

Abstract
XXYY syndrome occurs in approximately 1:18,000–1:40,000 males. Although the physical phenotype is similar to 47,XXY (tall stature, hypergonadotropic hypogonadism, and infertility), XXYY is associated with additional medical problems and more significant neurodevelopmental and psychological features. We report on the results of a cross-sectional, multicenter study of 95 males age 1–55 with XXYY syndrome (mean age 14.9 years), describing diagnosis, physical features, medical problems, medications, and psychological features stratified by age groups. The mean age of diagnosis was 7.7 years. Developmental delays and behavioral problems were the most common primary indication for genetic testing (68.4%). Physical and facial features varied with age, although hypertelorism, clinodactyly, pes planus, and dental problems were common across all age groups. Tall stature was present in adolescents and adults, with a mean adult stature of 192.4 cm (SD 7.5; n = 22). Common medical problems included allergies and asthma (>50%), congenital heart defects (19.4%), radioulnar synostosis (17.2%), inguinal hernia and/or cryptorchidism (16.1%), and seizures (15%). Medical features in adulthood included hypogonadism (100%), DVT (18.2%), intention tremor (71%) and type II diabetes (18.2%). Brain MRI (n = 35) showed white matter abnormalities in 45.7% of patients and enlarged ventricles in 22.8%. Neurodevelopmental and psychological difficulties were a significant component of the behavioral phenotype, with developmental delays and learning disabilities universal but variable in severity. Twenty-six percent had full-scale IQs in the range of intellectual disability (MR), and adaptive functioning was significantly impacted with 68% with adaptive composite scores <70. Rates of neurodevelopmental disorders, including ADHD (72.2%), autism spectrum disorders (28.3%), mood disorders (46.8%), and tic disorders (18.9%), were elevated with 55.9% on psychopharmacologic medication overall. Recommendations for evaluation and treatment are summarized.
Keywords: XXYY syndrome, Klinefelter syndrome, sex chromosome abnormality, autism spectrum disorder, tremor, ADHD
Introduction
48,XXYY syndrome was first described in 1964 and occurs in an estimated 1:18,000–1:50,000 males [Muldal and Ockey, 1960; Sorensen et al., 1978]. Approximately 100 cases have been reported to date, often characterizing XXYY syndrome as a variant of 47,XXY Klinefelter syndrome due to a shared physical and endocrinologic phenotype including tall stature, microorchidism, hypergonadotropic hypogonadism, and infertility [Borgaonkar et al., 1970]. Although original reports suggested that there were no phenotypic differences from 47,XXY males [Townes et al., 1965], it is now recognized that 48,XXYY differs in its medical, neurodevelopmental, and behavioral characteristics [Parker et al., 1970; Zack, 1980; Grammatico et al., 1990; Linden et al., 1995; Zelante et al., 2003; Tartaglia et al., 2005].
The last large review of the XXYY syndrome phenotype summarizing the first 53 described cases was published by Borgaonkar et al. [1970], with many subsequent individual case reports or short case series describing various medical and psychological features. In addition to the characteristic physical features of Klinefelter syndrome (tall stature, hypogonadism, gynecomastia), literature reports have described hypertelorism, cubitus varus, pes planus, clubfoot, clinodactyly, and scoliosis [Carr et al., 1961; Barr et al., 1964; Alter et al., 1966; Borgaonkar et al., 1970; Demirhan, 2003]. Neurological problems, including seizures, intention tremor, hypotonia, and tics have also been reported in multiple patients [Robinson et al., 1964; Alter et al., 1966; Sorensen et al., 1978; Garry, 1980; Vanyan, 1984; Donati et al., 1992; Izumi and Tsubahara, 2000; Zelante et al., 2003], along with occasional structural brain abnormalities including agenesis of the corpus callosum [Nyberg et al., 1994] and frontoparietal cortical atrophy [Demirhan, 2003]. One study estimated that congenital heart ddefects are present in approximately 8% of patients, with specific reports of tetralogy of Fallot, pulmonary stenosis, mitral valve prolapse, and septal defects [Schlegel et al., 1965; Alter et al., 1966; Meschede et al., 1995; Zelante et al., 2003]. Other reported medical problems in childhood include strabismus, inguinal hernia, cryptorchidism, asthma and recurrent respiratory infections, and dental problems [Townes et al., 1965; Waterman et al., 1966; Singer et al., 1973; Garvey and Kellett, 1975; Sorensen et al., 1978]. Lower extremity skin ulcers have been described in adults [Fuse et al., 1986; Grammatico et al., 1990; Izumi and Tsubahara, 2000], and increased mortality from non-Hodgkin lymphoma has also been reported [Swerdlow et al., 2005].
Speech delays and motor delays with poor motor coordination are commonly described in the first 3 years of life [Garvey and Mutton, 1973; Garvey and Kellett, 1975; Fryns et al., 1995]. In older children and adults, full-scale IQ reports have ranged from as low as 35 up to 111 [Laurence et al., 1963; Sachsse et al., 1967], although in most cases IQ has ranged from 70 to 80 [Borgaonkar et al., 1970]. IQ profiles generally show a verbal IQ significantly lower than performance IQ [Zack, 1980], similar to the cognitive profile seen in 47,XXY males [Netley, 1986]. Although most cases reported in the literature describe some degree of cognitive disability or behavioral problems, a few reports describe adolescents with “good social adjustment” ascertained because of pubertal delay or microorchidism [Loftus and Mellman, 1965]. Behavioral and psychiatric symptoms including hyperactivity, attention problems, impulsivity, aggression, mood instability, and “autistic-like” behaviors have also been described [Schlegel et al., 1965; Sorensen et al., 1978; Fryns et al., 1995; Hagerman, 1999].
In this cross-sectional, multi-institutional study, we describe the largest cohort of males with non-mosaic XXYY syndrome to date, including 95 males ranging from 1 to 55 years of age. We describe their ascertainment, physical findings, associated medical problems, developmental and psychological features, and include recommendations for medical evaluation and treatment.
Methods
Recruitment
Participants were recruited primarily from organizations supporting individuals and families with sex chromosome aneuploidy (SCA) to participate in an IRB-approved study on health and development in children and adults with SCA. Additional participants were identified from endocrinology, genetics, and pediatrics clinics at each site. Individuals from the US (n = 66) and Canada (n = 5) were seen at the UC-Davis Medical Center MIND Institute, The Children's Hospital Child Development Unit in Denver, and Thomas Jefferson University in Philadelphia. A group of participants from the United Kingdom and various European countries (n = 23), and Australia (n = 1) also participated through the UC-Davis site. All participants or their parents signed an IRB-approved consent form developed for each site prior to participation in research.
Subjects
The participants included 95 males with XXYY syndrome, ranging from 1 to 55 years of age, with a mean age of 14.9 years and median of 13.0 (see Table I). The majority of participants were in the 11–19 year age group (42%), and almost all (96.8%) were Caucasian. African-Americans and other minorities were not excluded from the study. The low percentage of minorities in this study is likely due to many factors, including underascertainment in minority populations, and recruitment through support groups that are largely Internet-based and thus less accessible to minorities of lower socioeconomic status. Two sets of twins with XXYY syndrome were participants in the study.
TABLE I. Age and Ethnicity of Participants.
| N = 95 | |
|---|---|
| Mean age/SD | 14.9 ± 9.2 |
| Age range | 1.8–55.0 |
| Age group | |
| 1–10 years | 33 (34.7%) |
| 11–19 years | 40 (42.1%) |
| 20–29 years | 15 (15.7%) |
| 30+ years | 7 (7.4%) |
| Ethnicity | |
| Caucasian (n = 92) | 96.8% |
| Hispanic (n = 1) | 1.0% |
| Asian (n = 2) | 2.1% |
Evaluation
The parent or primary caregiver of each participant first completed a comprehensive questionnaire detailing birth, medical, developmental, and psychological history. These features were then reviewed in a semi-structured direct interview and recorded on a data sheet completed for each participant. Medical and educational records were reviewed, including genetic testing results, to confirm a non-mosaic 48,XXYY karyotype. For participants from the U.S. and Canada, height, weight, occipitofrontal head circumference, and stretched penile length were measured directly and converted to Z-scores (standard deviation scores). Testicular volume was estimated using a Prader orchidometer. The presence or absence of common dysmorphic features and features selected based on review of the literature on XXYY syndrome and other sex chromosome aneuploidies were assessed by physical examination. Poor dentition was classified as four or more caries/fillings, significant malocclusion, current or previous orthodontia, the need for previous dental surgery, taurodontism, or edentulous. For the group from the UK, height and weight were extracted from recent medical records, and physical examination included only direct evaluation of facial features and extremities by one examiner (N.T.) at a family conference where medical facilities for a more comprehensive examination were not available.
