

Abstract
Despite the emergence of translational control pathways as mediators of nociceptive sensitization, effector molecules and mechanisms responsible for modulating activity in these pathways in pain conditions are largely unknown. We demonstrate that two major algogens, the cytokine interleukin 6 (IL-6) and the neurotrophin nerve growth factor (NGF), which are intimately linked to nociceptive plasticity across preclinical models and human pain conditions, signal primarily through two distinct pathways to enhance translation in sensory neurons by converging onto the eukaryotic initiation factor (eIF) eIF4F complex. We directly demonstrate that the net result of IL-6 and NGF signaling is an enhancement of eIF4F complex formation and an induction of nascent protein synthesis in primary afferent neurons and their axons. Moreover, IL-6- and NGF-induced mechanical nociceptive plasticity is blocked by inhibitors of general and cap-dependent protein synthesis. These results establish IL-6- and NGF-mediated cap-dependent translation of local proteins as a new model for nociceptive plasticity.
Introduction
Translational control of gene expression is rapid, reversible, and spatially controlled. Protein translation is comprised of three steps: initiation, elongation, and termination. The rate-limiting step, initiation (Sonenberg and Hinnebusch, 2009), is regulated by kinases that are critical for neuronal plasticity: the mammalian target of rapamycin (mTOR) and extracellular regulated kinase (ERK), which signal to eukaryotic initiation factors (eIF) eIF4E and eIF4G and the eIF4E binding protein (4EBP) (Sonenberg and Hinnebusch, 2009). These pathways are important factors in pain plasticity (Price and Géranton, 2009) as demonstrated by the antihyperalgesic activity of the mTOR inhibitor rapamycin administered either spinally (Price et al., 2007; Asante et al., 2009; Codeluppi et al., 2009; Géranton et al., 2009) or peripherally (Price et al., 2007; Jiménez-Díaz et al., 2008). Despite the clear relevance of translation control for enhanced pain states, endogenous factors responsible for modulating translation in primary sensory neurons are not known. In this study, we sought to identify these factors and their signaling mechanisms to gain insight into how translation regulation of gene expression is controlled in sensory neurons and how this process contributes to nociceptive plasticity.
Interleukin (IL)-6 and nerve growth factor (NGF) are critical mediators of inflammatory and neuropathic pain, but their potential role in controlling translation in sensory neurons is largely unexplored. IL-6 expression is increased after peripheral nerve injury (Bolin et al., 1995; Anderson and Rao, 2001) and skin incision (Clark et al., 2007). Injection of IL-6 induces long-lasting hyperalgesia (Oka et al., 1995; Dina et al., 2008), and IL-6 knock-out mice fail to develop thermal hyperalgesia and mechanical allodynia following chronic constriction of the sciatic nerve (Murphy et al., 1999b). Moreover, IL-6 neutralization therapy is effective for treating pain in humans (Nishimoto et al., 2009). Likewise, NGF levels are elevated in inflammatory and neuropathic pain states (Lewin et al., 1994; Woolf et al., 1994; Banik et al., 2005). NGF induces hyperalgesia and allodynia in rodents (Lewin et al., 1994) and humans (Rukwied et al., 2010). Additionally, NGF neutralization therapy alleviates pain in humans (Cattaneo, 2010). Hence, IL-6 and NGF are important mediators of pain across preclinical models and their importance for human pain conditions is clinically validated.
Here we demonstrate that IL-6 and NGF initiate signaling cascades that converge on the eIF4F complex to increase protein synthesis in primary sensory neurons and their axons. Moreover, IL-6- and NGF-induced mechanical sensitization is attenuated by inhibitors of general and cap-dependent translation. These findings elucidate molecular mechanisms through which rapid, local control of gene expression via translation can be achieved in these neurons and establish IL-6- and NGF-mediated, activity-dependent translation control as a new model for primary afferent nociceptive plasticity.
Materials and Methods
Primary sensory neuron cultures.
Male ICR mice (Harlan, 20–25 g) were used. All animal procedures were approved by the Institutional Animal Care and Use Committee of The University of Arizona and were in accordance with International Association for the Study of Pain guidelines. Dorsal root ganglia (DRGs) from all levels or trigeminal ganglia (TGs) were excised aseptically and placed in HBSS (Invitrogen) on ice. The ganglia were dissociated enzymatically with collagenase A (1 mg/ml, 25 min, Roche) and collagenase D (1 mg/ml, Roche) with papain (30 U/ml, Roche) for 20 min at 37°C. To eliminate debris, 70 μm (BD) cell strainers were used. The dissociated cells were resuspended in DMEM/F12 (Invitrogen) containing 1× pen-strep (Invitrogen), 1× GlutaMax, 3 μg/ml 5-FDU (Sigma), 7 μg/ml uridine (Sigma), and 10% fetal bovine serum (Hyclone). The cells were plated in either 6- or 96-well plates (BD Falcon) and incubated at 37°C in a humidified 95% air/5% CO2 incubator. On day 5, the cells were washed in DMEM/F12 media for 30 min followed by treatment with IL-6 and/or NGF for 15 min.
Behavioral testing.
Animals (male ICR mice, 20–25 g) were placed in acrylic boxes with wire mesh floors and allowed to habituate for 1 h. After predrug mechanical thresholds were recorded, animals received intraplantar injections in a volume of 20 μl. Calibrated von Frey filaments (Stoelting) were used for mechanical stimulation of the plantar surface of the left hindpaw and withdrawal thresholds were calculated using the up–down method (Chaplan et al., 1994).
Western blotting.
Protein was extracted from the cells in lysis buffer (50 mm Tris HCl, 1% Triton X-100, 150 mm NaCl, and 1 mm EDTA at pH 7.4) containing protease and phosphatase inhibitor mixtures (Sigma) with an ultrasonicator on ice, and cleared of cellular debris and nuclei by centrifugation at 14,000 × g for 15 min at 4°C. Fifteen micrograms of protein per well were loaded and separated by standard 7.5% or 10% SDS-PAGE. Proteins were transferred to Immobilon-P membranes (Millipore) and then blocked with 5% dry milk for 3 h at room temperature. The blots were incubated with primary antibody overnight at 4°C and detected the following day with donkey anti-rabbit antibody conjugated to horseradish peroxidase (Jackson ImmunoResearch). Signal was detected by ECL on chemiluminescent films. Each phosphoprotein was normalized to the expression of the corresponding total protein on the same membrane. Densitometric analyses were performed with ImageJ software (NIH).
5′ mRNA cap complex analysis.
After the protein extraction, 50 μg of protein was incubated with 7-methyl GTP Sepharose 4B beads (GE Healthcare) in the presence of 100 μm GTP for 2 h at 4°C. Unconjugated Sepharose 4B beads were used for the negative controls. The beads were then pelleted and washed twice with lysis buffer. After a final centrifugation, the pellet was suspended in 1× Laemmli sample buffer containing 5% v/v β-mercaptoethanol, and eIF4E, eIF4G, eIF4A, and 4EBP bound to the precipitated beads were analyzed by Western blotting.
High-throughput nascent protein synthesis assay.
DRG neurons were cultured in black, low-autofluorescence, transparent-bottom 96-well plates (Corning) for 5 d. Cells were incubated in methionine-free DMEM (Invitrogen) media for 30 min. The cells were pretreated with rapamycin (500 nm, LC Laboratories), U0126 (1 μm, Tocris Bioscience), anisomycin (50 μm, Sigma), or vehicle in methionine-free DMEM (Invitrogen) media for 15 min. The cells then received either vehicle or IL-6 and NGF with and without drug treatments along with 50 μm l-azidohomoalanine (AHA, Invitrogen) of 30 min. The cells were then washed with PBS and fixed with ice-cold acetone/methanol. This was followed by incubation in 3% bovine serum albumin (BSA) in PBS for 15 min. Proteins that incorporated AHA were labeled with Alexa 488–alkyne conjugate (Invitrogen) by incubating fixed cells with the conjugate for 30 min. The cells were then washed once with 3% BSA in PBS, followed by another wash with PBS. PBS was added to each well and fluorescence was read by a fluorometer (Synergy 2, Biotek) using excitation/emission filters 480/520 nm.
Nascent protein synthesis immunocytochemistry.
Primary sensory neurons were cultured on poly-d-lysine-coated coverslips (BD). The AHA labeling procedure followed the high-throughput nascent protein synthesis assay steps described above. Following the final wash, the coverslips were blocked with 5% normal goat serum (NGS) for 3 h. Neurons were identified by immunostaining for peripherin (1:200, Sigma) and neurofilament heavy chain (NFH) (1:300, Sigma) antibodies in 5% NGS and incubated overnight at 4°C. Alexa 555 anti-mouse secondary antibody (Invitrogen) was used to label the neurons. The coverslips were mounted onto slides.
Nascent protein synthesis Western blotting.
Primary afferent neurons were cultured in poly-d-lysine-coated 6-well plates for 5 d. Cells were pretreated with 4EGI1 (20 μm) or vehicle in methionine-free DMEM (Invitrogen) media for 30 min. The cells then received either vehicle or IL-6 and NGF with and without drug treatments along with 50 μm AHA (Invitrogen) of 30 min. The cells were lysed and protein was extracted by ultrasonication in RIPA buffer. AHA-incorporating proteins were labeled with biotin using Click-iT Biotin Protein Analysis Detection Kit (Invitrogen). The biotin-labeled proteins were assayed by Western blotting.
