

Abstract
Oxidative stress is implicated as a pathogenic factor in a spectrum of chronic diseases, notably, neurodegenerative disease. Noteworthy in this regard is that type 1 diabetes mellitus (T1DM) results in oxidative stress, leading to systemic complications of T1DM. We hypothesized that oxidative stress associated with diabetic ketoacidosis (DKA) of T1DM might have measurable brain sequelae. Consistent with this hypothesis are neurohistology and neuroradiologic studies of T1DM that suggest that oxidative insults are involved in the chronic complications of diabetic encephalopathy. To further address the role of oxidative stress in an acute setting, specifically in fatal brain edema (BE) associated with DKA, we studied neuronal localization and levels of oxidative stress markers reported to be increased in other neurodegenerative conditions. We demonstrated increased levels of 8-hydroxyguansine (8OHG), 4-hydroxynonenal (HNE) and hemeoxygenase-1 (HO-1) in the pyramidal neurons of the hippocampus of DKA BE in comparison to controls. However, in the cerebellum, only 8OHG was increased in the Purkinje cells and other cells of the molecular layer. These results indicate a role for oxidative stress in the pathogenesis of T1DM encephalopathy.
Keywords: Brain edema, Ceruloplasmin, Diabetic encephalopathy, Diabetic ketoacidosis, Hemeoxygenase-1, 8-Hydroxyguanosine, 4-Hydroxynoneal
1. Introduction
Limitations in the physiological control of type 1 diabetes mellitus (T1DM) result in varying degrees of hypoglycemia, hyperglycemia, and ketosis, each of which is a metabolic mediator of oxidative stress (Jain et al., 1999; Singh et al., 2004; Ceriello, 2006). As such it is not surprising that the deleterious effects of oxidative stress have a cumulative effect that is related to the duration of T1DM (Martin-Gallan et al., 2007). Such oxidative changes are recognized in the pathogenesis of the chronic vascular complications and peripheral neuropathy of diabetes (Baynes, 1991; Ceriello, 2006). Likewise, evidence for oxidative stress and neuroinflammation are also described in the pathogenesis of diabetic encephalopathy (Mastrocola et al. 2005; Hoffman et al. 2008; Sima et al. 2009 a,b). Additionally, the metabolic crisis of diabetic ketoacidosis (DKA) and its treatment accentuate systemic oxidative stress and a systemic inflammatory response (Jain et al., 1999; Vantyghem et al., 2000; Lee et al., 2002; Dalton et al., 2003; Hoffman et al., 2003 a,b; Jerath et al., 2005; Turk et al., 2006). The systemic insults associated with DKA and its treatment occur in parallel with the progression of subclinical brain edema (BE) and development of clinical BE (Hoffman et al., 1988; Durr et al., 1992). Clinical BE, a potentially fatal complication of DKA (Edge et al., 2001), is associated with disruption of tight junction proteins of the blood brain barrier (Hoffman et al., 2009).
Several characteristics make the brain susceptible to oxidative/nitrosative stress, including: a high metabolic rate and oxygen requirement (Kennedy and Sokoloff, 1957); a high concentration of polyunsaturated fatty acids that increase susceptibility to lipid peroxidation (Adibhatla and Hatcher, 2008); high levels of transition metals, capable of catalyzing reactive oxygen species and reactive nitrogen species (Halliwell, 1992; Smith et al., 1994a); and age-dependent differences in antioxidant reserves (Bayir et al., 2006). In TIDM, insulin and insulin growth factor-1 (IGF-1) deficits in the brain result in disordered insulin signaling, deficiencies in the protein kinases Akt and GSK3, compromised brain energy metabolism, and oxidative stress (Steen et al., 2005; Li et al., 2007; Jolivalt et al., 2008; Sima et al., 2009 a,b).
The role of oxidative stress in the pathogenesis of DKA/BE is incompletely understood. To address the cellular localization and levels of 8-hydroxyguanosine (8OHG), a marker of oxidative damage to RNA, the lipid peroxidation adduct 4-hydroxynonenal (HNE), the oxidatively-induced enzyme hemeoxygenase-1 (HO-1) and ceruloplasmin, the acute phase protein that is a transporter of iron and an anti-oxidant, were measured. In addition, phosphorylated p38, redox active iron, mitochondrial DNA (mtDNA) deletions, neuronal morphology and neuronal density were also studied.
2. Results
2.1 Immunohistochemistry
Within the hippocampus, products of oxidative damage accumulated at higher levels in the pyramidal neurons in both cases of DKA/BE compared to controls. 8OHG was localized to the neuronal cytoplasm and nucleoli at much higher levels in the two DKA/BE cases (Fig. 1 A, B), with the control cases showing only background levels (Fig. 1C). Consistent with this, quantification showed that the 8OHG was significantly higher in the DKA/BE cases than controls (p<0.001, Fig. 1D). In the cerebellum, differences of 8OHG were found between the DKA/BE cases and the control cases. Both DKA/BE cases showed high levels of 8OHG in the Purkinje cells and other cells within the molecular layer (Fig. 1 E, F). Comparatively, control cases had much lower amounts of 8OHG in those cells (Fig. 1G). Quantification revealed that the levels of 8OHG in both the Purkinje cells as well as the other cells within the molecular layer were significantly higher (p<0.001, Fig. 1H) in DKA/BE compared to the controls.
Figure 1.
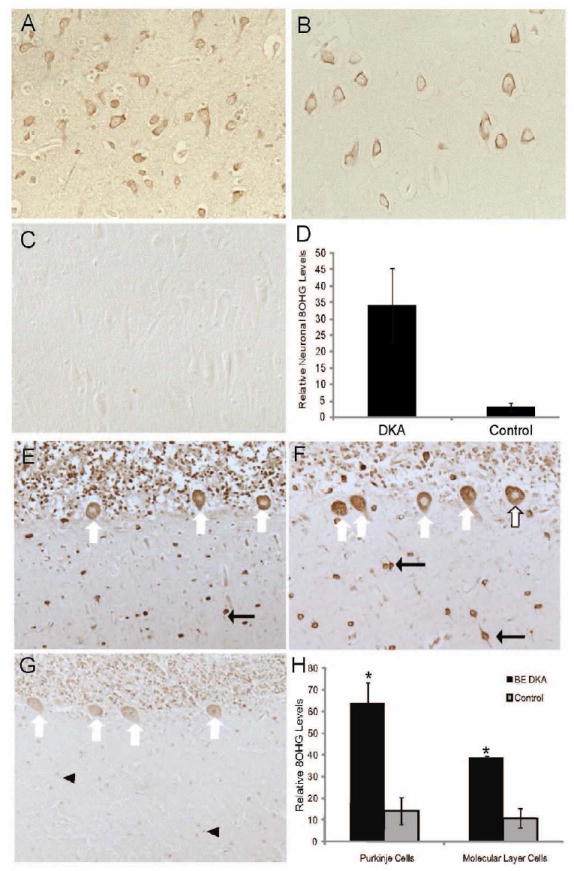
Within the hippocampus the pyramidal neurons in each of the DKA/BE cases accumulate strikingly increased levels of 8OHG (A,B) compared to controls (C). Image analysis of the intensity of neuronal staining revealed significantly higher levels of 8OHG in DKA/BE compared to controls ages 3, 17,17 years (D, error bars indicate +/- SEM, * p<0.001). The cerebellum demonstrated strong immunodetection of 8OHG in both the Purkinje cells (white arrows), other cells and cellular processes (black arrows) within the molecular layer of the DKA/BE cases (E,F) compared to controls (G), which showed weaker staining of the Purkinje cells and weakly stained nuclei (arrowheads). Upon quantification comparing the DKA/BE cases with controls ages 16, 17, 17 years, the cellular accumulation of 8OHG was significantly greater in both the Purkinje cells and other molecular layer cells (H, error bars indicate +/- SEM, * p<0.001).
