

Abstract
Notch receptors and their ligands have crucial roles in development and tumorigenesis. We present evidence demonstrating the existence of an antagonistic relationship between Notch 4 and Trp53, which is controlled by the Mdm2-dependent ubiquitylation and degradation of the Notch receptor. We show that this signal-controlling mechanism is mediated by physical interactions between Mdm2 and Notch 4 and suggest the existence of a trimeric complex between Trp53, Notch 4 and Mdm2, which ultimately regulates Notch activity. Functional studies indicate that Trp53 can suppress NICD4-induced anchorage-independent growth in mammary epithelial cells and present evidence showing that Trp53 has a pivotal role in the suppression of Notch-associated tumorigenesis in the mammary gland.
Key words: Trp53, Mdm2, Notch, Ubiquitylation, Tumorigenesis
Introduction
The Notch locus encodes a transmembrane receptor that is the central element of an evolutionarily conserved signaling pathway controlling a broad spectrum of cell-fate decisions during metazoan development. Signals through the Notch receptor couple cell-fate acquisition of an individual cell to the cell-fate choices made by its immediate neighbors (Artavanis-Tsakonas et al., 1999) affecting proliferation, differentiation and apoptotic decisions in development. Abnormal Notch signaling has profound consequences for normal development in metazoans and increasing evidence links the Notch signaling pathway with pathogenic conditions such as cancer (Callahan and Egan, 2004; Ellisen et al., 1991; Fre et al., 2009; Jhappan et al., 1992; Kiaris et al., 2004).
Our current mechanistic understanding of Notch signaling has the Notch receptor on the surface of one cell, interacting with membrane-bound ligands on the surface of a neighboring cell, triggering a cascade of proteolytic events that eventually cleave the entire intracellular domain of the receptor. The intracellular domain carries nuclear localization signals (Kopan et al., 1996; Stifani et al., 1992) and translocates into the nucleus, where it directly participates in a transcriptional complex, which drives Notch-dependent transcription. The complexity of the genetic circuitry controlling Notch signals is very high, and invariably the developmental outcome of modulating the activity of the Notch pathway depends on the cellular context (Hurlbut et al., 2009; Hurlbut et al., 2007; Kankel et al., 2007).
Mammals contain four Notch receptor paralogs: Notch 1, Notch 2, Notch 3 and Notch 4, all of which have been associated with tumorigenic events (Allenspach et al., 2002; Callahan and Egan, 2004; Capobianco et al., 1997; Kiaris et al., 2004). Notch can behave as a bona fide oncogene. For instance, somatic or viral-induced mutations that result in the constitutive activation of the Notch receptor have been shown to be oncogenic both in vitro and in vivo (Robbins et al., 1992; Smith et al., 1995; Talora et al., 2008). Importantly, activating mutations in Notch1 have been linked in humans to almost 50% of all cases of T-cell acute lymphoblastic leukemia (T-ALL) (Weng et al., 2004). Although the Notch receptor can behave as an oncogene, it is becoming increasingly clear that the Notch pathway can have a very significant role in oncogenesis via the synergy between Notch signals and other cellular elements, which, in a context-dependent manner, can create the conditions favoring tumor development (Fre et al., 2009; Kiaris et al., 2004). How Notch integrates its action with other cellular elements is of fundamental interest, both to understand the role of the pathway in development as well as to gain insights into its pathogenic action.
Several studies associated the Notch receptor and, indeed, differential Notch receptor paralog action, with the major tumor suppressor transformation-related protein 53 (henceforth we refer to the mouse gene as Trp53 and to the human counterpart as TP53), which is somatically mutated in almost half of all human cancers (Vogelstein et al., 2000). The levels of the Trp53 protein, which under normal circumstances are very low, are regulated by E3 ubiquitin-protein ligase Mdm2-dependent ubiquitylation (Fang et al., 2000; Haupt et al., 1997; Honda and Yasuda, 2000), which in turn is affected by an autoregulatory loop that directly targets expression of the Mdm2 gene by Trp53 (Gottlieb and Oren, 1996; Picksley and Lane, 1993).
In spite of the significant number of studies linking Notch and Trp53, the underlying molecular basis remains unclear (Beverly et al., 2005; Kim et al., 2007; Mao et al., 2004). Here, we examine the antagonistic relationship between Trp53 and Notch4, a Notch receptor paralog shown to be highly tumorigenic in the mouse mammary epithelium (Jhappan et al., 1992). We find that Trp53 directly affects Notch signaling through the Mdm2-dependent ubiquitylation of the receptor and present evidence indicating that this relationship is important for the oncogenic activity of Notch both in cell culture and in mammary tumors.
Results
Trp53 influences the levels of the Notch 4 protein
To probe the relationship between Notch 4 and Trp53, we compared either endogenous or exogenously delivered Notch 4 intracellular domain (NICD4) steady state protein levels. Several different cell lines, which have been well characterized and have mutant or wild-type TP53 genetic backgrounds, were used to test the generality of our observations. We first compared the endogenous NICD4 levels in TP53-deficient HCT116 human colon carcinoma cells, which were generated by targeted disruption of both TP53 alleles (Bunz et al., 1998), with those in the parental TP53 wild-type HCT116 cells. We found that the level of NICD4 was 20-fold higher in HCT116 TP53-null cells (Fig. 1A, lane 2) compared with the parental HCT116 cells (Fig. 1A, lane 1). This is consistent with the notion that Trp53 antagonizes the expression of NICD4 and corroborates observations involving mouse embryonic fibroblasts (MEFs) lacking Trp53 activity (Trp53−/−), where it was shown that Notch 4 levels were increased when compared with Trp53+/+ controls (Mao et al., 2004).
Fig. 1.

Trp53 antagonizes NICD4 steady-state protein levels. (A) Lysates from HCT116 TP53 wild-type cells (lane 1) and HCT116 TP53-null cells (lane 2) were analyzed for NICD4 protein levels by western blot using an anti-Notch-4 antibody and an anti-α-tubulin antibody (loading control). (B) Effects of Trp53 on NICD4 levels in transfected 293T/17 cells. Western blot analysis of lysates from 293T/17 cells mock transfected (lane 1) or transfected with V5–NICD4 (lane 2), FLAG–Trp53 (lane 3) or both expression vectors (lane 4). Relative transfection efficiencies were monitored by co-transfection with a plasmid encoding GFP (lanes 2–4). (C) Relative NICD4 RNA levels in HCT116 TP53 wild-type cells and HCT116 TP53-null cells were examined by qRT-PCR. Shown are NICD4 RNA levels normalized to HPRT (hypoxanthine phophoribosyltransferase) RNA levels. (D) Relative MDM2 RNA levels were examined by qRT-PCR in HCT116 TP53 wild-type cells and HCT116 TP53-null cells.
To examine further the relationship between Notch 4 and Trp53, co-expression experiments were carried out in 293T/17 cells, which can be readily transfected. As shown in Fig. 1B, when either FLAG-tagged Trp53 or a V5-tagged NICD4, was individually expressed, each of these proteins accumulated to readily detectable levels (Fig. 1B, lanes 2 and 3, respectively). However, when both Trp53 and NICD4 were simultaneously expressed, the detectable levels of the NICD4 protein were substantially reduced (Fig. 1B, lane 4 vs lane 2) whereas levels of Trp53 remained the same (Fig. 1B, lane 4 vs lane 3), suggesting that Trp53 is associated with the reduction of NICD4 protein levels.
To examine whether the observed Trp53-dependent reduction of the endogenous NICD4 protein levels in the HCT116 cells reflected transcriptional control, we compared levels of mRNA encoding NICD4 in the HCT116 parent cells versus TP53-null HCT116 cells, using qRT-PCR and found no significant differences (P>0.05, Fig. 1C). This observation indicated that the striking differences in NICD4 protein levels associated with the downregulation of Trp53 did not reflect Trp53-mediated transcriptional control.
NICD4 is a target of the Mdm2 E3 ligase
Given that the level and, consequently, the activity of Trp53 are controlled largely by the E3-ubiquitin ligase Mdm2 (Geyer et al., 2000) and, conversely, that Mdm2 is a transcriptional target of Trp53 (Haupt et al., 1997; Honda and Yasuda, 2000), we reasoned that the effect of Trp53 on NICD4 could be possibly influenced by Mdm2. We first examined MDM2 mRNA and protein expression in HCT116 cells and found, as expected (Haupt et al., 1997; Honda and Yasuda, 2000), that both were significantly lower in the HCT116 TP53-null cells compared with levels in the parental cells (Fig. 1D and data not shown). Consistent with the observations involving the HCT116 cells, a comparison of endogenous NICD4 protein levels in MEFs lacking Trp53 or lacking both Trp53 and Mdm2 revealed that the steady-state level of NICD4 in MEFs null for both Trp53 and Mdm2 was increased relative to MEFs null for Trp53 alone by approximately threefold (Fig. 2A,B). Since the Mdm2 E3 ligase activity depends on its RING domain (Fang et al., 2000; Honda and Yasuda, 2000), we investigated whether the observed differences in NICD4 expression levels could be directly linked to the ligase activity of Mdm2, by transfecting 293T/17 cells with either a transgene carrying a wild-type copy of Mdm2 or a mutant form lacking the RING domain (Mdm2 ΔR). Fig. 2C summarizes these results. The expression levels of HA-tagged NICD4 were reduced when co-expressed with wild-type Mdm2 (Fig. 2C, lane 2 vs lane 3), whereas they were essentially unaffected when co-expressed with Mdm2 ΔR (Fig. 2C, lane 2 vs lane 4).