Developmental assessment of children less than 6 years of age was completed using the Mullen Scales of Early Learning—AGS Edition [Mullen, 1989]. Cognitive evaluation of 46 participants 6 years of age or older was performed using the Wechsler Abbreviated Scales of Intelligence (WASI) [Wechsler, 1989]. For a small subset of participants (n = 4), cognitive scores were obtained by review of results of comparable standardized assessments administered by a licensed psychologist obtained within the previous 12 months. As per current practice, the term “intellectual disability” replaces the previous term “mental retardation” throughout this text. Adaptive functioning was assessed using either the Vineland-2 adaptive functioning interview [Sparrow et al., 2005] or the Adaptive Behavior Assessment System—2nd Edition [Harrison and Oakland, 2003].
Analysis of Results
Patients were divided into three age groups and features were evaluated separately in each age group due to differences in the natural history of various medical features such as hypogonadism, scoliosis, and gynecomastia. Incomplete data are noted in the tables, with data missing due to parents or primary caregivers being unsure or unable to recollect the information. Percentages reported include only data that was definitively present or absent, thus the total n included in calculations varies between fields but is noted in the tables. Ninety-five percent confidence intervals for frequency of associated medical features, with adjustments for frequencies <5%, were calculated as per Richardson et al. [2000]. Features were excluded if the confidence interval overlapped zero, unless that feature had been previously reported in the XXYY literature in more than one other case (talipes equinovarus, cleft palate/velopharyngeal insufficiency, and unilateral kidney). Height and BMI Z-scores (standard deviation scores) and cognitive scores were compared between age groups using ANOVA with post hoc Tukey analysis (SPSS), and results of these analyses are noted. Statistical significance was set at P < 0.05.
Results
Diagnosis of XXYY Syndrome
The age of diagnosis and the indication for genetic testing obtained by review of medical records from the time of diagnosis are presented in Tables II and III. Only two patients were diagnosed prenatally. Almost 10% (9/95) of the group was diagnosed at less than 1 year of age, and these cases were associated with more significant medical problems and dysmorphic features noted at the time of birth (i.e., cleft palate/Pierre Robin sequence, congenital heart malformation), or significant early motor delays. Approximately one-third of the patients (35/95) were diagnosed between 1 and 5 years of age, with the majority of these patients tested because of speech and/or motor delays. Four subjects in this age group received genetic testing following the diagnosis of an autism spectrum disorder. Approximately 25% (23/95) of patients were diagnosed between ages 6–10, primarily as part of the evaluation for cognitive delays and/or behavioral problems. Of the 27% (26/95) diagnosed after 11 years of age, 11 were because of microorchidism or hypogonadism, although all individuals in this group had a history of developmental delays, learning disorders, intellectual disability, or psychiatric problems that had not previously prompted genetic testing. Only two individuals were diagnosed after 21 years of age, both when evaluated by new physicians for a longstanding history of intellectual disability and mental health problems. The mean age of diagnosis in those diagnosed in the postnatal period across the entire group was 7.7 years (SD 5.62).
TABLE II. Age at Diagnosis of XXYY Syndrome.
| Age at diagnosisa | Percentage (n = 95) |
|---|---|
| Prenatal diagnosis | 2.1% (2) |
| Less than 1 year | 9.5% (9) |
| 1–5 years | 36.8% (35) |
| 6–10 years | 24.2% (23) |
| 11–19 years | 23.2% (22) |
| 20+ years | 4.2% (4) |
Mean age of diagnosis across the entire group was 7.7 years (SD 5.62).
TABLE III. Primary Indications for Genetic Testing.
| Ascertainment | N = 95 | % | Dx at birth–5 years, n = 44 (46.3%) |
Dx at 6–10 years, n = 23 (24.2%) |
Dx at ≥11 years, n = 26 (27.4%) |
|---|---|---|---|---|---|
| Prenatal | n = 2 | 2.1 | |||
| Advanced maternal age | 1 | 1.1 | |||
| Elevated maternal alpha fetoprotein | 1 | 1.1 | |||
| Development and behaviors | n = 65 | 68.4 | |||
| Speech and/or motor delays | 28 | 29.5 | 26 (59.1%) | 2 (8.7%) | 0 |
| Learning disability/intellectual disability | 21 | 22.1 | 2 (4.5%) | 11 (47.8%) | 8 (30.7%) |
| Autism spectrum disorder | 4 | 4.2 | 3 (6.8%) | 0 | 1 (3.8%) |
| Other behavior/psychiatric problemsa | 12 | 12.6 | 1 (2.2%) | 6 (26.0%) | 5 (19.2%) |
| Medical features | n = 28 | 29.5 | |||
| Dysmorphic featuresb | 7 | 7.4 | 5 (11.4%) | 2 (8.7%) | 0 |
| Microorchidism, delayed/incomplete puberty or testosterone deficiency | 11 | 11.6 | 0 | 0 | 11 (42.3%) |
| Tall stature | 2 | 2.1 | 0 | 1 (4.3%) | 1 (3.8%) |
| Other | 8.4 | ||||
| Radioulnar synostosis | 1 | 0 | 1 (4.3%) | 0 | |
| Congenital heart defectsc | 2 | 2 (4.5%) | 0 | 0 | |
| Cleft palate/Pierre-Robin | 1 | 1 (2.2%) | 0 | 0 | |
| Micropenis in infancy | 1 | 1 (2.2%) | 0 | 0 | |
| Hypotonia | 2 | 2 (4.5%) | 0 | 0 | |
| Intention tremor | 1 | 1 (2.2%) | 0 | 0 |
Behavioral/psychiatric problems included attention deficits/ADHD, tantrums, impulsivity, mood instability, and bipolar disorder.
Dysmorphic features listed include: plagiocephaly, hypertelorism, epicanthal folds, retrognathia, clinodactyly, and single palmar crease. One subject had dysplastic ears and a diagnosis of branchio-oto-renal syndrome.
Congenital heart defects included ventricular septal defect and L-transposition of the great arteries with ASD and VSD.
Physical Examination Findings
Results of physical examination findings are shown in Table IV, with auxologic measurements reported as Z-scores (standard deviation scores). Mean height Z-score significantly increased with age from +0.21 ± 1.45 in children less than 10 years old (n = 25), to +1.06 ± 1.17 in the 11–19 age group (n = 35) and +2.18 ± 1.08 in the 20+ age group (n = 22) (ANOVA F17.6, P < 0.0001). The mean stature of adult males (n = 22) was 192.4 cm (6 ft 3.7 in) with a standard deviation of 7.5 cm and a range from 180.1 cm (5 ft 11 in) to 216 cm (7 ft 1 in). In contrast to height, mean BMI Z-scores did not significantly differ between age groups (age 0–10 +0.84, age 11–19 +0.20, age 20+ 0.76; ANOVA F1.4, P = 0.25). Although mean BMI was stable across age groups, there was considerable variation in body habitus within each age group, and individuals ranged from underweight (childhood BMI <10th centile or adult BMI <20) to overweight (childhood BMI >85th centile or adult BMI >25). Mean adult BMI was 27.3 with a standard deviation of 6.7. Among the adults 18.2% were underweight while were 31.8% obese. As expected, testicular volume remained small with progression through adolescence, and microorchidism was present in 100% of patients over age 20 with an average adult testicular volume of 5.3 ml. Gynecomastia was present in 25% of adolescents and 41% of adults.
TABLE IV. Physical Examination Results*.
| Age ≤10 (n = 32) | Age 11–19 (n = 38) | Age 20+ (n = 22) | |
|---|---|---|---|
| Mean height (Z-score)a; height range (Z-score) | +0.21, n = 25; (−4.5 to +2.77) | +1.06, n = 35; (−0.74 to +4.26) | +2.18, n = 22; (+0.86 to +5.56) |
| Weight (Z-score) | +0.44, n = 24 | +0.67, n = 31 | +1.81, n = 21 |
| Body mass index (Z-score); body mass index range (Z-score) | +0.84, n = 24; (−2.16 to + 4.2) | +0.20, n = 31; (−2.53 to +2.3) | +0.76, n = 21; (−3.04 to +2.76) |
| Head circumference (Z-score) | −0.15, n = 17 | +0.81, n = 21 | +1.78, n = 11 |
| Phallus length (Z-score) | −1.12, n = 13 | −0.58, n = 21 | −0.99, n = 10 |
| Testicular volume (Z-score) | 0.16, n = 14 | −2.51, n = 21 | −6.61, n = 11 |
| Obesityb | 12.5%, n = 24 | 9.7%, n = 31 | 31.8%, n = 21 |
| Underweightc | 4.2%, n = 24 | 9.7%, n = 31 | 18.2%, n = 21 |
| Facial asymmetry | 52% | 34% | 45% |
| Long face | 9% | 32% | 68% |
| Epicanthal folds | 55% | 45% | 27% |
| Hypertelorism | 72% | 50% | 57% |
| Narrow palpebral fissures | 29% | 46% | 45% |
| Upslanting palpebral fissures | 58% | 31% | 18% |
| Hooded eyelid | 48% | 54% | 57% |
| Bitemporal narrowing | 19% | 31% | 64% |
| Prominent brow | 0% | 20% | 64% |
| Full lips | 37% | 37% | 45% |
| High arch palate | 26% | 36% | 29% |
| Poor dentitiond | 71% | 86% | 95% |
| Micrognathia | 48% | 21% | 5% |
| 5th digit clinodactyly | 80% | 62% | 75% |
| Cubitus varus | 36% | 61% | 71% |
| Pes planus | 79% | 78% | 81% |
| Pectus excavatum | 62%, n = 24 | 43%, n = 28 | 53%, n = 17 |
| Scoliosis | 0%, n = 24 | 25%, n = 28 | 24%, n = 17 |
| Gynecomastia | 0%, n = 24 | 25%, n = 28 | 41%, n = 17 |
| Microorchidism | 21%, n = 24 | 75%, n = 28 | 100%, n = 17 |
| Hypotonia | 65% | 16% | 24% |
| Intention tremor | 8% | 62% | 71% |
Number of subjects at top of column unless otherwise noted.