HNE adducts were also increased in the pyramidal neurons of the hippocampus in the DKA/BE cases compared to control cases (Fig. 2 A and B, respectively). As expected, quantification of the neuronal HNE levels revealed significantly higher levels in the DKA/BE cases compared to controls (p<0.05, Fig 2C). The cerebellum of the DKA/BE cases as well as controls demonstrated only background levels of HNE adducts (data not shown). In all cases, the large vessel walls, known sites of normally occurring HNE accumulation, were positively immunostained (data not shown).
Figure 2.
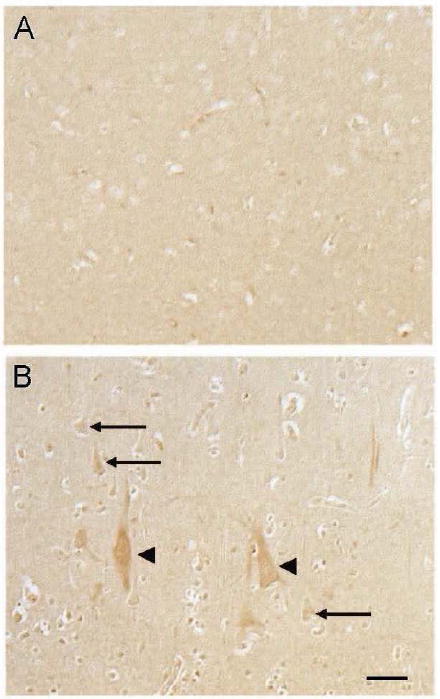
In the hippocampus, neuronal accumulation of HNE was higher in the DKA/BE cases (A) compared to controls (B). Quantification of the neuronal staining revealed significantly higher levels of HNE in DKA/BE cases compared to controls ages 9, 16 years (C, error bars indicate +/- SEM, * p<0.05).
HO-1 levels were also increased in the same population of neurons in the hippocampus of the DKA/ BE cases compared to controls (Fig. 3A and B, respectively). The HO-1 cytoplasmic localization in the pyramidal neurons was significant in the DKA/BE cases (p<0.001, Fig. 3C). In the cerebellum, HO-1 showed no differences between DKA/BE cases (Fig. 3 D) and controls (Fig. 3E), and upon quantification the Purkinje cells displayed similar levels of HO-1 (Fig. 3F).
Figure 3.
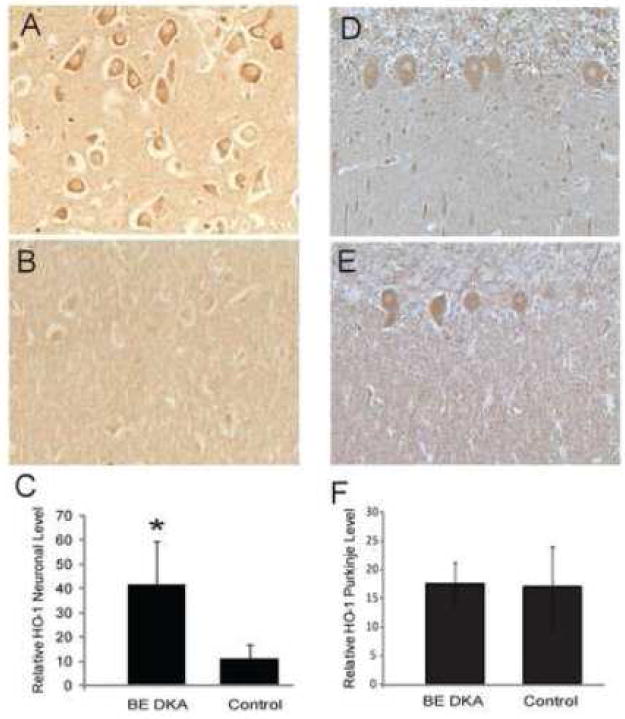
Neuronal levels of HO-1 were significantly higher in the pyramidal neurons of the hippocampus in the DKA/BE cases (A) compared to controls (B). Quantification of the neuronal staining revealed significantly higher levels of HO-1 in DKA/BE compared to controls ages 9, 20 years (C, error bars indicate +/- SD,* p<0.001). HO-1 was not consistently changed in the cerebellum between DKA/BE cases and controls. The Purkinje cells displayed similar levels of HO-1 in both the DKA/BE cases (D) and the controls (E), and quantification revealed no significant difference (F, error bars indicate +/- SD). Other cellular structures and processes were immunoreactive in some cases, but were not consistently found in any of the groups analyzed.
In the pons, no differences were noted with the oxidative stress markers between DKA/BE and controls.
In the occipital cortex, ceruloplasmin (Cp) was localized to pyramidal neurons (arrows) in the DKA/BE cases (Fig. 4B), while in controls Cp was not increased in the neuronal cell bodies (Fig. 4A). Pyramidal neurons in the occipital cortex from the cases of DKA/BE, showed the highest level of Cp (Fig. 4B, arrowheads), a finding that was not seen in the controls.
Figure 4.
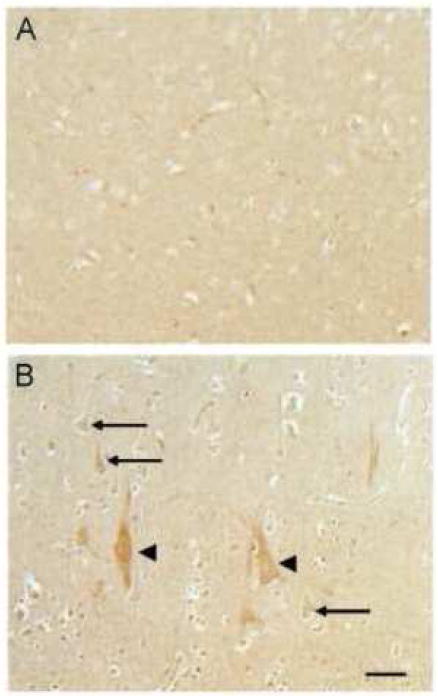
In the occipital cortex, ceruloplasmin was localized to pyramidal neurons (arrows) in the cases of DKA/BE (B), while in controls (ages 9, 16 years) ceruloplasmin was not found to be increased in the neuronal cell bodies (A). Of interest, large neuronal cells in the occipital cortex from the DKA/BE cases showed the highest level of ceruloplasmin (B, arrowheads). Scale bar = 50 μm.
