

Abstract
This study evaluated the role for 12/15-lipoxygenase, which converts arachidonic acid to 12(S)- and 15(S)-hydroxyeicosatetraenoic acids, in nitrosative stress in peripheral nervous system and peripheral prediabetic and diabetic neuropathies. The experiments were performed in C57Bl6/J mice made diabetic with streptozotocin or fed high-fat diet, and human Schwann cells cultured in 5.5 mM or 30 mM glucose. 12/15-lipoxygenase overexpression and activation were present in sciatic nerve and spinal cord of diabetic and high fat diet-fed mice, as well as in human Schwann cells cultured in high concentrations of D-, but not L-glucose. 12/15-lipoxygenase inhibition with cinnamyl-3,4-dihydroxy-α-cyanocinnamate (8 mgkg-1d-1 s.c., for 4 weeks after 12 weeks without treatment) alleviated accumulation of nitrated proteins in sciatic nerve and spinal cord, and large and small nerve fiber dysfunction, but not intraepidermal nerve fiber loss. 12/15-lipoxygenase gene deficiency alleviated nitrosative stress and nerve conduction deficit, but not small sensory fiber neuropathy, in high-fat diet fed mice. In conclusion, 12/15-lipoxygenase is implicated in nitrosative stress and peripheral neuropathy in mouse models of Type 1 and early Type 2 diabetes. Its presence in human Schwann cells and upregulation by high glucose suggest a potential involvement in human disease.
Keywords: human Schwann cell, 12(S)-hydroxyeicosatetraenoic acid, intraepidermal nerve fiber density, 12/15-lipoxygenase, nitrosative stress, nerve conduction, peripheral diabetic neuropathy, tactile allodynia, thermal algesia
A leukocyte-type 12/15-lipoxygenase (LO) is a cytosolic nonheme iron-containing dioxygenase that oxidizes esterified arachidonic acid in lipoproteins (cholesteryl esters) and phospholipids with formation of the bioactive lipids termed eicosanoids [1,2]. Those include primary unstable 12(S)- and 15(S)-hydropero-xytetraenoic acids which are rapidly transformed to 12(S)- and 15(S)-hydroxyeicosatetraenoic acids [12(S)HETE and 15(S)HETE] and a number of their derivatives. LO is expressed in multiple cell types including pancreatic β–cells [3], adipocytes [4], vascular endothelial [5,6] and smooth muscle [6] cells, renal podocytes [7], mesangial [8], and tubular [9] cells, monocytes/macrophages [10], as well as neurons [11], astrocytes [12], and oligodendrocytes [13] of the nervous system.
High glucose- and diabetes-induced LO overexpression and activation in vascular and renal cells have been found associated with increased cytosolic Ca++ [14], affected signal transduction mechanisms, including protein kinase C (PKC) and mitogen-activated protein kinase C (MAPK) signaling cascades [15,16], and a pro-inflammatory response [17]. LO-derived 12(S)HETE generates superoxide, a precursor of highly reactive oxidant peroxynitrite, known to play an important role in diabetes and diabetic complications [1,2,18,19].
Recent studies implicated the LO pathway in diabetes-induced β-cell degeneration and insulin resistance [3], as well as endothelial dysfunction [1,2,20], nephropathy [1,2,7-9,18,19,21], and atherosclerosis [1,2,20,22]. The contribution of this mechanism to diabetic retinopathy appeared minor [23]. The role for LO in peripheral diabetic neuropathy (PDN), a severe complication that affects ~ 50% of patients with diabetes mellitus, and is a leading cause of foot amputation [24,25], has never been explored. Here, we describe findings implicating LO in nitrosative stress in the sciatic nerve and spinal cord and neuropathic changes associated with prediabetes and overt diabetes, and demonstrating the presence of LO and its overexpression and activation in high glucose-exposed cultured human Schwann cells (HSC).
METHODS
A. Reagents
Unless otherwise stated, all chemicals were of reagent-grade quality, and were purchased from Sigma Chemical Co., St. Louis, MO, USA. Cinnamyl-3,4-dihydroxy-alpha-cyanocinnamate (CDC) was obtained from Enzo Life Sciences International, Plymouth Meeting, PA. Mouse monoclonal (clone 1A6) anti-nitrotyrosine (NT) antibody for Western blot analysis of nitrated proteins was purchased from Millipore, Billerica, MA, USA. Rabbit polyclonal (clone H-100) anti-12-lipoxygenase (LO) antibody for Western blot analysis was obtained from Santa Cruz Biotechnology, Santa Cruz, CA, USA. 12-lipoxygenase (murine leukocyte) polyclonal antiserum for immunohistochemistry was purchased from Cayman Chemical, Ann Arbor, MI, USA. For double immuno-histochemistry, mouse monoclonal anti-S-100 antibody was purchased from Santa Cruz, Santa Cruz, CA, USA, and isolectin GS-IB4 from Griffonia Simplicifolia, Alexa Fluor(R) 594 conjugate as well as goat anti-rabbit Alexa Fluor 488 and goat anti-mouse Alexa Fluor 594 from Molecular Probes, Eugene, OR, USA. For assessment of intraepidermal nerve fiber density (INFD), rabbit polyclonal anti-protein gene product 9.5 (PGP 9.5) antiserum was obtained from UltraClone, Isle of Wight, UK, Alexa Fluor 488 goat anti-rabbit highly cross-adsorbed IgG (H+L) from Invitrogen, Eugene, OR, USA, SuperBlock blocking buffer from Thermo Scientific, Rockford, IL, USA, and the optimum cutting temperature (O.C.T.) compound from Sakura Finetek USA, Torrance, CA, USA. VECTASHIELD Mounting Medium was obtained from Vector Laboratories, Burlingame, CA, USA. Other reagents for immunohistochemistry were purchased from Dako Laboratories, Inc., Santa Barbara, CA, USA. HSC and culture medium were obtained ScienCell Research Laboratories, Carlsbad, CA, USA.
B. Animals
The experiments were performed in accordance with regulations specified by The Guide for the Care and Handling of Laboratory Animals (NIH Publication No. 85-23) and Pennington Biomedical Research Center Protocol for Animal Studies. Mature C57Bl6/J mice were purchased from Jackson Laboratories. All the mice were fed standard mouse chow (PMI Nutrition International, Brentwood, MO, USA) and had ad libitum access to water. In experiment 1, several male mice were used for double fluorescent immunohistochemistry, to localize LO to endothelial and Schwann cells of sciatic nerve. In experiment 2, male mice were randomly divided into two groups. In one group, diabetes was induced by STZ as we described previously [26]. Blood samples for glucose measurements were taken from the tail vein three days after STZ injection and the day before the animals were killed. The mice with blood glucose ≥13.8 mM were considered diabetic. The control and diabetic mice were maintained for 12 weeks and then divided into three subgroups. One subgroup was euthanized for tissue harvest. Two other subgroups were maintained with or without treatment with CDC, 8 mg kg-1d-1 s.c., for another 4 weeks. Non-fasting blood glucose measurements were performed at induction of diabetes and at the end of the study. Physiological and behavioral measurements were taken at three time-points i.e., at the beginning of the study and before and after CDC treatment. MNCV and SNCV were measured in mice anaesthetized with a mixture of ketamine and xylazine (45 mgkg-1 body weight and 15 mgkg-1 body weight, respectively, i.p.). In experiment 3, a colony of LO-deficient mice (LO-/-, C57Bl6/J background, [3,23]) was established from several breeding pairs provided by Dr. Nadler’s laboratory. The female wild-type and LO-/- mice were randomly assigned to receive standard mouse chow (NC) or HFD (D12328, 10.5 kcal% fat, and D 12330, 58 kcal% fat with corn starch, respectively, Research Diets, New Brunswick, NJ, USA), for 16 weeks. We previously reported that C57Bl6/J female mice fed HFD for 16 weeks develop obesity, hyperinsulinemia, and impaired glucose tolerance, in the absence of overt hyperglycemia [27]. In the present study, non-fasting blood glucose measurements, and physiological and behavioral tests were performed prior to HFD feeding and at the end of the study.
C. Anesthesia, euthanasia and tissue sampling
The animals were sedated by CO2, and immediately sacrificed by cervical dislocation. Sciatic nerves and spinal cords were rapidly dissected and frozen in liquid nitrogen for subsequent assessment of LO and nitrated protein expression and 12(S)-HETE concentrations. Footpads were sampled for assessment of INFD.
D. Specific Methods
D.1. Double fluorescent immunohistochemistry
The sections of sciatic nerve were deparaffinized in xylene, hydrated in decreasing concentrations of ethanol, and washed in water. The anti-LO antiserum (1:3200 dilution) was applied in combination with isolectin Alexa Fluor(R) 594 conjugate or anti-S-100 antibody (1:100 dilution), for localization of LO to endothelial and Schwann cells, respectively. Then all the sections were washed in PBS containing 0.01% Triton X-100, and a cocktail of secondary Alexa Fluor 488 and 594 antibodies (both in 1:150 dilution) or secondary Alexa Fluor 488 antibody alone (1:150 dilution, for isolectin-stained sections only) were applied for 2 hr at room temperature. After this step, the sections were washed, coverslipped, and mounted in Kaiser’s glycerin-gelatine (Merck, Darmstadt, Germany). Low power observations were made using a Zeiss Axioplan 2 imaging microscope. Color images were captured with a Photometric CoolSNAP™ hq CCD camera at 1392 × 1040 resolution. Low power images were generated with a 40X acroplan objective using the RS Image™ 1.9.2 software. The Adobe Photoshop 7.0 program was used to merge the images. To verify specificity of all immunostainings, primary antibody (anti-S-100) or antiserum (LO) were omitted in negative controls.
