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Landgraf P, Rusu M, Sheridan R, Sewer A, Iovino N, Aravin A, Pfeffer S, Rice A, Kamphorst A, Landthaler M, Lin C, Socci N, Hermida L, Fulci V, Chiaretti S, Foà R, Schliwka J, Fuchs U, Novosel A, Müller R, Schermer B, Bissels U, Inman J, Phan Q, Chien M, Weir D, Choksi R, De Vita G, Frezzetti D, Trompeter H, Hornung V, Teng G, Hartmann G, Palkovits M, Di Lauro R, Wernet P, Macino G, Rogler C, Nagle J, Ju J, Papavasiliou F, Benzing T, Lichter P, Tam W, Brownstein M, Bosio A, Borkhardt A, Russo J, Sander C, Zavolan M, Tuschl T. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell. 2007;129:1401–1414. doi: 10.1016/j.cell.2007.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Saito Y, Friedman J, Chihara Y, Egger G, Chuang J, Liang G. Epigenetic therapy upregulates the tumor suppressor microRNA-126 and its host gene EGFL7 in human cancer cells. Biochem. Biophys. Res. Commun. 2009;379:726–731. doi: 10.1016/j.bbrc.2008.12.098. [DOI] [PubMed] [Google Scholar]
- [26].Tavazoie S, Alarcón C, Oskarsson T, Padua D, Wang Q, Bos P, Gerald W, Massagué J. Endogenous human microRNAs that suppress breast cancer metastasis. Nature. 2008;451:147–152. doi: 10.1038/nature06487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Chen CZ, Li L, Lodish HF, Bartel DP. MicroRNAs modulate hematopoietic lineage differentiation. Science. 2004;303:83–86. doi: 10.1126/science.1091903. [DOI] [PubMed] [Google Scholar]