Mean stature of adults 192.4 cm (6 ft 3.7 in), SD 7.5.
Obesity defined as BMI >95th centile for ages 1–18 and BMI >30 for adults.
Underweight defined as BMI <5th centile for ages 1–18 and BMI <20 for adults.
Poor dentition classified as four or more caries/fillings, significant malocclusion, current or previous orthodontia, or edentulous.
Some physical examination features were consistent across all three age groups, while some emerged with increasing age. Figures 1 and 2 illustrate common facial and physical findings in XXYY. The most common physical features in prepubertal children were 5th digit clinodactyly, pes planus, poor dentition, apparent hypertelorism, upslanting palpebral fissures, epicanthal folds and hypotonia. Micrognathia was most common in the youngest group, but decreased with age; older males had a long face with a prominent chin and brow. Limited supination and pronation of the forearm was present in 61% of cases. Intention tremor typically began at age 12–13, although it was present in some children as young as 6 years of age. Intention tremor increased significantly with advancing age, and was present in 71% of individuals in the adult age group.
Fig. 1.

Facial features in XXYY syndrome across different age groups. Note varying degree of dysmorphic features between individuals. Common facial features include mild hypertelorism, narrow and upslanting palpebral fissures, and full lips. In adulthood a long face, prominent brow, and bitemporal narrowing are common.
Fig. 2.

Common physical features in XXYY syndrome including clinodactyly of varying severity, hypotonia with genu valgum and pes planus with pronation at the ankles, and cubitus varus.
Medical Features and Medications
Eighty-six percent (80/95) of infants were born at term, with the average birthweight of term infants at 3,130 g (6.9 pounds). Early feeding problems including problems with latch-on, poor suck, and slow feeding were noted in 51% (39/77) of infants. Overall, the most common medical features were significant dental problems, intention tremor (in the adolescent and adult age groups), and asthma/reactive airway disease (Table V). Forty-six percent (43/93) had been hospitalized in their lifetime for respiratory infections or asthma (most commonly in the first 5 years of life), and 16.1% of the total group had required more than three hospitalizations for recurrent respiratory infections or asthma exacerbations. Seventeen percent (16/93) had confirmed radioulnar synostosis and/or congenital elbow dislocation by radiograph, although only 23 individuals had elbow radiographs completed. Eleven percent had obstructive sleep apnea. Notable medical problems of other study participants not included in the table include one child who died at age 13 from thrombotic thrombocytopenic purpura and one individual with panhypopituitarism. There is also one known male with XXYY who died from colorectal cancer at 29 years of age and was unable to participate in the current study.
TABLE V. Medical History in XXYY Syndrome.
| Medical feature | All ages | 95% CI | ||
|---|---|---|---|---|
| # | % | Lower | Upper | |
| Strabismus | 14/91 | 15.4 | 8.3 | 22.5 |
| Significant dental problemsa | 78/89 | 87.6 | 81.2 | 94.1 |
| Food/environmental allergies | 50/90 | 55.6 | 45.8 | 65.3 |
| Asthma/reactive airway disease | 55/92 | 59.8 | 50.2 | 69.4 |
| Recurrent otitis with tympanostomy tubes | 12/93 | 12.9 | 6.3 | 19.5 |
| Hospitalization for respiratory infection or asthmab | 43/93 | 46.2 | 36.5 | 56.0 |
| Gastroesophageal reflux | 17/88 | 19.3 | 11.6 | 27.1 |
| Constipation | 36/89 | 40.4 | 30.8 | 50.1 |
| Unilateral kidney | 3/91 | 3.3 | — | 6.8 |
| Cardiac abnormalitiesc | 18/93 | 19.4 | 11.6 | 27.1 |
| VSD | 10 | 10.8 | 4.7 | 16.8 |
| ASD | 1 | 1.1 | — | 3.1 |
| L-Transposition w/ASD and VSD | 1 | 1.1 | — | 3.1 |
| Mitral valve prolapse | 4 | 4.3 | 0.3 | 8.3 |
| Pulmonic stenosis | 1 | 1.1 | — | 3.1 |
| PDA | 1 | 1.1 | — | 3.1 |
| Radioulnar synostosis/congenital dislocation | 16/93 | 17.2 | 9.8 | 24.6 |
| Cleft palate | 2/93 | 2.2 | — | 5.0 |
| Palatoplasty for VPI (no cleft)d | 2/93 | 2.2 | — | 5.0 |
| Clubfoot | 2/94 | 2.1 | — | 5.0 |
| Inguinal hernia repair and/or orchipexy | 15/93 | 16.1 | 8.9 | 23.3 |
| Nonfebrile seizures | 14/93 | 15.0 | 8.0 | 22.1 |
| Obstructive sleep apnea | 5/44 | 11.4 | 5.1 | 17.6 |
| Medical feature | Age 11–19 (n = 40) | Age >20 (n = 22) | ||
| Scoliosis | 8/39 | 20.5% (12.6–28.4) | 7/22 | 31.8% (22.7–40.9) |
| Hypergonadotropic hypogonadisme | 18/40 | 45.0% (35.2–54.8) | 22/22 | 100% (100) |
| Hypothyroidism | 4/38 | 10.5% (4.5–16.5) | 2/22 | 9.1% (3.5–14.7) |
| Type II diabetes mellitus | 0/40 | 0% | 4/22 | 18.2% (10.6–25.7) |
| Deep venous thrombosis | 0/40 | 0% | 4/22 | 18.2% (10.6–25.7) |
| Lower extremity skin ulcer | 2/38 | 5.2% (0.9–9.6) | 4/22 | 18.2% (10.6–25.7) |
| Intention tremor | 23/38 | 60.5% (50.9–70.1) | 16/22 | 72.7% (64.0–81.5) |
| Psychiatric hospitalization | 2/38 | 5.3% (0.9–9.6) | 8/22 | 36.4% (26.9–45.8) |
| Brain MRI (n = 35) | ||||
| T2 white matter hyperintensities | 16/35 | 45.7% | ||
| Enlarged ventricles | 8/35 | 22.8% | ||
| Agenesis of corpus callosum | 2/35 | 5.7% | ||
| Corpus callosum lipoma | 3/35 | 8.6% | ||
| Cortical dysplasia | 3/35 | 8.6% | ||
| Pituitary adenoma | 1/35 | 2.8% | ||
Significant dental problems were defined as the need for three or more dental procedures (excluding common caries) or the need for orthodontia.
Fifteen of these individuals required more than three hospitalizations for respiratory infections or asthma exacerbations.
One patient with an isolated ASD required surgical repair. In all cases of VSD, the defect closed spontaneously without surgical intervention. The PDA was in a term infant without neonatal complications and it closed spontaneously. The case with L-transposition did not require surgical intervention.
VPI = velopharyngeal insufficiency.
Hypergonadotropic hypogonadism determined by history of elevated leutinizing hormone (LH) with decreased or low-normal testosterone levels.
Current medications are presented in Table VI. Psychopharmacologic medications were the most common medication class and 56% (52/93) of the subjects required one or more medications primarily targeting attention span, impulsivity, anxiety, and mood instability. Twenty of these 52 individuals were on more than one psychopharmacologic medication. In the adult group, approximately one-third had required hospitalization during their adolescence or later for psychiatric reasons.
TABLE VI. Current Medications in XXYY Participants.
| Medication class | # | % |
|---|---|---|
| All medications | 78/93 | 83.9 |
| Psychopharmacologic medicationsa | 52/93 | 55.9 |
| Stimulants | 29/93 | 31.2 |
| Serotonin specific reuptake inhibitors | 17/93 | 18.3 |
| Atypical neuroleptics | 21/93 | 22.6 |
| Mood stabilizers (lithium or depakote) | 10/93 | 10.8 |
| Otherb | 9/93 | 9.7 |
| Anticonvulsants | 6/93 | 6.5 |
| Tremorc | 4/93 | 4.3 |
| Respiratory medicationsd | 35/93 | 37.6 |
| Gastrointestinal medicationse | 12/93 | 12.9 |
| Current testosterone replacement | ||
| Age 11–14 | 8/23 | 34.8 |
| Age 15–19 | 11/17 | 64.7 |
| Age 20+ | 14/22 | 63.6 |
Includes all participants on one or more psychopharmacologic medications. Twenty out of 52 (38.5%) were on more than one psychopharmacologic medication.