D.2. Biochemical studies
D.2.1. Western blot analysis of LO and nitrated proteins
To assess LO and nitrated protein expressions by Western blot analysis, sciatic nerve and spinal cord materials (~ 10-20 mg) were placed on ice in 200 μl of RIPA buffer containing 50 mmol/l Tris-HCl, pH 7.2; 150 mmol/l NaCl; 0.1% sodium dodecyl sulfate; 1% NP-40; 5 mmol/l EDTA; 1 mmol/l EGTA; 1% sodium deoxycholate and the protease/ phosphatase inhibitors leupeptin (10 μg/ml), pepstatin (1 μg/ml), aprotinin (20 μg/ml), benzamidine (10 mM), phenylmethylsulfonyl fluoride (1 mM), sodium orthovanadate (1 mmol/l), and homogenized on ice. The homogenates were sonicated (4 × 10 s) and centrifuged at 14,000 g for 20 min. All the afore-mentioned steps were performed at 4 °C. The lysates (20 and 40 μg protein for sciatic nerve and spinal cord, respectively) were mixed with equal volumes of 2x sample-loading buffer containing 62.5 mmol/l Tris-HCl, pH 6.8; 2% sodium dodecyl sulfate; 5% β-mercaptoethanol; 10% glycerol and 0.025% bromophenol blue, and fractionated in 10 % (nitrated proteins) or 7.5% (LO) SDS-PAGE in an electrophoresis cell (Mini-Protean III; Bio-Rad Laboratories, Richmond, CA). Electrophoresis was conducted at 15 mA constant current for stacking, and at 25 mA for protein separation. Gel contents were electrotransferred (80 V, 2 hr) to nitrocellulose membranes using Mini Trans-Blot cell (Bio-Rad Laboratories, Richmond, CA) and western transfer buffer [25 mmol/l Tris-HCl, pH 8.3; 192 mmol/l glycine; and 20% (v/v) methanol] [27]. Free binding sites were blocked in 2% and 5% (w/v) BSA (for nitrated proteins and LO, respectively) in 20 mmol/l Tris-HCl buffer, pH 7.5, containing 150 mmol/l NaCl and 0.05% Tween 20, for 1 h, after which LO or nitrotyrosine antibodies were applied for 2 h, for detection of nitrated protein and LO expressions. The horseradish peroxidase-conjugated secondary antibody was then applied for 1 h. After extensive washing, protein bands detected by the antibodies were visualized with the Amersham ECL™ Western Blotting Detection Reagent (Little Chalfont, Buckinghamshire, UK). Membranes were then stripped in the 25 mmol/l glycine-HCl, pH 2.5 buffer containing 2% SDS, and reprobed with β-actin antibody to confirm equal protein loading.
D.2.2. 12(S)HETE measurements
For assessment of 12(S)HETE, sciatic nerve and spinal cord samples were homogenized on ice in 15 mM Tris-HCI buffer (1:100 w/v) containing 140 mM NaCl, pH 7.6, and centrifuged. 12(S)HETE was measured in supernatants with the 12(S)-hydroxyeicosatetraenoic acid [12(S)HETE] Enzyme Immuno Assay kit (Assay Designs, Ann Arbor, MI).
D.3. Physiological and behavioral tests
Sciatic MNCV, hind-limb digital SNCV, thermal algesia (paw withdrawal), and tactile response thresholds were measured as described previously [26,27].
D.4. INFD
Footpads were fixed in ice-cold Zamboni’s fixative for 3 hr, washed in 100 mM phosphate-buffered saline (PBS) overnight, and then in PBS containing increasing concentrations of sucrose i.e., 10%, 15%, and 20%, 3 hr in each solution. After washing, the samples were snap-frozen in O.C.T. compound and stored at -80°C. Three longitudinal 50 μm-thick footpad sections from each mouse were cut on Leica CM1950 cryostat (Leica Microsystems, Nussloch, Germany). Non-specific binding was blocked by 3% goat serum containing 0.5% porcine gelatin and 0.5% Triton X-100 in SuperBlock blocking buffer at room temperature, for 2 hr. The sections were incubated overnight with PGP 9.5 antiserum in 1:400 dilution, at 4°C, after which secondary Alexa Fluor 488 IgG (H+L) in 1:1000 was applied at room temperature, for 1 h. Sections were then coverslipped with VECTASHIELD mounting medium. Intraepidermal nerve fiber profiles were counted blindly by three independent investigators under Axioplan 2 microscope (Zeiss) at 40X magnification, and the average values were used. The length of epidermis was assessed on the microphotographs of stained sections taken at 5X magnification with a 3I Everest imaging system (Intelligent Imaging Innovations, Inc., Denver, CO, USA) equipped with Axioplan 2 microscope (Zeiss), using the NIH ImageJ software (version 1.42q). An average of 2.8 ± 0.3 mm of the sample length was investigated to calculate a number of nerve fiber profiles per mm of epidermis. Representative images of intraepidermal nerve fibers were obtained by confocal laser scanning microscopy at 40X magnification, using Leica TCS SP5 confocal system (Leica Microsystems, Mannheim, Germany).
D.5. Experiment 4: studies in HSC
D.5.1. HCS culture
Experimental evidence in support of the important role of SC in PDN is emerging. In addition to axonal atrophy and myelinated fiber loss characteristic for advanced PDN, SC contribute to early axonopathy and painful sensory neuropathy that develop in the absence of demyelination. The fact that diabetes-associated peripheral nerve oxidative-nitrosative stress originates primarily from SC is supported by several lines of evidence obtained in animal model studies [28-30]. Note, that cultured primary rat HS do not display increased superoxide production even after prolonged (up to 16 d) exposure to high (30 mM) glucose [31]. In contrast, cultured HSC (cell line cat. #1700, ScienCell, Carlsbad, CA) manifest increased superoxide production, accumulation of nitrated and poly(ADP-ribosyl)ated proteins and 4-hydroxynonenal adducts, inducible nitric oxide synthase overexpression, and downregulation of taurine transporter early (1-7 d) after exposure to high glucose [32-34], and therefore represent a good model for studying mechanisms of oxidative-nitrosative stress in the peripheral nerve. In experiment 4, HSC (passages 7-10) were cultured in 6-well plates in commercial media containing 5.5 mM D-glucose. At ~50% confluency, the commercial media were replaced with those containing either 5.5 mM D-glucose or 30 mM D-glucose with or without CDC, 10 μM (6-8 plates per condition). The cells cultured in 5.5 mM D-glucose + 24.5 mM L-glucose were used as the osmolarity control. After 24 hr, part of the cultures were used for Western blot analysis of LO. The parallel cultures were used for 12(S)HETE measurements.
E. Statistical analysis
The results are expressed as Mean ± SEM. Data were subjected to equality of variance F test, and then to log transformation, if necessary, before one-way analysis of variance. Where overall significance (p<0.05) was attained, individual between group comparisons for multiple groups were made using the Student-Newman-Keuls multiple range test. When between-group variance differences could not be normalized by log transformation (datasets for body weights and plasma glucose), the data were analyzed by the nonparametric Kruskal-Wallis one-way analysis of variance, followed by the Bonferroni/Dunn test for multiple comparisons. Individual pair-wise comparisons in experiments 3 and 4 were made using the unpaired two-tailed Student’s t-test or Mann-Whitney rank sum test where appropriate. Significance was defined at p ≤ 0.05.
RESULTS
In experiment 1, LO fluorescence was clearly identifiable in endothelial (Fig.1a) and Schwann (Fig.1b) cells of mouse sciatic nerve.
Fig.1.

Representative microphotographs of fluorescent immunostaining [or fluorescent staining with isolectin Alexa Fluor(R) 594 conjugate, (a, center)] of diabetic mouse sciatic nerve for (a) LO (left); vascular endothelium (center), LO and vascular endothelium (right), and (b) LO (left); S-100 (center), LO and S-100 (right). Magnification x 150.
In experiment 2, the initial (prior to STZ administration) body weights were similar in all experimental groups (Table 1). Weight gain during the 16-wk study was lower in both untreated and CDC-treated diabetic mice than in the non-diabetic control group. CDC treatment did not affect weight gain in either control or diabetic mice. Initial (after STZ administration) non-fasting blood glucose concentrations were 67% and 71% higher in untreated and CDC-treated diabetic mice than in the control group. Hyperglycemia progressed with the prolongation of diabetes, and the differences between final blood glucose concentrations in both diabetic groups and non-diabetic controls exceeded 3-fold. CDC treatment did not affect non-fasting blood glucose concentrations in either non-diabetic or diabetic mice. In experiment 3, the initial (prior to HFD feeding) body weights were similar in all experimental groups. A 16-week HFD feeding resulted in 30% and 28% differences in body weights between wild-type and LO-/- mice fed HFD and the corresponding groups fed NC. Non-fasting blood glucose concentrations were similar among wild-type and LO-/- mice fed NC or HFD, both at the beginning and at the end of the study.
Table 1.