Fig. 2.

Mdm2 expression affects NICD4 steady state protein levels. (A) Lysates from Trp53-null MEFs (lane 1) and MEFs null for both Trp53 and Mdm2 (lane 2) were analyzed for NICD4 protein levels by western blot. (B) Densitometric quantification of raw data in Fig. 2A. Plotted are NICD4 levels normalized to α-tubulin levels. (C) Lysates from H1299 cells expressing NICD4-HA and either wild-type MDM2 or a mutant MDM2 protein lacking the RING domain (Mdm2ΔR) were analyzed for NICD4 protein levels by western blot using the indicated antibodies. (D) Lysates from MEFs null for both Trp53 and Mdm2 (lane 1) and from MEFs reconstituted with Mdm2 (lane 2) were analyzed for NICD4 and MDM2 protein levels by western blot. (E) Lysates from H1299 cells expressing control shRNA (lane 1) or MDM2 shRNA (lane 2) were analyzed for NICD4 protein levels by western blot using the indicated antibodies. (F) H1299 cells were transfected with 2 μg of plasmid encoding V5-tagged NICD4 and 0.5 μg a GFP expression vector, which allowed us to monitor transfection efficiency, together with 0.2 μg (lane 3), 0.8 μg (lane 4), 3.2 μg (lane 5) and 6.4 μg (lane 6) of plasmid encoding Mdm2. Equal plasmid mass for each transfection was ensured by the addition of pcDNA3. The two characteristic MDM2 protein forms, long (L) and short (S) are indicated. Lysates were analyzed by western blot using the antibodies indicated to the right. For a densitometric quantification of the data see supplementary material Fig. S2.
This analysis was extended by transfecting a plasmid encoding wild-type Mdm2 into the MEFs null for both Trp53 and Mdm2, which displayed high NICD4 levels (see above), and resulted in the significant reduction of NICD4 (Fig. 2D, lane 2) protein levels compared with control cells (Fig. 2D, lane 1). Furthermore, when H1299 cells, which endogenously express Notch 4 but do not express Trp53 protein were treated with shRNA targeting MDM2, NICD4 levels increased (Fig. 2E, lane 2) above levels in the control (Fig. 2E, lane 1). Finally, to further probe the dependence of NICD4 levels on Mdm2, a series of H1299 cells were transfected with a constant amount of plasmid encoding NICD4 and increasing amounts of a plasmid encoding Mdm2. A clear dose response was observed, where NICD4 levels decreased with increasing levels of Mdm2 expression (Fig. 2F; and for densitometric evaluation, see supplementary material Fig. S1) corroborating an antagonistic link between Mdm2 and NICD4. The generality of this conclusion was corroborated by carrying out an analogous experiment with another cell line, 293T/17, in which we could also demonstrate the antagonism between Notch 4 and Mdm2 (data not shown).
Given that the antagonistic relationship between Mdm2 and NICD4 depends on the presence of the RING domain in Mdm2, we examined NICD4 ubiquitylation in 293T/17 cells overexpressing either wild-type Mdm2 or Mdm2ΔR (Fig. 3A). Cells expressing wild-type Mdm2 and treated with the MG132 proteasome inhibitor displayed robust NICD4 ubiquitylation (Fig. 3A, lane 3), whereas cells expressing the Mdm2ΔR protein did not show an appreciable increase in ubiquitylated NICD4 above background levels (Fig. 3A, compare lanes 2 and 4). We also examined the effect of overexpressing Sel-10 (also known as FBXW7), another E3 ubiquitin ligase reported to target NICD4 (Wu et al., 2001). Cells expressing Sel-10 also displayed, as expected, enhanced NICD4 ubiquitylation, albeit to an apparently lower degree, than cells expressing Mdm2 (Fig. 3A, lane 5).
Fig. 3.

Mdm2 targets NICD4 for ubiquitylation. (A) 293T/17 cells expressing HA-NICD4, ubiquitin and the indicated wild-type or mutant E3 ligases were treated with 30 μM MG-132 for 5 hours. NICD4 was immunoprecipitated from cell lysates with an anti-HA antibody, and immunoprecipitates were analyzed by western blot with anti-ubiquitin (top panel) and anti-HA (NICD4) antibodies (middle panel). Mdm2 expression was monitored by western blot of whole cell lysates using an anti-MDM2 antibody (lower panel). (B) 293T/17 cells expressing HA-NICD4, Myc-tagged ubiquitin and the indicated wild-type Mdm2 (lane 2) or the mutant Mdm2 E3 ligases (lane 3) were treated with 30 μM MG-132 overnight. Myc-tagged ubiquitin was immunoprecipitated from cell lysates with an anti-Myc antibody, and immunoprecipitates were analyzed by western blot with anti-HA (NICD4) antibodies (top panel). Mdm2 and Mdm2ΔR expression were monitored by western blot of whole-cell lysates using an anti-Mdm2 antibody (lower panel).
In a complementary strategy to monitor ubiquitylation of NICD4, we expressed HA-tagged NICD4 and Myc-tagged ubiquitin along with Mdm2 or Mdm2ΔR and treated cells with MG132 overnight. After immunoprecipitation with an anti-Myc antibody, immunoblotting was performed to detect HA–NICD4 expression. The result showed increased levels of higher molecular mass NICD4 in Mdm2-transfected cells (Fig. 3B, lane 2) compared with Mdm2ΔR-transfected cells (Fig. 3B, lane 3). On the basis of this analysis, we conclude that NICD4 is a substrate for Mdm2-mediated ubiquitylation and degradation.
Trp53 attenuates Notch 4 signaling
Because Trp53 controls the levels of Mdm2 protein, which can target NICD4, we expected that modulation of Trp53 should be consequential for Notch 4 signaling. To explore this possibility, we used a Notch signal reporter, HES-1–luc (Takebayashi et al., 1994), to monitor the effects of Trp53 on Notch-dependent transcription in H1299 cells, which are null for TP53, and in HC11 cells, in which both alleles of endogenous Trp53 are mutant (hypomorphic) (Merlo et al., 1994). As shown in Fig. 4A,B NICD4 expression significantly stimulated the activity of the HES-1–luc reporter transfected into both cell lines. This effect was suppressed when NICD4 was co-expressed with wild-type Trp53. Consistently, when parental wild-type HCT116 cells were treated with the DNA-damaging agent cisplatin, which upregulates Trp53 (Vikhanskaya et al., 1999) (Fig. 4C, bottom panel), we observed downregulation of levels of HES1 (an endogenous Notch target) mRNA as revealed by RT-PCR (Fig. 4C, top panel, lane 2 vs lane 1). This effect was not seen in the HCT116 TP53-null cells (Fig. 4C, top panel, lanes 3 and 4).
Fig. 4.
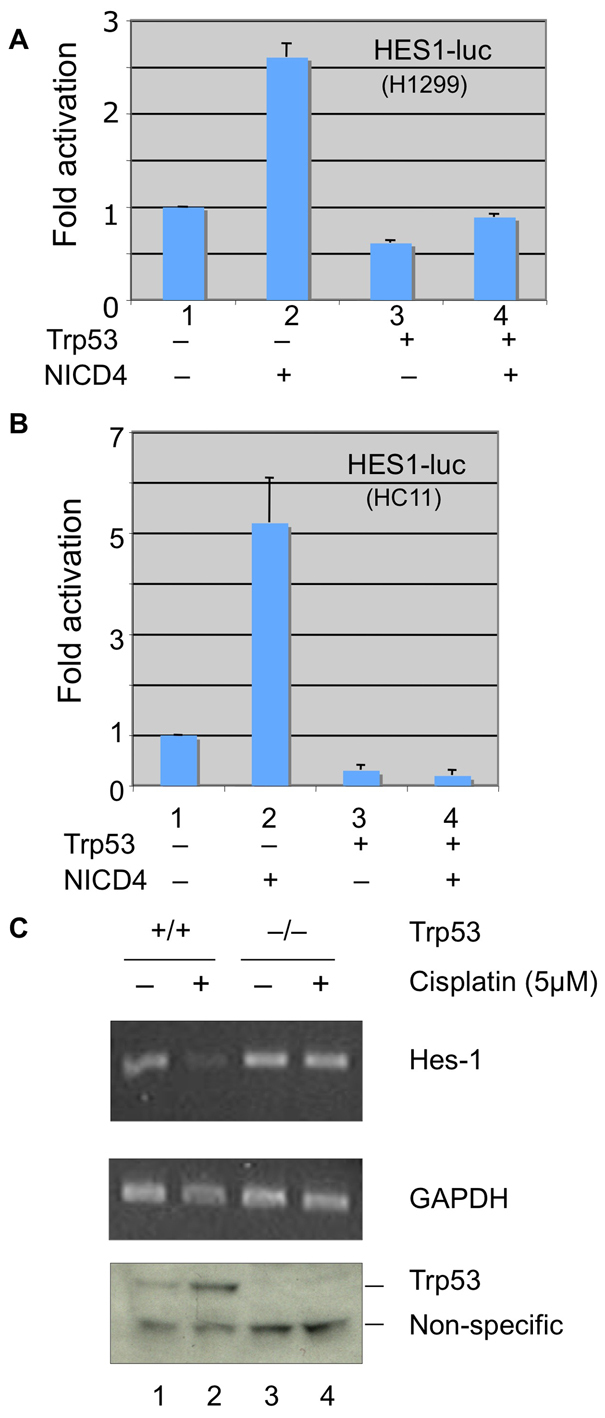
Trp53 modulates NICD4 signaling. (A) Luciferase assays in H1299 cells (TP53 null) expressing the HES-1–luc promoter reporter (0.5 μg) and the control Renilla luciferase reporter (0.05 μg) were either mock transfected (lane 1) or transfected with expression constructs encoding NICD4 alone (lane 2), Trp53 alone (lane 3) or both. Lane 4 shows reduced NICD4 activity when both NICD4 and Trp53 are expressed (compare lanes 2 and 4). Luciferase assays were performed according to the manufacturer's (Promega) protocol, as previously described (Sun et al., 2005a). (B) Luciferase assays of HC11 (Trp53 mutant) cells containing the HES-1–luc Notch signal reporter (Takebayashi et al., 1994). Cells were mock transfected (lane 1) or transfected with expression constructs encoding either NICD4 (lane 2) or Trp53 (lane 3) alone or both (lane 4). (C) RT-PCR assays of HES1 mRNA levels in HCT116 TP53 wild-type cells (lanes 1 and 2) and HCT116 TP53-null cells (lanes 3 and 4), either treated with cisplatin (5 μM) (lanes 2 and 4) for 5 hours, or untreated (lanes 1 and 3). Lower panel is a western blot showing the effects of cisplatin on Trp53 protein levels monitored with an anti-Trp53 antibody (Vikhanskaya et al., 1999) which, in addition to Trp53, crossreacts with another nonspecific protein (lower band), which was used as a loading control.