Other medications included: atomoxetine (2), clonidine (1), buprioprion (2), buspirone (2), clonazepam (2), amitriptylene (1).
Tremor medications included propranolol (2), amantadine (1) and primidone (1).
Respiratory medications included prescription medications for allergies or asthma/RAD.
Gastrointestinal medications included prescription medications for gastroesophageal reflux or constipation.
Hypergonadotropic hypogonadism was observed in pubertal-aged boys, and by adulthood all participants had LH/FSH and testosterone levels indicating hypogonadism and/or had been started on testosterone replacement therapy. The average age for initiation of testosterone therapy was 14.9 years (range 11–31). Despite microorchidism and laboratory evidence of hypogonadism in all, only 14/22 (63.6%) of the adult group were being actively treated with testosterone. Review of medical records of the eight subjects not receiving testosterone therapy showed: previous testosterone use with loss of follow-up in long-term care facility (3), concerns about worsening of behavioral symptoms upon initiation of testosterone treatment (3), one individual who disliked the effects, and one individual with total testosterone in the low-normal range and elevated LH who was advised that treatment was not indicated.
Neuroimaging
A subset of the sample had brain MRI performed (n = 35). The indications for MRI included cognitive or developmental problems, behavioral difficulties, hypotonia, and seizures. Review of the images showed nonspecific white matter abnormalities in 45.7% and enlarged ventricles in 22.8%. White matter abnormalities displayed a spectrum from tiny bifrontal (1 mm size) foci of increased T2/Flair signal to larger (3–5 mm size) confluent bifrontal and parietal foci. Abnormal signal involved both superficial and deep periventricular white matter, but spared the corpus callosum and internal capsule. Examples are shown in Figure 3. Four subjects had MRI imaging in their first 3 years of life, with white matter findings present at this time. Additional MRI findings are shown in Table V.
Fig. 3.
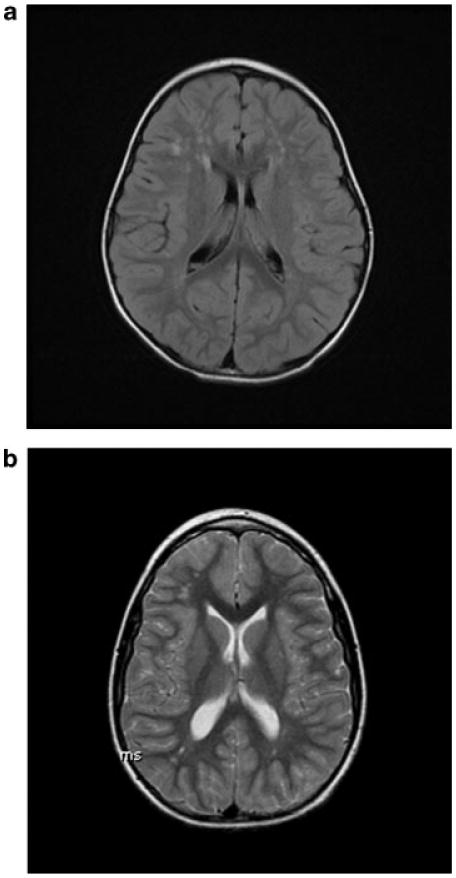
Brain MRI findings of frontal lobe white matter hyperintensities in XXYY. a: Axial flair MR image shows multiple rounded sub-centimeter foci of increased signal in bifrontal superficial and deep periventricular white matter in a 3-year-old male. b: 12-year-old male. Axial T2 MR image shows multiple foci of increased signal in bifrontal and biparietal superficial and deep periventricular white matter (smaller, but more diffusely distributed compared to image A).
Neurodevelopmental, Cognitive, and Psychiatric Conditions
Early developmental delays were almost universal, with 91.5% reporting delays in speech/language development and 74.5% reporting delays in motor development. The average age of walking independently was 18.0 months (n = 84, range 12–33 m) and the average age at first words was 23.6 months (n = 64, range 10–64 m). Direct developmental assessment of 5 children less than 6 years of age showed a wide range of developmental levels from significantly impaired to the average range, although relative weaknesses in gross motor skills, fine motor skills, and expressive language were universal. As shown in Table VII, the mean full-scale IQ (FSIQ) of individuals 6 years or older was 77.8, with a significantly lower mean verbal IQ (VIQ) (74.0) relative to mean performance IQ (PIQ) (87.4) (n = 47, t = 5.29, P < 0.0001). When IQ was compared between the three age groups, the VIQ decreased with increasing age, but PIQ remained unchanged (Fig. 4). The 20+ age group had a significantly lower verbal IQ compared to the younger age groups, with a mean VIQ for the oldest age group of 66.7 compared to a mean of 76.0 in children 11–19 and a mean of 80.2 in children 6–10 years. Despite significant differences across age groups in verbal IQ, there were not statistically significant differences between the three age groups in PIQ or FSIQ, although the FSIQ trended lower secondary to the decreasing VIQ (ANOVA VIQ F4.87, P < 0.05; PIQ F0.14, P = 0.9; FSIQ F1.47, P = 0.24).
TABLE VII. Cognitive Assessment and Adaptive Functioning in Participants Age 6–55.
| Mean | SD | Range | |
|---|---|---|---|
| VIQ (n = 47) | 74.0 | 11.9 | 55–100 |
| PIQ (n = 47) | 87.4 | 13.0 | 62–108 |
| FSIQ (n = 50) | 77.8 | 11.5 | 54–102 |
| Adaptive composite (n = 43) | 68.9 | 13.8 | 33–92 |
VIQ, verbal intelligence quotient; PIQ, performance intelligent quotient; FSIQ, full-scale IQ.
Fig. 4.

Mean verbal IQ, performance IQ, and full-scale IQ in different age groups. Verbal IQ decreases with age. There is no significant change in performance or full-scale IQ. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Cognitive problems were universal but variable in degree, and were most commonly language-based learning disabilities with deficits in reading and language comprehension. Interventions for learning difficulties varied from mainstream classrooms with individual tutoring to self-contained classrooms for children with intellectual disability (Tables VII and VIII). Of the 50 individuals for whom reliable cognitive testing was directly obtained or reviewed, 26% had a FSIQ in the ID range, with 48% in the borderline range of 70–84, and the remaining 26% with FSIQ of 85 or above. Adaptive functioning was significantly impaired when compared to IQ, especially in those over 18 where 68% fell below a score of 70. Adaptive functioning was most commonly impaired in the domains of communication, social skills, self-care, and self-direction.
TABLE VIII. Previous Neurodevelopmental and Psychiatric Diagnoses.
| Diagnosis | # | % |
|---|---|---|
| Speech delay | 86/94 | 91.5 |
| Motor delay | 70/94 | 74.5 |
| Autism spectrum disordera | 26/92 | 28.3 |
| Learning disability | 79/79 | 100 |
| Mental retardation/intellectual disability | 23/79 | 29.1 |
| ADHD | 57/79 | 72.2 |
| Oppositional defiant disorder | 8/79 | 10.1 |
| Mood disorder | 37/79 | 46.8 |
| Generalized anxiety disorder | 10/79 | 21.1 |
| Obsessive-compulsive disorder | 6/79 | 7.6 |
| Depression | 14/79 | 17.7 |
| Bipolar disorder | 6/79 | 7.6 |
| Cyclothymia | 1/79 | 1.2 |
| Schizoaffective disorder/schizophrenia | 3/79 | 3.8 |
| Impulse control disorder | 4/79 | 5.1 |
| Tourette syndrome/Tic disorder | 15/79 | 18.9 |
| Cognitive impairment | 79/79 | 100 |
| Learning disability | 56/79 | 70.9 |
| Intellectual disability | 23/79 | 29.1 |
Only participants age 6 or over (n = 79) were included in the analysis for learning disability/ID and other psychiatric diagnoses since these are not commonly diagnosed at less than 6 years of age.
Autism spectrum disorders included autistic disorder (n = 6) and PDD-NOS (n = 20).
Review of previous neurodevelopmental and psychiatric diagnoses included many DSM-IV diagnoses, most commonly attention deficit/hyperactivity disorder (ADHD), autism spectrum disorders (ASD), and mood disorders (Table VIII). Two individuals were being treated for psychotic disorders with paranoid delusions and auditory hallucinations. Other common diagnoses with less specific diagnostic criteria included sensory processing/sensory integration disorder (n = 16), developmental apraxia of speech (n = 9) and auditory processing disorder (n = 6). Parents also frequently reported sleep cycle disturbances, a worsening of behavioral symptoms at 8–9 years of age, sugar cravings, nail biting, and strong interests in computers and vehicles as part of the behavioral phenotype of XXYY.