Initial and final body weights and blood glucose concentrations in control and diabetic mice maintained with and without CDC inhibitor treatment (Experiment 2) or fed normal chow or high-fat diet (Experiment 3).
| Variable | Body weight (g) | Blood glucose (mmol/l) | |||
|---|---|---|---|---|---|
| Group | Initial | Final | Initial | Final | |
| EXPERIMENT 2 | |||||
| Control | 25.8 ± 0.6 | 38.5 ± 1.3 | 9.4 ± 0.7 | 8.7 ± 0.3 | |
| Control + CDC | 25.4 ± 0.5 | 36.6 ± 1.4 | 9.4 ± 0.4 | 9.3 ± 0.3 | |
| Diabetic | 25.1 ± 0.4 | 27.7 ± 0.5** | 15.7 ± 1.2** | 27.7 ± 1.1** | |
| Diabetic + CDC | 25.5 ± 0.5 | 26.8 ± 0.6** | 16.1 ± 1.5** | 28.9 ± 1.0** | |
| EXPERIMENT 3 | |||||
| Wild-type, NC | 24.8 ± 0.7 | 35.4 ± 1.0 | 8.4 ± 0.6 | 9.3 ± 0.6 | |
| Wild-type, HFD | 25.0 ± 0.6 | 46.0 ± 1.6** | 9.1 ± 0.7 | 9.5 ± 0.7 | |
| LO-/-, NC | 25.6 ± 0.7 | 34.9 ± 1.2 | 9.0 ± 0.5 | 9.4 ± 0.4 | |
| LO-/-, HFD | 25.3 ± 0.5 | 44.7 ± 1.5** | 8.6 ± 0.4 | 9.2 ± 0.5 | |
Data are expressed as Means ± SEM. n = 24 per group in Experiment 3 and at the beginning of the study in Experiment 2. n = 10-12 per group after the beginning of CDC treatment in Experiment 2.
p < 0.01 vs non-diabetic control group (Experiment 2) and vs mice fed normal chow (Experiment 3).
NC – normal chow, HFD- high-fat diet.
In experiment 2, diabetic mice displayed clearly manifest peripheral nerve and spinal cord LO overexpression and activation at the 12-wk time point, prior to CDC intervention. Sciatic nerve and spinal cord LO expressions were increased by 38% and 65%, respectively (Fig.2a-d). Sciatic nerve and spinal cord 12(S)HETE concentrations were increased 2.8-fold and 1.7-fold (Fig.2e,f).
Fig.2.
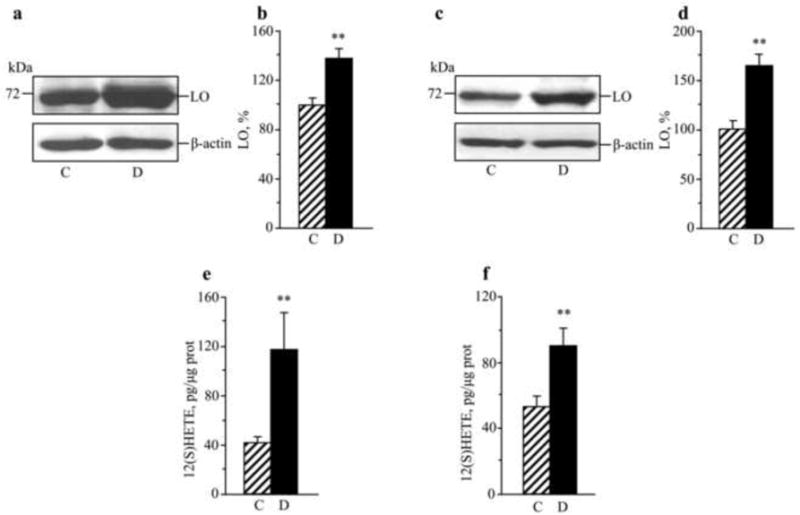
Representative Western blot analyses of LO expression, LO protein contents (densitometry, %), and 12(S)HETE concentrations in the sciatic nerve (a,b,e) and spinal cord (c,d,f) of control mice and those with STZ-diabetes of 12-wk duration. C – control; D – diabetic. Mean ± SEM, n = 6-8 per group. ** p <0.01 vs non-diabetic control group.
In a similar fashion, increased LO expression (Fig.3a,b and 3c,d) and 12(S)HETE concentrations (Fig.3,e,f) were found in sciatic nerve and spinal cord of untreated mice with 16-wk duration of diabetes (experiment 2). CDC treatment did not affect diabetes-induced sciatic nerve and spinal cord LO overexpression, but significantly reduced 12(S)HETE concentrations, indicative of a reduction of LO activity.
Fig.3.
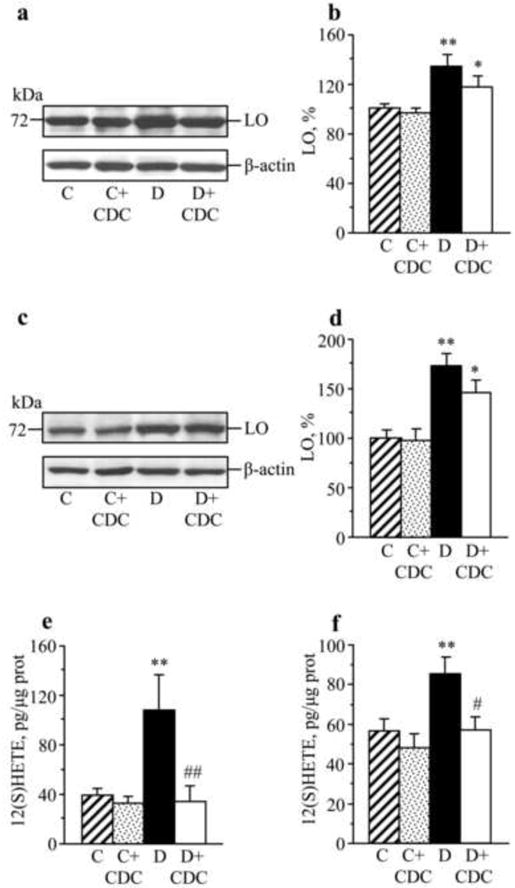
Representative Western blot analyses of LO expression, LO protein contents (densitometry, %), and 12(S)HETE concentrations in the sciatic nerve (a,b,e) and spinal cord (c,d,f) of control and STZ-diabetic mice maintained with or without CDC treatment. C – control; D – diabetic. Mean ± SEM, n = 8-12 per group. *, ** p < 0.05 and < 0.01 vs non-diabetic control group. #,## p < 0.05 and < 0.01 vs untreated diabetic group.
In experiment 3, a 16-wk HFD feeding increased LO expression by 29% and 30% in sciatic nerve and spinal cord of wild-type mice (Fig.4a,b and 4c,d, respectively). 12(S)HETE concentrations were increased by 113% and 27%, respectively (Fig.4e,f). 12(S)HETE was also identified in both sciatic nerve and spinal cord of LO-/- mice. However, 12(S)HETE concentrations, especially those in the sciatic nerve, were significantly lower in LO-/- mice than in the wild-type mice. No differences in either sciatic nerve or spinal cord 12(S)HETE concentrations were found between NC- and HFD-fed LO-/- mice.
Fig.4.

Upper and middle panels. Representative Western blot analyses of LO expression and LO protein contents (densitometry, %) in sciatic nerve (a,b) and spinal cord (c,d) of mice fed normal chow or high-fat diet. Lower panel. Sciatic nerve (e) and spinal cord (f) 12(S)HETE concentrations in wild-type and LO-/- fed normal chow or high-fat diet. NC – normal chow; HFD - high fat diet. Mean ± SEM, n = 6-9 per group. *, ** p <0.01 vs mice fed normal chow. §,§§ - p < 0.05 and < 0.01 vs wild-type mice fed NC; ## - p < 0.01 vs wild-type mice fed HFD.
In experiment 2, sciatic nerve and spinal cord nitrated protein expressions were increased by 56% and 32% and by 63% and 33%, in mice with 12-wk and 16-wk durations of STZ-diabetes, respectively, compared with non-diabetic controls (Fig.5). CDC treatment reduced nitrated protein expression in sciatic nerve, and normalized this variable in spinal cord of diabetic mice. LO inhibition was not associated with a significant decrease in nitrated protein expression in non-diabetic mice.
Fig.5.

Upper panel: Representative Western blot analyses of nitrated protein expression and their content (densitometry, %) in the sciatic nerve (a,b) and spinal cord (c,d) of control mice and those with STZ-diabetes of 12-wk duration. Lower panel: Representative Western blot analyses of nitrated protein expression and their content (densitometry, %) in the sciatic nerve (e,f) and spinal cord (g,h) of control and STZ-diabetic mice maintained with or without CDC treatment. Mean ± SEM, n = 7-12 per group. ** p < 0.01 vs non-diabetic control group. ## p < 0.01 vs untreated diabetic group.
In experiment 3, a 16-wk HFD feeding of wild-type mice increased sciatic nerve and spinal cord nitrated protein expressions by 38% and 68% (Fig.6a,b and Fig.6c,d, respectively). There was a tendency towards a modest increase in nitrated protein expression in HFD-fed LO-/- mice, but the difference with the corresponding group fed NC did not achieve statistical significance. Accumulation of nitrated proteins in the spinal cord of HFD-fed LO-/- mice was 31% lower than in the corresponding wild-type group.
Fig.6.
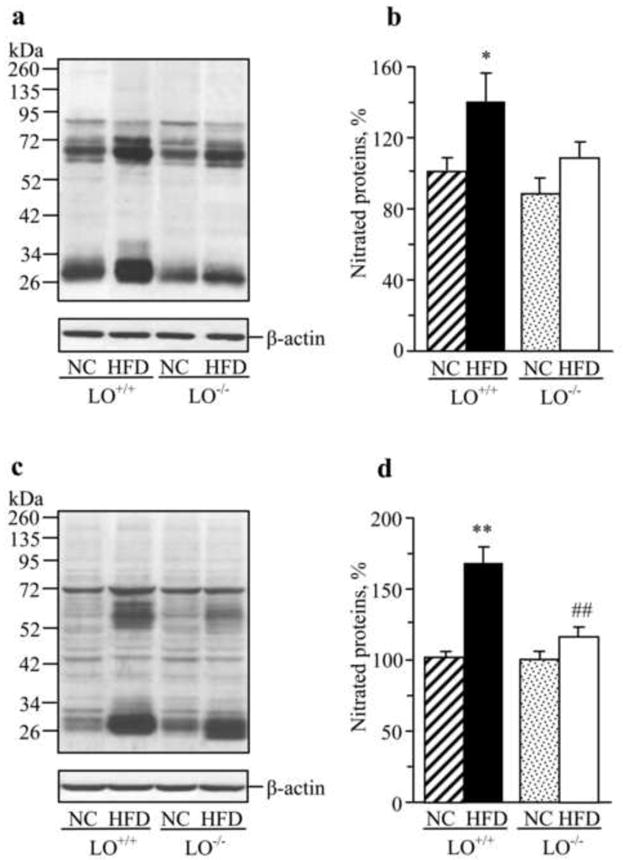
Representative Western blot analyses of nitrated protein expression and their content (densitometry, %) in the sciatic nerve (a,b) and spinal cord (c,d) of wild-type and LO-/- mice fed normal chow or high-fat diet. NC – normal chow; HFD-high-fat diet. Mean ± SEM, n = 7-12 per group. *, ** p < 0.05 and < 0.01 vs mice fed normal chow; ## p < 0.01 vs high-fat diet fed wild-type mice.