Other markers of oxidative damage were analyzed in some areas of the DKA/BE cases. Neither phosphorylated p38, redox active iron, nor mtDNA deletion in situ hybridization (data not shown) showed any specific accumulation in the DKA/BE cases with our methodologies.
2.2 Quantitative analysis
Morphological analysis of cresyl violet stained hippocampi sections revealed the mean neuronal densities were lower in the DKA/BE cases compared to controls confirming previous reports (Hoffman et al., 2008). Statistical analysis comparing all CA fields of DKA/BE cases with controls demonstrated the DKA/BE cases to have significantly lower neuronal density in the hippocampi than controls (187 +/- 53 versus 304 +/-59 neurons/mm2, p<0.01). However, no significant differences in the size of the pyramidal neurons in the hippocampus were found between the cases of DKA/BE and controls.
3. Discussion
There is convincing evidence that the bases of both messenger RNA and ribosomal RNA (Nunomura et al. 2009) are susceptible to sublethal oxidation insults (Nunomura et al., 1999; Nunomura et al., 2007; Castellani et al., 2008). 8OHG, an oxidized nucleoside derived from RNA, identifies vulnerable/damaged neurons at an early stage of several neurodegenerative diseases (Sayre et al., 2008). This study of well-recognized markers of oxidative stress in the brains of young patients with poorly controlled T1DM that included recurrent episodes of DKA, and the fatal BE of DKA supports and extends the vulnerability of the brain to oxidative stress and its potential importance in the neuronal dysfunction associated with severe abnormalities in glucose and insulin metabolism. The findings that oxidative damage mediated by insulin deficiency/ resistance and neuroinflammation as illustrated here raise the possibility that these mechanisms underlie the pathogenesis of both acute and chronic cerebral complications, including diabetic encephalopathy (Sima and Li, 2005; Li et al., 2007; Hoffman et al., 2008; Sima et al., 2009 a,b; Hoffman et al., 2010).
HNE, a biologically active carbonyl derived from polyunsaturated fatty acids peroxidation, is a signaling molecule at subtoxic levels (Dwivedi et al., 2007) and neurotoxic at higher levels resulting in perturbation of neuronal membranes with disruption of cellular energy metabolism and ion homeostasis (Mark et al., 1997); stress-mediated signaling and compromised activity of antioxidant glutathione (Raza and John, 2006); modification of DNA bases (Liu et al., 2006); inhibition of mitochondrial function (Picklo and Montine, 2007); caspase activation (Camandola et al., 2000); and apoptosis (Peng et al., 2007). A neurotoxic effect of HNE in the hippocampus of DKA/BE cases is supported by HNE conjugation of GLUT3, the high affinity isoform of type 1 glucose transporter specific to neurons, in the hippocampus of the streptozotocin rat, is an indication of compromised glucose utilization and a hypometabolic state (Reagan et al., 2000).
HO-1, a redox regulated enzyme, is induced with other heat shock proteins (HSP) as a protective antioxidant to various forms of stress (Syapin, 2008) and is also a mediator of the anti-apoptotic effect of insulin (Geraldes et al., 2008). The expression of HO-1 in the hippocampus of the DKA/BE cases is in keeping with the expression of the protective molecules HSP70 and IL-10 in these cases (Hoffman et al., 2007), as well as their increased systemic expression prior to treatment of severe DKA (Oglesbee et al., 2005) (Hoffman et al., 2003a).
Neuronal expression of Cp could be due to increased oxidative stress in T1DM (Cunningham et al., 1995). As with the systemic increase of the acute phase proteins C-reactive protein and von Willebrand factor in severe DKA (Dalton et al., 2003; Carl et al., 2003) Cp is likely to increase prior to and during treatment of DKA. While 8OHG, nitrotyrosine (NT) (Hoffman et al., 2009; Hoffman et al., 2010), HO-1and HNE are expressed in DKA/BE the absence of redox active iron and mtDNA deletions, a marker of mitochondrial injury (Hirai et al., 2001) suggest the oxidative damage is the result of a different mechanism than that found in neurodegenerative diseases such as Alzheimer’s disease where 8OHG, NT, HO-1, HNE, mtDNA deletions and redox active metals are all significantly increased (Castellani et al., 2007; Sayre et al., 2008).
A pattern of selective protection is evident in the cerebellum where 8OHG and NT (Hoffman et al., 2010) were expressed in the Purkinje cells; however HO-1, HNE, redox active iron, mtDNA deletions and p38 were not seen. The inverse relation between insulin and IGF-1 receptors with neuronal density in the cerebellum (Hoffman et al., 2010), and the selective neuronal adaptation of the cerebellum to chronic sublethal oxidative stress provided by altered cholesterol and sphingolipid (Clement et al., 2009), are possible explanations for the limited insult of oxidative stress in the cerebellum. Neither 8OHG, HO-1 nor HNE were consistently expressed in the epithelial cells of the choroid plexus, in contrast to the expression of NT (Hoffman et al., 2009; Hoffman et al., 2010) and the neuroinflammatory response, including RAGE, in the choroid plexus of the DKA/BE cases (Hoffman et al., 2007; Hoffman et al., 2008) This suggests that the choroid plexus selectively compensates for the oxidative stress of DKA/BE (Skinner et al., 2006; Hoffman et al., 2007; Marques et al., 2009)
The immunocytochemical evidence of insult to neuronal membrane lipids by peroxidation and magnetic resonance spectroscopy reports demonstrating a positive relation of both chronic and acute poor metabolic control in T1DM (Sarac et al., 2005; Wootton-Gorges et al., 2007; Hoffman et al., 2010) suggest perturbation of neuronal metabolism. Noteworthy is the evidence that insulin deficiency in T1DM results in variability of metabolic/immunologic insults involving hyperglycemia, dehydration, hyperlipidemia, ketoacidosis (MacGillivray et al., 1982; Durr et al., 1990; Hoffman et al., 2003a,b; Jerath et al., 2005; Edge et al., 2008) and peripheral (Barrett et al., 1982), and cerebral insulin resistance with reduced insulin signaling (Jolivalt et al., 2008) (Hoffman et al., 2010). The latter leading to enhanced activation of the AGE-RAGE system (Liu et al., 2010) and increased oxidative and inflammatory stress (Hoffman et al., 2008) (Sima et al., 2009a,b). Taken together, these data suggest that insulin deficiency results in chronic metabolic dysfunction associated with oxidative stress. In this respect, DKA in T1DM may be viewed as an accentuated and additive insult resulting in the metabolic brain disease of diabetic encephalopathy. The fluctuating metabolic state in T1DM makes the pathogenesis of the oxidative and inflammatory insults and compensatory mechanisms (Zhu et al., 2003; Su et al., 2008) more challenging to study.
