

Abstract
Collective cell migration is absolutely essential for a wide variety of physiological episodes including the re-epithelialisation component of tissue repair. However, the investigation of such processes has been frustrated by difficulties in quantitatively analysing the behaviours of a large body of cells within a migrating epithelial sheet, which previously required manually tracking a large number of individual cells, or using advanced computational techniques. Here, we describe a novel and simpler image subtraction method with which we can visualise and quantify collective cell mobilisation as a ‘white wave’ that propagates back from the leading edge of a scratch-wounded monolayer of cultured epithelial cells. Using this technique, we show that actomyosin constriction negatively regulates cell mobilisation and that the advancement of cell sheets and the mobilisation of rows of cells behind their leading edges are independently regulated. We also show that there is a finite limit to the number of rows of cells mobilised after wounding. Moreover, our data suggest that enhancing cell mobilisation, by release from myosin II contractility, accelerates the healing of large wounds in the long term, thus raising the possibility that the cell mobilisation ‘wave’ we reveal here might be a therapeutic target for improving wound healing.
Key words: Wound healing, Collective migration, Cell mobilisation, Myosin II
Introduction
During a wide variety of tissue-remodelling events, including embryonic morphogenesis, wound healing and tumour metastases, cohesive groups of cells move together as a unit (Friedl and Gilmour, 2009; Jacinto et al., 2001; Lecaudey and Gilmour, 2006; Rorth, 2009; Sahai, 2005). However, although locomotion of individual cells, such as leukocytes, has been extensively studied (Friedl and Weigelin, 2008; Ridley et al., 2003), the mechanisms that underpin collective cell migration, for example, how cells communicate with one another and regulate the numbers of cells that participate in concerted migration, are still largely unknown.
During the course of healing a wound, a finite number of rows of epithelial cells surrounding the wound become ‘mobilised’ and collectively migrate into the denuded area to repair the defect, whereas cells further back from the leading edge remain stationary. An appropriate number of cells needs to be mobilised depending on the size of the wound to be healed. If only the very front row of cells were mobilised, then they could only seal gaps of one or two cell diameters across. Equally, mobilisation of more than a few rows of cells is inappropriate to heal a tiny wound that is only a few cell diameters wide. This issue of cell mobilisation is also clinically important. Repair of large cutaneous wounds, greater than 2 or 3 cm across, requires skin grafting because of the inefficiency of long-distance re-epithelialisation (Singer and Dagum, 2009). However, these large wounds might be healed without skin grafting if the mobilisation of the cells surrounding the wound could be enhanced.
How is the number of mobilised cells determined, and is there a limit to the number of rows of cells that can be mobilised? These questions have been difficult to address, at least in part, because of the difficulty in quantitatively analysing the extent to which cells back from the wound edge are mobilised and how or when they participate in the collective migration of the epithelium. These issues have been, in part, addressed by tracking individual cells or by using other less traditional image analysis methods (Farooqui and Fenteany, 2005; Nikolic et al., 2006; Petitjean et al., 2010; Tanner et al., 2009; Vitorino and Meyer, 2008). Although these approaches are powerful, they require advanced computational techniques to track a large number of individual cells. We have designed a simpler method, which uses ImageJ and a time-lapse series of phase-contrast images, to analyse the mechanism underpinning collective cell mobilisation.
Results and Discussion
A novel computational method to visualise and quantify how epithelial cells collectively migrate
When a confluent monolayer of mIMCD3 mouse kidney epithelial cells (Rauchman et al., 1993) is scratch wounded, not only cells at the immediate wound margin, but also those located further back from the leading edge, collectively migrate into the denuded area (Fig. 1A and supplementary material Fig. S1A). This has also been previously shown for Madin-Darby canine kidney (MDCK) cells, which have been extensively used to analyse the collective migration of epithelial cells in vitro (Farooqui and Fenteany, 2005; Fenteany et al., 2000; Matsubayashi et al., 2004; Nikolic et al., 2006; Poujade et al., 2007; Trepat et al., 2009).
Fig. 1.