In experiment 2, diabetic mice displayed 20 % and 14% MNCV and SNCV deficits at the 12-wk time point i.e., prior to CDC intervention (Table 2). Note, that both MNCV and SNCV were similar in non-diabetic mice at the beginning of the study and at the 12-wk time point which indicates that diabetes-induced nerve conduction slowing did not develop due to affected peripheral nerve growth and maturation. CDC alleviated, but did not completely normalize, MNCV and SNCV in diabetic mice, without affecting those variables in non-diabetic controls. In experiment 3, MNCV and SNCV were similar in all experimental groups at the beginning of the study, prior to HFD feeding. The HFD-fed wild-type mice displayed 20% MNCV deficit and 16% SNCV deficit at the 16-wk time point. In contrast, HFD-fed LO-/- mice preserved essentially normal SNCV. They did develop MNCV deficit, but it was less severe (13%) than in the HFD-fed wild-type mice.
Table 2.
Motor and sensory nerve conduction velocities, thermal response latencies, and tactile response thresholds in control and diabetic mice maintained with or without CDC inhibitor treatment (Experiment 2) or fed normal chow or high-fat diet (Experiment 3).
| Group | Variable | MNCV | SNCV | Thermal response latency (s) | Tactile response threshold (g) | |
|---|---|---|---|---|---|---|
| EXPERIMENT 2 Baseline (prior to induction of diabetes) |
||||||
| Control | 51.2 ± 1.0 | 37.5 ± 0.3 | 9.4 ± 0.5 | 2.18 ± 0.14 | ||
| 12-wk time point (prior to CDC intervention) | ||||||
| Control | 50.2 ± 1.3 | 38.2 ± 0.4 | 9.3 ± 0.4 | 2.23 ± 0.13 | ||
| Diabetic | 40.1 ± 0.9** | 32.7 ± 0.85** | 18.9 ± 0.6** | 0.70 ± 0.1** | ||
| 16-wk time point | ||||||
| Control | 56.7 ± 1.9 | 37.8 ± 0.7 | 11.1 ± 0.5 | 2.54 ± 0.13 | ||
| Control +CDC | 54.5 ± 1.8 | 38.1 ± 0.8 | 11.4 ± 0.4 | 2.35 ± 0.18 | ||
| Diabetic | 41.2 ± 2.3** | 30.4 ± 0.8** | 18.0 ± 0.95** | 1.23 ± 0.06** | ||
| Diabetic+CDC | 47.1 ± 2.6**a | 34.0 ± 0.5**## | 15.3 ± 0.6**## | 1.91 ± 0.1**## | ||
| EXPERIMENT 3 Baseline (prior to high-fat diet feeding) |
||||||
| LO+/+, NC | 52.3 ± 1.3 | 36.7 ± 1.6 | 8.5 ± 0.3 | 2.31 ± 0.12 | ||
| LO-/-, NC | 52.9 ± 1.4 | 37.0 ± 1.3 | 8.1 ± 0.2 | 2.25 ± 0.13 | ||
| 16-wk time point | ||||||
| LO+/+, NC | 52.7 ± 2.0 | 36.2 ± 0.9 | 7.6 ± 0.2 | 2.36 ± 0.11 | ||
| LO+/+, HFD | 42.4 ± 1.5** | 30.5 ± 0.7** | 14.3 ± 0.2** | 1.22 ± 0.05** | ||
| LO-/-, NC | 54.5 ± 2.0 | 36.1 ± 0.5 | 8.7 ± 0.1 | 2.34 ± 0.08 | ||
| LO-/-, HFD | 47.5 ± 1.4*, # | 34.2 ± 1.3## | 14.1 ± 0.3** | 1.12 ± 0.04** | ||
Data are expressed as Means ± SEM, n = 8-12 per group.
p < 0.05 and < 0.01 vs non-diabetic control group (Experiment 2) and vs corresponding groups of mice fed normal chow (Experiment 3).
p < 0.05 and < 0.01 vs untreated diabetic group (Experiment 2) and vs HFD-fed wild-type mice (Experiment 3).
0.0599 vs untreated diabetic group.
NC – normal chow, HFD- high-fat diet.
In experiment 2, the sensitivities to both thermal and tactile noxious stimuli were similar in non-diabetic mice at the beginning of the study and at the 12-wk and 16-wk time points. Diabetic mice displayed thermal hypoalgesia and tactile allodynia at both 12 wks and 16 wks after induction of diabetes. CDC treatment partially reversed both sensory disorders in diabetic mice, without affecting either thermal response latencies or tactile response thresholds in non-diabetic controls. In experiment 3, thermal response latencies and tactile response thresholds were similar in all experimental groups at the beginning of the study. Both wild-type and LO-/- mice displayed thermal hypoalgesia and tactile allodynia after 16 wks of HFD feeding. LO gene deficiency did not alleviate either of these two sensory disorders in HFD-fed mice.
In experiment 2, diabetic mice displayed a 21% reduction in INFD at the 12-wk time point (Fig.7a,b). This reduction progressed to 40% at the 16-wk time point (Fig.7c,d). CDC treatment did not affect INFD in non-diabetic mice, and did not induce intraepidermal nerve fiber regeneration or slowed down their degeneration in diabetic mice. In experiment 3, a 16-wk HFD feeding did not affect INFD in either wild-type or LO-/- mice (Fig.7e,f). INFDs were similar in all experimental groups.
Fig.7.
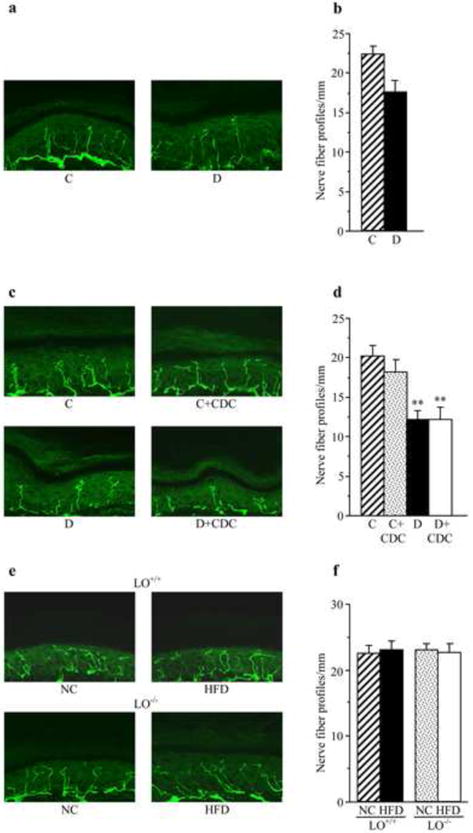
Intraepidermal nerve fiber profiles in (a,b) control and diabetic mice at the 12-wk time point (prior to CDC intervention); (c,d) control and diabetic mice maintained with or without CDC treatments (a 16-wk time point), and (e,f) LO+/+ and LO-/- mice after 16 wks of feeding with normal chow or high-fat diet. (a,c,e) Representative images of intraepidermal nerve fiber profiles, magnification x 40; (b,d,f) Skin fiber density. Mean ± SEM, n = 10-12 per group. C – control mice, D – diabetic mice, NC- normal chow, HFD-high-fat diet. ** – p < 0.01 vs non-diabetic control group.
In experiment 4, LO was identified in HSC. LO expression (Fig.8a,b) and 12(S)HETE accumulation (Fig.8c) were 29% and 3.5-fold greater in HSC cultured in 30 mM D-glucose, than in those cultured in 5.5 mM glucose (Fig.7a,b). In contrast, HSC cultured in 5.5 mM D-glucose plus 24.5 mM L-glucose (osmolarity control) did not display increased LO expression or 12(S)HETE accumulation.
Fig.8.
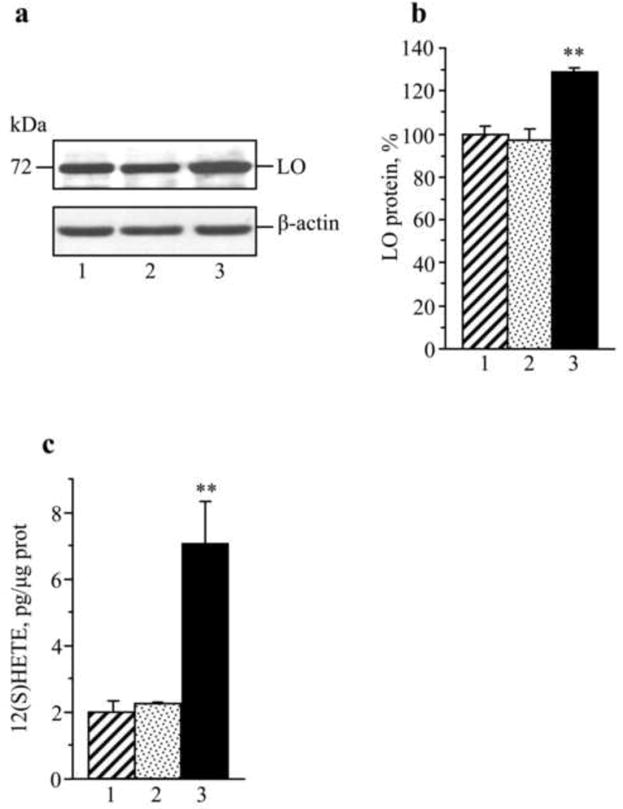
LO expression and 12(S)HETE concentrations in human Schwann cells cultured in 5.5 mM D-glucose (1), 5,5 mM D-glucose plus 24.5 mM L-glucose (2), and 30 mM D-glucose (3). (a) Representative Western blot analysis of LO expression, (b) LO protein content, and (c) 12(S)HETE concentrations. Mean ± SEM, n = 6 per group. ** p < 0.01 vs cells cultured in 5.5 mM D-glucose.