The neurons in the DKA/BE cases showed no evidence of morphologic degeneration and no evidence of apoptosis (Hoffman et al., 2006). Also, despite insulin deficiency/ resistance and likely metabolic neuronal perturbation we found no difference in the size of the hippocampal neurons in DKA/BE. The basis for the decreased neuronal density in DKA/BE cases maybe related to recurrent episodes of DKA with subclinical BE and endothelial perturbation (Hoffman et al., 1988; Isales et al., 1999; Hoffman et al., 2002) contributing to varying degrees of brain injury (Sarac et al., 2007; Wootton-Gorges et al., 2007; Hoffman et al, 2008; Hoffman et al., 2009; Hoffman et al., 2010).
In summary, we describe two cases of DKA associated with fatal clinical BE with increased expression of 8OHG, HNE, HO-1, and Cp in vulnerable brain regions. These findings of substantial oxidative stress in the clinical BE of DKA in addition to insulin deficiency and increased neuroinflammation (Hoffman et al., 2007; Hoffman et al. 2008; Hoffman et al., 2009; Sima et al. 2009 a,b; Hoffman et al., 2010;) support a pathogenesis for neurologic impairment. We further suggest that the basic feature of insulin deficiency initiates a similar metabolic/immunologic dysfunction in poorly controlled T1DM associated with DKA and subclinical BE that could lead to chronic sequelae of oxidative stress, such as diabetic encephalopathy.
4. Experimental procedures
4.1 Subjects
4.1.1 Case 1
An adolescent girl had a four-year history of poorly controlled T1DM which resulted in recurrent hospitalization for DKA. There was no history of other medical conditions or microvascular disease. There was no history of recent fever or enteritis. The final admission was preceded by a 12 hour history of abdominal pain and several episodes of emesis. On physical examination she was oriented but drowsy. Her height was 163 cm; weight was 68.5 kg; blood pressure was 140/70 mm Hg; pulse was 140/min; respirations were 30/min; and temperature was 98.5°F. There were no signs of infection. The mucous membranes were dry, the abdomen was diffusely tender, and bowel pattern sounds were hypoactive. The Tanner stage was thelarche stage 3 and pubarche stage 2 (Marshall and Tanner, 1969). Admission laboratory tests consisted of: pH 7.10; pCO2 15 mmHg; pO2 106 mm Hg; glucose 810 mg/dl; Na 132 meq/L; K 5.7 meq/L; Cl 93meq/L; HCO3 5 meq/L; BUN 30 mg/dl, and calculated effective plasma osmolality of 309 Osm. Treatment was in a Pediatric Intensive Care Unit and correction of the hyperglycemia and metabolic acidosis was unremarkable. Twelve hours into treatment she developed sudden onset of labored respirations and within 20 min had a cardio-respiratory arrest. An emergency CT scan showed sulcal effacement, cerebral and pontine edema with evidence of herniation. Efforts at resuscitation were unsuccessful, and she was pronounced dead 1½ hours after the cardiorespiratory arrest.
4.1.2 Case 2
An adolescent girl had an eight year history of poorly controlled T1DM which had resulted in recurrent hospitalizations for DKA. There was no history of other medical conditions or microvascular disease. The final admission was preceded by an 18-hour history of capillary blood glucoses of over 300 mg/dl, ketonuria, a 4-hour history of headache and several episodes of emesis. There was no history of fever or enteritis. On physical examination she was slightly confused and lethargic. Her height was 154 cm; weight was 45 kg; blood pressure was 135/68 mm/Hg; pulse was 132/min; respirations were 26/min; and temperature was 97°F. There were no signs of infection. Diffuse abdominal tenderness and decreased bowel sounds were present. The Tanner stage was thelarche stage 2 and pubarche stage 1 (Marshall and Tanner, 1969). Admission laboratory tests consisted of: pH 7.16; pCO2 17 mmHg; pO2 100 mmHg; blood glucose 581 mg/dl; Na 130 meq/L; K 4.8 meq/L; Cl 89 meq/L; HCO3 6 meq/L; BUN 28 mg/dl and calculated effective plasma of 292 Osm. Treatment was in a Pediatric Intensive Care Unit and correction of the hyperglycemia and metabolic acidosis was unremarkable. Ten hours into treatment, she became unresponsive, and was treated with mannitol and hyperventilation and placed on mechanical ventilation. A CT head scan showed diffuse cerebral edema and decreased intercaudate diameter. She was pronounced dead approximately 10 hours after the cardiorespiratory arrest.
4.1.3 Controls
Control brain tissue consisted of six non-diabetic cases as stated in the Legends.
4.2 Immunohistochemistry
Paraffin embedded tissue samples from two cases of fatal DKA/BE were examined simultaneously with controls.
Brain regions examined include hippocampus, cerebellum, occipital cortex, and pons. For immunohistochemistry, sections were deparaffinized in 2 changes of xylene and rehydrated through graded ethanol to Tris buffered saline (TBS, 50 mM Tris, 150 mM Na Cl, pH=7.6). Endogenous peroxidase was removed with a 30 min incubation in 3% H2O2. Sections were then incubated in 10% normal goat serum for 30 min, and primary antibodies applied for 16 hr at 4°C. The peroxidase-anti-peroxidase method (Sternberger, 1986) was used with 3’-3’-diaminobenzidine as chromagen (Dako, Carpinteria, CA). Primary antibodies used included rabbit polyclonal antisera to HO-1 (Smith et al. 1994b), lipid peroxidation adduct HNE (Sayre et al. 1997), anti-ceruloplasmin (Dako, Carpinteria, CA) (Castellani et al. 1999), phosphorylated38 (Cell Signaling Technology, Inc., Beverly, MA) (Zhu et al., 2000), and monoclonal antibody against 8OHG (clone 15A3, Trevigen) (Nunomura et al. 1999; Nunomura et al. 2001). Sections immunostained for 8OHG were pretreated with 10 μg/ml proteinase K (Sigma Chemical Co., St. Louis) for 20 min at 37°C before blocking in the 10% normal goat serum. For each antibody, sections from all cases available were immunostained simultaneously and developed with 3’-3’-diaminobenzidine for the same amount of time. Other assays performed included the in situ hybridization using digoxigen labeled probes specific for mtDNA deletion (Hirai et al. 2001), a modified Perls stain for detecting in situ redox active iron (Smith et al. 1997), and cresyl violet histochemistry.