Confocal microscopy.
Immunofluorescent micrographs were acquired on Zeiss LSM 510 META NLO upright microscope using a 40×, 1.3 numerical aperture oil-immersion objective.
Intensity correlation analysis.
Intensity correlation analysis (ICA) was performed using a plug-in for ImageJ provided by Li et al. (2004) at the Wright Cell Imaging Facility, University Health Network Research, Canada (http://www.uhnresearch.ca/facilities/wcif/fdownload.html). To determine the extent of colocalization of AHA with NFH/peripherin in axons, ICA was used (Li et al., 2004) to calculate an intensity correlation quotient (ICQ) for each pair of images. By excluding the cell bodies of the neurons from the analysis by deselecting from the region of interest, we removed investigator bias from our assessment of nascent protein synthesis in axons following various treatments. ICA postulates that colocalized signals covary in their intensity. Pixels intensities that covary result in a positive product difference from the mean (PDM) value, while pixels that are exclusive result in negative PDM value. A PDM value of 0 results when a pixel intensity is around the average of the pixel intensity of the whole image. The PDM values were used to generate an image that visualizes colocalization of AHA with NFH/peripherin in the axons. ICQ values were calculated by dividing the number of positive PDM pixels by the total PDM pixels and then subtracting by 0.5. A positive ICQ value indicates colocalization, while a negative value indicates counterlocalization. Random distribution would result in an ICQ value around 0. A similar analysis was used on entire images to determine ICQ values for all neurons, axons and somas. To evaluate localization of nascently synthesized proteins along the length of axons, regions of interest from IL-6- and NGF-treated images proximal to the soma and extending distally up to 10 μm were selected. More distal sites were also selected starting at 50 μm from the soma and extending an additional 10 μm (60 μm from the soma). Only axons that were clearly distinguishable as extending from a particular soma were selected. ICQ values were calculated for both regions of interest (proximal and distal).
Drugs and primary antibodies.
U0126 and CGP57380 were from Tocris Bioscience; 4EGI1 was from Axxora; mouse and human recombinant IL-6 were from R&D Systems; mouse 2.5S NGF was from Millipore. The following rabbit polyclonal antibodies were obtained from Cell Signaling Technology: p-ERK (Thr202/Tyr204, cat# 4377), total ERK, p-eIF4E (Ser209, cat# 9741), total eIF4E, p-mTOR (Ser2448, cat# 2971), total mTOR, p-4EBP (Thr37/46, cat # 9459), total 4EBP, p-eIF4G (Ser1108, cat# 2441), total eIF4G, p-AKT (Ser473, cat# 4058), total AKT, and eIF4A. Anisomycin and actinomycin D were from Sigma, and rapamycin was from LC Laboratories.
Statistical analysis.
Data are shown as means ± SEMs of eight independent cell culture wells for all in vitro experiments or six animals for behavioral assays. Graph plotting and statistical analysis used GraphPad Prism Version 5.03 (Graph Pad Software). Statistical evaluation was performed by one-way ANOVA, followed by post hoc Dunnett's test, or by two-way ANOVA (behavioral data), and the a priori level of significance at 95% confidence level was considered at p < 0.05.
Results
IL-6 signals to the translational machinery through the ERK-Mnk1-eIF4E pathway
We first asked whether IL-6 signals to the translational machinery in adult mouse sensory neurons. We used IL-6 because multiple lines of evidence indicate that IL-6 is a major mediator of nociceptive plasticity in the periphery (Bolin et al., 1995; Murphy et al., 1995, 1999a,b; Anderson and Rao, 2001; Vardanyan et al., 2010). Along with the canonical pathway of Janus kinase (JAK)/signal transducers and activators of transcription (Stat) activation, IL-6 is known to activate the ERK MAP kinase pathway (Akira et al., 1995). The ERK pathway has been implicated in the development and maintenance of pain conditions (Karim et al., 2001; Ji et al., 2009). Changes in ERK activity modulate translation through MAP kinase-interacting kinase 1 (Mnk1) signaling to eIF4E (Waskiewicz et al., 1999). To assess whether IL-6 engages ERK-mediated signaling to eIF4E in primary sensory neurons, we treated adult DRG (Fig. 1A) and TG (Fig. 1B) neurons with escalating doses of IL-6 for 15 min. This resulted in a dose-dependent increase in the phosphorylation of ERK (for DRG: 0 ng/ml, 100 ± 9.8%; 1 ng/ml, 128.8 ± 31.5%; 10 ng/ml, 183 ± 43%; and 50 ng/ml, 207 ± 24.5%) and eIF4E (for DRG: 0 ng/ml, 100 ± 15%; 1 ng/ml, 119.1 ± 24.5%; 10 ng/ml, 166 ± 49%; and 50 ng/ml, 273 ± 74%). To verify that activation of eIF4E is due to IL-6 signaling through the ERK pathway (rather than the p38 pathway), we used the MEK inhibitor U0126. Pretreatment of primary afferent neurons in culture with U0126 for 30 min followed by cotreatment with IL-6 (50 ng/ml, 15 min) blocked IL-6-mediated phosphorylation of eIF4E (Fig. 1C). We then assessed whether IL-6-induced changes in eIF4E phosphorylation are dependent on Mnk1 using the Mnk1 inhibitor CGP57380. Pretreatment of the DRG or TG cultures with CGP57380 (50 μm, 30 min) and subsequent cotreatment with IL-6 (50 ng/ml, 15 min) blocked IL-6-mediated phosphorylation of eIF4E in DRG (Fig. 1D) and TG (Fig. 1E) neurons. There was no statistically significant difference between the groups that received CGP57380 and the vehicle. This finding indicates that IL-6 signals to the translational machinery via the ERK/Mnk1 pathway. The mTOR pathway also signals to the translational machinery and promotes cap-dependent translation (Beretta et al., 1996). In addition to phosphorylating eIF4G, which allows for the efficient assembly of the eIF4F complex, the mTOR pathway inhibits the negative regulator of cap-dependent translation, 4EBP, through phosphorylation (Mathews et al., 2007). Treatment of DRG neurons in culture with IL-6 (50 ng/ml, 15 min) and/or CGP57380 (30 min) did not induce a significant change in mTOR phosphorylation (Fig. 1F), showing that IL-6 fails to signal through the mTOR pathway in sensory neurons at these concentrations. Hence, IL-6-mediated signaling to the translational machinery occurs through ERK and Mnk1 to enhance eIF4E phosphorylation in DRG and TG neurons.
Figure 1.

Interleukin-6 signals to the translational machinery through ERK and Mnk1. A, Western blot and quantification for p-ERK, total ERK, p-eIF4E, and total eIF4E following 15 min of treatment of DRG cultures with increasing doses of IL-6. B, Western blot and quantification for p-ERK, total ERK, p-eIF4E, and total eIF4E following 15 min of treatment of TG cultures with escalating doses of IL-6. C, Western blot and quantification for p-eIF4E and total eIF4E following pretreatment of DRG neurons in culture with the MEK inhibitor U0126 (10 μm) for 30 min and subsequent treatment with IL-6 (50 ng/ml) for 15 min. D, Western blot and quantification for p-eIF4E and total eIF4E following pretreatment of DRG cultures with the Mnk1 inhibitor CGP57380 (CGP, 50 μm) for 30 min and subsequent treatment with IL-6 (50 ng/ml) for 15 min. E, The Mnk1 inhibitor CGP57380 also inhibited IL-6-induced phosphorylation of eIF4E in TG neurons. F, IL-6 (50 ng/ml) did not stimulate mTOR phosphorylation in DRG neurons, and this was also not changed by CGP57380 (50 μm). Phosphorylated proteins were standardized relative to their respective totals and shown as percentage of vehicle. *p < 0.05, **p < 0.01.
NGF signals to the translational machinery through the mTOR pathway
We next assessed whether NGF engages signaling to the translational machinery in adult sensory neurons. Like IL-6, NGF is well established as a sensitizer of nociceptors and plays an important role in preclinical models of chronic pain and human pain conditions (Lewin et al., 1994; Woolf et al., 1994; Banik et al., 2005; Watson et al., 2006; Cattaneo, 2010). NGF is known to increase activity of the PI3 kinase/AKT pathway in sensory neurons (Zhang et al., 2005), but its potential role in signaling to the translational machinery in adult sensory neurons is not known. In developing DRG neurons, NGF stimulates local synthesis of CREB, leading to retrograde transport to the neuronal soma (Cox et al., 2008). To demonstrate that NGF can rapidly signal to the translational machinery through the mTOR pathway in adult DRG (Fig. 2A) and TG (Fig. 2B) neurons, cultures were treated with NGF for 15 min. Treatment with NGF induced a significant increase in the phosphorylation of AKT (for DRG: 0 ng/ml, 100 ± 14%; 0.1 ng/ml, 160 ± 38%; 1 ng/ml, 192 ± 43%; and 10 ng/ml, 252 ± 30%) and the serine 2448 residue of mTOR at 10 ng/ml (for DRG: 0 ng/ml, 100 ± 7.4% and 10 ng/ml, 185 ± 14%). An NGF-mediated, dose-dependent increase in the phosphorylation of 4EBP (for DRG: 0 ng/ml, 100 ± 4.3% and 10 ng/ml, 125 ± 3.2%) and eIF4G (for DRG: 0 ng/ml, 100 ± 17%; 0.1 ng/ml, 169 ± 36%; 1 ng/ml, 370 ± 63%; and 10 ng/ml, 485 ± 119%) was also observed, suggesting that NGF increases the assembly of the eIF4F cap complex by signaling though the PI3 kinase/AKT pathway. Treatment with 10 ng/ml NGF was not sufficient to activate the ERK pathway (Fig. 2C); however, at 100 ng/ml, NGF did activate the ERK pathway (Fig. 2C). NGF may activate the ERK pathway at higher doses through cross talk between PI3K and Ras pathways (Moelling et al., 2002; Carracedo et al., 2008; Kinkade et al., 2008). NGF-induced (15 min treatment) phosphorylation of 4EBP (Fig. 2D), mTOR (Fig. 2E), and eIF4G (Fig. 2F) was blocked by the mTOR inhibitor rapamycin (500 nm, 30 min pretreatment) in sensory neurons. While there was a nonsignificant trend toward increased mTOR phosphorylation with NGF in the presence of rapamycin, signaling to downstream targets (eIF4G and 4EBP) was completely abrogated by rapamycin treatment. Therefore, these data indicate that NGF is a potent activator of signaling through the mTOR pathway to cap-dependent translational machinery in adult DRG and TG neurons.