DISCUSSION
It has been known for at least a decade, that diabetes is associated with disturbances of arachidonic acid metabolism in peripheral nerve [35]. Arachidonic acid is metabolized by cyclooxygenases (COX)-1 and -2, 5- and 12/15-LOs, and cytochrome p450 epoxygenase. Diabetes does not affect rat sciatic nerve COX-1 expression, and the findings with nonselective COX inhibitor flurbiprofen suggest that COX-1 is essentially required for normal peripheral nerve function [36]. In contrast, sciatic nerve COX-2 overexpression was identified as a pathogenetic factor in PDN [36,37]. The roles for 5-lipoxygenase and cytochrome p450 epoxygenase in PDN have never been explored, although evidence for cell toxicity of their products, leukotrienes and epoxyeicosatrienoc acids, is emerging [23]. The findings reported herein demonstrate an important role of 12/15-LO in MNCV and SNCV deficits and small sensory nerve fiber dysfunction in mouse models of Type 1 diabetes and Type 2 prediabetes and obesity.
Sciatic nerve and spinal cord LO overexpression and 12(S)HETE accumulation were observed in STZ-diabetic and HFD-fed mice. LO inhibition with CDC did not reduce LO protein expression, but normalized 12(S)HETE concentrations, a measure of LO activity, in both tissue targets for PDN. Note, that 12(S)HETE was present in sciatic nerve and spinal cord of both NC- and HFD-fed LO-/- mice, probably due to activities of other, non-leukocyte type, LO(s).
Studies with conventional antioxidants an metal chelators [38-46], inhibitors of superoxide-generating enzymes and superoxide dismutase mimetics [47-49], and so called “indirect antioxidants” such as aldose reductase [50], COX-2 [37], poly(ADP-ribose) ribose polymerase [32] inhibitors, suggest a key role for oxidative stress in neurovascular dysfunction, nerve conduction deficits, axonal atrophy of large myelinated fibers, and small fiber neuropathy. The role for oxidative stress in PDN has also been confirmed in several transgenic mouse model studies [37,51,52]. The potent oxidant peroxynitrite [53,54], previously shown to play an important role in neurodegenerative synucleinopathies such as Parkinson’s disease, dementia with Lewy bodies, and multiple system atrophy [55], as well as Alzheimer disease and other tauopathies [56,57], has recently been implicated in both peripheral [58-60] and autonomic [61] neuropathies. Accumulation of a foot-print of peroxynitrite injury, nitrotyrosine, has been identified in peripheral nerve, vasa nervorum, spinal cord, and dorsal root ganglia in animal models of prediabetes and overt diabetes [27,29,30,32,43,47,58-60,62], and in microvasculature of diabetic human subjects [63]. In our recent study in STZ-diabetic rat model [64], nitrotyrosine accumulation was present in endothelial and Schwann cells of the peripheral nerve, spinal cord neurons, astrocytes, and oligodendrocytes, and neuron (perikarya) and glial cells of dorsal root ganglia, and nerve NT concentrations inversely correlated with MNCV, SNCV, and myelin thickness. Others identified correlations between plasma NT content and endothelial dysfunction [65], an important factor in the pathogenesis of PDN [38,40-45,47-50], in human subjects with diabetes. Furthermore, evaluation of pholasin as a probe for the determination of plasma total antioxidant capacity, revealed a correlation between quenching of pholasin chemiluminescence with peroxynitrite and total neurological impairment score-lower limbs [66]. Mechanisms underlying nitrotyrosine accumulation in PNS and other tissue-sites for diabetic complications are not completely understood, although previous studies suggest involvement of increased AR [67-69], inducible and neuronal nitric oxide synthases [29,30], PARP [32,64,69,70], as well as oxidized lipoproteins [62]. In the present study, nitrated protein accumulation was reduced (sciatic nerve) or normalized (spinal cord) in diabetic mice treated with CDC. HFD-fed LO-/- mice developed less severe nitrosative stress in both tissue targets for PDN, than the HFD-fed wild-type mice. Thus, our findings identify LO as an important contributor to nitrosative stress associated with both prediabetic and diabetic neuropathies.
The afore-mentioned data are consistent with a beneficial effect of LO inhibition or gene deficiency on peripheral nerve function. HFD-fed LO-/- mice were partially protected from MNCV and SNCV deficits that were clearly manifest in HFD-fed wild-type mice. LO inhibition with CDC alleviated motor and sensory nerve conduction slowing in STZ-diabetic mice, and this was not due to correction or amelioration of hyperglycemia. The incomplete correction of MNCV and SNCV by CDC could result from a relatively short duration of treatment after quite extended (12 wks) presence of a severe diabetes. Also note that although CDC treatment normalized 12(S)HETE concentrations in the peripheral nerve in toto, it is unclear whether the selected dose was sufficient for vasa nervorum. Endothelial cells do contain LO and accumulate 12(S)HETE [1,2,5,6], and endothelial dysfunction leading to decreased nerve blood flow and endoneurial hypoxia is known to play an important role in diabetes-induced MNCV and SNCV deficits [38,40-45,47-50,72-74].
In addition to MNCV and SNCV deficits, human subjects with advanced PDN display increased vibration and thermal perception thresholds [24]. A significant proportion of patients also have tactile allodynia i.e., a condition in which a light touch is perceived as painful [24]. Whereas most investigators agree on the presence of MNCV and SNCV deficits in C57Bl6/J mice made diabetic with STZ [28-30,51,58,59,75,76], the data on thermal and tactile sensitivity in this and other rodent models of PDN are extremely contradictory (reviewed in [25]). Therefore, the behavioral test data should be interpreted with caution and can not be used as reliable endpoints for drug discovery, until the controversies are resolved. We consistently observe tactile allodynia (consistent with findings of C.Toth, personal communication) and thermal hypoalgesia in C57Bl6/J mice with different durations of STZ-diabetes. In the current study, LO inhibition with CDC alleviated thermal hypoalgesia and tactile allodynia in diabetic mice which implicates LO in small sensory nerve fiber dysfunction in PDN. Alleviation of thermal hypolalgesia was achieved despite the absence of intraepidermal nerve fiber regeneration. Together with corneal confocal microscopy, intraepidermal nerve fiber density is emerging as a new approach for detection of small sensory nerve fiber degeneration in human subjects with diabetes and impaired glucose tolerance [24,25,75,76]. Both variables have shown excellent correlations with sensory function [75,76]. In contrast, several animal studies including those of our group revealed dissociations between loss of sensitivity to thermal noxious stimuli and intraepidermal nerve fiber loss ([77], reviewed in [25]). Note, that LO gene deficiency did not protect from development of either thermal hypoalgesia or tactile allodynia in HFD-fed mice.
It has previously been shown [78] that high glucose induced depletion of glycerol-phospholipid arachidonoyl-containing molecular species in HSC, a widely used cell culture model of PDN [32-34,78]. The present study demonstrating LO overexpression and 12(S)HETE accumulation in cultured HSC as early as 24 h after exposure to high glucose suggests that such depletion is at least partially due to increased arachidonate metabolism through the LO pathway. These findings combined with evidence of the contribution of LO to neuropathic changes obtained in two animal models suggest that the LO mechanism may be implicated in human PDN.
In conclusion, LO overexpression and activation contribute to nitrosative stress in peripheral nervous system and development of neuropathic changes associated with prediabetes and overt diabetes. The findings support the rationale for development and future studies of LO inhibitors and LO-inhibitor-containing combination therapies. The presence of LO in HSC and its upregulation early after exposure to high glucose suggest a potential involvement in human disease.
Acknowledgments
The study was supported by the National Institutes of Health Grants DK074517 and DK077141 and the American Diabetes Association Research Grant 7-08-RA-102 (both to I.G.O.) The Cell Biology and Bioimaging Core utilized in this work is supported in part by COBRE (NIH P20 RR021945) and CNRU (NIH 1P30-DK072476) center grants from the National Institutes of Health. The authors thank Drs. Douglas E.Wright from the University of Kansas Medical Center, Kansas City, KS, USA and Gary L. Pittenger from Eastern Virginia Medical School, Norfolk, VA, USA for valuable recommendations regarding intraepidermal nerve fiber density measurements.