4.3. Quantitative analyses
The cellular accumulation of some of the oxidative damage markers was quantified. High resolution images were acquired using an Axiocam (Zeiss, Thornwood, NY) from 3 fields of the CA1/CA2 region of the hippocampus or of the cerebellum inclusive of the Purkinje and molecular layers. The immunoreactive intensity was measured with computer-assisted image analysis (Axiovision Rel 4.5, Zeiss) of all neurons or Purkinje cells in the fields with the background levels of the surrounding neuropil subtracted. The neuronal levels of the oxidative markers were analyzed using the General Linear Model method with reported measures using SAS statistics software. Quantitative analysis was performed to compare the neuronal density and the neuronal size in the hippocampus from the cases of DKA/BE and 4 controls stained with cresyl violet to distinguish neuronal cell bodies. Using the 5X objective, representative fields from each of the CA1, CA2, CA3, and CA4 areas was imaged using a Zeiss Axiocam. The Axiovision software allows for detection of all stained cells and a size threshold was used to remove any small stained structures that were not pyramidal cells, or partial cell bodies that did not contain a nucleus. Additionally, any vascular structures as well as cells stained that were not within the stratum pyramidal layer were removed manually. Finally, the entire stratum pyramidal area (Duvernoy 1988) was encircled to determine the total area analyzed in mm2, the number of pyramidal neurons per mm2 determined and the data analyzed using the student’s t-test. Additionally the size of the neurons in each of the CA1, CA2, CA3, and CA4 regions was measured. Between 65-245 cells were analyzed for each CA field in each of the 2 DKA/BE cases and 4 controls.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adibhatla RM, Hatcher JF. Altered lipid metabolism in brain injury and disorders. Subcell Biochem. 2008;49:241–268. doi: 10.1007/978-1-4020-8831-5_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett EJ, DeFronzo RA, Bevilacqua S, Ferrannini E. Insulin resistance in diabetic ketoacidosis. Diabetes. 1982;10:923–928. doi: 10.2337/diab.31.10.923. [DOI] [PubMed] [Google Scholar]
- Bayir H, Kochanek PM, Kagan VE. Oxidative stress in immature brain after traumatic brain injury. Dev Neurosci. 2006;28(4-5):420–431. doi: 10.1159/000094168. [DOI] [PubMed] [Google Scholar]
- Baynes JW. Role of oxidative stress in development of complications in diabetes. Diabetes. 1991;40(4):405–412. doi: 10.2337/diab.40.4.405. [DOI] [PubMed] [Google Scholar]
- Camandola S, Poli G, Mattson MP. The lipid peroxidation product 4-hydroxy-2,3-nonenal increases AP-1-binding activity through caspase activation in neurons. J Neurochem. 2000;74(1):159–168. doi: 10.1046/j.1471-4159.2000.0740159.x. [DOI] [PubMed] [Google Scholar]
- Carl GF, Hoffman WH, Passmore GG, Truemper EJ, Lightsey AL, Cornwell PE, Jonah MH. Diabetic ketoacidosis promotes a prothrombotic state. Endocr Res. 2003;29(1):73–82. doi: 10.1081/erc-120018678. [DOI] [PubMed] [Google Scholar]
- Castellani RJ, Moreira PI, Liu G, Dobson J, Perry G, Smith MA, Zhu X. Iron: the Redox-active center of oxidative stress in Alzheimer disease. Neurochem Res. 2007;32(10):1640–1645. doi: 10.1007/s11064-007-9360-7. [DOI] [PubMed] [Google Scholar]
- Castellani RJ, Nunomura A, Rolston RK, Moreira PI, Takeda A, Perry G, Smith MA. Sublethal RNA oxidation as a mechanism for neurodegenerative disease. Int J Mol Sci. 2008;9(5):789–806. doi: 10.3390/ijms9050789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellani RJ, Smith MA, Nunomura A, Harris PL, Perry G. Is increased redox-active iron in Alzheimer disease a failure of the copper-binding protein ceruloplasmin? Free Radic Biol Med. 1999;26(11-12):1508–1512. doi: 10.1016/s0891-5849(99)00016-7. [DOI] [PubMed] [Google Scholar]
- Ceriello A. Oxidative stress and diabetes-associated complications. Endocr Pract. 2006;12(Suppl 1):60–62. doi: 10.4158/EP.12.S1.60. [DOI] [PubMed] [Google Scholar]
- Clement AB, Gamerdinger M, Tamboli IY, Lutjohann D, Walter J, Greeve I, Gimpl G, Behl C. Adaptation of neuronal cells to chronic oxidative stress is associated with altered cholesterol and sphingolipid homeostasis and lysosomal function. J Neurochem. 2009;111(3):669–682. doi: 10.1111/j.1471-4159.2009.06360.x. [DOI] [PubMed] [Google Scholar]
- Cunningham J, Leffell M, Mearkle P, Harmatz P. Elevated plasma ceruloplasmin in insulin-dependent diabetes mellitus: evidence for increased oxidative stress as a variable complication. Metabolism. 1995;44(8):996–999. doi: 10.1016/0026-0495(95)90095-0. [DOI] [PubMed] [Google Scholar]
- Dalton RR, Hoffman WH, Passmore GG, Martin SL. Plasma C-reactive protein levels in severe diabetic ketoacidosis. Ann Clin Lab Sci. 2003;33(4):435–42. [PubMed] [Google Scholar]
- Dogan Y, Akarsu S, Ustundag B, Yilmaz E, Gurgoze MK. Serum IL-1beta, IL-2, and IL-6 in insulin-dependent diabetic children. Mediators Inflamm. 2006;2006(1):59206. doi: 10.1155/MI/2006/59206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durr JA, Hoffman WH, Hensen J, Sklar AH, el Gammal T, Steinhart CM. Osmoregulation of vasopressin in diabetic ketoacidosis. Am J Physiol. 1990;259(5 Pt 1):E723–8. doi: 10.1152/ajpendo.1990.259.5.E723. [DOI] [PubMed] [Google Scholar]
- Durr JA, Hoffman WH, Sklar AH, el Gammal T, Steinhart CM. Correlates of brain edema in uncontrolled IDDM. Diabetes. 1992;41(5):627–32. doi: 10.2337/diab.41.5.627. [DOI] [PubMed] [Google Scholar]
- Duvernoy HM. The Human Hippocampus: An Atlas of Applied Anatomy. Munchen: Springer-Verlag; 1988. [Google Scholar]
- Dwivedi S, Sharma A, Patrick B, Sharma R, Awasthi YC. Role of 4-hydroxynonenal and its metabolites in signaling. Redox Rep. 2007;12(1):4–10. doi: 10.1179/135100007X162211. [DOI] [PubMed] [Google Scholar]