Figure 2.

NGF signals to the translational machinery by activating the mTOR pathway. A, Western blots and quantification for p-AKT, total AKT, p-mTOR, total mTOR, p-eIF4G, total eIF4G, p-4EBP, and total 4EBP following 15 min of treatment of DRG cultures with increasing doses of NGF. B, Western blot and quantification for p-mTOR, total mTOR, p-eIF4G, total eIF4G, p-4EBP, and total 4EBP following 15 min of treatment of TG cultures with NGF (10 ng/ml). C, Western blot and quantification for p-ERK and total ERK following 15 min of treatment of TG cultures with escalating doses of NGF. D–F, Western blots and quantification for p-4EBP and total 4EBP (D), p-mTOR and total mTOR (E), and p-eIF4G and total eIF4G (F) following pretreatment of DRG cultures with rapamycin (Rap, 500 nm) for 30 min and subsequent treatment with NGF (10 ng/ml) for 15 min. *p < 0.05, **p < 0.01.
Cotreatment with IL-6 and NGF signal to the eIF4F complex through the ERK and mTOR pathways
We have demonstrated that IL-6 signals through the MEK/ERK/Mnk1 pathway to eIF4E and NGF signals through the mTOR pathway to eIF4G and 4EBP. All of the preceding phosphorylation events enhance formation of the eIF4F complex and increase cap-dependent translation. Given that IL-6 and NGF are both upregulated in neuropathic and inflammatory pain conditions, we assessed whether cotreatment (15 min) with IL-6 (50 ng/ml) and NGF (10 ng/ml) would simultaneously activate ERK and mTOR signaling to the cap-dependent translational machinery (Fig. 3A). Cotreatment of DRG cultures with IL-6 (50 ng/ml) and NGF (10 ng/ml) for 15 min led to a significant increase in the phosphorylation of ERK (vehicle 100 ± 45% and IL-6 + NGF 242 ± 28%), eIF4E (vehicle 100 ± 5% and IL-6 + NGF 124 ± 7%), AKT (vehicle 100 ± 17% and IL-6 + NGF 339 ± 28%), mTOR (vehicle 100 ± 12% and IL-6 + NGF 197 ± 38%), 4EBP (vehicle 100 ± 4% and IL-6 + NGF 125 ± 10%), and eIF4G (vehicle 100 ± 19% and IL-6 + NGF 170 ± 18%) (Fig. 3A). Thus, combined treatment with IL-6 and NGF engages signaling through the ERK and mTOR pathways, leading to convergence onto the primary components responsible for control of cap-dependent translation.
Figure 3.

Cotreatment with IL-6 and NGF signal through both ERK and mTOR pathways to stabilize the formation of eIF4F complex and dissociate 4EBP from eIF4E. A, Western blots and quantification for p-ERK, total ERK, p-eIF4E, total eIF4E, p-4EBP, total 4EBP, p-AKT, total AKT, p-mTOR, total mTOR, p-eIF4G, and total eIF4G following 15 min of treatment of DRG cultures with IL-6 (50 ng/ml) and NGF (10 ng/ml). B, A schematic diagram demonstrating that, in vehicle conditions (left panel), 7-methyl-GTP-conjugated Sepharose beads are associated with both the positive and negative regulators of cap-dependent translation, eIF4F (eIF4E, eIF4A, and eIF4G) and 4EBP, respectively. After cotreatment with IL-6 and NGF (right panel) the balance shifts toward the assembly of the eIF4F complex on the Sepharose beads. C, Western blots of eIF4G, eIF4A, 4EBP, and eIF4E following treatment of DRG neurons with IL-6 (50 ng/ml) and NGF (10 ng/ml) and coprecipitation with 7-methyl-GTP-conjugated Sepharose beads or control Sepharose beads. D, Western blots of eIF4G, eIF4A, 4EBP, and eIF4E following treatment of TG neurons with IL-6 (50 ng/ml) and NGF (10 ng/ml) and coprecipitation with 7-methyl-GTP-conjugated Sepharose beads or control Sepharose beads. E, eIF4G, eIF4A, and 4EBP coprecipitation from DRG were standardized relative to the levels of eIF4E and shown as percentage of vehicle. F, eIF4G, eIF4A, and 4EBP coprecipitation from TG were standardized relative to the levels of eIF4E and shown as percentage of vehicle. Cotreatment with IL-6 and NGF increased the association eIF4E to eIF4G and eIF4A while decreasing its association with 4EBP in both DRG and TG neurons. *p < 0.05.
IL-6 and NGF induce assembly of eIF4F complex
The biochemical changes induced by IL-6 and NGF observed above predict that there would be enhanced formation of the mRNA 5′-cap complex eIF4F, leading to increased cap-dependent translation. To provide direct evidence for increased formation of eIF4F complex, we used 7-methyl-GTP-coated Sepharose beads to coprecipitate the proteins that are bound to 7-methyl-GTP following IL-6 and NGF cotreatment (Fig. 3B). Western blot analysis of the proteins coprecipitated with the 7-methyl-GTP-conjugated Sepharose beads revealed that there is an increased association of eIF4G (for DRG: vehicle 100 ± 9% and IL-6 + NGF 160 ± 20%) and eIF4A (for DRG: vehicle 100 ± 15% and IL-6 + NGF 174 ± 32%) following cotreatment of DRG (Fig. 3C,E) and TG neurons (Fig. 3D,F) with IL-6 (50 ng/ml) and NGF (10 ng/ml) for 15 min. Moreover cotreatment resulted in decreased coprecipitation of 4EBP (for DRG: vehicle 100 ± 15% and IL-6 + NGF 57 ± 11%), which is the negative regulator of the cap complex formation and cap-dependent translation, in DRG (Fig. 3C,E) and TG (Fig. 3D,F) neurons. These data provide direct evidence for enhanced formation of the eIF4F complex following cotreatment of primary sensory neurons with the IL-6 and NGF.
IL-6 and NGF increase nascent protein synthesis
Increased formation of the eIF4F complex on the 7-methyl-GTP cap of mRNA suggests increased protein synthesis through cap-dependent translation. To directly determine the effects of IL-6 and/or NGF on translation of nascent proteins, we used the methionine analog AHA to label the newly translated proteins in DRG neurons cultured in 96-well plates. By using click chemistry, these AHA-incorporating nascent proteins were then labeled with a fluorescent dye (Alexa 488) conjugated to an alkyne group (Dieterich et al., 2006, 2007). The fluorescence intensity was determined with a fluorimeter where the increased conjugation of the fluorescent dye to the proteins indicates increased protein synthesis (Fig. 4A). Treatment of DRG neurons in culture with IL-6 (50 ng/ml) and/or NGF (10 ng/ml) for 30 min results in significant increase (vehicle 100 ± 38%, NGF 501 ± 36%, IL-6 321 ± 54%, and IL-6 + NGF 562 ± 85%) in nascent protein synthesis (Fig. 4B). We did not observe an additive effect with combined IL-6 and NGF treatment. The reasons for this are not clear but could be related to limitations of the present assay or, alternatively, might indicate that signaling through either pathway is sufficient to maximally engage protein synthesis over the time course examined here. The increase in protein synthesis was partially blocked by 30 min pretreatment with U0126 (1 μm). Pretreatment with anisomycin (50 μm) or rapamycin (500 nm) (with or without U0126, 1 μm) for 30 min (rapamycin 105 ± 53%, U0126 329 ± 33%, rapamycin + U0126 150 ± 28%, and anisomycin 206 ± 48%) blocked IL-6- and NGF-induced increases in nascent protein synthesis (Fig. 4B). To determine whether direct interference with eIF4F formation would block IL-6 + NGF-dependent protein synthesis, we used 4EGI1, a small molecule demonstrated to interrupt eIF4E/eIF4G binding through either stabilization of 4EBP binding to eIF4E or molecularly mimicking this interaction (Moerke et al., 2007). For this assay, we used conventional Western blotting to serve as a control for AHA incorporation in the 96-well plate format assay. Pretreatment with 4EGI1 (20 μm) for 15 min and cotreatment with IL-6 (50 ng/ml) and NGF (10 ng/ml) for 30 min (vehicle 100 ± 7, 4EGI1 + IL-6 + NGF 108 ± 26, and IL-6 + NGF 204 ± 29) blocked the IL-6- and NGF-induced AHA incorporation (Fig. 4C). Together, these results indicate that IL-6 and NGF signal through the ERK and mTOR pathways, respectively, to enhance eIF4F assembly, thus increasing nascent, cap-dependent translation in primary sensory neurons.