Visualisation of collective cell mobilisation by the ‘white wave’ method. (A) In vitro scratch-wound healing assay using mIMCD3 cells. Representative images taken at the indicated time points post wounding (p.w.) are displayed. (B) Scheme of the ‘white wave’ method. Cartoons of a sheet of cells moving into the denuded region (gray) (a), and the same cell sheet a short time interval later (b). The first four rows of cells from the wound move forward, whereas the next two rows remain stationary. We calculate the absolute value of the difference between a and b by subtracting individual pixel differences, and display the result in a new ‘difference’ image (c). (C) Application of the ‘white wave’ method to an image of an mIMCD3 cell sheet 1 hour p.w. and one image from 15 minutes later. Bracket shows the white area in which cells are moving within this 15 minute interval. (D) Mobilisation of the cells in the experiment shown in A at the indicated time points, visualised using the ‘white wave’ method. The ‘difference image at time T’ is derived by subtracting the phase-contrast image at time T from that at T+ΔT (here, ΔT=15 minutes – this is true for all the other data, unless otherwise stated). Dashed lines show the propagation of the ‘white wave’ of cell mobilisation; bracket shows the ‘black area’ where cells are starting to halt at 18 hours p.w. The bare substrate seen in the denuded area appears black, because it remains unchanged during the experiments. However, occasionally some white pixels appear because of cell debris from the wound drifting in the medium. (E) Schematic of d and μ. For details, see the Results section and supplementary material Fig. S2. To avoid the complexities associated with the immediate white difference readout (D), measurement of μ commences 1 hour after wounding for all our experiments. (F) Representative difference images of E1 cells at the indicated time points after wounding. Here, ΔT=45 minutes (see also supplementary material Fig. S1). Note that the ‘white wave’ does not propagate back into this ‘fibroblast-like’ cell sheet. Scale bars: 200 μm (A,D), 100 μm (C,F).
To analyse the mechanism of collective cell mobilisation, we have designed a novel computational method to highlight the moving cells in migrating cell sheets. Subtracting one still image of the moving sheet from a similar image taken a short time later, and displaying the absolute value of the difference (|difference|) in a new image (Fig. 1B) renders the territory in which cells are actively moving white (i.e. |difference|>0), whereas regions where cells are not moving turn black (i.e. |difference|=0). This method is very effective for phase-contrast images of real epithelial sheets because of the dark nucleus and paler cytoplasm of cells (Fig. 1C). This analysis reveals how cells become mobilised as a white zone spreading back from the wound edge (Fig. 1C).
It is now simple to generate a series of ‘difference’ images from a movie that captures the entirety of a closing ~400 μm scratch wound and from these images to make a ‘difference movie’ (Fig. 1D; supplementary material Movie 1). At the initial time point, the whole cell sheet reproducibly turns white (Fig. 1D, top panel). Because this occurs even in fixed cell sheets (data not shown), it must reflect a phenomenon not related to active cell movement, possibly gaping back of the whole cell sheet after wounding. This immediate white ‘difference’ readout has almost resolved after 15 minutes (the earliest frame of supplementary material Movie 1). At this time point, the ‘difference’ movie indicates that an area extending about five cell diameters from wound edge has mobilised (i.e. turns white). Subsequently, the white area extends back as a ‘wave’ of cell mobilisation (Fig. 1D dotted lines and supplementary material Movie 1). Over a period of 18 hours, this ‘white wave’ propagates up to 400 μm (>30 cell diameters) back from the initial position of the wound edge and continues spreading even after the wound edges have met, about 6 hours after wounding. A further 6 hours after the opposing epithelial fronts have met, the cell sheet now returns to ‘black’ indicating that cells are becoming static again (Fig. 1D, 18 hours and supplementary material Movie 1). By 24 hours, the entire cell sheet has returned to black, indicating no further cell movement (Fig. 1D and supplementary material Movie 1). In this way, we can visualise collective cell mobilisation upon wounding and cessation of migration after the wound fronts have met. We found that any cell movements associated with division contribute little to the global white wave at any single time point, because the percentage of cells that are in M phase at each time point is <2.5% (supplementary material Fig. S3) during the 24 hours immediately after wounding.