- Edge JA. Cerebral oedema during treatment of diabetic ketoacidosis: are we any nearer finding a cause? Diabetes Metab Res Rev. 2000;16(5):316–24. doi: 10.1002/1520-7560(2000)9999:9999<::aid-dmrr143>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- Edge JA, Hawkins MM, Winter DL, Dunger DB. The risk and outcome of cerebral oedema developing during diabetic ketoacidosis. Arch Dis Child. 2001;85(1):16–22. doi: 10.1136/adc.85.1.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edge JA, James T, Shine B. Longitudinal screening of serum lipids in children and adolescents with Type 1 diabetes in a UK clinic population. Diabet Med. 2008;25(8):942–8. doi: 10.1111/j.1464-5491.2008.02518.x. [DOI] [PubMed] [Google Scholar]
- Geraldes P, Yagi K, Ohshiro Y, He Z, Maeno Y, Yamamoto-Hiraoka J, Rask-Madsen C, Chung SW, Perrella MA, King GL. Selective regulation of heme oxygenase-1 expression and function by insulin through IRS1/phosphoinositide 3-kinase/Akt-2 pathway. J Biol Chem. 2008;283(49):34327–36. doi: 10.1074/jbc.M807036200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaser N, Barnett P, McCaslin I, Nelson D, Trainor J, Louie J, Kaufman F, Quayle K, Roback M, Malley R, Kuppermann N. Risk factors for cerebral edema in children with diabetic ketoacidosis. The Pediatric Emergency Medicine Collaborative Research Committee of the American Academy of Pediatrics. N Engl J Med. 2001;344(4):264–269. doi: 10.1056/NEJM200101253440404. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Cuyar LF, Perry G, Miyajima H, Atwood CS, Riveros-Angel M, Lyons PF, Siedlak SL, Smith MA, Castellani RJ. Redox active iron accumulation in aceruloplasminemia. Neuropathology. 2008;28(5):466–471. doi: 10.1111/j.1440-1789.2008.00901.x. [DOI] [PubMed] [Google Scholar]
- Halliwell B. Reactive oxygen species and the central nervous system. J Neurochem. 1992;59(5):1609–1623. doi: 10.1111/j.1471-4159.1992.tb10990.x. [DOI] [PubMed] [Google Scholar]
- Hirai K, Aliev G, Nunomura A, Fujioka H, Russell RL, Atwood CS, Johnson AB, Kress Y, Vinters HV, Tabaton M, Shimohama S, Cash AD, Siedlak SL, Harris PL, Jones PK, Petersen RB, Perry G, Smith MA. Mitochondrial abnormalities in Alzheimer’s disease. J Neurosci. 2001;21(9):3017–3023. doi: 10.1523/JNEUROSCI.21-09-03017.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isales CM, Leilin M, Hoffman WH. Acetoacetate and B-Hydroxybutyrate differentially regulate endothelin-1 and vascular endothelial growth factor in mouse brain microvascular endothelial cells. J Diabetes and Complications. 1999;13:91–97. doi: 10.1016/s1056-8727(99)00030-6. [DOI] [PubMed] [Google Scholar]
- Hoffman WH, Steinhart CM, el Gammal T, Steele S, Cuadrado AR, Morse PK. Cranial CT in children and adolescents with diabetic ketoacidosis. AJNR Am J Neuroradiol. 1988;9(4):733–739. [PMC free article] [PubMed] [Google Scholar]
- Hoffman WH, Cheng C, Passmore GG, Carroll JE, Hess D. Acetoacetate increases expression of intercellular adhesion molecule-1 (ICAM-1) in human brain microvascular endothelial cells. Neurosci Lett. 2002;334:71–74. doi: 10.1016/s0304-3940(02)00816-9. [DOI] [PubMed] [Google Scholar]
- Hoffman WH, Burek CL, Waller JL, Fisher LE, Khichi M, Mellick LB. Cytokine response to diabetic ketoacidosis and its treatment. Clin Immunol. 2003a;108(3):175–181. doi: 10.1016/s1521-6616(03)00144-x. [DOI] [PubMed] [Google Scholar]
- Hoffman WH, Kappler F, Passmore GG, Mehta R. Diabetic ketoacidosis and its treatment increase plasma 3-deoxyglucosone. Clin Biochem. 2003b;36(4):269–273. doi: 10.1016/s0009-9120(03)00030-4. [DOI] [PubMed] [Google Scholar]
- Hoffman WH, Cudrici CD, Zafranskaia E, Rus H. Complement activation in diabetic ketoacidosis brains. Exp Mol Pathol. 2006;80(3):283–288. doi: 10.1016/j.yexmp.2005.12.007. [DOI] [PubMed] [Google Scholar]
- Hoffman WH, Casanova MF, Cudrici CD, Zakranskaia E, Venugopalan R, Nag S, Oglesbee MJ, Rus H. Neuroinflammatory response of the choroid plexus epithelium in fatal diabetic ketoacidosis. Exp Mol Pathol. 2007;83(1):65–72. doi: 10.1016/j.yexmp.2007.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman WH, Artlett CM, Zhang W, Kreipke CW, Passmore GG, Rafols JA, Sima AA. Receptor for advanced glycation end products and neuronal deficit in the fatal brain edema of diabetic ketoacidosis. Brain Res. 2008;1238:154–162. doi: 10.1016/j.brainres.2008.08.041. [DOI] [PubMed] [Google Scholar]
- Hoffman WH, Stamatovic SM, Andjelkovic AV. Inflammatory mediators and blood brain barrier disruption in fatal brain edema of diabetic ketoacidosis. Brain Res. 2009;1254:138–148. doi: 10.1016/j.brainres.2008.11.100. [DOI] [PubMed] [Google Scholar]
- Hoffman WH, Andjelkovic AV, Zhang W, Passmore GG, Sima AAF. Insulin and IGF-1 receptors, nitrotyrosin and cerebral neuronal deficits in two young patients with diabetic ketoacidosis and fatal brain edema. Brain Res. 1343:168–177. doi: 10.1016/j.brainres.2010.04.042. [DOI] [PubMed] [Google Scholar]
- Jain SK, McVie R, Jackson R, Levine SN, Lim G. Effect of hyperketonemia on plasma lipid peroxidation levels in diabetic patients. Diabetes Care. 1999;22(7):1171–1175. doi: 10.2337/diacare.22.7.1171. [DOI] [PubMed] [Google Scholar]
- Jerath RS, Burek CL, Hoffman WH, Passmore GG. Complement activation in diabetic ketoacidosis and its treatment. Clin Immunol. 2005;116(1):11–17. doi: 10.1016/j.clim.2005.03.004. [DOI] [PubMed] [Google Scholar]
- Jolivalt CG, Lee CA, Beiswenger KK, Smith JL, Orlov M, Torrance MA, Masliah E. Defective insulin signaling pathway and increased glycogen synthase kinase-3 activity in the brain of diabetic mice: parallels with Alzheimer’s disease and correction by insulin. J Neurosci Res. 2008;86(15):3265–3274. doi: 10.1002/jnr.21787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy C, Sokoloff L. An adaptation of the nitrous oxide method to the study of the cerebral circulation in children; normal values for cerebral blood flow and cerebral metabolic rate in childhood. J Clin Invest. 1957;36(7):1130–1137. doi: 10.1172/JCI103509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DM, Hoffman WH, Carl GF, Khichi M, Cornwell PE. Lipid peroxidation and antioxidant vitamins prior to, during, and after correction of diabetic ketoacidosis. J Diabetes Complications. 2002;16(4):294–300. doi: 10.1016/s1056-8727(01)00215-x. [DOI] [PubMed] [Google Scholar]