These observations were extended with a second Notch reporter assay in HCT116 cells. We determined whether Trp53 modulation affects Notch-induced expression of the viral TP1 promoter, using a TP1–luc reporter assay (Strobl et al., 1997). In HCT116 TP53-null cells, NICD4 activated the TP1–luc reporter as expected (supplementary material Fig. S2A); however, TP1–luc reporter activity was reduced to 29% when Trp53 was coexpressed (supplementary material Fig. S2A). Significantly, and consistent with the analysis described above, we found that Mdm2 expression reduced Notch-induced TP1 reporter activity by 42% (supplementary material Fig. S2B). Taken together, these results indicate that modulation of Trp53 attenuates Notch 4 signaling through Mdm2 and that this property is general, because it is observed across different cell lines.
NICD4 physically interacts with Trp53 and Mdm2
To gain further insight into the antagonistic relationship between NICD4, Trp53 and Mdm2, we sought to examine the possibility that these proteins physically interact, as is known to be the case between Trp53 and Mdm2 (Barak and Oren, 1992; Chen et al., 1993; Momand et al., 1992). We first examined the relationship between Trp53 and NICD4 and included in these experiments NICD1, which has been previously reported to interact with Trp53 (Kim et al., 2007), as well as NICD3. HA-tagged forms of the intracellular domains of these Notch receptors were either independently expressed or co-expressed with FLAG-tagged Trp53 in 293T/17 cells (Fig. 5A). Forty-eight hours after transfection, cells were treated with MG132 for an additional 5 hours and co-immunoprecipitation assays were performed using an anti-FLAG antibody. We note that treatment with MG132 was crucial for these experiments. As judged by co-immunoprecipitation, interactions were observed between Trp53 and all three Notch receptors (Fig. 5A) after treatment with MG132. We examined whether the Trp53–NICD4 interaction can be corroborated by assessing endogenous protein interactions and, indeed, we detected NICD4 in immunoprecipitates using an anti-Trp53 antibody (Fig. 5B) in NIH3T3 cells.
Fig. 5.

Protein interactions between Trp53, Mdm2 and NICD. (A) Co-immunoprecipitation of Trp53 with different NICD paralogs. 293T/17 cells were transfected with plasmids encoding HA-tagged NICD1 (lanes 2 and 3), NICD3 (lanes 4 and 6) or NICD4 (lanes 5 and 7) either alone (lanes 3, 6, and 7) or with Trp53 (lanes 1, 2, 4 and 5). Forty-eight hours after transfection, cells were treated with 30 μM MG-132 for 5 hours. Co-immunoprecipitations were performed on cell lysates with an anti-FLAG antibody (Trp53), and protein interactions were monitored by western blots using anti-HA and anti-FLAG (Trp53) antibodies (upper two panels). Relative levels of protein expression were monitored by western blot of cell lysates (lower two panels). (B) Western blot analysis of endogenous NICD4–Trp53 interaction in NIH3T3 cells treated with 30 μM MG-132 for 5 hours. Immunoprecipitations were performed with an anti-Trp53 antibody (Trp53) (lane 3) or control serum (lane 2), and proteins were detected by western blot using the indicated antibodies. (C) Interaction between NICD4 and Mdm2 Lysates from H1299 cells (TP53-null) treated with 30 μM MG-132 for 5 hours were immunoprecipitated with an anti-Mdm2 antibody or control serum. NICD4 and Mdm2 coimmunoprecipitation was assessed by western blot using antibodies against Mdm2 and NICD4 (see also supplementary material Fig. S1).
To map the region(s) of NICD4 that mediate interaction with Trp53, we generated a series of deletion constructs (supplementary material Fig. S3A). These HA-tagged polypeptides were expressed in 293T/17 cells with or without FLAG-tagged Trp53, and interactions were monitored by co-immunoprecipitation. We found that both the Ankyrin repeats and RAM domains of NICD4 appear to be involved in the Trp53 interaction, whereas the C-terminal PEST-containing domain does not show detectable interactions (supplementary material Fig. S3B).
We also explored the possibility that Mdm2 and NICD4 could physically interact. Immunoprecipitation experiments using extracts of TP53-null H1299 cells, which express endogenously both NICD4 and Mdm2, revealed interactions between these two proteins (Fig. 5C, lane 3). These findings are consistent with the notion that NICD4 is a direct target of the Mdm2 E3 ligase. Using the Notch 4 deletion mutants (supplementary material Fig. S3A), we examined which domain of Notch 4 is responsible for the interaction between NICD4 and Mdm2. The relevant deletion constructs were expressed in 293T/17 cells with or without Mdm2 coexpression (supplementary material Fig. S3C). Interactions were detected with full-length NICD4 (construct F, supplementary material Fig. S3A) and the C-terminal region (construct C, supplementary material Fig. S3A) but not with the NICD4 deletion construct N, which contains the RAM-Ankyrin domains, but lacks the C-terminal region, in contrast to the NICD4–Trp53 interactions. A reciprocal experiment was performed using the wild-type Mdm2 or the Mdm2ΔR deletion, which lacks the RING domain. These constructs were co-transfected with a V5-tagged NICD4 in 293T/17 cells. After 48 hours, the cells were treated with MG132 for 5 hours and the lysates were immunoprecipitated with antibodies against the V5 NICD4 tag. Both Mdm2 constructs displayed interactions with NICD4, suggesting that the interaction is independent of the RING domain (supplementary material Fig. S3D).
Trp53 suppresses NICD4-induced anchorage-independent growth in mammary epithelial cells
The observations described above clearly indicate a mechanistic link between Notch 4 and Trp53 and suggest that this Mdm2-dependent relationship could be relevant in tumorigenesis. To explore this possibility, we took advantage of an assay for anchorage-independent growth in soft agar, using HC11 and C57MG mammary epithelial cells that express NICD4 (Robbins et al., 1992). Both alleles of Trp53 in HC11 cells are mutated (Merlo et al., 1994), whereas in C57MG cells both alleles are wild type (our unpublished data).
Introduction of a NICD4 expression vector into the HC11 (Robbins et al., 1992) and C57MG cell lines confers on them the ability to form colonies in soft agar (Fig. 6A,B). This is consistent with the well-documented role of NICD4 as an oncogene in the mouse mammary gland (Jhappan et al., 1992). Significantly, however, the oncogenic activity of NICD4, as revealed by the soft agar assay, was reduced by the cadmium (Cd2+)-induced expression (Merlo et al., 1994) of wild-type Trp53 (P<0.001) in HC11-NICD4 cells (Fig. 6A) consistent with the notion that Trp53 regulates the protein level of NICD4. Similarly, C57MG-NICD4 cells, in which Trp53 is not mutated but is only weakly expressed, colony formation was inhibited by the expression of exogenous wild-type Trp53 (Fig. 6B). The ability of Trp53 to block oncogene-related colony formation in HC11 cells is not universal. When we introduced the Cd2+-inducible wild-type Trp53 retroviral vector into HC11 cells stably expressing the FGF-3, ErbB2 or Wnt1 oncogenes, anchorage-independent growth by these cells in soft agar was not blocked (Fig. 6C). Thus Trp53 is able to suppress the tumorigenic activity of NICD4, as predicted from the aforementioned analysis, but not that of FGF-3, Wnt1 or ErbB2.
Fig. 6.