To quantify the extent of cell mobilisation at any one time point, we measured the value of μ, the distance to which the white wave propagates back from the initial position of the wound edge (Fig. 1E and supplementary material Fig. S2B–D), rather than the entire width of the zone that turns white (i.e. d+μ in Fig. 1E), because we find that μ better emphasises the difference between poor and extensive cell mobilisation (Fig. 1E, middle and bottom, respectively), and because μ can be measured without complication, even after the opposing wound edges have first confronted each other, which is not true for d+μ. Indeed, we found that μ increases as the white wave propagates back through the cell sheet (supplementary material Fig. S1C, ‘C12’), indicating that more and more cells are continuously mobilised over time, even beyond the time when the wound fronts have met.
By contrast, when monolayers of fibroblast-like ‘E1’ cells (supplementary material Fig. S1B, see also Movies 2 and 3) were wounded, cell mobilisation appeared to be considerably restricted. No increase in μ over time beyond its initial value of ~100 μm was observed (Fig. 1F and supplementary material Fig. S1C). This suggests that no new cell mobilisation occurs in these wounded cell sheets after the first several rows of cells have commenced migration, and is consistent with previous reports that mesenchymal cells do not exhibit coordinated sheet migration (Dipasquale, 1975; Matsubayashi et al., 2004). This validates the robustness of our ‘white wave’ technique to quantify and distinguish mobilisation of different cell types.
This method clearly offers advantages over the pre-existing methods to analyse cell mobilisation as mentioned in the Introduction: primarily, it is very much simpler to perform and it also offers a clearer picture of the temporal sequence of cell mobilisations extending back from a wound edge; however, it has weaknesses, including its inability to distinguish the directionality of individual cell migrations. In our experiments, most of the individual cells appeared to migrate directly towards the denuded area (supplementary material Fig. S1A and Movie 1), but other studies have shown that some cells can migrate and extend protrusions ‘unproductively’, in directions different from the main direction of cell sheet advancement (Farooqui and Fenteany, 2005; Poujade et al., 2007; Vitorino and Meyer, 2008). Our analysis would miss such behaviour. Similarly, this method cannot discriminate between a true displacement of a cell and a change of cell area or shape.
A finite number of cells are mobilised in a migrating epithelial sheet
The wounds discussed so far generated a gap of ~400 μm and closed within 6–8 hours. Next, we made larger wounds and analysed how cells were mobilised at much later timepoints (Fig. 2). In these experiments the wounds were so large that their opposing edges failed to reach one another during the period of our observation (Fig. 2B, inset). Initially, the value of μ increased with time. However, ~24 hours after wounding μ began to diminish, from a maximum of ~500 μm (Fig. 2A,B). This suggests that the mobilisation of cells back from the wound edge is finite, even though the wound has still not closed. Thus the limit of cell mobilisation might define the limit of the wound size that can be healed without intervention.
Fig. 2.

Only a finite number of cells can be mobilised after wounding. ‘White wave’ analyses of cell sheets with large wounds whose opposing edges did not meet, even by 40 hours after wounding. (A) Representative ‘difference’ images at the indicated time points. (B) The μ values are plotted against time post wounding. n=3. Inset shows wound at 40 hours. Scale bars: 200 μm.
Myosin II negatively regulates the collective mobilisation of epithelial cell groups
We then wondered how the collective mobilisation of cells (propagation of the white wave) back from an epithelial wound edge, is regulated. We focused on non-muscle myosin II, because this motor protein is reported to negatively regulate some epithelial sheet expansions both in vivo and vitro (Farooqui and Fenteany, 2005; Gutzman and Sive, 2010; Mizuno et al., 2002). If the same were true for cell mobilisation back from the wound edge, then inhibition of myosin II contractility would enhance it, whereas activation of myosin II would reduce its extent. We examined these possibilities using a combination of pharmacological and molecular experiments:
First, we treated the cells with blebbistatin (Bb), an inhibitor of myosin II ATPase activity. At concentrations of up to 50 μM, Bb did not significantly alter the speed of wound edge advancement (Fig. 3A,B, left; P=0.1616). Nevertheless, Bb did increase the number of rows of cells mobilised by wounding, causing the white wave to propagate further back, in a dose-dependent manner (Fig. 3A,B, right; P<0.01 for DMSO vs 25 μM Bb and DMSO vs 50 μM Bb). These results suggest that inhibition of myosin II activity within the epithelial sheet enhances the potential for cell mobilisation. Moreover, ROCK inhibitors Y27632 and H1152, which indirectly inhibit myosin II contractility, both cause a similar increase in μ (Fig. 3C, right). In contrast to Bb, however, Y27632 and H1152 both enhanced the forward migration of wound edges (Fig. 3C). This is presumably because targets of ROCK other than myosin II, such as LIMK (Hopkins et al., 2007; Maekawa et al., 1999), are negative regulators of some aspect of epithelial sheet movement. Notably, both of these ROCK inhibitors enhanced cell mobilisation 1 hour after wounding (P<0.01 for water vs Y27632; P<0.05 for water vs H1152), which was some hours before we saw enhanced migration of cells at the leading edge (P=0.3995), suggesting that enhancement of cell mobilisation by the ROCK inhibitors is not simply a consequence of faster migration by cells in the front row.