- Lee KH, Yun SJ, Nam KN, Gho YS, Lee EH. Activation of microglia cells by ceruloplasmin. Brain Res. 2007;1171:1–8. doi: 10.1016/j.brainres.2007.07.053. [DOI] [PubMed] [Google Scholar]
- Li ZG, Zhang W, Sima AA. Alzheimer-like changes in rat models of spontaneous diabetes. Diabetes. 2007;56(7):1817–1824. doi: 10.2337/db07-0171. [DOI] [PubMed] [Google Scholar]
- Liu X, Lovell MA, Lynn BC. Detection and quantification of endogenous cyclic DNAadducts derived from trans-4-hydroxy-2-nonenal in human brain tissue by isotope dilution capillary liquid chromatography nanoelectrospray tandem mass spectrometry. Chem Res Toxicol. 2006;19(5):710–718. doi: 10.1021/tx0502903. [DOI] [PubMed] [Google Scholar]
- Liu X, Luo D, Zheng M, Hao Y, Hou L, Zhang S. Effect of pioglitazone on insulin resistance in fructose-drinking rats correlates with AGEs/RAGE inhibition and block of NADPH oxidase and NF kappa B activation. Eur J Pharmacol. 2010;629(1-3):153–158. doi: 10.1016/j.ejphar.2009.11.059. [DOI] [PubMed] [Google Scholar]
- MacGillivray MH, Voorhess ML, Putnam TI, Li PK, Schaefer PA, Bruck E. Hormone and metabolic profiles in children and adolescents with type I diabetes mellitus. Diabetes Care. 1982;5(Suppl 1):38–47. [PubMed] [Google Scholar]
- Marcus DL, Thomas C, Rodriguez C, Simberkoff K, Tsai JS, Strafaci JA, Freedman ML. Increased peroxidation and reduced antioxidant enzyme activity in Alzheimer’s disease. Exp Neurol. 1998;150(1):40–44. doi: 10.1006/exnr.1997.6750. [DOI] [PubMed] [Google Scholar]
- Mark RJ, Lovell MA, Markesbery WR, Uchida K, Mattson MP. A role for 4-hydroxynonenal, an aldehydic product of lipid peroxidation, in disruption of ion homeostasis and neuronal death induced by amyloid beta-peptide. J Neurochem. 1997;68(1):255–264. doi: 10.1046/j.1471-4159.1997.68010255.x. [DOI] [PubMed] [Google Scholar]
- Markesbery WR, Kryscio RJ, Lovell MA, Morrow JD. Lipid peroxidation is an early event in the brain in amnestic mild cognitive impairment. Ann Neurol. 2005;58(5):730–735. doi: 10.1002/ana.20629. [DOI] [PubMed] [Google Scholar]
- Marshall WA, Tanner JM. Variationsin the pattern of pubertal change in girls. Arch Dis Child. 1969;44:291–303. doi: 10.1136/adc.44.235.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques F, Falcao AM, Sousa JC, Coppola G, Geschwind D, Sousa N, Correia-Neves M, Palha JA. Altered iron metabolism is part of the choroid plexus response to peripheral inflammation. Endocrinology. 2009;150(6):2822–2828. doi: 10.1210/en.2008-1610. [DOI] [PubMed] [Google Scholar]
- Martin-Gallan P, Carrascosa A, Gussinye M, Dominguez C. Oxidative stress in childhood type 1 diabetes: Results from a study covering the first 20 years of evolution. Free Radic Res. 2007;41(8):919–928. doi: 10.1080/10715760701435228. [DOI] [PubMed] [Google Scholar]
- Mastrocola R, Restivo F, Vercellinatto I, Danni O, Brignardello E, Aragno M, Boccuzzi G. Oxidative and nitrosative stress in brain mitochondria of diabetic rats. J Endocrinol. 2005;187(1):37–44. doi: 10.1677/joe.1.06269. [DOI] [PubMed] [Google Scholar]
- Nunomura A, Hofer T, Moreira PI, Castellani RJ, Smith MA, Perry G. RNA oxidation in Alzheimer disease and related neurodegenerative disorders. Acta Neuropathol. 2009;118(1):151–166. doi: 10.1007/s00401-009-0508-1. [DOI] [PubMed] [Google Scholar]
- Nunomura A, Moreira PI, Lee HG, Zhu X, Castellani RJ, Smith MA, Perry G. Neuronal death and survival under oxidative stress in Alzheimer and Parkinson diseases. CNS Neurol Disord Drug Targets. 2007;6(6):411–423. doi: 10.2174/187152707783399201. [DOI] [PubMed] [Google Scholar]
- Nunomura A, Perry G, Aliev G, Hirai K, Takeda A, Balraj EK, Jones PK, Ghanbari H, Wataya T, Shimohama S, Chiba S, Atwood CS, Petersen RB, Smith MA. Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol. 2001;60(8):759–767. doi: 10.1093/jnen/60.8.759. [DOI] [PubMed] [Google Scholar]
- Nunomura A, Perry G, Pappolla MA, Wade R, Hirai K, Chiba S, Smith MA. RNA oxidation is a prominent feature of vulnerable neurons in Alzheimer’s disease. J Neurosci. 1999;19(6):1959–1964. doi: 10.1523/JNEUROSCI.19-06-01959.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oglesbee MJ, Herdman AV, Passmore GG, Hoffman WH. Diabetic ketoacidosis increases extracellular levels of the major inducible 70-kDa heat shock protein. Clin Biochem. 2005;38(10):900–904. doi: 10.1016/j.clinbiochem.2005.05.011. [DOI] [PubMed] [Google Scholar]
- Peng ZF, Koh CH, Li QT, Manikandan J, Melendez AJ, Tang SY, Halliwell B, Cheung NS. Deciphering the mechanism of HNE-induced apoptosis in cultured murine cortical neurons: transcriptional responses and cellular pathways. Neuropharmacology. 2007;53(5):687–698. doi: 10.1016/j.neuropharm.2007.07.016. [DOI] [PubMed] [Google Scholar]
- Perry G, Nunomura A, Raina AK, Aliev G, Siedlak SL, Harris PL, Casadesus G, Petersen RB, Bligh-Glover W, Balraj E, Petot GJ, Smith MA. A metabolic basis for Alzheimer disease. Neurochem Res. 2003;28(10):1549–1552. doi: 10.1023/a:1025678510480. [DOI] [PubMed] [Google Scholar]
- Picklo MJ, Sr, Montine TJ. Mitochondrial effects of lipid-derived neurotoxins. J Alzheimers Dis. 2007;12(2):185–193. doi: 10.3233/jad-2007-12209. [DOI] [PubMed] [Google Scholar]
- Prins ML. Cerebral metabolic adaptation and ketone metabolism after brain injury. J Cereb Blood Flow Metab. 2008;28(1):1–16. doi: 10.1038/sj.jcbfm.9600543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raza H, John A. 4-hydroxynonenal induces mitochondrial oxidative stress, apoptosis and expression of glutathione S-transferase A4-4 and cytochrome P450 2E1 in PC12 cells. Toxicol Appl Pharmacol. 2006;216(2):309–318. doi: 10.1016/j.taap.2006.06.001. [DOI] [PubMed] [Google Scholar]