Figure 4.
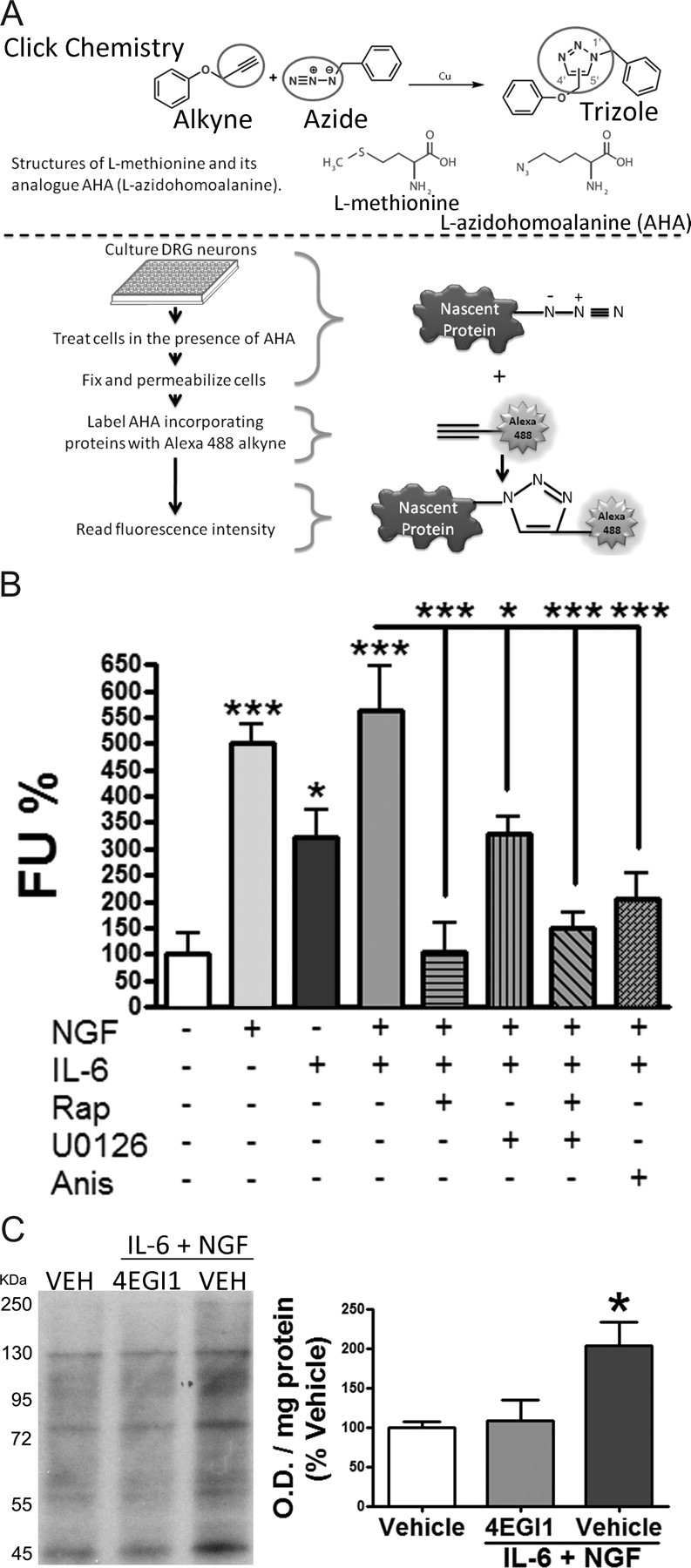
IL-6 and NGF induce cap-dependent translation of nascent proteins in primary sensory neurons by activating ERK and mTOR pathways. A, A schematic diagram illustrating the experimental procedure. B, DRG neurons pretreated with rapamycin (500 nm, Rap), U0126 (10 μm), and anisomycin (50 μm, Anis) for 30 min followed by treatment with vehicle, IL-6 (50 ng/ml), and/or NGF (10 ng/ml) with and without inhibitors for 30 min. Fluorescence units were reported as percentage of vehicle. Treatment with IL-6 and/or NGF increases the incorporation of AHA, indicative of increased nascent protein synthesis, and this effect was inhibited by mTOR (rapamycin), MEK (U0126), and general protein synthesis (anisomycin) inhibitors. C, Western blot and quantification of primary sensory neurons in culture pretreated with vehicle or the inhibitor of eIF4F complex formation, 4EGI1 (20 μm), for 30 min followed by treatment with vehicle or IL-6 (50 ng/ml) and NGF (10 ng/ml) in AHA-containing media 30 min. *p < 0.05, ***p < 0.001.
In acute and chronic pain conditions, changes in IL-6 and NGF expression are observed within the ganglia (Lee et al., 1998; Vardanyan et al., 2010), but the most profound changes are found at distal sites (e.g., painful skin areas or at the site of nerve injury) from the DRG or TG (Lewin et al., 1994; Woolf et al., 1994; Bolin et al., 1995; Anderson and Rao, 2001; Ji et al., 2002; Banik et al., 2005; Clark et al., 2007). This suggests that these mediators may lead to local changes in nociceptor sensitivity resulting from an IL-6- and NGF-induced enhancement of translational control of gene expression in the axons of primary sensory neurons. To test this, we used confocal microcopy to visualize the colocalization of AHA incorporation in sensory neuron axons following treatment with IL-6 and NGF with markers for primary sensory neurons (NFH + peripherin). ICA of the pair (AHA incorporation and neuronal markers) of micrographs, with somas excluded, demonstrated an increased PDM value in the axons of neurons treated with IL-6 and NGF. This effect was blocked by pretreatment (15 min) and subsequent cotreatment (30 min) with anisomycin and rapamycin (Fig. 5A). The range of color-coded PDM values is shown next to scale bars in Figure 5A. More positive PDM values (e.g., 0.69 maximum PDM with IL-6 and NGF) indicate greater intensity of AHA incorporation along axons colabeled with NFH and peripherin. ICQ values were derived from the ICA analysis, and in every treatment group, the values were positive, indicating that AHA colocalized with the neuronal markers in the axons (Fig. 5B). Moreover, we observed a significant increase in the ICQ values of the cultures that were treated with IL-6 and NGF relative to vehicle and groups cotreated with anisomycin or rapamycin (vehicle 0.29 ± 0.008, anisomycin + IL-6 + NGF 0.28 ± 0.014, rapamycin + IL-6 + NGF 0.29 ± 0.012, and IL-6 + NGF 0.33 ± 0.004). This finding supports the hypothesis that IL-6 and NGF regulate local changes in translation in the axons of sensory neurons. Similar to results obtained in a 96-well plate format, when soma and axons were used in the analysis, there was also a significant increase in ICQ values in IL-6- and NGF-treated cultures (Fig. 5C). This indicates that enhanced nascent protein synthesis observed in the high-throughput format (Fig. 4B) as well as in Western blots (Fig. 4C) is of a neuronal origin.
Figure 5.
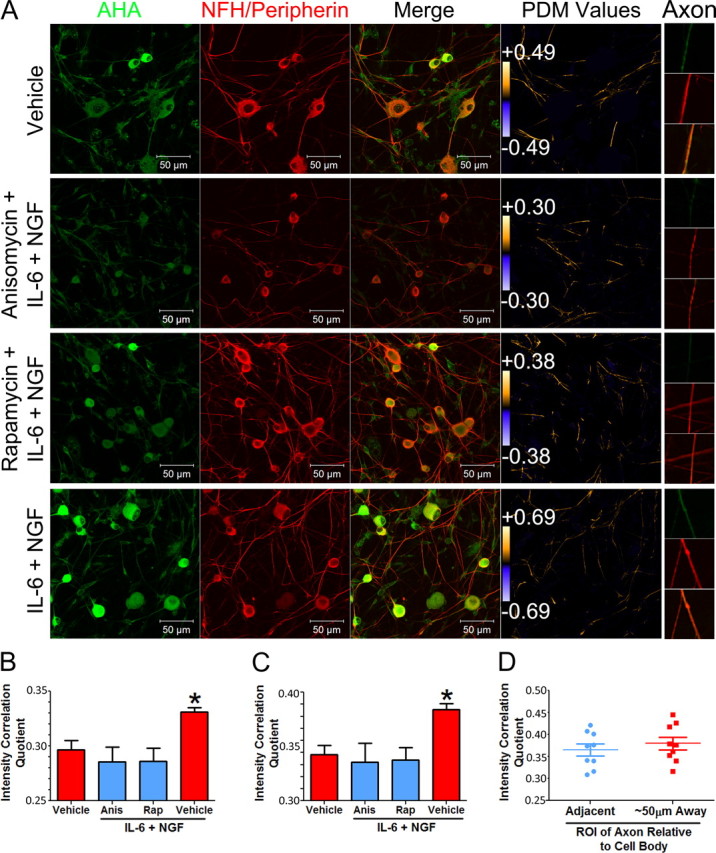
IL-6 and NGF induce cap-dependent translation of nascent proteins in the axons of primary sensory neurons. A, Representative micrographs of intensity correlation analysis of fluorescently labeled AHA with neuronal markers (NF200 and peripherin). Neurons in cultures were treated with vehicle or with IL-6 (50 ng/ml) and NGF (10 ng/ml) with or without anisomycin (50 μm) or rapamycin (500 nm) pretreatment (30 min) and cotreatment. PDM values were used to generate an image that visualizes the extent of colocalization of AHA with NFH/peripherin in the axons (PDM Values panel). Intensity correlation analysis that excluded the cell bodies of the neurons resulted in higher PDM values with IL-6 and NGF treatment than in the vehicle group. PDM values are shown on the color bar on the far right. While the color bar scales are equivalent for all images, PDM values show the range of PDMs within that scale. Higher PDM values indicate higher intensity of colocalization between neuronal markers and AHA incorporation (more nascent protein synthesis). This increase was inhibited with anisomycin and rapamycin. High-magnification images of individual axons are shown on the far right (Axon). B, ICQ of fluorescently labeled AHA with neuronal markers (NF200 and peripherin) in axons quantified by ICA. Treatment with IL-6 (50 ng/ml) and NGF (10 ng/ml) results in a signification increase in the ICQ value in axons of primary sensory neurons in culture relative to the vehicle group. This increase was blocked with anisomycin (Anis) and rapamycin (Rap). C, ICQ analysis of the entire image (axons not excluded) showing that there is also an increase in nascent protein synthesis with IL-6 and NGF when axons and somas are considered. D, Analysis of proximal (adjacent to soma to 10 μm distal—Adjacent) versus distal (50–60 μm from the soma—∼50 μm Away) axon segments from IL-6- and NGF-treated cultures showing that the distribution of ICQ values are equivalent regardless of distance from the soma. *p < 0.05.