Abbreviations
- CDC
cinnamyl-3,4-dihydroxy-alpha-cyanocinnamate
- COX-1 and COX-2
cyclooxygenases-1 and –2
- DAB
diaminobenzidine
- DRG
dorsal root ganglion
- 12(S)HETE
12(S)-hydroxyeicosatetraenoic acid
- HSC
human Schwann cells
- INFD
intraepidermal nerve fiber density
- LO
12/15-lipoxygenase
- MNCV
motor nerve conduction velocity
- NC
normal chow
- NT
nitrotyrosine
- PBS
phosphate-buffered saline
- PDN
peripheral diabetic neuropathy
- PGP 9.5
protein gene product 9.5
- PKC
protein kinase C
- MAPK
mitogen-activated protein kinase
- SNCV
sensory nerve conduction velocity
- STZ
streptozotocin
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Natarajan R, Nadler JL. Lipoxygenases and lipid signaling in vascular cells in diabetes. Front Biosci. 2003;8:783–795. doi: 10.2741/1144. [DOI] [PubMed] [Google Scholar]
- 2.Natarajan R, Nadler JL. Lipid inflammatory mediators in diabetic vascular disease. Arterioscler Thromb Vasc Biol. 2004;24:1542–1548. doi: 10.1161/01.ATV.0000133606.69732.4c. [DOI] [PubMed] [Google Scholar]
- 3.Nunemaker CS, Chen M, Pei H, Kimble SD, Keller SR, Carter JD, Yang Z, Smith KM, Wu R, Bevard MH, Garmey JC, Nadler JL. 12-Lipoxygenase-knockout mice are resistant to inflammatory effects of obesity induced by Western diet. Am J Physiol Endocrinol Metab. 2008;295:E1065–E1075. doi: 10.1152/ajpendo.90371.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chakrabarti SK, Cole BK, Wen Y, Keller SR, Nadler JL. 12/15-lipoxygenase products induce inflammation and impair insulin signaling in 3T3-L1 adipocytes. Obesity (Silver Spring) 2009;17:1657–1663. doi: 10.1038/oby.2009.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bolick DT, Orr AW, Whetzel A, Srinivasan S, Hatley ME, Schwartz MA, Hedrick CC. 12/15-lipoxygenase regulates intercellular adhesion molecule-1 expression and monocyte adhesion to endothelium through activation of RhoA and nuclear factor-kappaB. Arterioscler Thromb Vasc Biol. 2005;25:2301–2307. doi: 10.1161/01.ATV.0000186181.19909.a6. [DOI] [PubMed] [Google Scholar]
- 6.Sasson S, Eckel J. Disparate effects of 12-lipoxygenase and 12-hydroxyeicosatetraenoic acid in vascular endothelial and smooth muscle cells and in cardiomyocytes. Arch Physiol Biochem. 2006;112:119–129. doi: 10.1080/13813450600712035. [DOI] [PubMed] [Google Scholar]
- 7.Kang SW, Natarajan R, Shahed A, Nast CC, LaPage J, Mundel P, Kashtan C, Adler SG. Role of 12-lipoxygenase in the stimulation of p38 mitogen-activated protein kinase and collagen alpha5(IV) in experimental diabetic nephropathy and in glucose-stimulated podocytes. J Am Soc Nephrol. 2003;14:3178–3187. doi: 10.1097/01.asn.0000099702.16315.de. [DOI] [PubMed] [Google Scholar]
- 8.Kim YS, Xu ZG, Reddy MA, Li SL, Lanting L, Sharma K, Adler SG, Natarajan R. Novel interactions between TGF-{beta}1 actions and the 12/15-lipoxygenase pathway in mesangial cells. J Am Soc Nephrol. 2005;16:352–362. doi: 10.1681/ASN.2004070568. [DOI] [PubMed] [Google Scholar]
- 9.González-Núñez D, Solé M, Natarajan R, Poch E. 12-Lipoxygenase metabolism in mouse distal convoluted tubule cells. Kidney Int. 2005;67:178–186. doi: 10.1111/j.1523-1755.2005.00068.x. [DOI] [PubMed] [Google Scholar]
- 10.Dioszeghy V, Rosas M, Maskrey BH, Colmont C, Topley N, Chaitidis P, Kühn H, Jones SA, Taylor PR, O’Donnell VB. 12/15-Lipoxygenase regulates the inflammatory response to bacterial products in vivo. J Immunol. 2008;181:6514–6524. doi: 10.4049/jimmunol.181.9.6514. [DOI] [PubMed] [Google Scholar]
- 11.van Leyen K, Arai K, Jin G, Kenyon V, Gerstner B, Rosenberg PA, Holman TR, Lo EH. Novel lipoxygenase inhibitors as neuroprotective reagents. J Neurosci Res. 2008;86:904–909. doi: 10.1002/jnr.21543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Prasad VV, Nithipatikom K, Harder DR. Ceramide elevates 12-hydroxyeicosatetraenoic acid levels and upregulates 12-lipoxygenase in rat primary hippocampal cell cultures containing predominantly astrocytes. Neurochem Int. 2008;53:220–229. doi: 10.1016/j.neuint.2008.07.002. [DOI] [PubMed] [Google Scholar]
- 13.Li J, Wang H, Rosenberg PA. Vitamin K prevents oxidative cell death by inhibiting activation of 12-lipoxygenase in developing oligodendrocytes. J Neurosci Res. 2009;87:1997–2005. doi: 10.1002/jnr.22029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rao GN, Baas AS, Glasgow WC, Eling TE, Runge MS, Alexander RW. Activation of mitogen-activated protein kinases by arachidonic acid and its metabolites in vascular smooth muscle cells. J Biol Chem. 1994;269:32586–32591. [PubMed] [Google Scholar]
- 15.Natarajan R, Reddy MA, Malik KU, Fatima S, Khan BV. Signaling mechanisms of nuclear factor-kappab-mediated activation of inflammatory genes by 13-hydroperoxyoctadecadienoic acid in cultured vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2001;21:1408–1413. doi: 10.1161/hq0901.095278. [DOI] [PubMed] [Google Scholar]
- 16.Reddy MA, Thimmalapura PR, Lanting L, Nadler JL, Fatima S, Natarajan R. The oxidized lipid and lipoxygenase product 12(S)-hydroxyeicosatetraenoic acid induces hypertrophy and fibronectin transcription in vascular smooth muscle cells via p38 MAPK and cAMP response element-binding protein activation. Mediation of angiotensin II effects. J Biol Chem. 2002;277:9920–9928. doi: 10.1074/jbc.M111305200. [DOI] [PubMed] [Google Scholar]
- 17.Kühn H, O’Donnell VB. Inflammation and immune regulation by 12/15-lipoxygenases. Prog Lipid Res. 2006;45:334–356. doi: 10.1016/j.plipres.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 18.Kim YS, Reddy MA, Lanting L, Adler SG, Natarajan R. Differential behavior of mesangial cells derived from 12/15-lipoxygenase knockout mice relative to control mice. Kidney Int. 2003;64:1702–1714. doi: 10.1046/j.1523-1755.2003.00286.x. [DOI] [PubMed] [Google Scholar]
- 19.Li SL, Dwarakanath RS, Cai Q, Lanting L, Natarajan R. Effects of silencing leukocyte-type 12/15-lipoxygenase using short interfering RNAs. J Lipid Res. 2005;46:220–229. doi: 10.1194/jlr.M400328-JLR200. [DOI] [PubMed] [Google Scholar]
- 20.Reilly KB, Srinivasan S, Hatley ME, Patricia MK, Lannigan J, Bolick DT, Vandenhoff G, Pei H, Natarajan R, Nadler JL, Hedrick CC. 12/15-Lipoxygenase activity mediates inflammatory monocyte/endothelial interactions and atherosclerosis in vivo. J Biol Chem. 2004;279:9440–9450. doi: 10.1074/jbc.M303857200. [DOI] [PubMed] [Google Scholar]
- 21.Yuan H, Lanting L, Xu ZG, Li SL, Swiderski P, Putta S, Jonnalagadda M, Kato M, Natarajan R. Effects of cholesterol-tagged small interfering RNAs targeting 12/15-lipoxygenase on parameters of diabetic nephropathy in a mouse model of type 1 diabetes. Am J Physiol Renal Physiol. 2008;295:F605–F617. doi: 10.1152/ajprenal.90268.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huo Y, Zhao L, Hyman MC, Shashkin P, Harry BL, Burcin T, Forlow SB, Stark MA, Smith DF, Clarke S, Srinivasan S, Hedrick CC, Praticò D, Witztum JL, Nadler JL, Funk CD, Ley K. Critical role of macrophage 12/15-lipoxygenase for atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2004;110:2024–2031. doi: 10.1161/01.CIR.0000143628.37680.F6. [DOI] [PubMed] [Google Scholar]
- 23.Gubitosi-Klug RA, Talahalli R, Du Y, Nadler JL, Kern TS. 5-Lipoxygenase, but not 12/15-lipoxygenase, contributes to degeneration of retinal capillaries in a mouse model of diabetic retinopathy. Diabetes. 2008;57:1387–1393. doi: 10.2337/db07-1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boulton AJ, Vinik AI, Arezzo JC, Bril V, Feldman EL, Freeman R, Malik RA, Maser RE, Sosenko JM, Ziegler D. American Diabetes Association. Diabetic neuropathies: a statement by the American Diabetes Association. Diabetes Care. 2005;28:956–962. doi: 10.2337/diacare.28.4.956. [DOI] [PubMed] [Google Scholar]
- 25.Obrosova IG. Diabetic painful and insensate neuropathy: pathogenesis and potential treatments. Neurotherapeutics. 2009;6:638–647. doi: 10.1016/j.nurt.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Obrosova IG, Li F, Abatan OI, Forsell MA, Komjáti K, Pacher P, Szabó C, Stevens MJ. Role of poly(ADP-ribose) polymerase activation in diabetic neuropathy. Diabetes. 2004;53:711–720. doi: 10.2337/diabetes.53.3.711. [DOI] [PubMed] [Google Scholar]