Fig. 3.

Actomyosin contraction negatively regulates cell mobilisation. (A–C) Effects of blebbistatin (Bb, 10–50 μM), Y27632 (50 μM) and H1152 (25 μM). Cell sheets were treated with each drug, or control DMSO (1:2000 v/v) or water (1:1000 v/v), wounded and subjected to time-lapse imaging. The values of d and μ are measured and plotted against time. (A) Representative difference images of cells treated with DMSO or 25 μM Bb at the indicated time points. The pink line indicates the initial position of the wound edges and the yellow dashed line, the extent of white wave propagation. (B) Quantification of d and μ in cell sheets treated with DMSO (control) or with various concentrations of Bb. n=4 per treatment condition. (C) Quantification of d and μ in cell sheets treated with Y27632, H1152 or water (control). n=8 per treatment condition. (D) Effect of knockdown of Mypt1. Cells were transfected with control RNA or siRNAs against Mypt1 (si-A or si-B), and then subjected to scratch-wound assays to measure d and μ. The wounds were so large that they did not close during the period of our observation. n=4 for each transfected siRNA. Scale bar: 100 μm.
We next examined the effect of hyperactivating myosin II. For this purpose, we knocked down expression of Mypt1 (protein phosphatase 1 regulatory subunit 12A), a negative regulator of myosin II contraction (Ito et al., 2004), by small interfering RNA (siRNA)-mediated gene silencing. Neither of the siRNAs we used reduced the expression of Mypt1 protein by greater than 50% (supplementary material Fig. S4), but both significantly reduced μ by up to ~30% (Fig. 3D, P<0.01 for control vs Mypt1 si-A and P<0.001 for control vs Mypt1 si-B), thus complementing our pharmacological experiments above. Both siRNAs exerted only minimal effects on the speed of the advancing cell sheet (Fig. 3D), indicating that the inhibition of cell mobilisation by the siRNAs is not simply an indirect effect of slowing down of leading edge migration.
Taken together, these results support our hypothesis that actomyosin contractility functions as a negative regulator of cell mobilisation during the epithelial sheet migrations that occur during wound healing. Intracellular actomyosin fibres, such as those located beneath the adherens junctions, presumably provide a constitutive restrictive force on cells in an intact epithelium. We speculate that, as cells spread forward to heal a wound, this constriction must be overcome by signals and/or opposing forces to enable cell mobilisation. We suppose that the suppression of cell mobilisation by myosin II serves as a rheostat to ensure efficient tissue repair by braking unnecessarily intense cell mobilisation.
Moreover, our observations that the mobilisation of cells located back in the sheet can be altered without affecting the forward migration of leading edge cells, indicate that these two components of the re-epithelialisation process are independently regulated. This is consistent with a previous analysis of vascular endothelial cells, which also indicated that the migration of cells at the wound edge and the behaviour of the cells behind them are independently regulated (Vitorino and Meyer, 2008).