While increased AHA labeling in the axons of sensory neurons strongly suggests enhanced translation in the axonal compartment upon IL-6 and NGF exposure, it is possible that transport of newly synthesized proteins from the soma also contributes to this effect. We predicted that if anterograde transport contributes significantly to the axonal presence of AHA, then a gradient of AHA incorporation should be observed from the proximal axon (higher ICQ values) to more distal sites (lower ICQ values). In IL-6- and NGF-treated samples, we observed no difference in ICQ values (Fig. 5D) between proximal 10 μm lengths of axon (10 μm lengths directly adjacent to the soma) and distal 10 μm lengths of axon (50–60 μm distal from the soma). The lack of a gradient of newly synthesized proteins in the axons of sensory neurons after IL-6 and NGF exposure supports the conclusion that local, axonal protein synthesis is induced by IL-6 and NGF.
IL-6- and IL-6 + NGF-induced allodynia is translation dependent
To determine whether IL-6- and NGF-induced regulation of cap-dependent translation plays a role in local nociceptive plasticity, we assessed whether locally applied inhibitors of translation would attenuate mechanical allodynia produced by these mediators. Intraplantar injection of IL-6 (0.1 ng) induced a profound mechanical allodynia that lasted at least 48 h. Coinjection with anisomycin [25 μg (Jiménez-Díaz et al., 2008)] attenuated this IL-6-induced allodynia (Fig. 6A), suggesting that local translation of proteins in the axons of sensory neurons mediates IL-6-induced nociceptive plasticity. We have demonstrated that IL-6 signals to the cap-dependent translational machinery through the ERK/Mnk1/eIF4E pathway. Hence, we assessed whether local ERK inhibition abrogated IL-6-mediated allodynia. Coinjection of IL-6 (0.1 ng) with U0126 (10 μg) resulted in the attenuation of IL-6-induced allodynia (Fig. 6B). The groups that received U0126 (regardless of IL-6 cotreatment) demonstrated an acute reduction of withdrawal thresholds that returned to baseline levels by 24 h. The reasons for this intrinsic effect of MEK inhibition are not clear. Cap-dependent translation requires stabilization of eIF4F cap complex. To verify that IL-6-induced allodynia is dependent on the stable interaction of eIF4G with eIF4E, we coinjected 4EGI1 with IL-6. This treatment resulted in a dose-dependent attenuation of IL-6-mediated mechanical allodynia (Fig. 6C,D). Therefore, disruption of general translation (anisomycin), blockade of ERK signaling (U0126), and destabilization of eIF4F complex formation (4EGI1) all attenuate IL-6-induced mechanical allodynia. Because IL-6-mediated nociceptive plasticity is dependent on direct actions of IL-6 on sensory nerves (Dina et al., 2008; Andratsch et al., 2009), we conclude that IL-6 controls mechanical nociceptive plasticity via cap-dependent translation mechanisms in the peripheral endings of sensory neurons.
Figure 6.
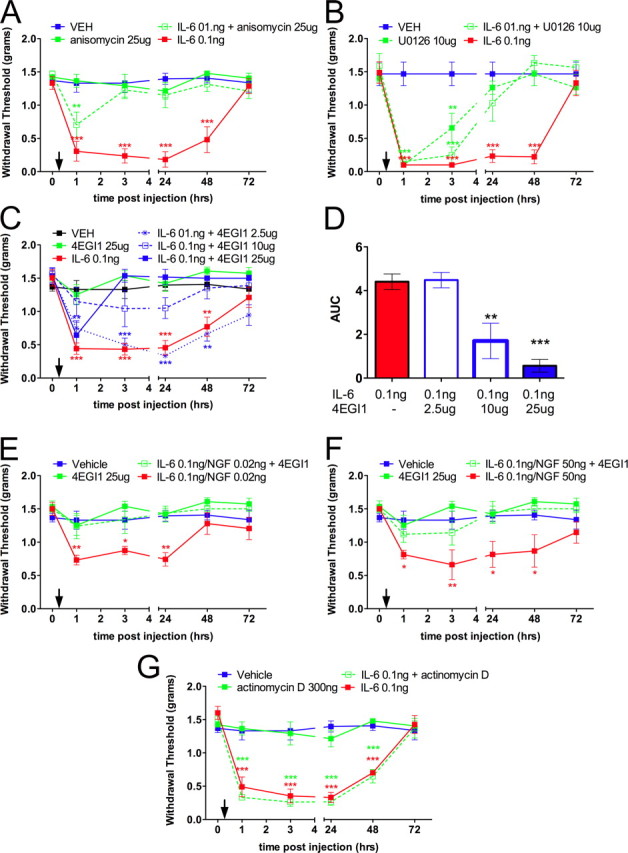
Intraplantar injection of IL-6 or coinjection of IL-6 and NGF into the hindpaw of mice induces long-lasting mechanical allodynia blocked by cotreatment with anisomycin, U0126, or 4EGI1. Mechanical thresholds were measured at the indicated time points before and after intraplantar injection (indicated by downward arrows). A, IL-6 (0.1 ng) induced a significant decrease in withdrawal thresholds that was blocked by coinjection of the general protein synthesis inhibitor anisomycin (25 μg). B, IL-6 (0.1 ng)-induced decreases in withdrawal thresholds were blocked by coinjection of the MEK inhibitor U0126 (10 μg). C, D, IL-6 (0.1 ng)-mediated allodynia was blocked with the inhibitor of eIF4F complex formation, 4EGI1 (2.5, 10, and 25 μg) in a dose-dependent manner. E, F, Coinjection of IL-6 (0.1 ng) and NGF [0.02 (E) and 50 (F) ng] resulted in a significant decrease in the mechanical thresholds at the indicated time points, and these effects were blocked by coinjection of 4EGI1 (25 μg). G, Coinjection of the transcription inhibitor actinomycin D (300 ng) had no effect on IL-6 (0.1 ng)-mediated allodynia. *p < 0.05, **p < 0.01, ***p < 0.001.
Given that we have shown that IL-6 and NGF signaling converge onto the eIF4F complex to induce cap-dependent translation, we explored whether allodynia elicited by the coinjection of these algogens is dependent on eIF4F complex formation. Because 4EGI-1 is capable of blocking eIF4F complex formation regardless of pathway activation, this pharmacological tool is ideal for testing whether IL-6 and NGF require eIF4F complex formation to induce allodynia. Coinjection of IL-6 (0.1 ng) with NGF at two different doses induced mechanical allodynia that lasted for at least 24 h with 0.02 ng of NGF (dose chosen based on mixed dose ratios from in vitro experiments) and for 48 h with 50 ng of NGF (Amaya et al., 2006) (Fig. 6E,F). For both dose ratios of IL-6 and NGF, mechanical allodynia was completely blocked by coinjection of 4EGI1 (25 μg). At the higher dose of NGF (50 ng), NGF-mediated ERK activation may also be expected to occur as we observed ERK activation with higher doses of NGF in vitro (Fig. 2C). This finding supports the hypothesis that the generation of IL-6 and NGF-mediated mechanical allodynia requires local formation of the eIF4F complex.
While the behavioral pharmacology results above support the notion that IL-6 and IL-6 and NGF promote mechanical hypersensitivity through translation-dependent mechanisms, it is not clear whether this occurs via the translation of preexisting pools of mRNA. To assess this directly, we coinjected IL-6 (0.1 ng) with the transcription inhibitor actinomycin D [300 ng (Ferreira et al., 2008)]. Actinomycin D had no effect on IL-6-induced allodynia at any time point (Fig. 6G). This supports the conclusion that allodynia induced by IL-6 and NGF is dependent on the translation of preexisting pools of mRNA, likely present in the axons of sensory nerve fibers.