- 27.Obrosova IG, Ilnytska O, Lyzogubov VV, Pavlov IA, Mashtalir N, Nadler JL, Drel VR. High-fat diet induced neuropathy of pre-diabetes and obesity: effects of “healthy” diet and aldose reductase inhibition. Diabetes. 2007;56:2598–2608. doi: 10.2337/db06-1176. [DOI] [PubMed] [Google Scholar]
- 28.Song Z, Fu DT, Chan YS, Leung S, Chung SS, Chung SK. Transgenic mice overexpressing aldose reductase in Schwann cells show more severe nerve conduction velocity deficit and oxidative stress under hyperglycemic stress. Mol Cell Neurosci. 2003;23:638–647. doi: 10.1016/s1044-7431(03)00096-4. [DOI] [PubMed] [Google Scholar]
- 29.Vareniuk I, Pavlov IA, Obrosova IG. Inducible nitric oxide synthase gene deficiency counteracts multiple manifestations of peripheral neuropathy in a streptozotocin-induced mouse model of diabetes. Diabetologia. 2008;51:2126–2133. doi: 10.1007/s00125-008-1136-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vareniuk I, Pacher P, Pavlov IA, Drel VR, Obrosova IG. Peripheral neuropathy in mice with neuronal nitric oxide synthase gene deficiency. Int J Mol Med. 2009;23:571–580. doi: 10.3892/ijmm_00000166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang L, Yu C, Vasquez FE, Galeva N, Onyango I, Swerdlow RH, Dobrowsky RT. Hyperglycemia alters the schwann cell mitochondrial proteome and decreases coupled respiration in the absence of superoxide production. J Proteome Res. 2010;9:458–71. doi: 10.1021/pr900818g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Obrosova IG, Drel VR, Pacher P, Ilnytska O, Wang ZQ, Stevens MJ, Yorek MA. Oxidative-nitrosative stress and poly(ADP-ribose) polymerase (PARP) activation in experimental diabetic neuropathy: the relation is revisited. Diabetes. 2005;54:3435–3441. doi: 10.2337/diabetes.54.12.3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stevens MJ, Li F, Drel VR, Abatan OI, Kim H, Burnett D, Larkin D, Obrosova IG. Nicotinamide reverses neurological and neurovascular deficits in streptozotocin diabetic rats. J Pharmacol Exp Ther. 2007;320:458–464. doi: 10.1124/jpet.106.109702. [DOI] [PubMed] [Google Scholar]
- 34.Askwith T, Zeng W, Eggo MC, Stevens MJ. Oxidative stress and dysregulation of the taurine transporter in high-glucose-exposed human Schwann cells: implications for pathogenesis of diabetic neuropathy. Am J Physiol Endocrinol Metab. 2009;297:620–628. doi: 10.1152/ajpendo.00287.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kuruvilla R, Peterson RG, Kincaid JC, Eichberg J. Evening primrose oil treatment corrects reduced conduction velocity but not depletion of arachidonic acid in nerve from streptozotocin-induced diabetic rats. Prostaglandins Leukot Essent Fatty Acids. 1998;59:195–202. doi: 10.1016/s0952-3278(98)90063-1. [DOI] [PubMed] [Google Scholar]
- 36.Pop-Busui R, Marinescu V, Van Huysen C, Li F, Sullivan K, Greene DA, Larkin D, Stevens MJ. Dissection of metabolic, vascular, and nerve conduction interrelationships in experimental diabetic neuropathy by cyclooxygenase inhibition and acetyl-L-carnitine administration. Diabetes. 2002;51:2619–2628. doi: 10.2337/diabetes.51.8.2619. [DOI] [PubMed] [Google Scholar]
- 37.Kellogg AP, Wiggin TD, Larkin DD, Hayes JM, Stevens MJ, Pop-Busui R. Protective effects of cyclooxygenase-2 gene inactivation against peripheral nerve dysfunction and intraepidermal nerve fiber loss in experimental diabetes. Diabetes. 2007;56:2997–3005. doi: 10.2337/db07-0740. [DOI] [PubMed] [Google Scholar]
- 38.Nagamatsu M, Nickander KK, Schmelzer JD, Raya A, Wittrock DA, Tritschler H, Low PA. Lipoic acid improves nerve blood flow, reduces oxidative stress, and improves distal nerve conduction in experimental diabetic neuropathy. Diabetes Care. 1995;18:1160–1167. doi: 10.2337/diacare.18.8.1160. [DOI] [PubMed] [Google Scholar]
- 39.Sagara M, Satoh J, Wada R, Yagihashi S, Takahashi K, Fukuzawa M, Muto G, Muto Y, Toyota T. Inhibition of development of peripheral neuropathy in streptozotocin-induced diabetic rats with N-acetylcysteine. Diabetologia. 1996;39:263–269. doi: 10.1007/BF00418340. [DOI] [PubMed] [Google Scholar]
- 40.Love A, Cotter MA, Cameron NE. Effects of the sulphydryl donor N-acetyl-L-cysteine on nerve conduction, perfusion, maturation and regeneration following freeze damage in diabetic rats. Eur J Clin Invest. 1996;26:698–706. doi: 10.1111/j.1365-2362.1996.tb02156.x. [DOI] [PubMed] [Google Scholar]
- 41.Stevens MJ, Obrosova I, Cao X, Van Huysen C, Greene DA. Effects of DL-alpha-lipoic acid on peripheral nerve conduction, blood flow, energy metabolism, and oxidative stress in experimental diabetic neuropathy. Diabetes. 2000;49:1006–1015. doi: 10.2337/diabetes.49.6.1006. [DOI] [PubMed] [Google Scholar]
- 42.Cameron NE, Jack AM, Cotter MA. Effect of alpha-lipoic acid on vascular responses and nociception in diabetic rats. Free Radic Biol Med. 2001;31:125–135. doi: 10.1016/s0891-5849(01)00564-0. [DOI] [PubMed] [Google Scholar]
- 43.Coppey LJ, Gellett JS, Davidson EP, Dunlap JA, Lund DD, Yorek MA. Effect of antioxidant treatment of streptozotocin-induced diabetic rats on endoneurial blood flow, motor nerve conduction velocity, and vascular reactivity of epineurial arterioles of the sciatic nerve. Diabetes. 2001;50:1927–1937. doi: 10.2337/diabetes.50.8.1927. [DOI] [PubMed] [Google Scholar]
- 44.Cameron NE, Tuck Z, McCabe L, Cotter MA. Effect of the hydroxyl radical scavenger, dimethylthiourea, on peripheral nerve tissue perfusion, conduction velocity and nociception in experimental diabetes. Diabetologia. 2001;44:1161–1169. doi: 10.1007/s001250100626. [DOI] [PubMed] [Google Scholar]
- 45.Inkster ME, Cotter MA, Cameron NE. Effects of trientine, a metal chelator, on defective endothelium-dependent relaxation in the mesenteric vasculature of diabetic rats. Free Radic Res. 2002;36:1091–1099. doi: 10.1080/1071576021000028325. [DOI] [PubMed] [Google Scholar]
- 46.Schmeichel AM, Schmelzer JD, Low PA. Oxidative injury and apoptosis of dorsal root ganglion neurons in chronic experimental diabetic neuropathy. Diabetes. 2003;52:165–171. doi: 10.2337/diabetes.52.1.165. [DOI] [PubMed] [Google Scholar]
- 47.Coppey LJ, Gellett JS, Davidson EP, Dunlap JA, Lund DD, Salvemini D, Yorek MA. Effect of M40403 treatment of diabetic rats on endoneurial blood flow, motor nerve conduction velocity and vascular function of epineurial arterioles of the sciatic nerve. Br J Pharmacol. 2001;134:21–29. doi: 10.1038/sj.bjp.0704216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cotter MA, Cameron NE. Effect of the NAD(P)H oxidase inhibitor, apocynin, on peripheral nerve perfusion and function in diabetic rats. Life Sci. 2003;73:1813–1824. doi: 10.1016/s0024-3205(03)00508-3. [DOI] [PubMed] [Google Scholar]
- 49.Inkster ME, Cotter MA, Cameron NE. Treatment with the xanthine oxidase inhibitor, allopurinol, improves nerve and vascular function in diabetic rats. Eur J Pharmacol. 2007;561:63–71. doi: 10.1016/j.ejphar.2006.12.029. [DOI] [PubMed] [Google Scholar]
- 50.Obrosova IG, Van Huysen C, Fathallah L, Cao XC, Greene DA, Stevens MJ. An aldose reductase inhibitor reverses early diabetes-induced changes in peripheral nerve function, metabolism, and antioxidative defense. FASEB J. 2002;16:123–125. doi: 10.1096/fj.01-0603fje. [DOI] [PubMed] [Google Scholar]
- 51.Ho EC, Lam KS, Chen YS, Yip JC, Arvindakshan M, Yamagishi S, Yagihashi S, Oates PJ, Ellery CA, Chung SS, Chung SK. Aldose reductase-deficient mice are protected from delayed motor nerve conduction velocity, increased c-Jun NH2-terminal kinase activation, depletion of reduced glutathione, increased superoxide accumulation, and DNA damage. Diabetes. 2006;55:1946–1953. doi: 10.2337/db05-1497. [DOI] [PubMed] [Google Scholar]
- 52.Vincent AM, Russell JW, Sullivan KA, Backus C, Hayes JM, McLean LL, Feldman EL. SOD2 protects neurons from injury in cell culture and animal models of diabetic neuropathy. Exp Neurol. 2007;208:216–227. doi: 10.1016/j.expneurol.2007.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Szabó C, Ischiropoulos H, Radi R. Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nat Rev Drug Discov. 2007;6:662–680. doi: 10.1038/nrd2222. [DOI] [PubMed] [Google Scholar]