For the two children who were diagnosed prenatally, there were no significant differences in IQ or in the presence of other neurodevelopmental disorders compared to those ascertained postnatally. Both had early speech and motor delays requiring therapy, both were in special education for language-based learning disabilities, and one had been diagnosed with ASD. The mean IQ of individuals ascertained at puberty for microorchidism (n = 11) was slightly higher when compared to those ascertained due to cognitive or psychological problems, however this did not reach statistical significance (Mean FSIQ 84 vs. 76, t = 1.57, P = 0.12). However, none of the 11 boys ascertained due to microorchidism had a full-scale IQ of less than 70.
Discussion
This is the largest study to date of medical and psychological features of children and adults with XXYY syndrome. This cohort was primarily Caucasian, and all but two individuals were ascertained in the postnatal period due to a variety of neurodevelopmental problems or physical features. In contrast to XXY, where it is estimated that only approximately 3.6% of individuals diagnosed in the postnatal period are identified prior to age 10 [Abramsky and Chapple, 1997], over 70% of individuals with XXYY in this cohort were diagnosed prior to age 10. This difference is due to the greater severity of developmental delays, dysmorphisms, medical problems, and neurodevelopmental disorders in children with XXYY compared to XXY leading to genetic testing. It is also an interesting contrast to the 11.3% of individuals with XXYY syndrome diagnosed prior to age 10 described in the last major case series published in 1970 [Borgaonkar et al., 1970], most likely due to the increased practice of genetic testing in children with developmental delays and neurodevelopmental disorders over the past 35 years.
Stature in childhood is variable, with some children falling in the lower centiles and others having tall stature, consistent with previous reports [Borgaonkar et al., 1970]. However, stature above the mean increases with age, and tall stature in adulthood is a characteristic feature. This is consistent with growth patterns in XXY [Aksglaede et al., 2008]; however, the mean final adult height in XXYY (192.4 cm) is slightly taller than in XXY (188 cm). Our findings of a mean adult height of 192.4 cm (75.7 in) and almost two thirds (62%) of the adults falling above the 90th centile are more remarkable than the previously reported mean of 72 inches and only 30% above the 90th centile [Hunter, 1966; Borgaonkar et al., 1970]. Previous reports estimate gynecomastia in 62% of adult cases [Borgaonkar et al., 1970]; however, in our study only 25% of adolescents and 41% of adults had palpable gynecomastia. Again, this difference is likely due to our current ascertainment of a larger percentage of individuals with a milder phenotype due to increased genetic testing for early developmental delays or behavioral problems that were not screened in the 1960s and 1970s.
In contrast to XXY, there are characteristic facial features in XXYY syndrome (Fig. 1). The most common facial features in childhood included hypertelorism with epicanthal folds, and narrow, upslanting palpebral fissures. Although our study is cross-sectional, some facial features seem to emerge with age, with the development of a more prominent brow and jaw. It is interesting that this progression has also been described in previous individual case reports [Spencer et al., 1969; Davies, 1970; Yamane et al., 1993]. Dental problems including late or atypical dental eruption, thin enamel, taurodontism, crowding, malocclusion, multiple caries, and the need for dental surgery and orthodontia were almost universal in this population. Clinodactyly, flat feet, and tremor were also very common. The discrepancy between those with limited supination and pronation on physical examination (61%), and those with confirmed radioulnar synostosis or congenital elbow dislocation (17%) on radiograph suggests undiagnosed cases, especially since 70% of those who had been radiographed (n = 23) were found to have abnormalities. The high rates of asthma, allergies, and hospitalizations for respiratory infections are also higher than expected for the general pediatric population and raise the possibility of immunologic and/or pulmonary function abnormalities in this group.
Tremor has been previously reported in XXYY, as well as in other males with SCA [Baughman, 1969; Boltshauser et al., 1978; Telfeian et al., 2000], and has generally been classified as essential tremor. Although essential tremor is common in the general adult population (4%) [Benito-Leon and Louis, 2006], it is rare in children and it is interesting that essential tremor is estimated to present in 1/500 children [Jankovic et al., 2004], the same general estimated prevalence of SCA in the population [Hook and Hamerton, 1978]. Although clearly not all children with essential tremor have SCA, the presence of essential tremor in a child with a history of learning disabilities, developmental delays, or associated physical features should prompt genetic testing. The etiology, underlying neurophysiology, and treatment of tremor in this population needs further research and may shed light onto genes contributing to essential tremor in the general population.
This group of individuals with XXYY syndrome showed the expected development of hypogonadism seen in males with X chromosome polysomy. There have been no studies directly comparing hypogonadism in XXY and XXYY, although clinical and laboratory characteristics of hypergonadotropic hypogonadism, and studies of testicular histology show similar findings to those seen in XXY [Leisti et al., 1964; Tabata et al., 1964]. Thus, for now we presume that clinical endocrinologic monitoring and initiation of testosterone treatment should be similar to adolescents with XXY syndrome until further research is completed.
Three of the adult subjects with XXYY had laboratory evidence and physical symptoms of hypogonadism, however they were not on testosterone due to concerns of worsening behavioral problems with a history of mood disorders and impulsivity. The behavioral effect of testosterone treatment in XXYY was a common question asked by families and physicians of study participants. There is one report describing the initiation of androgen replacement in a 32-year-old with XXYY with a history of behavioral problems, emphasizing that androgen treatment did not worsen behaviors, aggression or hypersexuality, and significantly improved fatigue, bone density, and apathy [Heuser et al., 1999]. Sourial and Fenton [1988] also described a case of an adult with XXYY in which testosterone replacement significantly improved aggression and atypical sexual behaviors [Sourial and Fenton, 1988]. However, Lee described a male with XXYY and schizophrenia treated with testosterone at age 31 who developed agitation and aggression [Lee, 1996]. Our clinical experience with this group supports the view that testosterone treatment at standard doses generally improves or has no significant impact on behavior, although treatment was discontinued in two older individuals with significant mental heath problems due to increased agitation. Unfortunately, in both cases changes to other psychiatric medications occurred at the same time, making it difficult to determine if the testosterone indeed led to the agitation.
Interestingly, review of brain MR imaging showed a spectrum of nonspecific T2/Flair white matter hyperintensities in almost half of those who had brain MR performed. Some patients had a few tiny (1–2 mm) abnormal white matter foci whereas others had more diffuse and confluent (4–5 mm) abnormal white matter foci. The differential for these findings is broad and includes developmental, dysmyelination/demyelination, and gliosis or scar from remote insult (inflammation or trauma). This finding may also be underreported as neuroradiologists with predominantly adult practices may consider these to be normal findings. Warwick et al. [1999] described similar findings of “high-intensity signal foci” in 5/10 XXY, 3/11 XXX, and 1/10 XYY adult subjects compared to none in controls, postulating the increased susceptibility of developmentally abnormal brains to injury associated with aging. However, Boettger et al. [2004] describe similar findings in a 5-year-old male with XXY, and in our series these findings were present as early as 10 months of age and in three children 3 years of age. Thus, they may represent the sequelae of abnormal neurodevelopment and can provide clues to the pathology of the neurological/neurodevelopmental abnormalities seen in XXYY and other sex chromosome aneuploidies. In our series, the individuals with T2/Flair white matter hyperintensities did not differ in the presence of ID, seizures or tremor compared with those without reported MR findings, although larger sample sizes are needed.
Cognitive profiles with strengths in visuoperceptual skills relative to language-based tasks are consistent with previous reports, although the mean FSIQ of this group is almost 10 points higher than previously reported [Borgaonkar et al., 1970]. This is likely related to increased ascertainment of higher-functioning individuals with the increasing genetic testing in children with developmental delays over the past 30 years. We also found that VIQ significantly decreased with age, and it is important to point out that this is a cross-sectional study in which most individuals in this older age group were ascertained 20–30 years ago when genetic testing was only obtained for significant dysmorphology or severe cognitive deficits. There was also a discrepancy between overall cognitive measures and adaptive functioning, with adaptive skills almost one standard deviation less than full-scale IQ scores. The striking rates of neurodevelopmental disorders such as ADHD and autism spectrum disorders, other psychological diagnoses, psychiatric hospitalizations, and psychopharmacologic medication use emphasize the significance of the behavioral phenotype in XXYY syndrome and the need for further study in etiologies and treatments for these associated problems.