Discussion
The important role of translation regulation in synaptic plasticity has led to the recognition of translation control as a critical factor for learning and memory (Costa-Mattioli et al., 2009). In the area of nociceptive plasticity, multiple lines of evidence suggest that translation control at the level of the primary afferent neuron is crucial for the establishment and maintenance of enhanced pain states (Price and Géranton, 2009). Local hindpaw injection of the mTOR inhibitor rapamycin reduces formalin-induced pain (Price et al., 2007). Group I mGluR-mediated thermal hyperalgesia fails to develop in mice lacking the RNA-binding and translation regulating protein, fragile X mental retardation protein [FMRP (Price et al., 2007)]. Rapamycin treatment reduces injury-induced changes in nociceptive hypersensitivity by acting on both peripheral terminals (Jiménez-Díaz et al., 2008) and the central projections of sensory afferents (Géranton et al., 2009). Furthermore, sustained mTOR activity is required to maintain the mechanical sensitivity of a subset of primary afferent fibers (Jiménez-Díaz et al., 2008). Finally, nerve injury induces a redistribution of Nav1.8 mRNA to the axons of sensory neurons (Thakor et al., 2009), suggesting that local translation of Nav1.8 may contribute to hyperexcitability of sensory neurons following injury. Together, these findings make a clear case for the importance of translation control in pain plasticity (Price and Géranton, 2009); however, a critical gap in knowledge exists, as the factors involved in regulating translational activity in these neurons in association with pain states are completely unknown. Hence, the major finding of the present work is the identification of two such factors, IL-6 and NGF. Since these are both well known sensitizers of nociceptors (Murphy et al., 1999b; Pezet and McMahon, 2006; Dina et al., 2008; Andratsch et al., 2009), the present findings tie our emerging understanding of the role of translation control in nociceptive plasticity to definitive molecular mechanisms responsible for modulating these pathways and broaden our knowledge of the mechanisms of action of IL-6 and NGF in regulating nociceptive plasticity.
IL-6 binds to IL-6R (gp80), which exists as either soluble or transmembrane protein, forming a composite ligand capable of binding to gp130, which is the signal-transducing component of the IL-6/IL-6R/gp130 complex. Similar to other cytokines, IL-6/IL-6R/gp130 recruits JAKs, leading to the activation of STATs (Boulanger et al., 2003; Kamimura et al., 2003). The ERK pathway, an established mediator of nociceptive potentiation (Karim et al., 2001; Obata et al., 2004a,b; Ji et al., 2009; Stamboulian et al., 2010), also mediates the actions of IL-6. Given that many pathologies associated with pain involve an increase in IL-6 levels (Bolin et al., 1995; Murphy et al., 1999b; Anderson and Rao, 2001; Clark et al., 2007; Nishimoto et al., 2009) and that pathological pain states depend on IL-6 receptor expression in sensory neurons (Andratsch et al., 2009), we assessed the ability of IL-6 to signal to the translational machinery in DRG and TG neurons. We show that treatment with IL-6 leads to the phosphorylation of eIF4E via MEK and Mnk1 and increased nascent protein synthesis. Hence, our experiments show that IL-6 signals to the translational machinery in primary afferents, leading to enhanced cap-dependent translation by activating the ERK/Mnk1/eIF4E pathway (Fig. 7). Furthermore, we have demonstrated that IL-6-mediated, mechanical nociceptive plasticity is crucially dependent on this pathway, as it is blocked by inhibition of ERK and/or eIF4F complex formation. Together, these findings demonstrate that IL-6 controls translation in sensory neurons and that IL-6-mediated events leading to mechanical allodynia require nascent protein synthesis.
Figure 7.

Schematic summarizing the present findings. IL-6 signals through the MEK/ERK/Mnk pathway to phosphorylate the cap-binding protein eIF4E. NGF signals primarily through the PI3 kinase/AKT/mTOR pathway to phosphorylate both 4EBP and eIF4G. Phosphorylation of 4EBP, which is a negative regulator of eIF4F complex formation, results in its dissociation from eIF4E, thus allowing the binding of eIF4E to eIF4G to occur. Phosphorylation of eIF4E and eIF4G enhances the formation of the eIF4F cap-binding complex. The outcome of IL-6- and NGF-mediated eIF4F cap complex formation is the enhancement of cap-dependent translation, leading to rapid changes in translational control of gene expression in sensory neurons, and this effect is linked to mechanical allodynia evoked by these algogens.
NGF-mediated effects are central to inflammatory, surgical, visceral, and neuropathic pain (Lewin et al., 1994; Woolf et al., 1994; McMahon et al., 1995; Anderson and Rao, 2001; Banik et al., 2005; Wild et al., 2007). The canonical NGF signal transduction pathway commences at the trkA receptor, activating PI3 kinase and AKT (Kimpinski and Mearow, 2001). We also demonstrate that this pathway activates mTOR in DRG and TG neurons through phosphorylation of serine 2448, a site directly phosphorylated by AKT (Navé et al., 1999). In addition to stimulation of AKT/mTOR, NGF is known to stimulate ERK in DRG neurons (Obata et al., 2003; Zhuang et al., 2004), which we also observed, albeit at higher doses. Hence, our findings demonstrate that NGF's most potent activity in primary afferents is via the AKT/mTOR pathway. Activation of mTOR inhibits the negative regulator of translation, 4EBP, and activates a key component of the eIF4F complex, eIF4G (Sonenberg and Hinnebusch, 2009). As with IL-6, treatment of DRG neurons with NGF was sufficient to enhance nascent protein synthesis in these neurons. Therefore, NGF is a key regulator of translation regulation in primary afferent neurons via stimulation of the mTOR pathway (Fig. 7). Sustained mTOR activity is required to maintain the sensitivity of a subset of nociceptive A-type fibers (Jiménez-Díaz et al., 2008; Géranton et al., 2009). Because NGF is important for regulating nociceptor function across developmental stages (Pezet and McMahon, 2006), it is possible that constitutive NGF-dependent maintenance of mTOR activity underlies this effect.
Cotreatment of primary afferent neurons with IL-6 and NGF results in the activation of both ERK and mTOR pathways. We provide direct evidence that eIF4F complex assembly rapidly results from cotreatment with IL-6 and NGF. Formation of the eIF4F complex leads to nascent protein synthesis in these neurons as demonstrated by Western blotting and via a high-throughput compatible, fluorescence-based assay. Importantly, using confocal microscopy and ICA, our data support the conclusion that NGF and IL-6 stimulate nascent protein synthesis in the axons of sensory neurons. These data suggest that IL-6- and NGF-induced cap-dependent translation in adult primary sensory neurons may also occur in the axons of these afferents in vivo, providing a mechanistic rationale for local changes in translational control of gene expression in conditions where NGF and IL-6 are increased at peripheral sites. This observation is consistent with NGF-mediated CREB translation in the axons of embryonic DRG neurons (Cox et al., 2008) and the effects of NGF on RNA trafficking into the axons of DRG neurons with a preconditioning crush lesion (Willis et al., 2005).
Although IL-6 and NGF are known to sensitize the thermal sensitivity of nociceptors through posttranslational modification of ion channels (Chuang et al., 2001; Ji et al., 2002; Obreja et al., 2005), mechanisms underlying mechanical sensitization by these factors have remained enigmatic. We show that IL-6 and NGF-mediated mechanical allodynia is completely blocked by inhibition of eIF4F complex formation with 4EGI1. While we cannot exclude the possibility that protein synthesis in non-neuronal cells contributes to this effect, two points support the hypothesis that convergent signaling to eIF4F in the axons of sensory neurons is critical for nociceptive sensitization: (1) we found strong evidence for IL-6- and NGF-induced nascent protein synthesis in the axons of these neurons in vitro, and (2) IL-6-mediated nociceptive sensitization requires gp130 expression in sensory afferents (Dina et al., 2008; Andratsch et al., 2009), ruling out effects on other local cell types in the skin to IL-6-mediated allodynia. Moreover, since IL-6-induced allodynia was not blocked by local injection of transcription inhibitors, it is likely that these translation events are dependent on preexisting pools of mRNAs. Hence, our findings support a novel and rapid mechanism of action for IL-6 and NGF on sensory neurons: enhanced cap-dependent translation leading to prolonged mechanical hypersensitivity.
In conclusion, we have elucidated specific mechanisms through which the pain promoting compounds IL-6 and NGF control translation in primary sensory neurons. Our findings point toward a model in which the injury-induced release of IL-6 and/or NGF alters local gene expression via cap-dependent translation, leading to sustained changes in the mechanical sensitivity of primary afferent neurons. This model suggests a therapeutic framework in which pathological changes in translational control of gene expression accompanying the development of pain conditions might be controlled with specific inhibitors of cap-dependent translation.
Footnotes
This work was supported by startup funds from The University of Arizona School of Medicine, The American Pain Society, The Rita Allen Foundation, and National Institutes of Health (NIH) Grant R01NS065926 to T.J.P. K.A.P. was supported by NIH Grant DA023513. N.E. was supported by the University of Arizona Medical Student Research Program (T35HL007479). T.J.P. is a Rita Allen Foundation Scholar in Pain. We thank Ning Qu for expert technical assistance and Frank Porreca for useful discussions in the preparation of the manuscript.