The efficiency of cell mobilisation affects the healing of large wounds
We have shown that the mobilisation wave extends back only a finite distance (Fig. 2). In the longer term, is this extent of cell mobilisation rate limiting for cell sheet migration and can it be increased by modulation of the efficiency of cell mobilisation? To address these questions, we treated cell sheets with drugs that specifically enhance or impair cell mobilisation, without affecting leading edge migration, at least at early time points after wounding. As a mobilisation enhancer we used Bb (Fig. 3). As a mobilisation inhibitor, we used 2,3-butadione monoxime (BDM). BDM is a relatively non-specific myosin II inhibitor (Ostap, 2002), which we first expected might be another enhancer of cell mobilisation, for confirmation of our Bb data. But in fact, we found that BDM blocked cell mobilisation without altering the rate of the forward migration of wound edges for the first 5 hours after wounding (supplementary material Fig. S5). It is unclear why BDM has such a contrasting effect to Bb, although BDM is known to be highly promiscuous and have effects on several non-myosin targets (Ostap, 2002). We therefore used BDM as a tool to specifically inhibit cell mobilisation. This effect of BDM lends credence to the idea that cell mobilisation and leading edge migration are independently regulated.
We challenged large wounds with Bb, BDM or control DMSO. The wounds were 5 mm across and so did not close for at least 30 hours after wounding. We plotted the values of ‘d’, the distance to which the wound edge migrated into the denuded area, against time (Fig. 4). At time points beyond 5 hours, the migration of BDM-treated cell sheets was retarded, whereas Bb-treated sheets travelled further than control DMSO-treated cells (P<0.05 for DMSO vs Bb; P<0.01 for DMSO vs BDM). These results suggest that, in the longer term, enhancement or inhibition of cell mobilisation increases or decreases forward migration of the cell sheet, respectively, raising the possibility that we might accelerate the healing of large wounds by modulating the activity of myosin II.
Fig. 4.
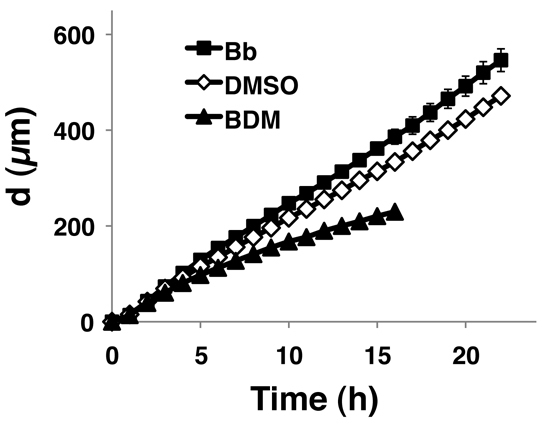
Long-term effects of Bb and BDM on the migration of cell sheets. Cell sheets were pretreated with either Bb (25 μM), BDM (10 mM) or control DMSO (1:2000 v/v), wounded and subjected to time-lapse imaging. The extent of leading edge migration (d) is plotted against time. n=3 per treatment condition.
Materials and Methods
Cell culture
mIMCD3 cells were cultured as previously described (Shaw and Martin, 2009). Subclones C12 and E1 were established by amplifying single colonies obtained from the parental mIMICD3 cells. Unless otherwise stated, C12 was used for the experiments in this study. For both subclones, cells were plated into 35 mm plastic tissue culture dishes (Nunc, cat. No. 153066), except for the experiment in supplementary material Fig. S3. Unless otherwise stated, cells were plated at an initial density of 3×105 cells per 35 mm dish 3.5–4 days before wounding, and replenished with fresh medium 1.5–2 days before wounding.
Drug treatment
(±)-Blebbistatin (Tocris) was diluted to 20 or 100 mM stock in DMSO. Y27632 and H1152 from Calbiochem and 2,3-butadion monoxime (BDM; Sigma) were diluted to 10 mM, 10 mM and 500 mM stock in water, respectively. For pharmacological treatments, cells were incubated with each drug at the concentrations described in the figure legends for 30 minutes before wounding, and scratch-wound assays were carried out in the continuous presence of the drug.
Transfection
One day before transfection, cells were split at a density of 1×105 cells/35 mm dish. siRNAs were transfected using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Mypt1 si-A and si-B (Stealth siRNA No. MSS206919 and MSS275914, respectively) and control siRNA (Cat. No. 12935-300) were purchased from Invitrogen. The final concentrations of Lipofectamine 2000 and siRNAs were 0.17% v/v and 66 nM, respectively. Two days after transfection, cells were replenished with fresh medium, incubated for further 1.5 days and subjected to wounding or immunoblot assays.
Immunofluorescence and immunoblot analysis
Immunofluorescence and immunoblot detection was carried out using established methods. Anti-phospho-histone H3 (Ser10) (Cell Signaling) was used at 1:1000; anti-Mypt1 (Covance) at 1:10,000; and anti-α-tubulin (Serotec) at 1:30,000.