An ideal study of this population would include prospective studies of a cohort identified prior to or at birth to eliminate ascertainment bias and to study changes in health, cognition and adaptive functioning related to age. Cases identified in the prenatal period and carried to term are rare, with only two such cases in our cohort. There are no studies describing termination rates in early XXYY pregnancies; however, considering the recently reported termination rate of 70% in XXY fetuses by women electing prenatal genetic testing [Shaffer et al., 2006], it is most likely higher in XXYY due to the association with more significant cognitive and psychological problems in the current medical literature. Our two cases identified in the prenatal period did not have significant differences in physical features, medical problems, or cognitive/psychological features compared to the rest of the group. Screening of newborns to identify unselected cases would require a very large collaborative study to obtain sufficient samples sizes due to the low prevalence of XXYY.
It is posited that XXYY syndrome results from a double nondisjunction during meiosis in spermatogenesis. Very few studies have reported on further molecular investigations in XXYY males. The parent-of-origin of the extra chromosomes has been evaluated in six cases, and all have identified the extra chromosomes as being paternal in origin [Rinaldi et al., 1979; Leal et al., 1994; Iitsuka et al., 2001]. It is believed that the phenotypic differences in XXYY syndrome and other SCAs likely result from gene dosage effects of genes in the pseudoautosomal regions (PAR) of the X and Y chromosomes that escape X-inactivation. Thus, in XXYY males, genes in the PAR regions would be expressed from the four sex chromosomes, compared to the two sex chromosomes in 46,XY males. Overexpression of other X chromosome genes that escape X-inactivation or Y chromosome genes may also be involved. The phenotypic differences may be also be related to polymorphisms in selected X and Y chromosome genes, and other recent research has suggested that the presence of supernumerary sex chromosomes may lead to alterations in DNA methylation at various loci in the genome, leading to altered gene expression and subsequent phenotypic differences [Coffee et al., 2007].
To date, only two specific genes on the X and Y chromosomes have been described that may be associated with the phenotype of SCA. It is postulated that the tall stature of individuals with supernumerary sex chromosomes is related to overexpression of the SHOX gene, a pseudoautosomal gene that escapes X-inactivation and is highly expressed in growth plates of long bones where it plays a role in bone growth and maturation [Rao et al., 1997; Munns et al., 2004]. Haploinsufficiency of SHOX has been shown to be related to short stature in Turner syndrome [Clement-Jones et al., 2000], microdeletions of SHOX have been shown to be associated with short stature [Rao at al., 1997], and duplications of SHOX have been associated with tall stature [Ogata et al., 2000]. Thus, the tall stature in XXYY may be related to an overexpression of SHOX. Gene dosage effects of a potential growth control gene on the Y chromosome (GCY) has also been proposed and may be involved in the additional height in XXYY [Kirsch et al., 2004].
A polymorphism in the androgen receptor (AR) gene on the X chromosome gene has also been found to be related to phenotypic differences in XXY in some studies. The CAG-repeat length of the AR gene is associated with taller adult height and more gynecomastia in adult males [Zitzmann et al., 2004], and with shorter penile length in children with XXY [Zinn et al., 2005]. The longer CAG-repeat polymorphism codes for a receptor that is less responsive to androgen, thus perhaps low receptor responsiveness coupled with mild-moderate androgen deficiency contributes to the more severe phenotype in XXY. Studies of the genotype–phenotype relationship of the AR gene polymorphism in a sample of this XXYY population are currently underway. Also, future study of the two sets of twins with XXYY in this report comparing DNA methylation patterns and gene expression to their phenotypes may provide interesting data about the role of epigenetics in XXYY and other forms of SCA.
In summary, there are many important comparisons between the phenotypes of XXYY syndrome and XXY. In most cases, the number of congenital malformations, dysmorphic features, and associated medical problems are more significant in XXYY, along with a taller final adult height. However, features of hypergonadotropic hypogonadism are similar, and testosterone treatment is indicated when laboratory evidence or clinical symptoms of androgen deficiency present. The most important distinctions lay in the differences in the behavioral phenotype, where males with XXYY have more significant developmental delays, language-based cognitive deficits, and problems with adaptive functioning. Neurodevelopmental disorders such as autism spectrum disorders and ADHD, as well as other psychological comorbidities are also more common in XXYY compared to XXY. However, as in XXY, there is considerable variation in the phenotype, with the genetic and environmental factors underlying this variation yet to be identified.
Treatment Recommendations
Medical evaluation at the time of diagnosis depends on the age of the patient, but should include a complete medical history and examination with an emphasis on features requiring monitoring and intervention outlined in this study. In infants and children, cardiac evaluation for congenital heart defects and mitral valve prolapse, renal ultrasound, ophthalmologic evaluation, and orthopedic evaluation for flat feet, scoliosis, and elbow dysplasia are recommended. Aggressive preventative dental care and hygiene should also be implemented. Brain MRI, EEG, and sleep study should be strongly considered depending on clinical symptoms. Adolescents and adults should also be screened for other associated medical problems including Type 2 diabetes, hypothyroidism, osteoporosis, and obesity. A high index of suspicion for deep vein thrombosis, pulmonary embolus, and non-Hodgkin lymphoma should be maintained for various presenting symptoms.
Endocrinology consultation should be obtained upon diagnosis to inform parents and patients about options and timing of androgen treatments and updated practices in the treatment of hypogonadism and infertility [see reviews Simpson et al., 2005; Richmond and Rogol, 2007]. If hypogonadism is present, testosterone treatment should be considered in all individuals regardless of cognitive abilities due to positive effects on bone health, muscle strength, fatigue, and endurance, with possible mental health/behavioral benefits as well. For those with significant mental health problems such as bipolar or psychotic disorders, testosterone treatment should be initiated slowly with close observation by the mental health team and avoidance of concurrent changes to other psychotropic medications or environmental settings.
Since almost all children with XXYY have developmental delays and learning disabilities, a comprehensive neurodevelopmental evaluation is warranted at the time of diagnosis, including assessments by psychology (cognitive and social–emotional development), speech/language therapy, occupational therapy, and physical therapy. Consultation with a developmental pediatrician, psychiatrist, or neurologist to develop a treatment plan including therapies, behavioral interventions, educational supports, and psychotropic medications for behavioral and psychiatric symptoms should be arranged. Common diagnoses such as learning disability/ID, ADHD, autism spectrum disorders, mood disorders, tic disorders, and other mental health problems should be considered, screened for, and treated. Good responses to standard medication treatments for inattention, impulsivity, anxiety, and mood instability are seen in this group and such treatment can positively impact academic progress, emotional wellbeing and long-term outcome.
Poor fine motor coordination and the development of intention tremor can make handwriting slow and laborious, and occupational therapy and keyboarding should be introduced at an early age to facilitate schoolwork and self-help skills. Educational difficulties should be evaluated with a full psychological evaluation to identify discrepancies between verbal and performance skills and to identify individual academic needs. Expressive language skills are often affected throughout the lifespan and speech therapy interventions targeting expressive language skills, dyspraxia, and language pragmatics may be needed into adulthood. Adaptive skills (life skills) are a significant area of weakness necessitating community-based supports for almost all individuals in adulthood. Additional treatment recommendations based on the individual strengths and weaknesses in XXYY syndrome are further detailed by Visootsak et al. [2007]. Families should also be referred for support and further information to The XXYY Project (www.xxyysydrome.org), the international support organization for XXYY syndrome.
Acknowledgments
We would like to acknowledge all families and participants in the study, and appreciate support from the Bonfils-Stanton Foundation, The MIND Institute at University of California-Davis, The XXYY Project, and NIH LRP grant for Dr. Tartaglia.