- Reagan LP, Magarinos AM, Yee DK, Swzeda LI, Van Bueren A, McCall AL, McEwen BS. Oxidative stress and HNE conjugation of GLUT3 are increased in the hippocampus of diabetic rats subjected to stress. Brain Res. 2000;862(1-2):292–300. doi: 10.1016/s0006-8993(00)02212-5. [DOI] [PubMed] [Google Scholar]
- Sarac K, Akinci A, Alkan A, Aslan M, Baysal T, Ozcan C. Brain metabolite changes on proton magnetic resonance spectroscopy in children with poorly controlled type 1 diabetes mellitus. Neuroradiology. 2005;47(7):562–565. doi: 10.1007/s00234-005-1387-3. [DOI] [PubMed] [Google Scholar]
- Sayre LM, Zelasko DA, Harris PL, Perry G, Salomon RG, Smith MA. 4-Hydroxynonenal-derived advanced lipid peroxidation end products are increased in Alzheimer’s disease. J Neurochem. 1997;68(5):2092–2097. doi: 10.1046/j.1471-4159.1997.68052092.x. [DOI] [PubMed] [Google Scholar]
- Sayre LM, Perry G, Smith MA. Oxidative stress and neurotoxicity. Chem Res Toxicol. 2008;21(1):172–188. doi: 10.1021/tx700210j. [DOI] [PubMed] [Google Scholar]
- Sima AA, Li ZG. The effect of C-peptide on cognitive dysfunction and hippocampal apoptosis in type 1 diabetic rats. Diabetes. 2005;54(5):1497–1505. doi: 10.2337/diabetes.54.5.1497. [DOI] [PubMed] [Google Scholar]
- Sima AAF, Zhang W, Kreipke CW, Rafols JA, Hoffman WH. Inflammation in diabetic encephalopathy is prevented by C-peptide. Rev Diab Stud. 2009a;6:37–42. doi: 10.1900/RDS.2009.6.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sima AAF, Zhang W, Muzik O, Kreipke CW, Rafols JA, Hoffman WH. Sequential abnormalities in type 1 diabetic encephalopathy and the effects of C-Peptide. Rev Diabet Stud. 2009b;6(3):211–222. doi: 10.1900/RDS.2009.6.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh P, Jain A, Kaur G. Impact of hypoglycemia and diabetes on CNS: correlation of mitochondrial oxidative stress with DNA damage. Mol Cell Biochem. 2004;260:153–159. doi: 10.1023/b:mcbi.0000026067.08356.13. [DOI] [PubMed] [Google Scholar]
- Skinner SJ, Geaney MS, Rush R, Rogers ML, Emerich DF, Thanos CG, Vasconcellos AV, Tan PL, Elliott RB. Choroid plexus transplants in the treatment of brain diseases. Xenotransplantation. 2006;13(4):284–288. doi: 10.1111/j.1399-3089.2006.00310.x. [DOI] [PubMed] [Google Scholar]
- Smith MA, Taneda S, Richey PL, Miyata S, Yan SD, Stern D, Sayre LM, Monnier VM, Perry G. Advanced Maillard reaction end products are associated with Alzheimer disease pathology. Proc Natl Acad Sci U S A. 1994a;91(12):5710–5714. doi: 10.1073/pnas.91.12.5710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MA, Kutty RK, Richey PL, Yan SD, Stern D, Chader GJ, Wiggert B, Petersen RB, Perry G. Heme oxygenase-1 is associated with the neurofibrillary pathology of Alzheimer’s disease. Am J Pathol. 1994b;145(1):42–47. [PMC free article] [PubMed] [Google Scholar]
- Smith MA, Harris PL, Sayre LM, Perry G. Iron accumulation in Alzheimer disease is a source of redox-generated free radicals. Proc Natl Acad Sci U S A. 1997;94(18):9866–9868. doi: 10.1073/pnas.94.18.9866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MA, Rottkamp CA, Nunomura A, Raina AK, Perry G. Oxidative stress in Alzheimer’s disease. Biochim Biophys Acta. 2000;1502(1):139–144. doi: 10.1016/s0925-4439(00)00040-5. [DOI] [PubMed] [Google Scholar]
- Steen E, Terry BM, Rivera EJ, Cannon JL, Neely TR, Tavares R, Xu XJ, Wands JR, de la Monte SM. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease--is this type 3 diabetes? J Alzheimers Dis. 2005;7(1):63–80. doi: 10.3233/jad-2005-7107. [DOI] [PubMed] [Google Scholar]
- Sternberger LA. Immunocytochemistry. New York: Wiley; 1986. [Google Scholar]
- Su B, Wang X, Nunomura A, Moreira PI, Lee HG, Perry G, Smith MA, Zhu X. Oxidative stress signaling in Alzheimer’s disease. Curr Alzheimer Res. 2008;5(6):525–532. doi: 10.2174/156720508786898451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syapin PJ. Regulation of haeme oxygenase-1 for treatment of neuroinflammation and brain disorders. Br J Pharmacol. 2008;155(5):623–640. doi: 10.1038/bjp.2008.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turk Z, Nemet I, Varga-Defteardarovic L, Car N. Elevated level of methylglyoxal during diabetic ketoacidosis and its recovery phase. Diabetes Metab. 2006;32(2):176–180. doi: 10.1016/s1262-3636(07)70266-5. [DOI] [PubMed] [Google Scholar]
- Vantyghem MC, Balduyck M, Zerimech F, Martin A, Douillard C, Bans S, Degand PM, Lefebvre J. Oxidative markers in diabetic ketoacidosis. J Endocrinol Invest. 2000;23(11):732–736. doi: 10.1007/BF03345062. [DOI] [PubMed] [Google Scholar]
- Wang J, Xiong S, Xie C, Markesbery WR, Lovell MA. Increased oxidative damage in nuclear and mitochondrial DNA in Alzheimer’s disease. J Neurochem. 2005;93(4):953–962. doi: 10.1111/j.1471-4159.2005.03053.x. [DOI] [PubMed] [Google Scholar]
- Wootton-Gorges SL, Buonocore MH, Kuppermann N, Marcin JP, Barnes PD, Neely EK, DiCarlo J, McCarthy T, Glaser NS. Cerebral proton magnetic resonance spectroscopy in children with diabetic ketoacidosis. AJNR Am J Neuroradiol. 2007;28(5):895–899. [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Rottkamp CA, Boux H, Takeda A, Perry G, Smith MA. Activation of p38 kinase links tau phosphorylation, oxidative stress, and cell cycle-related events in Alzheimer disease. J Neuropathol Exp Neurol. 2000;59(10):880–888. doi: 10.1093/jnen/59.10.880. [DOI] [PubMed] [Google Scholar]
- Zhu X, Raina AK, Lee HG, Chao M, Nunomura A, Tabaton M, Petersen RB, Perry G, Smith MA. Oxidative stress and neuronal adaptation in Alzheimer disease: the role of SAPK pathways. Antioxid Redox Signal. 2003;5(5):571–576. doi: 10.1089/152308603770310220. [DOI] [PubMed] [Google Scholar]