Trp53 suppresses NICD4-induced anchorage-independent growth of mammary epithelial cells in soft agar. Representative colony formation assay for (A) HC11, HC11-NICD4 and (B) C57MG, C57MG-NICD4 cell pools. Wild-type Trp53 was introduced into the cell lines using the inducible retroviral vector 1529-neo containing the metallothionine promoter sensitive to cadmium ions. Growth medium was supplemented with cadmium ions (cd) to induce Trp53 expression (v is empty vector control). The original magnification was 40×. Numbers of counted colonies are shown in the graphs. (C) Representative colony formation assay for HC11 cells stably overexpressing oncogenes FGF-3, ErbB2 and Wnt1. Wild-type Trp53 was introduced into cell lines through transient transfection with wt Trp53 vector. Soft agar assays were performed 48 hours after transfection. Cells were placed in six-well plates in 0.3% soft agar at a density of 30,000 cells/well. After 21 days, they were visualized by overnight staining with Nitroblue Tetrazolium and counted for size ≥0.2 mm.
Trp53 status associated with NICD4 mouse mammary tumors
To examine the status of Trp53 locus in NICD4 mammary tumors, we determined the nucleotide sequence of exons 3–7 of the Trp53 gene from mouse mammary tumors induced by NICD4 driven by the whey acidic protein (WAP) (Michelsen et al., 2007) or the MMTV LTR promoters. The Trp53 status was initially examined using SSCP (single-strand conformation polymorphisms) analysis. Using this technique, a single nucleotide substitution in DNA can be detected as a mobility shift during gel electrophoresis (indicated with arrows in Fig. 7A). The nucleotide sequences of amplified products derived from the Trp53 exons 3–7 from the WAP-NICD4 and MMTV-NICD4 mammary tumors exhibiting different mobilities were determined. We found a remarkable frequency of Trp53 mutations in the NICD4 tumors (41%, 6/14) and NICD1 (14%, 1/7) in comparison with that in tumors generated by the expression of Cripto-1 (0%, 0/4) or Int6 (0%, 0/7) oncogenes (Fig. 7B). The characteristics of these mutations are shown in Fig. 7C. These results clearly show an unexpectedly high frequency of Trp53 mutations in the NICD4 tumors, strengthening the relevance of a crucial functional link between Notch 4 and Trp53 in oncogenesis.
Fig. 7.

Trp53 status associated with NICD4 mammary tumors. (A) Representative examples of SSCP polymorphism. Exons 3–7 of Trp53 were amplified by PCR, separated by 10% TBE polyacrylamide gel electrophoresis and silver stained. Differences of mobility shift were observed among the PCR products of samples 2 and 3 obtained from WAP-Int3 (NICD4) transgenic tumors (indicated with arrows). (B) Frequencies and (C) characteristics of mutations found in all analyzed transgenic models. The mutations of Trp53 DNA binding domain status were checked in several transgenic models: WAP–Int3 (NICD4) (Gallahan and Callahan, 1987), MMTV LTR-Int3 (NICD4) (Jhappan et al., 1992), Wap-Int3sh (Wap-Int3short) (Raafat et al., 2004), Wap-CBF1 KO (Wap-NICD4/CBF1 knockout) (Raafat et al., 2009) Wap-Cripto-1 (Sun et al., 2005b), Wap-Int6 (Mack et al., 2007) and MMTV-NICD1 (Kiaris et al., 2004). PCR products showing different SSCP patterns were purified by electrophoresis on 1% agarose gel, cloned into TA-cloning system or directly sequenced.
Discussion
The Notch pathway is one of a handful of fundamental signaling mechanisms controlling metazoan cell fate (Gerhart, 1999; Hurlbut et al., 2007). The fundamental nature of the pathway is reflected by the striking conservation across species, as well as the pleiotropic action across tissues and developmental processes. Molecular evidence indicates that, in spite of the numerous effects of Notch activity, there is a unitary, basic underlying molecular mechanism that governs Notch signaling. However, genetic studies have uncovered a complex circuitry that is capable of modulating Notch activity and a diversity of mechanisms that can ultimately control signaling at distinct cellular levels (Kankel et al., 2007).
An important and unique aspect of the Notch pathway is its exquisite sensitivity to dosage, first revealed by the haplo-insufficient genetic behavior of the Notch receptor and its ligands (Artavanis-Tsakonas et al., 1995; Artavanis-Tsakonas et al., 1999). Moreover, the developmental outcome of Notch signals might differ, depending on the level of receptor activity, such that the quantity of ligand-competent receptors in a cell defines a crucial developmental parameter. Not surprisingly, therefore, mechanisms that affect the trafficking and the stability of the Notch receptor have emerged as pivotal signal-controlling devices (Baron et al., 2002; Fortini, 2009) and might also serve as integration nodes between Notch and other cellular processes (Mukherjee et al., 2005).
Here, we uncovered a molecular mechanism linking Notch protein stability to oncogenic events driven by the tumor suppressor Trp53. We demonstrate that the intracellular domain of Notch 4 is targeted for ubiquitylation and hence degradation by the ubiquitin ligase Mdm2. Mdm2, however, is involved in a feedback mechanism with Trp53, being on one hand the transcriptional target of Trp53 and on the other hand, a protein interaction partner (Kussie et al., 1996; Schon et al., 2002), eventually targeting the Trp53 protein for degradation through ubiquitylation (Fang et al., 2000; Haupt et al., 1997; Honda and Yasuda, 2000). The data we gathered indicate that although the Mdm2 E3 ligase can target Notch 4 in the absence of Trp53, its presence seems to significantly accelerate this process, given that we found higher levels of NICD4 in Trp53−/− Mdm2−/− MEFs than in Trp53−/− Mdm2+/+ MEFs. We thus propose a model where the Trp53–Mdm2 feedback loop is linked to the regulation of Notch protein levels (Fig. 8). Moreover, given that our biochemical analyses support the existence of physical interactions between NICD4 and both Mdm2 and Trp53, our model suggests that a tripartite interaction between Notch, Mdm2 and Trp53 modulates the degradation of NICD4 and hence NICD4-mediated signaling (Fig. 8).
Fig. 8.

A model for the Trp53-mediated downregulation of Notch signaling. The Mdm2 E3 ubiquitin ligase is a transcriptional target of Trp53. The Trp53 protein associates with and is ubiquitylated (red dots) by Mdm2 (Fang et al., 2000; Honda and Yasuda, 2000). Mdm2 can also associate with the intracellular domain of Notch 4 (NICD4) and cause its ubiquitylation (dashed line). However, in the presence of Trp53, which can also associate with NICD4, a trimeric complex (solid bold lines) is formed (NICD–Trp53–Mdm2), and as a result NICD4 becomes highly ubiquitylated and, subsequently, rapidly degraded. This results in a loss of Notch signaling and might provide a selective environment for fixation of Trp53 mutations during the evolution of mammary tumor development in the Notch transgenic mammary tumor models.
The inverse correlation between Trp53 expression and Notch 4 protein levels was, to our knowledge, first documented by Mao and co-workers using fibroblasts lacking Trp53 (Mao et al., 2004). Our biochemical analysis indicating physical interactions not only between NICD4 and Trp53 but also with the intracellular domain of other Notch receptor paralogs raises the possibility that the Trp53 and, by extension, Mdm2-directed degradation is a general mechanism that controls Notch signalling, at least in certain developmental contexts. Indeed, the general features of the mechanism for the antagonistic relationship between Notch and Trp53 we propose is compatible and, in some cases, explains a diverse set of observations that have suggested links between Notch and Trp53 over the years. Kim and co-workers (Kim et al., 2007) have previously shown that the N-terminal region of Trp53 binds to the RAM-Ank domain of NICD1. Beverly and colleagues observed suppression of Trp53 levels by NICD1 and suggested that this might reflect Mdm2-dependent events (Beverly et al., 2005). Secchiero and colleagues (Secchiero et al., 2009) found that, in cells lacking active Trp53, Notch1 protein levels were stable in the presence or absence of the Mdm2 and Trp53 inhibitor Nutlin, consistent with the notion that binding of Trp53 to Notch 1 is important for Mdm2-mediated degradation of Notch1.
Our study indicates that NICD4 physically interacts with the Mdm2 ubiquitin ligase, but the notion of direct interactions between Notch and ubiquitin ligases is not unique, because there have been other documented examples. The C-terminal domain of Notch 1 and Notch 4 have been shown to interact with another nuclear E3 ligase, Sel-10 (FBXW7) (Mao et al., 2004; Wu et al., 2001). Another example is the E3 ligase Deltex in Drosophila, which interacts physically with the Ankyrin domain region of Notch and results in the ubiquitylation of the receptor (Matsuno et al., 1995). This latter example offers some noteworthy analogies with the Trp53–Mdm2–Notch tripartite mechanism we propose here. The efficiency of Deltex to ubiquitylate Notch is highly enhanced through a third molecule, Kurz, which is the single Drosophila homolog of non visual β-arrestin, which interacts physically with deltex, and as a result, influences signaling (Mukherjee et al., 2005). The topology of this trimeric complex is different from what we suggest here between Notch–Trp53 and Mdm2 in that Notch does not seem to interact directly with Kurz. Nevertheless, the notion that sequences in the intracellular domain of Notch act as adaptors to recruit its ubiquitin ligases, whose activity, and hence effects on Notch signaling, might be modulated by a third molecular partner could extend to other cellular circumstances (Mazaleyrat et al., 2003).