References
- Akira S, Yoshida K, Tanaka T, Taga T, Kishimoto T. Targeted disruption of the IL-6 related genes: gp130 and NF-IL-6. Immunol Rev. 1995;148:221–253. doi: 10.1111/j.1600-065x.1995.tb00100.x. [DOI] [PubMed] [Google Scholar]
- Amaya F, Wang H, Costigan M, Allchorne AJ, Hatcher JP, Egerton J, Stean T, Morisset V, Grose D, Gunthorpe MJ, Chessell IP, Tate S, Green PJ, Woolf CJ. The voltage-gated sodium channel Na(v)1.9 is an effector of peripheral inflammatory pain hypersensitivity. J Neurosci. 2006;26:12852–12860. doi: 10.1523/JNEUROSCI.4015-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson LC, Rao RD. Interleukin-6 and nerve growth factor levels in peripheral nerve and brainstem after trigeminal nerve injury in the rat. Arch Oral Biol. 2001;46:633–640. doi: 10.1016/s0003-9969(01)00024-3. [DOI] [PubMed] [Google Scholar]
- Andratsch M, Mair N, Constantin CE, Scherbakov N, Benetti C, Quarta S, Vogl C, Sailer CA, Uceyler N, Brockhaus J, Martini R, Sommer C, Zeilhofer HU, Müller W, Kuner R, Davis JB, Rose-John S, Kress M. A key role for gp130 expressed on peripheral sensory nerves in pathological pain. J Neurosci. 2009;29:13473–13483. doi: 10.1523/JNEUROSCI.1822-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asante CO, Wallace VC, Dickenson AH. Formalin-induced behavioural hypersensitivity and neuronal hyperexcitability are mediated by rapid protein synthesis at the spinal level. Mol Pain. 2009;5:27. doi: 10.1186/1744-8069-5-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banik RK, Subieta AR, Wu C, Brennan TJ. Increased nerve growth factor after rat plantar incision contributes to guarding behavior and heat hyperalgesia. Pain. 2005;117:68–76. doi: 10.1016/j.pain.2005.05.017. [DOI] [PubMed] [Google Scholar]
- Beretta L, Gingras AC, Svitkin YV, Hall MN, Sonenberg N. Rapamycin blocks the phosphorylation of 4E-BP1 and inhibits cap-dependent initiation of translation. EMBO J. 1996;15:658–664. [PMC free article] [PubMed] [Google Scholar]
- Bolin LM, Verity AN, Silver JE, Shooter EM, Abrams JS. Interleukin-6 production by Schwann cells and induction in sciatic nerve injury. J Neurochem. 1995;64:850–858. doi: 10.1046/j.1471-4159.1995.64020850.x. [DOI] [PubMed] [Google Scholar]
- Boulanger MJ, Chow DC, Brevnova EE, Garcia KC. Hexameric structure and assembly of the interleukin-6/IL-6 alpha-receptor/gp130 complex. Science. 2003;300:2101–2104. doi: 10.1126/science.1083901. [DOI] [PubMed] [Google Scholar]
- Carracedo A, Ma L, Teruya-Feldstein J, Rojo F, Salmena L, Alimonti A, Egia A, Sasaki AT, Thomas G, Kozma SC, Papa A, Nardella C, Cantley LC, Baselga J, Pandolfi PP. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest. 2008;118:3065–3074. doi: 10.1172/JCI34739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattaneo A. Tanezumab, a recombinant humanized mAb against nerve growth factor for the treatment of acute and chronic pain. Curr Opin Mol Ther. 2010;12:94–106. [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Chuang HH, Prescott ED, Kong H, Shields S, Jordt SE, Basbaum AI, Chao MV, Julius D. Bradykinin and nerve growth factor release the capsaicin receptor from PtdIns(4,5)P2-mediated inhibition. Nature. 2001;411:957–962. doi: 10.1038/35082088. [DOI] [PubMed] [Google Scholar]
- Clark JD, Shi X, Li X, Qiao Y, Liang D, Angst MS, Yeomans DC. Morphine reduces local cytokine expression and neutrophil infiltration after incision. Mol Pain. 2007;3:28. doi: 10.1186/1744-8069-3-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Codeluppi S, Svensson CI, Hefferan MP, Valencia F, Silldorff MD, Oshiro M, Marsala M, Pasquale EB. The Rheb-mTOR pathway is upregulated in reactive astrocytes of the injured spinal cord. J Neurosci. 2009;29:1093–1104. doi: 10.1523/JNEUROSCI.4103-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa-Mattioli M, Sossin WS, Klann E, Sonenberg N. Translational control of long-lasting synaptic plasticity and memory. Neuron. 2009;61:10–26. doi: 10.1016/j.neuron.2008.10.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox LJ, Hengst U, Gurskaya NG, Lukyanov KA, Jaffrey SR. Intra-axonal translation and retrograde trafficking of CREB promotes neuronal survival. Nat Cell Biol. 2008;10:149–159. doi: 10.1038/ncb1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieterich DC, Link AJ, Graumann J, Tirrell DA, Schuman EM. Selective identification of newly synthesized proteins in mammalian cells using bioorthogonal noncanonical amino acid tagging (BONCAT) Proc Natl Acad Sci U S A. 2006;103:9482–9487. doi: 10.1073/pnas.0601637103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieterich DC, Lee JJ, Link AJ, Graumann J, Tirrell DA, Schuman EM. Labeling, detection and identification of newly synthesized proteomes with bioorthogonal non-canonical amino-acid tagging. Nat Protoc. 2007;2:532–540. doi: 10.1038/nprot.2007.52. [DOI] [PubMed] [Google Scholar]
- Dina OA, Green PG, Levine JD. Role of interleukin-6 in chronic muscle hyperalgesic priming. Neuroscience. 2008;152:521–525. doi: 10.1016/j.neuroscience.2008.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira J, Trichês KM, Medeiros R, Cabrini DA, Mori MA, Pesquero JB, Bader M, Calixto JB. The role of kinin B1 receptors in the nociception produced by peripheral protein kinase C activation in mice. Neuropharmacology. 2008;54:597–604. doi: 10.1016/j.neuropharm.2007.11.008. [DOI] [PubMed] [Google Scholar]
- Géranton SM, Jiménez-Díaz L, Torsney C, Tochiki KK, Stuart SA, Leith JL, Lumb BM, Hunt SP. A rapamycin-sensitive signaling pathway is essential for the full expression of persistent pain states. J Neurosci. 2009;29:15017–15027. doi: 10.1523/JNEUROSCI.3451-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Samad TA, Jin SX, Schmoll R, Woolf CJ. p38 MAPK activation by NGF in primary sensory neurons after inflammation increases TRPV1 levels and maintains heat hyperalgesia. Neuron. 2002;36:57–68. doi: 10.1016/s0896-6273(02)00908-x. [DOI] [PubMed] [Google Scholar]
- Ji RR, Gereau RW, 4th, Malcangio M, Strichartz GR. MAP kinase and pain. Brain Res Rev. 2009;60:135–148. doi: 10.1016/j.brainresrev.2008.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiménez-Díaz L, Géranton SM, Passmore GM, Leith JL, Fisher AS, Berliocchi L, Sivasubramaniam AK, Sheasby A, Lumb BM, Hunt SP. Local translation in primary afferent fibers regulates nociception. PLoS One. 2008;3:e1961. doi: 10.1371/journal.pone.0001961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamimura D, Ishihara K, Hirano T. IL-6 signal transduction and its physiological roles: the signal orchestration model. Rev Physiol Biochem Pharmacol. 2003;149:1–38. doi: 10.1007/s10254-003-0012-2. [DOI] [PubMed] [Google Scholar]
- Karim F, Wang CC, Gereau RW., 4th Metabotropic glutamate receptor subtypes 1 and 5 are activators of extracellular signal-regulated kinase signaling required for inflammatory pain in mice. J Neurosci. 2001;21:3771–3779. doi: 10.1523/JNEUROSCI.21-11-03771.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimpinski K, Mearow K. Neurite growth promotion by nerve growth factor and insulin-like growth factor-1 in cultured adult sensory neurons: role of phosphoinositide 3-kinase and mitogen activated protein kinase. J Neurosci Res. 2001;63:486–499. doi: 10.1002/jnr.1043. [DOI] [PubMed] [Google Scholar]
- Kinkade CW, Castillo-Martin M, Puzio-Kuter A, Yan J, Foster TH, Gao H, Sun Y, Ouyang X, Gerald WL, Cordon-Cardo C, Abate-Shen C. Targeting AKT/mTOR and ERK MAPK signaling inhibits hormone-refractory prostate cancer in a preclinical mouse model. J Clin Invest. 2008;118:3051–3064. doi: 10.1172/JCI34764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SE, Shen H, Taglialatela G, Chung JM, Chung K. Expression of nerve growth factor in the dorsal root ganglion after peripheral nerve injury. Brain Res. 1998;796:99–106. doi: 10.1016/s0006-8993(98)00335-7. [DOI] [PubMed] [Google Scholar]
- Lewin GR, Rueff A, Mendell LM. Peripheral and central mechanisms of NGF-induced hyperalgesia. Eur J Neurosci. 1994;6:1903–1912. doi: 10.1111/j.1460-9568.1994.tb00581.x. [DOI] [PubMed] [Google Scholar]