References
- Abramsky L, Chapple J. 47,XXY (Klinefelter syndrome) and 47,XYY: Estimated rates of and indication for postnatal diagnosis with implications for prenatal counselling. Prenat Diagn. 1997;17:363–368. doi: 10.1002/(sici)1097-0223(199704)17:4<363::aid-pd79>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- Aksglaede L, Skakkebaek NE, Juul A. Abnormal sex chromosome constitution and longitudinal growth: Serum levels of IGF-1, IGFBP-3, LH, and testosterone in 109 males with 47,XXY, 47,XYY or SRY-positive 46,XX karyotypes. J Clin Endocrinol Metab. 2008;93:169–176. doi: 10.1210/jc.2007-1426. [DOI] [PubMed] [Google Scholar]
- Alter M, Gorlin R, Yunis J, Peagler F, Bruhl H. Dermatoglyphics in XXYY Klinefelter's syndrome. Am J Hum Genet. 1966;18:507–513. [PMC free article] [PubMed] [Google Scholar]
- Barr ML, Carr DH, Soltan HC, Wiens RG, Plunkett ER. The XXYY variant of Klinefelter's syndrome. Can Med Assoc J. 1964;90:575–580. [PMC free article] [PubMed] [Google Scholar]
- Baughman F. Klinefelter syndrome and essential tremor. Lancet. 1969;2:545. doi: 10.1016/s0140-6736(69)90244-x. [DOI] [PubMed] [Google Scholar]
- Benito-Leon J, Louis E. Essential tremor: Emerging views of a common disorder. Nat Clin Pract Neurol. 2006;2:666–678. doi: 10.1038/ncpneuro0347. [DOI] [PubMed] [Google Scholar]
- Boettger M, Kirchhof K, Sergi C, Sakmann CPM. Colobomas of the iris and choroid and high signal intensity cerebral foci on T2-weighted magnetic resonance images in Klinefelter's syndrome. J Pediatric Ophthalmol Strabismus. 2004;41:247–248. doi: 10.3928/0191-3913-20040701-17. [DOI] [PubMed] [Google Scholar]
- Boltshauser E, Meyer M, Deonna T. Klinefelter syndrome and neurological disease. J Neurol. 1978;219:253–259. doi: 10.1007/BF00312978. [DOI] [PubMed] [Google Scholar]
- Borgaonkar D, Mules E, Char F. Do the males with 48,XXYY have a characteristic phenotype? Clin Genet. 1970;1:272–293. [Google Scholar]
- Carr DH, Barr ML, Plunkett ER. A probable XXYY sex determining mechanism in a mentally defective male with Klinefelter's syndrome. Can Med Assoc J. 1961;84:873–878. [PMC free article] [PubMed] [Google Scholar]
- Clement-Jones M, Schiller S, Rao E, Blaschke RJ, Zuniga A, Zeller R, Robson SC, Binder G, Glass I, Strchan T, Lindsay S, Rappold GA. The short stature homeobox gene SHOX is involved in skeletal abnormalities in Turner syndrome. Hum Mol Genet. 2000;9:695–702. doi: 10.1093/hmg/9.5.695. [DOI] [PubMed] [Google Scholar]
- Coffee B, Albizua I, Warren S. DNA methylation alterations in males with Klinefelter syndrome [Abstract 715]. Presented at the annual meeting of The American Society of Human Genetics; October 25, 2007; San Diego, California. 2007. Available at http://www.ashg.org/genetics/ashg07s/index.shtml. [Google Scholar]
- Davies TS. Bone abnormalities and XXYY. Lancet. 1970;1:92. doi: 10.1016/s0140-6736(70)91891-x. [DOI] [PubMed] [Google Scholar]
- Demirhan O. Clinical findings and phenotype in a toddler with 48,XXYY syndrome. Am J Med Genet Part A. 2003;119A:393–394. doi: 10.1002/ajmg.a.20015. [DOI] [PubMed] [Google Scholar]
- Donati F, Gasser S, Mullis P, Braga S, Vassella F. 48,XXYY syndrome in a boy with essential tremor. Comparison with 120 cases from the literature. Monatsschr Kinderheilkd. 1992;140:216–219. [PubMed] [Google Scholar]
- Fryns JP, Kleczkowska A, Kubien E, Van den Berghe H. XYY syndrome and other Y chromosome polysomies. Mental status and psychosocial functioning. Genet Couns. 1995;6:197–206. [PubMed] [Google Scholar]
- Fuse H, Takahara M, Ito H, Shimazaki J. A case of 48,XXYY Klinefelter's syndrome with incurable skin ulcer. Urol Int. 1986;41:235–237. doi: 10.1159/000281207. [DOI] [PubMed] [Google Scholar]
- Garry MB. Two cases of 48,XXYY: With discussion on the behaviour of prepubertal and postpubertal patients. NZ Med J. 1980;92:49–51. [PubMed] [Google Scholar]
- Garvey M, Kellett B. Case studies of three “XXYY” children. Br J Disord Commun. 1975;10:17–30. doi: 10.3109/13682827509011271. [DOI] [PubMed] [Google Scholar]
- Garvey M, Mutton DE. Sex chromosome aberrations and speech development. Arch Dis Child. 1973;48:937–941. doi: 10.1136/adc.48.12.937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grammatico P, Bottoni U, De Sanctis S, Sulli N, Tonanzi T, Onorio AC, Del Porto G. A male patient with 48,XXYY syndrome: Importance of distinction from Klinefelter's syndrome. Clin Genet. 1990;38:74–78. doi: 10.1111/j.1399-0004.1990.tb03550.x. [DOI] [PubMed] [Google Scholar]
- Hagerman RJ. Neurodevelopmental disorders: Diagnosis and treatment. New York: Oxford University Press; 1999. [Google Scholar]
- Harrison PL, Oakland T. Adaptive behavior assessment system. 2nd. San Antonio: The Psychological Corporation; 2003. [Google Scholar]
- Heuser I, Hartmann A, Oertel H. Androgen replacement in a 48, XXYY-male patient. Arch Gen Psychiatry. 1999;56:194–195. doi: 10.1001/archpsyc.56.2.194. [DOI] [PubMed] [Google Scholar]
- Hook E, Hamerton J. Analyses of data on rates of cytogenetic disorders in live births. Am J Hum Genet. 1978;30:330–331. [PMC free article] [PubMed] [Google Scholar]
- Hunter H. YY chromosomes and Klinefelter's syndrome. Lancet. 1966;1:984. doi: 10.1016/s0140-6736(66)90991-3. [DOI] [PubMed] [Google Scholar]
- Iitsuka Y, Boack A, Nguyen DD, Samango-Sprouse CA, Simpson JL, Bischoff FZ. Evidence of skewed X-inactivation in 47, XXY and 48,XXYY Klinefelter patients. Am J Med Genet. 2001;98:25–31. [PubMed] [Google Scholar]
- Izumi S, Tsubahara A. Improvement of peripheral neuropathy by testosterone in a patient with 48,XXYY syndrome. Tokai J Exp Clin Med. 2000;25:39–44. [PubMed] [Google Scholar]
- Jankovic J, Madisetty J, Vuong K. Essential tremor among children. Pediatrics. 2004;114:1203–1205. doi: 10.1542/peds.2004-0031. [DOI] [PubMed] [Google Scholar]
- Kirsch S, Weiss B, Zumbach K, Rappold G. Molecular and evolutionary analysis of the growth-controlling region on the human Y chromosome. Hum Genet. 2004;114:173–181. doi: 10.1007/s00439-003-1028-z. [DOI] [PubMed] [Google Scholar]
- Laurence KM, Ishmael J, Davies TS. A case of a mentally defective male with XXYY sex chromosome constitution. Cytogenetics. 1963;19:50–54. doi: 10.1159/000129765. [DOI] [PubMed] [Google Scholar]
- Leal CA, Belmont JW, Nachtman R, Cantu JM, Medina C. Parental origin of the extra chromosomes in polysomy X. Hum Genet. 1994;94:423–426. doi: 10.1007/BF00201605. [DOI] [PubMed] [Google Scholar]
- Lee JW. An XXYY male with schizophrenia. Aust NZ J Psychiatry. 1996;30:553–556. doi: 10.3109/00048679609065032. [DOI] [PubMed] [Google Scholar]
- Leisti JT, Aula PP, Hjelt LH. Two cases of (Prepubertal) Klinefelter's syndrome with XXYY sex chromosomes. Ann Hum Genet. 1964;28:71–76. doi: 10.1111/j.1469-1809.1964.tb00461.x. [DOI] [PubMed] [Google Scholar]
- Linden MG, Bender BG, Robinson A. Sex chromosome tetrasomy and pentasomy. Pediatrics. 1995;96:672–682. [PubMed] [Google Scholar]
- Loftus J, Mellman WJ. Chromosome studies performed at cytogenetics laboratory, Children's Hospital of Philadelphia, PA. Hum Chromosome Newslett. 1965;18:12. [Google Scholar]
- Meschede D, Nekarda T, Kececioglu D, Loser H, Vogt J, Miny P, Horst J. Congenital heart disease in the 48,XXYY syndrome. Clin Genet. 1995;48:100–102. doi: 10.1111/j.1399-0004.1995.tb04064.x. [DOI] [PubMed] [Google Scholar]
- Muldal S, Ockey CH. Double male: New chromosome constitution in Klinefelter's syndrome. Lancet. 1960;2:492–493. doi: 10.1530/acta.0.0390183. [DOI] [PubMed] [Google Scholar]
- Mullen E. Mullen scales of early learning. Cranston, RI: TOTAL Child, Inc.; 1989. [Google Scholar]