Notch signal modulation has profound developmental and pathogenic consequences, and the link with Trp53 is potentially relevant for tumorigenesis given the importance of Trp53 in these events. The ultimate consequences of lower Notch receptor levels is downregulation of the Notch signal, whereas stabilization of the Notch protein might, in the right developmental context, lead to signal upregulation (Mukherjee et al., 2005). Here, in the context of all the cell types we used in our in vitro experiments, we demonstrated that the Trp53–Mdm2-mediated regulation of Notch has direct consequences on Notch signaling, as measured by the expression of the Notch transcriptional target HES-1. Importantly, our study revealed that the Trp53–Mdm2–Notch relationship influences anchorage-independent growth in soft agar, which is a measure of malignant transformation. Thus, the ability of wild-type Trp53 to inhibit anchorage-independent growth of HC11 and C57MG-NICD4 cells in soft agar, but not of HC11 cells expressing Wnt1, FGF-3 and ErbB2, is consistent with a functionally significant relationship between Trp53 and NICD4. This finding has in vivo implications on NICD4-induced mammary tumor development in mice and suggests that an early event in tumorigenesis is the loss or mutation of Trp53.
It is well documented that analogous Trp53 mutations in the solid tumors of mice are far less frequent than in humans (Blackburn and Jerry, 2002). For instance, no Trp53 mutations have been found in MMTV-Ras (Hundley et al., 1997), MMTV-Wnt1 (Donehower et al., 1995) or WAP-DES(1–3) IGF-induced mammary tumors (Hadsell et al., 2000). Remarkably, we found that 41% (6/14) of the mammary tumors in NICD4 transgenic mice, as well as 14% (1/7) of mammary tumors in NICD1 transgenic mice had Trp53 mutations. Although we have not identified any Trp53 mutations in four WAP-Cripto-1 and seven WAP-Int6sh mammary tumors, others have found loss of heterozygosity (LOH) for Trp53 in transgenic MMTV–c-Myc and MMTV-Wnt1 mammary tumors (Blackburn and Jerry, 2002). The mechanism by which LOH for Trp53 synergizes or collaborates with these transgenes has not been established.
The implication our findings have for human mammary oncogenesis is yet to be determined, but the apparent generality of the antagonistic relationship between Notch and Mdm2 or Trp53 raises some significant hypotheses. Moreover, recent evidence that is compatible with our model indicates that expansion of stem or progenitor cells, cell populations that are crucial for carcinogenesis in the mouse mammary gland, might be influenced by Trp53 through its ability to influence Notch activity (Tao et al., 2010). Notch can act as an oncogene, as demonstrated by the association of activating mutations with T-ALL in humans (Ellisen et al., 1991; Weng et al., 2004). However, as we have argued before on the basis of tumor models in the mouse mammary gland and the intestine (Fre et al., 2009; Kiaris et al., 2004), the oncogenic activity of Notch, unlike what has been documented in the case of T-ALL, might not be manifested by oncogenic Notch mutations per se, but rather by an oncogenic synergy of Notch signals with other cellular elements. It is for this reason that the modulation of Notch signaling through the Notch–Mdm2–Trp53 axis defined here might be consequential in human tumorigenesis.
Materials and Methods
Cell culture, retroviral production and transduction
The murine mammary epithelial cell line HC11 was maintained in RPMI-1640 medium, supplemented with 10% fetal bovine serum (FBS), 5 mg/ml insulin, and 10 ng/ml EGF (Invitrogen; Carlsbad, CA). The murine mammary epithelial cell line C57MG (Jue et al., 1992) was grown in DMEM, supplemented with 10% FBS and 10 mg/ml insulin. Phoenix293/Ecotropic and HEK293T/17 cells were purchased from the ATCC (Mannassas, VA) and EcoPack 293T cells were purchased from Clontech (Valencia, CA). These cells were grown in DMEM medium, supplemented with 10% FBS. HCT116 and HCT116 TP53-null cells (kindly supplied by Bert Vogelstein, Johns Hopkins University, Baltimore, MD) were cultured with McCoy's 5a medium with 10% FBS. H1299 cells were purchased from the ATCC (Manassas, VA) and cultured with RPMI-1640 medium, supplemented with 10% FBS. HeLa cells were grown in DMEM, supplemented with 10% FBS. Wild-type, Trp53-null and Mdm2-null MEFs (Montes de Oca Luna et al., 1995) were kindly provided by Guillermina Lozano (University of Texas M.D. Anderson Cancer Center, Houston, TX).
For the production of Trp53 and Int2/FGF3 retroviral constructs, 10 mg of vector DNA were transfected into EcoPack 293T cells using FuGene (Roche Applied Science; Indianapolis, IN). All other retroviral vectors were transfected into Phoenix293/Ecotropic cells using LipoD293 DNA transfection reagent (SignaGen Laboratories; Gaithersburg, MD) according to the manufacturer's suggested procedures. Viral supernatants were collected at 48 and 72 hours after transfection, pooled, filtered (0.45 mm) and stored at −80°C.
Recipient HC11 and C57MG cells were transduced with retroviral particles at approximately 40% confluence for 8–12 hours in the presence of polybrene (5 μg/ml), followed by replacement with fresh medium. Cells were selected with appropriate antibiotics 36 hours after infection. Transgene expression was confirmed by RT-PCR and immunoblotting. All viral work conformed to accepted Biosafety Level 2+ guidelines as described by the National Institutes of Health (http://bmbl.od.nih.gov/contents.htm).
HC11 cells were transfected with 2 mg pCEV29-ErbB2 (gift from S. Aaronson, Mount Sinai School of Medicine, New York, NY) or HA-tagged Wnt-1 (Upstate Biotech; Lake Placid, NY) using FuGene6 (Roche Applied Science) reagent in a 3:1 ratio according to the manufacturer's instructions. Cells were selected with the appropriate antibiotics under standard conditions.
Construction of Trp53 and NICD4 vectors
FLAG-tagged Trp53 (Zhao et al., 2004) obtained from Daiqing Liao (University of Florida, Gainsville, FL) and HA–tagged human NICD1 and human NICD3 from Lizi Wu (Wu et al., 2000), Ha-tagged Mouse NICD4 expression vectors were made through PCR using Pfu polymerase from Stratagene and NICD4 sequences were cloned into pcDNA3 vector (Invitrogen; Carlsbad, CA), NICD4 was also cloned into pcDNA 3.1 pEF1/V5His TOPO TA expression vector (Sun et al., 2005a) (Invitrogen). The 1529-neo retroviral vector (McGeady et al., 1989) was used to introduce the wild-type TP53 cDNA into HC11 and C57MG cells. This vector contains an internal metallothionine (MT) promoter that can be induced by low levels of cadmium sulfate (3 μM). Mouse Trp53 was cloned into the 1529 vector using BamHI and EcoRI restriction sites.
The murine NICD4 cDNA corresponding to a truncated Notch4 cDNA (residues 4382–6043) has been described (Gallahan and Callahan, 1987; Jhappan et al., 1992). An oligonucleotide encoding hemagglutinin (HA) tag was appended to the 3′ end of NICD4 cDNA. The HA-tagged NICD4 expression vector was generated through PCR using Pfu polymerase from Stratagene and cloned into the eukaryotic expression vector pcDNA3. The forward primers, A1: 5′-GGAATTCCGCCACCATGGCAGCAGTGGGAGCTCTGGAGCCCCTGCTGC-3′ was used to generate an EcoRI site and B1: 5′-CCGCTCGAGTCAAGCGTATCTGGAACATCGTATGGGTTCAGAT TTCTTACAACCGAGTTTAAG-3′ to create an XhoI site and an HA tag. pc DNA3 was cut by both EcoRI and XhoI and the ligations were done as described previously (Sun et al., 2005a). The RAM domain (4382-4861) was amplified by the forward primer A1: 5′-GGAATTCCGCCACCATGGCAGCAGTGGGAGCTCTGGAGCCCCTGCTGC-3′ that created a EcoRI site and the reverse primer B2: 5′-CCGCTCGAGTCAAGCGTAATCTGGAACATCGTATGGCAGAACCTCCGATTCACACTCCTGAG-3′, which created an XhoI site as well as HA tag. The Ankyrin repeat domain (4862–5512) was amplified by forward primer A2: 5′-CGCGGATCCGCCACCATGGATGTGGACACCTGTGGACCTGATGGGGTGACACCCCTGATGTC-3′, which created a EcoRI site and the reverse primer B3: 5′-CCGCTCGAGTCAAGCGTAATCTGGAACATCGTATGGGCCGCCCCGAGCTCCAGCAACAGCTG-3′, which created an XhoI site as well as HA tag. The RAM plus Ankyrin repeats domain (4382–5512) (RAM_Ank) was amplified using primer A1 and primer B3. The C-terminal domain (5513–6043) was amplified using the forward primer A3: 5′-GGAATTCCGCCACCATGCCGGGGACTGCGAGACCAGGCCGGGCTGGCCCCAGGAGATGTGG-3′, which created a EcoRI site and the reverse primer B1. All NICD4 and its deletion plasmids have sequenced and verified. Mdm2 and Mdm2 RING deletion (ΔR) vectors were obtained from Carl G. Maki (University of Chicago).