Scratch-wound assay
For the scratch-wound healing assay, linear scratches were made in cell sheets with either a pipette tip or a cotton bud. The former produced wounds ~400 μM across, and the latter ~5 mm.
‘White wave’ analysis
After wounding, phase-contrast images of migrating cell sheets were acquired every 15 minutes at 5× magnification as previously described (Mori et al., 2006). Images were processed using Volocity (Improvision) and ImageJ (http://rsb.info.nih.gov/ij/). The rate of forward migration of the leading edge cells and the mobilisation of cells behind the wound edge were analysed from images of dimensions of 1344×1024 pixels (1706×1299 μm). For ‘white wave analyses’ of multiple images, we used a newly developed ImageJ macro (supplementary material Macro 1). For detail, see supplementary material Fig. S2.
Statistical analysis
Quantified data are presented as mean ± s.e.m. and the numbers of samples (at least three) are indicated in the figure legends. The data were statistically analysed by Friedman's tests followed by Dunn's post tests except in Fig. 3C, where we analysed the values of d and μ at 1 hour post wounding by Kruskal–Wallis test followed by Dunn's post tests. All the statistical analyses were carried out using PRISM4 (GraphPad Software).
Supplementary Material
Acknowledgments
We thank all the members of the Martin and Nobes labs, Mark Bass and colleagues in the Japanese Society for Quantitative Biology for their helpful discussions and advice. This work was supported by the Wellcome Trust (Y.M., W.R. and P.M.) and Yamada Science Foundation (Y.M.). Deposited in PMC for release after 6 months.
Footnotes
Supplementary material available online at http://jcs.biologists.org/cgi/content/full/124/7/1017/DC1
References
- Dipasquale A. (1975). Locomotory activity of epithelial cells in culture. Exp. Cell Res. 94, 191-215 [DOI] [PubMed] [Google Scholar]
- Farooqui R., Fenteany G. (2005). Multiple rows of cells behind an epithelial wound edge extend cryptic lamellipodia to collectively drive cell-sheet movement. J. Cell Sci. 118, 51-63 [DOI] [PubMed] [Google Scholar]
- Fenteany G., Janmey P. A., Stossel T. P. (2000). Signaling pathways and cell mechanics involved in wound closure by epithelial cell sheets. Curr. Biol. 10, 831-838 [DOI] [PubMed] [Google Scholar]
- Friedl P., Gilmour D. (2009). Collective cell migration in morphogenesis, regeneration and cancer. Nat. Rev. Mol. Cell Biol. 10, 445-457 [DOI] [PubMed] [Google Scholar]
- Friedl P., Weigelin B. (2008). Interstitial leukocyte migration and immune function. Nat. Immunol. 9, 960-969 [DOI] [PubMed] [Google Scholar]
- Gutzman J. H., Sive H. (2010). Epithelial relaxation mediated by the myosin phosphatase regulator Mypt1 is required for brain ventricle lumen expansion and hindbrain morphogenesis. Development 137, 795-804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins A. M., Pineda A. D. A., Winfree L. M., Brown G. T., Laukoetter M. G., Nusrat A. (2007). Organized migration of epithelial cells requires control of adhesion and protrusion through Rho kinase effectors. Am. J. Physiol. Gastrointest. Liver Physiol. 292, G806-G817 [DOI] [PubMed] [Google Scholar]
- Ito M., Nakano T., Erdodi F., Hartshorne D. J. (2004). Myosin phosphatase: structure, regulation and function. Mol. Cell. Biochem. 259, 197-209 [DOI] [PubMed] [Google Scholar]
- Jacinto A., Martinez-Arias A., Martin P. (2001). Mechanisms of epithelial fusion and repair. Nat. Cell Biol. 3, E117-E123 [DOI] [PubMed] [Google Scholar]
- Lecaudey V., Gilmour D. (2006). Organizing moving groups during morphogenesis. Curr. Opin. Cell Biol. 18, 102-107 [DOI] [PubMed] [Google Scholar]