- Munns CJ, Haase HR, Crowther LM, Hayes MT, Blaschke R, Rappold G, Glass IA, Batch JA. Expression of SHOX in human fetal and childhood growth plate. J Clin Endocrinol Metab. 2004;89:4130–4135. doi: 10.1210/jc.2003-032230. [DOI] [PubMed] [Google Scholar]
- Netley CT. Summary overview of behavioural development in individuals with neonatally identified X and Y aneuploidy. Birth Defects Orig Artic Ser. 1986;22:293–306. [PubMed] [Google Scholar]
- Nyberg RH, Karhu R, Karikoski R, Simola KO. The 48,XXYY syndrome: A case detected by maternal serum alpha-fetoprotein screening. Prenat Diagn. 1994;14:644–645. doi: 10.1002/pd.1970140724. [DOI] [PubMed] [Google Scholar]
- Ogata T, Kosho T, Wakui K, Fukushima Y, Yoshimoto M, Miharu N. Short stature homeobox-containing gene duplication on the der(x) chromosome in a female with 45,X/46,X,der(X), gonadal dysgenesis and tall stature. J Clin Endocrinol Metab. 2000;85:2927–2930. doi: 10.1210/jcem.85.8.6745. [DOI] [PubMed] [Google Scholar]
- Parker CE, Mavalwala J, Melnyk J, Fish CH. The 48,XXYY syndrome. Am J Med. 1970;48:777–781. doi: 10.1016/s0002-9343(70)80013-4. [DOI] [PubMed] [Google Scholar]
- Rao E, Weiss B, Fukami M, Rump A, Niesler B, Mertz A, Muroya K, Binder G, Kirsch S, Winkelmann M, Nordsiek G, Heinrich U, Breuning MH, Ranke MB, Rosenthal A, Ogata T, Rappold GA. Pseudoautosomal deletions encompassing a novel homeobox gene cause growth failure in idiopathic short stature and Turner syndrome. Nat Genet. 1997;16:54–63. doi: 10.1038/ng0597-54. [DOI] [PubMed] [Google Scholar]
- Richardson W, Wilson M, Williams J, Moyer V, Naylor C. Users' guide to the medical literature. XXIV. How to use an article on the clinical manifestations of disease. J Am Med Assoc. 2000;284:869–875. doi: 10.1001/jama.284.7.869. [DOI] [PubMed] [Google Scholar]
- Richmond E, Rogol A. Male pubertal development and the role of androgen therapy. Nat Clin Pract Endocrinol Metab. 2007;3:338–344. doi: 10.1038/ncpendmet0450. [DOI] [PubMed] [Google Scholar]
- Rinaldi A, Archidiacono N, Rocchi M, Filippi G. Additional pedigree supporting the frequent origin of XXYY from consecutive meiotic non-disjunction in paternal gametogenesis. J Med Genet. 1979;16:225–226. doi: 10.1136/jmg.16.3.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson GC, Miller JR, Dill FJ, Kamburoff TD. Klinefelter's syndrome with the XXYY sex chromosome complex. With particular reference to prepubertal diagnosis. J Pediatr. 1964;65:226–232. doi: 10.1016/s0022-3476(64)80524-2. [DOI] [PubMed] [Google Scholar]
- Sachsse W, Overzier C, Knolle J. A special form of Klinefelter's syndrome: Mosaic with doubling of the Y chromosome (2 X-2 Y form) Dtsch Med Wochenschr. 1967;92:1213–1220. doi: 10.1055/s-0028-1105711. [DOI] [PubMed] [Google Scholar]
- Schlegel RJ, Aspillaga MJ, Neu R, Gardner LI. Studies on a boy with XXYY chromosome constitution. Pediatrics. 1965;36:113–119. [PubMed] [Google Scholar]
- Shaffer BL, Caughey AB, Norton ME. Variation in the decision to terminate pregnancy in the setting of fetal aneuploidy. Prenat Diagn. 2006;26:667–671. doi: 10.1002/pd.1462. [DOI] [PubMed] [Google Scholar]
- Simpson JL, Graham JM, Jr, Samango-Sprouse CA, Swerdloff RS. Klinefelter syndrome. In: Cassidy SB, Allanson JE, editors. Management of genetic syndromes. 2nd. New Jersey: John Wiley & Sons; 2005. pp. 323–334. [Google Scholar]
- Singer H, Zankl H, Rodewald-Rudescu A. Combined Klinefelter-Down syndrome or XXYY syndrome? Humangenetik. 1973;19:261–264. doi: 10.1007/BF00278399. [DOI] [PubMed] [Google Scholar]
- Sorensen K, Nielsen J, Jacobsen P, Rolle T. The 48,XXYY syndrome. J Ment Defic Res. 1978;22:197–205. [PubMed] [Google Scholar]
- Sourial N, Fenton F. Testosterone treatment of an XXYY male presenting with aggression: A case report. Can J Psychiatry. 1988;33:846–850. doi: 10.1177/070674378803300912. [DOI] [PubMed] [Google Scholar]
- Sparrow S, Cicchetti D, Balla D. Vineland adaptive behavior scales. 2nd. Circle Pines, MN: American Guidance Service; 2005. [Google Scholar]
- Spencer DA, Eyles JW, Mason MK. XYY syndrome, and XYY-XXYY mosaicism also showing features of Klinefelter's syndrome. J Med Genet. 1969;6:159–165. doi: 10.1136/jmg.6.2.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow AJ, Schoemaker MJ, Higgins CD, Wright AF, Jacobs PA. Cancer incidence and mortality in men with Klinefelter syndrome: A cohort study. J Natl Cancer Inst. 2005;97:1204–1210. doi: 10.1093/jnci/dji240. [DOI] [PubMed] [Google Scholar]
- Tabata T, Fedoroff S, Gerrard JW. Klinefelter's syndrome due to XXYY chromosome anomaly. Can Med Assoc J. 1964;90:590–592. [PMC free article] [PubMed] [Google Scholar]
- Tartaglia N, Reynolds A, Visootsak J, Davis S, Hansen R, Hagerman RJ. Behavioral phenotypes of males with sex chromosome aneuploidy—XXYY syndrome: Not just a variant of Klinefelter syndrome. J Dev Behav Pediatr. 2005;26:464–465. [Google Scholar]
- Telfeian A, Boockvar J, Simuni T, Jaggi J, Skolnick B, Baltuch G. Efficacy of unilateral deep brain stimulation of the thalamic ventralis intermedius nucleus in a patient with bipolar disorder associated with Klinefelter syndrome and essential tremor. Case report. J Neurosurg. 2000;93:127–128. doi: 10.3171/jns.2000.93.1.0127. [DOI] [PubMed] [Google Scholar]
- Townes PL, Ziegler NA, Scheiner AP. An XXYY variant of the Klinefelter syndrome in a prepubertal body. J Pediatr. 1965;67:410–414. doi: 10.1016/s0022-3476(65)80401-2. [DOI] [PubMed] [Google Scholar]
- Vanyan M. Mental illness, epilepsy and hypothyroidism in XXYY syndrome. Br J Psychiatry. 1984;144:668. [PubMed] [Google Scholar]
- Visootsak J, Rosner B, Dykens E, Tartaglia N, Graham JM., Jr Behavioral phenotype of sex chromosome aneuploidies: 48, XXYY, 48, XXXY, and 49,XXXXY. Am J Med Genet Part A. 2007;143A:1198–1203. doi: 10.1002/ajmg.a.31746. [DOI] [PubMed] [Google Scholar]
- Warwick MM, Doody GA, Lawrie SM, Kestelman JN, Best JJ, Johnstone EC. Volumetric magnetic resonance imaging study of the brain in subjects with sex chromosome aneuploidies. J Neurol Neurosurg Psychiatry. 1999;66:628–632. doi: 10.1136/jnnp.66.5.628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterman DF, London J, Valdmanis A, Mann JD. The XXYY chromosome constitution. Deoxyribonucleic acid replication and the Xg blood group. Am J Dis Child. 1966;111:421–425. doi: 10.1001/archpedi.1966.02090070119019. [DOI] [PubMed] [Google Scholar]
- Wechsler D. Weschler abbreviated scale of intelligence. San Antonio: The Psychological Corporation; 1989. [Google Scholar]
- Yamane Y, Okamoto S, Fukui H, Matsumura Y, Yoshikawa M, Tsujita S, Tsujii T. 48,XXYY syndrome associated with acromegaloidism. Intern Med. 1993;32:160–165. doi: 10.2169/internalmedicine.32.160. [DOI] [PubMed] [Google Scholar]
- Zack BG. XXYY syndrome discovered on routine physical examination. J Adolesc Health Care. 1980;1:60–62. doi: 10.1016/s0197-0070(80)80012-x. [DOI] [PubMed] [Google Scholar]
- Zelante L, Piemontese MR, Francioli G, Calvano S. Two 48, XXYY patients: Clinical, cytogenetic and molecular aspects. Ann Genet. 2003;46:479–481. doi: 10.1016/s0003-3995(03)00030-3. [DOI] [PubMed] [Google Scholar]
- Zinn A, Ramos P, Elder F, Kowal K, Samango-Sprouse C, Ross J. Androgen receptor CAGn repeat length influences phenotype of 47,XXY (Klinefelter) syndrome. J Clin Endocrinol Metab. 2005;90:5041–5046. doi: 10.1210/jc.2005-0432. [DOI] [PubMed] [Google Scholar]
- Zitzmann M, Depenbusch M, Gromoll JEN. X-chromosome inactivation patterns and androgen receptor functionality influence phenotype and social characteristics as well as pharmacogenetics of testosterone therapy in Klinefelter patients. J Clin Endocrinol Metab. 2004;89:6208–6217. doi: 10.1210/jc.2004-1424. [DOI] [PubMed] [Google Scholar]