Protein extraction, antibodies and immunoprecipitation
Cells were lysed as previously described (Sun et al., 2005b). The antibodies used were: anti-V5 (Invitrogen; 1:5000), anti-FLAG-HRP (Sigma; 1:1000), anti-Notch 4 are from Upstate (Charlottesville, VA; 1:1000) and AVIVA System Biology (San Diego, CA; 1:2000), anti-HA (Roche), anti-Mdm2 (Santa Cruz, N-20 and SMP14), anti-α-HSP70, anti-NICD4 (AVIVA, 1:4000), anti-Trp53 protein (CM5) (Vector Laboratories).
For the Notch and Trp53 interaction analysis, 293T/17 cells (1×106 cells on 60-mm-diameter plates) were transfected with 2 μg HA–NICD1, HA–NICD3, HA–NICD4 expression vectors with, or without, 2 μg FLAG–Trp53. Alternatively, for the Notch–MDM2 interaction analysis, the cells were transfected with 2 μg HA-NICD4 mutant expression vectors with or without the addition of 2 μg Mdm2 expression vector. Tranfections were performed using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer's instruction. Seventy-two hours after transfection, the cells were lysed for 20 minutes with 0.5 ml lysis buffer as previously described (Sun et al., 2005a). Before lysis, the cells were treated for 5 hours with 30 μM MG-132. Immunoprecipitations were carried out as previously described (Sun et al., 2005a) by incubating 1.0 mg of total protein with 30 μl anti-FLAG antibody conjugated to resin beads (Aguila et al., 2007) overnight at 4°C. The resin was washed four times with 0.5 ml lysis buffer and eluted with high concentration of FLAG peptide according to the procedure from Sigma. Immunoprecipitates were visualized by western blotting with anti-HA (Roche) or anti-FLAG horseradish peroxidase-conjugated antibody (Aguila et al., 2007).
The antibodies against Trp53 (Santa Cruz, sc-99 AC) and Mdm2 (Santa Cruz, sc-965 AC) and control mouse serum, conjugated to agarose beads (Santa Cruz, sc-2343) were used in immunoprecipitations involving NIH3T3 and H1299 cell lysates.
Western blots
Cells were harvested in RIPA buffer (150 mM NaCl, 10 mM Tris-HCl, pH 7.2, 0.1% SDS, 1.0% Triton X-100, 1% sodium deoxycholate, 5 mM EDTA) supplemented with the protease inhibitor cocktail set 1 (Calbiochem). Total protein concentration was determined with the BCA Protein Assay Kit (Pierce Biotechnology). Protein (50 μg total) was loaded on gradient 4–20% SDS-PAGE gels and transferred to nitrocellulose membrane using the iBlot Transfer System (Invitrogen). Membranes were blocked with blocking buffer (5% non-fat milk in TBS with 0.1% Tween20 (Tris-buffered saline Tween-20, TBS/T) for 1 hour at room temperature and then incubated with primary α-tubulin (Aguila et al., 2007) antibodies. After washing with TBS/T (Tris-Buffered Saline tween-20) membranes were incubated for 1 hour at room temperature with the appropriate secondary antibody conjugated to HRP, diluted in TBS/T. Proteins were visualized using the ECL Western Blotting Detection System (Amersham, GE Healthcare).
In vivo ubiquitylation assay
In vivo ubiquitylation assays were carried out essentially as described (Wu et al., 2001; Yang et al., 2005) with minor modifications. Briefly, 293T/17 cells or Bosc23 cells were transfected with 2 μg HA-NICD4, 1 μg ubiquitin and 2 μg indicated wild-type or mutant Mdm2 E3 ligases or Sel-10 E3 ligase expression vectors, for 48 hours, using Lipofectamine 2000 and then treated with MG-132 for 5 hours before harvesting. NICD4 was immunoprecipitated from cell lysates with an anti-HA antibody, and the immunoprecipitates were analyzed by western blot with anti-ubiquitin (Yang et al., 2005).
Quantitative real-time PCR
Quantitative real-time polymerase chain reaction (qRT-PCR) was performed using the SYBR Green Master Mix (Bio-Rad Laboratories). The following primers were used for qRT-PCR: Notch 4 Forward primer; 5′ AGT CCA GGC CTT GCC AGA ACG-3′; Notch 4 Reverse primer; 5′ GTA GAA GGC ATT GGC CAG AGA G-3′; Mdm2-Forward primer; GCA AAT GTG CAA TAC CAA CAT GTC; Mdm2-Reverse primer; 5′-GCC AAA CAA ATC TCC TAG AAG ATC-3′; HPRT Forward primer; 5′-GACACTGGCAAAACAATGCAGAC-3′ and HPRT Reverse primer; 5′-CAGTTTCACTAATGACACATTCATG-3′. Normalization of NICD4 and Mdm2 mRNA levels to HPRT were done according to the manufacturer's protocol. Mdm2 vectors were purchased from Open Biosystem. Lentivirus was produced following the manufacturer's protocol and viral infections were carried out according to Sun et al. (Sun et al., 2005a).
PCR, SSCP, and Trp53 nucleotide sequence analysis
The primers for Exon 3 are F, 5′-CCATCACCTCACTGCATGGACGAT-3′ and R, 5′-CGTGCACATAACAGACTTGGCTG-3′; exon 4 F, 5′-TACTCTCCTCCCCTCAATAAGC-3′ and R, 5′-CATCACCATCGGAGCAGCGCTC-3′; exon 5 F, 5′-GCCTGGCTCCTCCCCAGCATCTTATC-3′ and R, 5′-CTCGGGTGGCTCATAAGGTACCACC-3′; exons 6 and 7 F, 5′-CTCTCTTTGCGCTCCCTGGGGGC-3′ and R, 5′-GCCGGCTCTGAGTATACCACCATCC-3′. Amplified products were screened for sequence variations using single strand conformation polymorphism (SSCP) analysis (Merlo et al., 1994). The PCR product was heat-denatured at 95°C for 5 minutes and immediately placed on ice until it was loaded onto the gel. The denatured products were resolved in 10% polyacrylamide gels. Gels were subsequently fixed and stained with silver nitrate and photographed. Direct sequencing was done on selected samples as follows: after PCR amplification, products were purified by electrophoresis on 1% agarose gel. The DNA fragment was excised from the gel, purified with an Extraction Kit (QIAquick Spin; Qiagen, Hilden, Germany), and ligated into a plasmid TOPO TA Cloning (Invitrogen, Carlsbad, CA). The constructs were transformed into Escherichia coli strain DH5alpha and plated. Plasmid DNA from single cell clones was isolated using EcoRI to assay for an insert. The clones to be sequenced were purified with QIAprep Miniprep (Qiagen). The sequence was obtained using one of the original primers in a sequencing reaction performed on an ABI PRISM 310 automated DNA sequencer with the BigDye terminator sequencing kit (Applied Biosystems, Foster City, CA). Sequences were compared with the GenBank sequence number U94788 for the mouse Trp53 gene.
The Trp53 DNA binding domain status was checked in cellular DNA from mammary tumors of several transgenic models: WAP-Int3 (Gallahan and Callahan, 1987), MMTV LTR-Int3 (Jhappan et al., 1992), WAP-Int3sh (WAP-Int3short) (Raafat et al., 2004), WAP-CBF1 KO (Wap-NICD4/CBF1 knock out) (Raafat et al., 2009), WAP-Cripto-1 (Sun et al., 2005b), WAP-Int6 (Mack et al., 2007) and MMTV-NICD1 (Kiaris et al., 2004).
RNA isolation, cDNA synthesis and RT-PCR
RNA isolation, cDNA synthesis and RT-PCR were performed as previously described (Sun et al., 2005a; Sun et al., 2002). Briefly, total cellular RNA was isolated using TRIzol (Invitrogen, Carlsbad, CA). cDNA was synthesized from 1 mg total RNA, after treatment with DNaseI (Invitrogen), using oligo(dT)12-15 primers and the SuperScript II kit, according to the manufacturer's protocol (Invitrogen, Carlsbad, CA). Primers used were GAPDH, 5′-CCCTTCATTGACCTCAACTAC-3′ and 5′-CCACCTTCTTGATGTCATCAT-3′, and HES-1, 5′-ATGAGATCAGTACTGCGGATGCCATCT-3′ and 5′-GCACAGAGACGGTGCTGCCATCAGACT-3′ (Accession Number, M16006)
Colony formation in soft agar
The soft agar assay was performed as described previously (Raafat et al., 2007). Briefly, tissue culture cells in an agar mixture were seeded at 1.5×104 or 3×104 cells/well in triplicate. The plates were incubated at 37°C with 5% CO2 for 21 days. Colonies measuring 0.2 μm or more were counted using the AccuCount 1000 colony counter (Biologics, Inc., Manassas, VA, USA). For Trp53 induction 3 μM CdSO4 was added to growth media.
Statistics
Quantitative values are represented as the mean value of at least three repeats of one representative experiment. All experiments were repeated at least three times. The statistical significance of the difference between groups was determined by the Student t-test. Comparisons resulting in P<0.05 were considered statistically significant and identified in the figures with an asterisk.