- 55.Souza JM, Giasson BI, Chen Q, Lee VM, Ischiropoulos H. Dityrosine cross-linking promotes formation of stable alpha -synuclein polymers. Implication of nitrative and oxidative stress in the pathogenesis of neurodegenerative synucleinopathies. J Biol Chem. 2000;275:18344–18349. doi: 10.1074/jbc.M000206200. [DOI] [PubMed] [Google Scholar]
- 56.Zhang YJ, Xu YF, Liu YH, Yin J, Li HL, Wang Q, Wang JZ. Peroxynitrite induces Alzheimer-like tau modifications and accumulation in rat brain and its underlying mechanisms. FASEB J. 2006;20:1431–1442. doi: 10.1096/fj.05-5223com. [DOI] [PubMed] [Google Scholar]
- 57.Horiguchi T, Uryu K, Giasson BI, Ischiropoulos H, LightFoot R, Bellmann C, Richter-Landsberg C, Lee VM, Trojanowski JQ. Nitration of tau protein is linked to neurodegeneration in tauopathies. Am J Pathol. 2003;163:1021–1031. doi: 10.1016/S0002-9440(10)63462-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Obrosova IG, Mabley JG, Zsengellér Z, Charniauskaya T, Abatan OI, Groves JT, Szabó C. Role for nitrosative stress in diabetic neuropathy: evidence from studies with a peroxynitrite decomposition catalyst. FASEB J. 2005;19:401–403. doi: 10.1096/fj.04-1913fje. [DOI] [PubMed] [Google Scholar]
- 59.Drel VR, Pacher P, Vareniuk I, Pavlov IA, Ilnytska O, Lyzogubov VV, Bell SR, Groves JT, Obrosova IG. Evaluation of the peroxynitrite decomposition catalyst Fe(III) tetra-mesitylporphyrin octasulfonate on peripheral neuropathy in a mouse model of type 1 diabetes. Int J Mol Med. 2007;20:783–792. [PMC free article] [PubMed] [Google Scholar]
- 60.Obrosova IG, Drel VR, Oltman CL, Mashtalir N, Tibrewala J, Groves JT, Yorek MA. Role of nitrosative stress in early neuropathy and vascular dysfunction in streptozotocin-diabetic rats. Am J Physiol Endocrinol Metab. 2007;293:E1645–E1655. doi: 10.1152/ajpendo.00479.2007. [DOI] [PubMed] [Google Scholar]
- 61.Nangle MR, Cotter MA, Cameron NE. Effects of the peroxynitrite decomposition catalyst, FeTMPyP, on function of corpus cavernosum from diabetic mice. Eur J Pharmacol. 2004;502:143–148. doi: 10.1016/j.ejphar.2004.08.033. [DOI] [PubMed] [Google Scholar]
- 62.Vincent AM, Hayes JM, McLean LL, Vivekanandan-Giri A, Pennathur S, Feldman EL. Dyslipidemia-induced neuropathy in mice: the role of oxLDL/LOX-1. Diabetes. 2009;58:2376–2385. doi: 10.2337/db09-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Szabó C, Zanchi A, Komjáti K, Pacher P, Krolewski AS, Quist WC, LoGerfo FW, Horton ES, Veves A. Poly(ADP-Ribose) polymerase is activated in subjects at risk of developing type 2 diabetes and is associated with impaired vascular reactivity. Circulation. 2002;106:2680–2686. doi: 10.1161/01.cir.0000038365.78031.9c. [DOI] [PubMed] [Google Scholar]
- 64.Drel VR, Lupachyk S, Shevalye H, Vareniuk I, Xu W, Zhang J, Delamere NA, Shahidullah M, Slusher B, Obrosova IG. New therapeutic and biomarker discovery for peripheral diabetic neuropathy: PARP inhibitor, nitrotyrosine, and tumor necrosis factor-{alpha} Endocrinology. 2010;151:2547–2555. doi: 10.1210/en.2009-1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ceriello A, Esposito K, Piconi L, Ihnat MA, Thorpe JE, Testa R, Boemi M, Giugliano D. Oscillating glucose is more deleterious to endothelial function and oxidative stress than mean glucose in normal and type 2 diabetic patients. Diabetes. 2008;57:1349–1354. doi: 10.2337/db08-0063. [DOI] [PubMed] [Google Scholar]
- 66.Nourooz-Zadeh J, Ziegler D, Sohr C, Betteridge JD, Knight J, Hothersall J. The use of pholasin as a probe for the determination of plasma total antioxidant capacity. Clin Biochem. 2006;39:55–61. doi: 10.1016/j.clinbiochem.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 67.El-Remessy AB, Abou-Mohamed G, Caldwell RW, Caldwell RB. High glucose-induced tyrosine nitration in endothelial cells: role of eNOS uncoupling and aldose reductase activation. Invest Ophthalmol Vis Sci. 2003;44:3135–3143. doi: 10.1167/iovs.02-1022. [DOI] [PubMed] [Google Scholar]
- 68.Obrosova IG, Pacher P, Szabó C, Zsengeller Z, Hirooka H, Stevens MJ, Yorek MA. Aldose reductase inhibition counteracts oxidative-nitrosative stress and poly(ADP-ribose) polymerase activation in tissue sites for diabetes complications. Diabetes. 2005;54:234–242. doi: 10.2337/diabetes.54.1.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cheung AK, Fung MK, Lo AC, Lam TT, So KF, Chung SS, Chung SK. Aldose reductase deficiency prevents diabetes-induced blood-retinal barrier breakdown, apoptosis, and glial reactivation in the retina of db/db mice. Diabetes. 2005;54:3119–3125. doi: 10.2337/diabetes.54.11.3119. [DOI] [PubMed] [Google Scholar]
- 70.Drel VR, Xu W, Zhang J, Kador PF, Ali TK, Shin J, Julius U, Slusher B, El-Remessy AB, Obrosova IG. Poly(ADP-ribose)polymerase inhibition counteracts cataract formation and early retinal changes in streptozotocin-diabetic rats. Invest Ophthalmol Vis Sci. 2009;50:1778–1790. doi: 10.1167/iovs.08-2191. [DOI] [PubMed] [Google Scholar]
- 71.Drel VR, Xu W, Zhang J, Pavlov IA, Shevalye H, Slusher B, Obrosova IG. Poly(Adenosine 5’-diphosphate-ribose) polymerase inhibition counteracts multiple manifestations of experimental type 1 diabetic nephropathy. Endocrinology. 2009;150:5273–5283. doi: 10.1210/en.2009-0628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nakamura J, Kato K, Hamada Y, Nakayama M, Chaya S, Nakashima E, Naruse K, Kasuya Y, Mizubayashi R, Miwa K, Yasuda Y, Kamiya H, Ienaga K, Sakakibara F, Koh N, Hotta N. A protein kinase C-beta-selective inhibitor ameliorates neural dysfunction in streptozotocin-induced diabetic rats. Diabetes. 1999;48:2090–2095. doi: 10.2337/diabetes.48.10.2090. [DOI] [PubMed] [Google Scholar]
- 73.Obrosova IG, Van Huysen C, Fathallah L, Cao X, Stevens MJ, Greene DA. Evaluation of alpha(1)-adrenoceptor antagonist on diabetes-induced changes in peripheral nerve function, metabolism, and antioxidative defense. FASEB J. 2000;14:1548–1558. doi: 10.1096/fj.14.11.1548. [DOI] [PubMed] [Google Scholar]
- 74.Nakae M, Kamiya H, Naruse K, Horio N, Ito Y, Mizubayashi R, Hamada Y, Nakashima E, Akiyama N, Kobayashi Y, Watarai A, Kimura N, Horiguchi M, Tabata Y, Oiso Y, Nakamura J. Effects of basic fibroblast growth factor on experimental diabetic neuropathy in rats. Diabetes. 2006;55:1470–1477. doi: 10.2337/db05-1160. [DOI] [PubMed] [Google Scholar]
- 75.McGuire JF, Rouen S, Siegfreid E, Wright DE, Dobrowsky RT. Caveolin-1 and altered neuregulin signaling contribute to the pathophysiological progression of diabetic peripheral neuropathy. Diabetes. 2009;58:2677–2686. doi: 10.2337/db09-0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yagihashi S, Yamagishi SI, Wada Ri R, Baba M, Hohman TC, Yabe-Nishimura C, Kokai Y. Neuropathy in diabetic mice overexpressing human aldose reductase and effects of aldose reductase inhibitor. Brain. 2001;124:2448–2458. doi: 10.1093/brain/124.12.2448. [DOI] [PubMed] [Google Scholar]
- 77.Pittenger GL, Ray M, Burcus NI, McNulty P, Basta B, Vinik AI. Intraepidermal nerve fibers are indicators of small-fiber neuropathy in both diabetic and nondiabetic patients. Diabetes Care. 2004;27:1974–1979. doi: 10.2337/diacare.27.8.1974. [DOI] [PubMed] [Google Scholar]
- 78.Quattrini C, Tavakoli M, Jeziorska M, Kallinikos P, Tesfaye S, Finnigan J, Marshall A, Boulton AJ, Efron N, Malik RA. Surrogate markers of small fiber damage in human diabetic neuropathy. Diabetes. 2007;56:2148–2154. doi: 10.2337/db07-0285. [DOI] [PubMed] [Google Scholar]
- 79.Brussee V, Guo G, Dong Y, Cheng C, Martinez JA, Smith D, Glazner GW, Fernyhough P, Zochodne DW. Distal degenerative sensory neuropathy in a long-term type 2 diabetes rat model. Diabetes. 2008;57:1664–1673. doi: 10.2337/db07-1737. [DOI] [PubMed] [Google Scholar]
- 80.Kuruvilla R, Eichberg J. Depletion of phospholipid arachidonoyl-containing molecular species in a human Schwann cell line grown in elevated glucose and their restoration by an aldose reductase inhibitor. J Neurochem. 1998;71:775–783. doi: 10.1046/j.1471-4159.1998.71020775.x. [DOI] [PubMed] [Google Scholar]