- Maekawa M., Ishizaki T., Boku S., Watanabe N., Fujita A., Iwamatsu A., Obinata T., Ohashi K., Mizuno K., Narumiya S. (1999). Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science 285, 895-898 [DOI] [PubMed] [Google Scholar]
- Matoltsy A. G., Viziam C. B. (1970). Further observations on epithelialization of small wounds: an autoradiographic study of incorporation and distribution of 3H-thymidine in the epithelium covering skin wounds. J. Invest. Dermatol. 55, 20-25 [DOI] [PubMed] [Google Scholar]
- Matsubayashi Y., Ebisuya M., Honjoh S., Nishida E. (2004). ERK activation propagates in epithelial cell sheets and regulates their migration during wound healing. Curr. Biol. 14, 731-735 [DOI] [PubMed] [Google Scholar]
- Mizuno T., Tsutsui K., Nishida Y. (2002). Drosophila myosin phosphatase and its role in dorsal closure. Development 129, 1215-1223 [DOI] [PubMed] [Google Scholar]
- Mori R., Power K. T., Wang C. H. M., Martin P., Becker D. L. (2006). Acute downregulation of connexin43 at wound sites leads to a reduced inflammatory response, enhanced keratinocyte proliferation and wound fibroblast migration. J. Cell Sci. 119, 5193-5203 [DOI] [PubMed] [Google Scholar]
- Nikolic D. L., Boettiger A. N., Bar-Sagi D., Carbeck J. D., Shvartsman S. Y. (2006). Role of boundary conditions in an experimental model of epithelial wound healing. Am. J. Physiol. Cell Physiol. 291, C68-C75 [DOI] [PubMed] [Google Scholar]
- Ostap E. M. (2002). 2,3-Butanedione monoxime (BDM) as a myosin inhibitor. J. Muscle Res. Cell Motil. 23, 305-308 [DOI] [PubMed] [Google Scholar]
- Petitjean L., Reffay M., Grasland-Mongrain E., Poujade M., Ladoux B., Buguin A., Silberzan P. (2010). Velocity fields in a collectively migrating epithelium. Biophys. J. 98, 1790-1800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poujade M., Grasland-Mongrain E., Hertzog A., Jouanneau J., Chavrier P., Ladoux B., Buguin A., Silberzan P. (2007). Collective migration of an epithelial monolayer in response to a model wound. Proc. Natl. Acad. Sci. USA 104, 15988-15993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauchman M. I., Nigam S. K., Delpire E., Gullans S. R. (1993). An osmotically tolerant inner medullary collecting duct cell-line from an Sv40 transgenic mouse. Am. J. Physiol. 265, F416-F424 [DOI] [PubMed] [Google Scholar]
- Ridley A. J., Schwartz M. A., Burridge K., Firtel R. A., Ginsberg M. H., Borisy G., Parsons J. T., Horwitz A. R. (2003). Cell migration: integrating signals from front to back. Science 302, 1704-1709 [DOI] [PubMed] [Google Scholar]
- Rorth P. (2009). Collective cell migration. Annu. Rev. Cell Dev. Biol. 25, 407-429 [DOI] [PubMed] [Google Scholar]
- Sahai E. (2005). Mechanisms of cancer cell invasion. Curr. Opin. Genet. Dev. 15, 87-96 [DOI] [PubMed] [Google Scholar]
- Shaw T., Martin P. (2009). Epigenetic reprogramming during wound healing: loss of polycomb-mediated silencing may enable upregulation of repair genes. EMBO Rep. 10, 881-886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer A. J., Dagum A. B. (2009). Current management of acute cutaneous wounds. N. Eng. J. Med. 359, 1037-1046 [DOI] [PubMed] [Google Scholar]
- Tanner K., Ferris D. R., Lanzano L., Mandefro B., Mantulin W. W., Gardiner D. M., Rugg E. L., Gratton E. (2009). Coherent movement of cell layers during wound healing by image correlation spectroscopy. Biophys. J. 97, 2098-2106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trepat X., Wasserman M. R., Angelini T. E., Millet E., Weitz D. A., Butler J. P., Fredberg J. J. (2009). Physical forces during collective cell migration. Nat. Phys. 5, 426-430 [Google Scholar]
- Vitorino P., Meyer T. (2008). Modular control of endothelial sheet migration. Genes Dev. 22, 3268-3281 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.