Acknowledgments
We thank our colleagues R. A. Obar, A. Mukherjee, K. G. Guruharsha and A. Louvi (Yale University) for their help. We also thank Guillermina Lozano for providing Trp53-null, Mdm2-null MEFs and wild-type MEFs; Bert Vogelstein for supplying HCT116 TP53-null and parental cells; S. Aaronson for crucial reagents and Daiqing Liao for pcDNA3 FLAG–Trp53 expression vectors; Lizi Wu for pcDNA3 HA–NICD1 and pcDNA3 HA–NICD3; and Carl G. Maki for pcDNA3 Mdm2 and pcDNA3 Mdm2ΔR vectors. This research was supported in part by the National Institutes of Health Grants NS26084 and CA098402 (to S.A.-T.) and partly by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. The authors declare no competing interests. Deposited in PMC for release after 12 months.
Footnotes
Supplementary material available online at http://jcs.biologists.org/cgi/content/full/124/7/1067/DC1
References
- Aguila B., Coulbault L., Boulouard M., Liotaveilliota F., Davis A., Tsigmath G., Borsodi A., Balboni G., Salvadori S., Jauzac P., et al. (2007). In vitro and in vivo pharmacological profile of UFP-512, a novel selective delta-opioid receptor agonist; correlations between desensitization and tolerance. Br. J. Pharmacol. 152, 1312-1324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allenspach E. J., Maillard I., Aster J. C., Pear W. S. (2002). Notch signaling in cancer. Cancer Biol. Ther. 1, 466-476 [DOI] [PubMed] [Google Scholar]
- Artavanis-Tsakonas S., Matsuno K., Fortini M. E. (1995). Notch signaling. Science 268, 225-232 [DOI] [PubMed] [Google Scholar]
- Artavanis-Tsakonas S., Rand M. D., Lake R. J. (1999). Notch signaling: cell fate control and signal integration in development. Science 284, 770-776 [DOI] [PubMed] [Google Scholar]
- Barak Y., Oren M. (1992). Enhanced binding of a 95 kDa protein to p53 in cells undergoing p53-mediated growth arrest. EMBO J. 11, 2115-2121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron M., Aslam H., Flasza M., Fostier M., Higgs J. E., Mazaleyrat S. L., Wilkin M. B. (2002). Multiple levels of Notch signal regulation (review). Mol. Membr. Biol. 19, 27-38 [DOI] [PubMed] [Google Scholar]
- Beverly L. J., Felsher D. W., Capobianco A. J. (2005). Suppression of p53 by Notch in lymphomagenesis: implications for initiation and regression. Cancer Res. 65, 7159-7168 [DOI] [PubMed] [Google Scholar]
- Blackburn A. C., Jerry D. J. (2002). Knockout and transgenic mice of Trp53: what have we learned about p53 in breast cancer? Breast Cancer Res. 4, 101-111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunz F., Dutriaux A., Lengauer C., Waldman T., Zhou S., Brown J. P., Sedivy J. M., Kinzler K. W., Vogelstein B. (1998). Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 282, 1497-1501 [DOI] [PubMed] [Google Scholar]
- Callahan R., Egan S. E. (2004). Notch signaling in mammary development and oncogenesis. J. Mammary Gland Biol. Neoplasia 9, 145-163 [DOI] [PubMed] [Google Scholar]
- Capobianco A. J., Zagouras P., Blaumueller C. M., Artavanis-Tsakonas S., Bishop J. M. (1997). Neoplastic transformation by truncated alleles of human NOTCH1/TAN1 and NOTCH2. Mol. Cell. Biol. 17, 6265-6273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J., Marechal V., Levine A. J. (1993). Mapping of the p53 and mdm-2 interaction domains. Mol. Cell. Biol. 13, 4107-4114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donehower L. A., Godley L. A., Aldaz C. M., Pyle R., Shi Y. P., Pinkel D., Gray J., Bradley A., Medina D., Varmus H. E. (1995). Deficiency of p53 accelerates mammary tumorigenesis in Wnt-1 transgenic mice and promotes chromosomal instability. Genes Dev. 9, 882-895 [DOI] [PubMed] [Google Scholar]
- Ellisen L. W., Bird J., West D. C., Soreng A. L., Reynolds T. C., Smith S. D., Sklar J. (1991). TAN-1, the human homolog of the Drosophila notch gene, is broken by chromosomal translocations in T lymphoblastic neoplasms. Cell 66, 649-661 [DOI] [PubMed] [Google Scholar]
- Fang S., Jensen J. P., Ludwig R. L., Vousden K. H., Weissman A. M. (2000). Mdm2 is a RING finger-dependent ubiquitin protein ligase for itself and p53. J. Biol. Chem. 275, 8945-8951 [DOI] [PubMed] [Google Scholar]
- Fortini M. E. (2009). Notch signaling: the core pathway and its posttranslational regulation. Dev. Cell 16, 633-647 [DOI] [PubMed] [Google Scholar]
- Fre S., Pallavi S. K., Huyghe M., Lae M., Janssen K. P., Robine S., Artavanis-Tsakonas S., Louvard D. (2009). Notch and Wnt signals cooperatively control cell proliferation and tumorigenesis in the intestine. Proc. Natl. Acad. Sci. USA 106, 6309-6314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallahan D., Callahan R. (1987). Mammary tumorigenesis in feral mice: identification of a new int locus in mouse mammary tumor virus (Czech II)-induced mammary tumors. J. Virol. 61, 66-74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhart J. (1999). 1998 Warkany lecture: signaling pathways in development. Teratology 60, 226-239 [DOI] [PubMed] [Google Scholar]
- Geyer R. K., Yu Z. K., Maki C. G. (2000). The MDM2 RING-finger domain is required to promote p53 nuclear export. Nat. Cell Biol. 2, 569-573 [DOI] [PubMed] [Google Scholar]
- Gottlieb T. M., Oren M. (1996). p53 in growth control and neoplasia. Biochim. Biophys. Acta 1287, 77-102 [DOI] [PubMed] [Google Scholar]
- Hadsell D. L., Murphy K. L., Bonnette S. G., Reece N., Laucirica R., Rosen J. M. (2000). Cooperative interaction between mutant p53 and des(1-3)IGF-I accelerates mammary tumorigenesis. Oncogene 19, 889-898 [DOI] [PubMed] [Google Scholar]
- Haupt Y., Maya R., Kazaz A., Oren M. (1997). Mdm2 promotes the rapid degradation of p53. Nature 387, 296-299 [DOI] [PubMed] [Google Scholar]
- Honda R., Yasuda H. (2000). Activity of MDM2, a ubiquitin ligase, toward p53 or itself is dependent on the RING finger domain of the ligase. Oncogene 19, 1473-1476 [DOI] [PubMed] [Google Scholar]
- Hundley J. E., Koester S. K., Troyer D. A., Hilsenbeck S. G., Subler M. A., Windle J. J. (1997). Increased tumor proliferation and genomic instability without decreased apoptosis in MMTV-ras mice deficient in p53. Mol. Cell. Biol. 17, 723-731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurlbut G. D., Kankel M. W., Lake R. J., Artavanis-Tsakonas S. (2007). Crossing paths with Notch in the hyper-network. Curr. Opin. Cell Biol. 19, 166-175 [DOI] [PubMed] [Google Scholar]
- Hurlbut G. D., Kankel M. W., Artavanis-Tsakonas S. (2009). Nodal points and complexity of Notch-Ras signal integration. Proc. Natl. Acad. Sci. USA 106, 2218-2223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhappan C., Gallahan D., Stahle C., Chu E., Smith G. H., Merlino G., Callahan R. (1992). Expression of an activated Notch-related int-3 transgene interferes with cell differentiation and induces neoplastic transformation in mammary and salivary glands. Genes Dev. 6, 345-355 [DOI] [PubMed] [Google Scholar]
- Jue S. F., Bradley R. S., Rudnicki J. A., Varmus H. E., Brown A. M. (1992). The mouse Wnt-1 gene can act via a paracrine mechanism in transformation of mammary epithelial cells. Mol. Cell. Biol. 12, 321-328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kankel M. W., Hurlbut G. D., Upadhyay G., Yajnik V., Yedvobnick B., Artavanis-Tsakonas S. (2007). Investigating the genetic circuitry of mastermind in Drosophila, a notch signal effector. Genetics 177, 2493-2505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiaris H., Politi K., Grimm L. M., Szabolcs M., Fisher P., Efstratiadis A., Artavanis-Tsakonas S. (2004). Modulation of notch signaling elicits signature tumorsand inhibits hras1-induced oncogenesis in the mouse mammary epithelium. Am. J. Pathol. 165, 695-705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S. B., Chae G. W., Lee J., Park J., Tak H., Chung J. H., Park T. G., Ahn J. K., Joe C. O. (2007). Activated Notch1 interacts with p53 to inhibit its phosphorylation and transactivation. Cell Death Differ. 14, 982-991 [DOI] [PubMed] [Google Scholar]
- Kopan R., Schroeter E. H., Weintraub H., Nye J. S. (1996). Signal transduction by activated mNotch: importance of proteolytic processing and its regulation by the extracellular domain. Proc. Natl. Acad. Sci. USA 93, 1683-1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kussie P. H., Gorina S., Marechal V., Elenbaas B., Moreau J., Levine A. J., Pavletich N. P. (1996). Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 274, 948-953 [DOI] [PubMed] [Google Scholar]