- Li Q, Lau A, Morris TJ, Guo L, Fordyce CB, Stanley EF. A syntaxin 1, Gαo, and N-type calcium channel complex at a presynaptic nerve terminal: analysis by quantitative immunocolocalization. J Neurosci. 2004;24:4070–4081. doi: 10.1523/JNEUROSCI.0346-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews M, Sonenberg N, Hershey JWB. Ed 3. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 2007. Translational control in biology and medicine. [Google Scholar]
- McMahon SB, Bennett DL, Priestley JV, Shelton DL. The biological effects of endogenous nerve growth factor on adult sensory neurons revealed by a trkA-IgG fusion molecule. Nat Med. 1995;1:774–780. doi: 10.1038/nm0895-774. [DOI] [PubMed] [Google Scholar]
- Moelling K, Schad K, Bosse M, Zimmermann S, Schweneker M. Regulation of Raf-Akt cross-talk. J Biol Chem. 2002;277:31099–31106. doi: 10.1074/jbc.M111974200. [DOI] [PubMed] [Google Scholar]
- Moerke NJ, Aktas H, Chen H, Cantel S, Reibarkh MY, Fahmy A, Gross JD, Degterev A, Yuan J, Chorev M, Halperin JA, Wagner G. Small-molecule inhibition of the interaction between the translation initiation factors eIF4E and eIF4G. Cell. 2007;128:257–267. doi: 10.1016/j.cell.2006.11.046. [DOI] [PubMed] [Google Scholar]
- Murphy PG, Grondin J, Altares M, Richardson PM. Induction of interleukin-6 in axotomized sensory neurons. J Neurosci. 1995;15:5130–5138. doi: 10.1523/JNEUROSCI.15-07-05130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy PG, Borthwick LS, Johnston RS, Kuchel G, Richardson PM. Nature of the retrograde signal from injured nerves that induces interleukin-6 mRNA in neurons. J Neurosci. 1999a;19:3791–3800. doi: 10.1523/JNEUROSCI.19-10-03791.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy PG, Ramer MS, Borthwick L, Gauldie J, Richardson PM, Bisby MA. Endogenous interleukin-6 contributes to hypersensitivity to cutaneous stimuli and changes in neuropeptides associated with chronic nerve constriction in mice. Eur J Neurosci. 1999b;11:2243–2253. doi: 10.1046/j.1460-9568.1999.00641.x. [DOI] [PubMed] [Google Scholar]
- Navé BT, Ouwens M, Withers DJ, Alessi DR, Shepherd PR. Mammalian target of rapamycin is a direct target for protein kinase B: identification of a convergence point for opposing effects of insulin and amino-acid deficiency on protein translation. Biochem J. 1999;344:427–431. [PMC free article] [PubMed] [Google Scholar]
- Nishimoto N, Miyasaka N, Yamamoto K, Kawai S, Takeuchi T, Azuma J. Long-term safety and efficacy of tocilizumab, an anti-IL-6 receptor monoclonal antibody, in monotherapy, in patients with rheumatoid arthritis (the STREAM study): evidence of safety and efficacy in a 5-year extension study. Ann Rheum Dis. 2009;68:1580–1584. doi: 10.1136/ard.2008.092866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obata K, Yamanaka H, Dai Y, Tachibana T, Fukuoka T, Tokunaga A, Yoshikawa H, Noguchi K. Differential activation of extracellular signal-regulated protein kinase in primary afferent neurons regulates brain-derived neurotrophic factor expression after peripheral inflammation and nerve injury. J Neurosci. 2003;23:4117–4126. doi: 10.1523/JNEUROSCI.23-10-04117.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obata K, Yamanaka H, Dai Y, Mizushima T, Fukuoka T, Tokunaga A, Noguchi K. Differential activation of MAPK in injured and uninjured DRG neurons following chronic constriction injury of the sciatic nerve in rats. Eur J Neurosci. 2004a;20:2881–2895. doi: 10.1111/j.1460-9568.2004.03754.x. [DOI] [PubMed] [Google Scholar]
- Obata K, Yamanaka H, Kobayashi K, Dai Y, Mizushima T, Katsura H, Fukuoka T, Tokunaga A, Noguchi K. Role of mitogen-activated protein kinase activation in injured and intact primary afferent neurons for mechanical and heat hypersensitivity after spinal nerve ligation. J Neurosci. 2004b;24:10211–10222. doi: 10.1523/JNEUROSCI.3388-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obreja O, Biasio W, Andratsch M, Lips KS, Rathee PK, Ludwig A, Rose-John S, Kress M. Fast modulation of heat-activated ionic current by proinflammatory interleukin 6 in rat sensory neurons. Brain. 2005;128:1634–1641. doi: 10.1093/brain/awh490. [DOI] [PubMed] [Google Scholar]
- Oka T, Oka K, Hosoi M, Hori T. Intracerebroventricular injection of interleukin-6 induces thermal hyperalgesia in rats. Brain Res. 1995;692:123–128. doi: 10.1016/0006-8993(95)00691-i. [DOI] [PubMed] [Google Scholar]
- Pezet S, McMahon SB. Neurotrophins: mediators and modulators of pain. Annu Rev Neurosci. 2006;29:507–538. doi: 10.1146/annurev.neuro.29.051605.112929. [DOI] [PubMed] [Google Scholar]
- Price TJ, Géranton SM. Translating nociceptor sensitivity: the role of axonal protein synthesis in nociceptor physiology. Eur J Neurosci. 2009;29:2253–2263. doi: 10.1111/j.1460-9568.2009.06786.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price TJ, Rashid MH, Millecamps M, Sanoja R, Entrena JM, Cervero F. Decreased nociceptive sensitization in mice lacking the fragile X mental retardation protein: role of mGluR1/5 and mTOR. J Neurosci. 2007;27:13958–13967. doi: 10.1523/JNEUROSCI.4383-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rukwied R, Mayer A, Kluschina O, Obreja O, Schley M, Schmelz M. NGF induces non-inflammatory localized and lasting mechanical and thermal hypersensitivity in human skin. Pain. 2010;148:407–413. doi: 10.1016/j.pain.2009.11.022. [DOI] [PubMed] [Google Scholar]
- Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. 2009;136:731–745. doi: 10.1016/j.cell.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamboulian S, Choi JS, Ahn HS, Chang YW, Tyrrell L, Black JA, Waxman SG, Dib-Hajj SD. ERK1/2 mitogen-activated protein kinase phosphorylates sodium channel Na(v)1.7 and alters its gating properties. J Neurosci. 2010;30:1637–1647. doi: 10.1523/JNEUROSCI.4872-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakor DK, Lin A, Matsuka Y, Meyer EM, Ruangsri S, Nishimura I, Spigelman I. Increased peripheral nerve excitability and local NaV1.8 mRNA up-regulation in painful neuropathy. Mol Pain. 2009;5:14. doi: 10.1186/1744-8069-5-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vardanyan M, Melemedjian OK, Price TJ, Ossipov MH, Lai J, Roberts E, Boos TL, Deschamps JR, Jacobson AE, Rice KC, Porreca F. Reversal of pancreatitis-induced pain by an orally available, small molecule interleukin-6 receptor antagonist. Pain. 2010;151:257–265. doi: 10.1016/j.pain.2010.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waskiewicz AJ, Johnson JC, Penn B, Mahalingam M, Kimball SR, Cooper JA. Phosphorylation of the cap-binding protein eukaryotic translation initiation factor 4E by protein kinase Mnk1 in vivo. Mol Cell Biol. 1999;19:1871–1880. doi: 10.1128/mcb.19.3.1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson JJ, Fahey MS, van den Worm E, Engels F, Nijkamp FP, Stroemer P, McMahon S, Allen SJ, Dawbarn D. TrkAd5: a novel therapeutic agent for treatment of inflammatory pain and asthma. J Pharmacol Exp Ther. 2006;316:1122–1129. doi: 10.1124/jpet.105.095844. [DOI] [PubMed] [Google Scholar]
- Wild KD, Bian D, Zhu D, Davis J, Bannon AW, Zhang TJ, Louis JC. Antibodies to nerve growth factor reverse established tactile allodynia in rodent models of neuropathic pain without tolerance. J Pharmacol Exp Ther. 2007;322:282–287. doi: 10.1124/jpet.106.116236. [DOI] [PubMed] [Google Scholar]
- Willis D, Li KW, Zheng JQ, Chang JH, Smit A, Kelly T, Merianda TT, Sylvester J, van Minnen J, Twiss JL. Differential transport and local translation of cytoskeletal, injury-response, and neurodegeneration protein mRNAs in axons. J Neurosci. 2005;25:778–791. doi: 10.1523/JNEUROSCI.4235-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf CJ, Safieh-Garabedian B, Ma QP, Crilly P, Winter J. Nerve growth factor contributes to the generation of inflammatory sensory hypersensitivity. Neuroscience. 1994;62:327–331. doi: 10.1016/0306-4522(94)90366-2. [DOI] [PubMed] [Google Scholar]
- Zhang X, Huang J, McNaughton PA. NGF rapidly increases membrane expression of TRPV1 heat-gated ion channels. EMBO J. 2005;24:4211–4223. doi: 10.1038/sj.emboj.7600893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang ZY, Xu H, Clapham DE, Ji RR. Phosphatidylinositol 3-kinase activates ERK in primary sensory neurons and mediates inflammatory heat hyperalgesia through TRPV1 sensitization. J Neurosci. 2004;24:8300–8309. doi: 10.1523/JNEUROSCI.2893-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]