- Mack D. L., Boulanger C. A., Callahan R., Smith G. H. (2007). Expression of truncated Int6/eIF3e in mammary alveolar epithelium leads to persistent hyperplasia and tumorigenesis. Breast Cancer Res. 9, R42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao J. H., Perez-Losada J., Wu D., Delrosario R., Tsunematsu R., Nakayama K. I., Brown K., Bryson S., Balmain A. (2004). Fbxw7/Cdc4 is a p53-dependent, haploinsufficient tumour suppressor gene. Nature 432, 775-779 [DOI] [PubMed] [Google Scholar]
- Matsuno K., Diederich R. J., Go M. J., Blaumueller C. M., Artavanis-Tsakonas S. (1995). Deltex acts as a positive regulator of Notch signaling through interactions with the Notch ankyrin repeats. Development 121, 2633-2644 [DOI] [PubMed] [Google Scholar]
- Mazaleyrat S. L., Fostier M., Wilkin M. B., Aslam H., Evans D. A., Cornell M., Baron M. (2003). Down-regulation of Notch target gene expression by Suppressor of deltex. Dev. Biol. 255, 363-372 [DOI] [PubMed] [Google Scholar]
- McGeady M. L., Kerby S., Shankar V., Ciardiello F., Salomon D., Seidman M. (1989). Infection with a TGF-alpha retroviral vector transforms normal mouse mammary epithelial cells but not normal rat fibroblasts. Oncogene 4, 1375-1382 [PubMed] [Google Scholar]
- Merlo G. R., Venesio T., Taverna D., Marte B. M., Callahan R., Hynes N. E. (1994). Growth suppression of normal mammary epithelial cells by wild-type p53. Oncogene 9, 443-453 [PubMed] [Google Scholar]
- Michelsen K., Schmid V., Metz J., Heusser K., Liebel U., Schwede T., Spang A., Schwappach B. (2007). Novel cargo-binding site in the {beta} and {delta} subunits of coatomer. J. Cell Biol. 179, 209-217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momand J., Zambetti G. P., Olson D. C., George D., Levine A. J. (1992). The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 69, 1237-1245 [DOI] [PubMed] [Google Scholar]
- Montes de Oca Luna R., Wagner D. S., Lozano G. (1995). Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature 378, 203-206 [DOI] [PubMed] [Google Scholar]
- Mukherjee A., Veraksa A., Bauer A., Rosse C., Camonis J., Artavanis-Tsakonas S. (2005). Regulation of Notch signalling by non-visual beta-arrestin. Nat. Cell Biol. 7, 1191-1201 [DOI] [PubMed] [Google Scholar]
- Picksley S. M., Lane D. P. (1993). The p53-mdm2 autoregulatory feedback loop: a paradigm for the regulation of growth control by p53? BioEssays 15, 689-690 [DOI] [PubMed] [Google Scholar]
- Raafat A., Bargo S., Anver M. R., Callahan R. (2004). Mammary development and tumorigenesis in mice expressing a truncated human Notch4/Int3 intracellular domain (h-Int3sh). Oncogene 23, 9401-9407 [DOI] [PubMed] [Google Scholar]
- Raafat A., Zoltan-Jones A., Strizzi L., Bargo S., Kimura K., Salomon D., Callahan R. (2007). Kit and PDGFR-alpha activities are necessary for Notch4/Int3-induced tumorigenesis. Oncogene 26, 662-672 [DOI] [PubMed] [Google Scholar]
- Raafat A., Lawson S., Bargo S., Klauzinska M., Strizzi L., Goldhar A. S., Buono K., Salomon D., Vonderhaar B. K., Callahan R. (2009). Rbpj conditional knockout reveals distinct functions of Notch4/Int3 in mammary gland development and tumorigenesis. Oncogene 28, 219-230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins J., Blondel B. J., Gallahan D., Callahan R. (1992). Mouse mammary tumor gene int-3: a member of the notch gene family transforms mammary epithelial cells. J. Virol. 66, 2594-2599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schon O., Friedler A., Bycroft M., Freund S. M., Fersht A. R. (2002). Molecular mechanism of the interaction between MDM2 and p53. J. Mol. Biol. 323, 491-501 [DOI] [PubMed] [Google Scholar]
- Secchiero P., Melloni E., di Iasio M. G., Tiribelli M., Rimondi E., Corallini F., Gattei V., Zauli G. (2009). Nutlin-3 up-regulates the expression of Notch1 in both myeloid and lymphoid leukemic cells, as part of a negative feedback antiapoptotic mechanism. Blood 113, 4300-4308 [DOI] [PubMed] [Google Scholar]
- Smith G. H., Gallahan D., Diella F., Jhappan C., Merlino G., Callahan R. (1995). Constitutive expression of a truncated INT3 gene in mouse mammary epithelium impairs differentiation and functional development. Cell Growth Differ. 6, 563-577 [PubMed] [Google Scholar]
- Stifani S., Blaumueller C. M., Redhead N. J., Hill R. E., Artavanis-Tsakonas S. (1992). Human homologs of a Drosophila Enhancer of split gene product define a novel family of nuclear proteins. Nat. Genet. 2, 119-127 [DOI] [PubMed] [Google Scholar]
- Strobl L. J., Hofelmayr H., Stein C., Marschall G., Brielmeier M., Laux G., Bornkamm G. W., Zimber-Strobl U. (1997). Both Epstein-Barr viral nuclear antigen 2 (EBNA2) and activated Notch1 transactivate genes by interacting with the cellular protein RBP-J kappa. Immunobiology 198, 299-306 [DOI] [PubMed] [Google Scholar]
- Sun Y., Nonobe E., Kobayashi Y., Kuraishi T., Aoki F., Yamamoto K., Sakai S. (2002). Characterization and expression of L-amino acid oxidase of mouse milk. J. Biol. Chem. 277, 19080-19086 [DOI] [PubMed] [Google Scholar]
- Sun Y., Lowther W., Kato K., Bianco C., Kenney N., Strizzi L., Raafat D., Hirota M., Khan N. I., Bargo S., et al. (2005a). Notch4 intracellular domain binding to Smad3 and inhibition of the TGF-beta signaling. Oncogene 24, 5365-5374 [DOI] [PubMed] [Google Scholar]
- Sun Y., Strizzi L., Raafat A., Hirota M., Bianco C., Feigenbaum L., Kenney N., Wechselberger C., Callahan R., Salomon D. S. (2005b). Overexpression of human Cripto-1 in transgenic mice delays mammary gland development and differentiation and induces mammary tumorigenesis. Am. J. Pathol. 167, 585-597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takebayashi K., Sasai Y., Sakai Y., Watanabe T., Nakanishi S., Kageyama R. (1994). Structure, chromosomal locus, and promoter analysis of the gene encoding the mouse helix-loop-helix factor HES-1. Negative autoregulation through the multiple N box elements. J. Biol. Chem. 269, 5150-5156 [PubMed] [Google Scholar]
- Talora C., Campese A. F., Bellavia D., Felli M. P., Vacca A., Gulino A., Screpanti I. (2008). Notch signaling and diseases: an evolutionary journey from a simple beginning to complex outcomes. Biochim. Biophys. Acta 1782, 489-497 [DOI] [PubMed] [Google Scholar]
- Tao L., Roberts A. L., Dunphy K. A., Bigelow C., Yan H., Jerry D. J. (2010). Repression of mammary stem/progenitor cells by P53 is mediated by Notch and separable from apoptotic activity. Stem Cells 29, 119-127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vikhanskaya F., Colella G., Valenti M., Parodi S., D'Incalci M., Broggini M. (1999). Cooperation between p53 and hMLH1 in a human colocarcinoma cell line in response to DNA damage. Clin. Cancer Res. 5, 937-941 [PubMed] [Google Scholar]
- Vogelstein B., Lane D., Levine A. J. (2000). Surfing the p53 network. Nature 408, 307-310 [DOI] [PubMed] [Google Scholar]
- Weng A. P., Ferrando A. A., Lee W., Morris J. P. T., Silverman L. B., Sanchez-Irizarry C., Blacklow S. C., Look A. T., Aster J. C. (2004). Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 306, 269-271 [DOI] [PubMed] [Google Scholar]
- Wu G., Lyapina S., Das I., Li J., Gurney M., Pauley A., Chui I., Deshaies R. J., Kitajewski J. (2001). SEL-10 is an inhibitor of notch signaling that targets notch for ubiquitin-mediated protein degradation. Mol. Cell. Biol. 21, 7403-7415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L., Aster J. C., Blacklow S. C., Lake R., Artavanis-Tsakonas S., Griffin J. D. (2000). MAML1, a human homologue of Drosophila mastermind, is a transcriptional co-activator for NOTCH receptors. Nat. Genet. 26, 484-489 [DOI] [PubMed] [Google Scholar]
- Yang Y., Lorick K. L., Jensen J. P., Weissman A. M. (2005). Expression and evaluation of RING finger proteins. Methods Enzymol. 398, 103-112 [DOI] [PubMed] [Google Scholar]
- Zhao L. Y., Liu J., Sidhu G. S., Niu Y., Liu Y., Wang R., Liao D. (2004). Negative regulation of p53 functions by Daxx and the involvement of MDM2. J. Biol. Chem. 279, 50566-50579 [DOI] [PubMed] [Google Scholar]