

Abstract
The duck hepatitis B virus model system was used to elucidate the characteristics of receptor (carboxypeptidase D, gp180) interaction with polypeptides representing the receptor binding site in the preS part of the large viral surface protein. We demonstrate the pivotal role of carboxypeptidase D for virus entry and show its C-domain represents the virus attachment site, which binds preS with extraordinary affinity. Combining results from surface plasmon resonance spectroscopy and two-dimensional NMR analysis we resolved the contribution of preS sequence elements to complex stability and show that receptor binding potentially occurs in two steps. Initially, a short α–helix in the C–terminus of the receptor binding domain facilitates formation of a primary complex. This complex is stabilized sequentially, involving ∼60 most randomly structured amino acids preceding the helix. Thus, hepadnaviruses exhibit a novel mechanism of high affinity receptor interaction by conserving the potential to adapt structure during binding rather than to preserve it per se. We propose that this process represents an alternative strategy to escape immune surveillance and the evolutionary pressure inherent in the compact hepadnaviral genome organization.
Keywords: carboxypeptidase D/gp180/hepatitis B virus/surface plasmon resonance/virus–receptor interaction
Introduction
Identification of virus receptors and characterization of their interaction with the virus is a major goal in molecular virology. So far, for some viruses, e.g. influenza virus, picornaviruses, measles virus or HIV, substantial knowledge has been accumulated, allowing some mechanisms of host recognition by the virus to be understood and, consequently, opening ways for possible therapeutic interventions (Wimmer, 1994). However, there is a lack of knowledge for hepadnaviruses, which are small, enveloped DNA viruses found in mammals and birds, where they cause an acute or chronic liver infection. Regarding the human hepatitis B virus (HBV), chronic infection, estimated to occur in ∼300 million people worldwide, increases the risk of developing hepatocellular carcinoma. Thus, HBV is a major health problem today (Hildt et al., 1996). Despite considerable understanding of the details of hepadnaviral genome replication (Ganem and Varmus, 1987; Nassal and Schaller, 1993, 1996), we lack fundamental insights into the early steps of an HBV infection, particularly viral attachment to cellular receptor(s). This situation reflects the difficulties in sustaining the only currently available in vitro infection system: cultured primary human hepatocytes (Gripon et al., 1993). With respect to this impediment, the duck hepatitis B virus (DHBV) model has gained prominent attention as an experimental system to study the initial steps of the hepadnaviral life cycle (Tuttleman et al., 1986). DHBV infection of primary duck hepatocytes can be inhibited by non-infectious subviral particles (SVP) consisting of only the virus membrane shell with the embedded large (L-) and small (S-) envelope proteins or with recombinant particles containing only the L-protein (Klingmüller and Schaller, 1993). While both viral surface proteins share the hydrophobic S-moiety anchoring them into the membrane, the L-protein additionally has an N–terminal hydrophilic sequence of 161 amino acids, termed preS. Recombinant DHBV-preS (DpreS) from Escherichia coli is sufficient to interfere with infection and therefore essential for receptor recognition. A mutational analysis of DpreS allowed the identification of an extended internal sequence (amino acid residues 30–115) as the receptor binding site of DHBV (Urban et al., 1998).
Following the hypothesis that preS binds a cellular receptor, Kuroki et al. (1994) discovered a glycoprotein of 180 kDa (gp180) that interacts with DHBV particles in a species-specific manner via the preS region of the viral L-protein. They showed that binding was inhibited by neutralizing anti-preS antibodies, providing circumstantial evidence for gp180 as a possible entry factor for DHBV. gp180 turned out to be the prototype of a new class of basic carboxypeptidases (Kuroki et al., 1995; Tong et al., 1995), which have been described later in other species, including man, and are now termed carboxypeptidase D (CPD). CPDs are type I transmembrane proteins consisting of three carboxypeptidase E-like luminal/extracellular domains (called A, B and C), the hydrophobic transmembrane anchor and a highly conserved cytoplasmic tail that is involved in vesicular retrieval (Eng et al., 1999). While the two distal carboxypeptidase-like domains (A and B) are enzymatically active and function in the processing of preproprotein substrates, the evolutionarily conserved C-domain does not possess carboxypeptidase activity, but in the case of duck CPD (duCPD) is required for preS binding (Eng et al., 1998).
gp180/duCPD, being a trans-Golgi network-resident protein that cycles to the plasma membrane and back, fulfills the expectations for a DHBV receptor (Breiner et al., 1998; Breiner and Schaller, 2000). Although attempts to prove this hypothesis directly either by rendering non-permissive cell lines infectable by transfection with duCPD cDNA or by blocking DHBV infection with anti-duCPD antibodies have failed so far, increasing evidence points to CPD as a universal receptor for avian hepadnaviruses. First, transfection of various cell lines with duCPD expression plasmids promotes uptake of DpreS and viral particles (Breiner et al., 1998). Secondly, soluble duCPD (sduCPD) efficiently blocks infection of duck hepatocytes with DHBV (Urban et al., 1999). Thirdly, the receptor binding site of preS, which has been mapped by an infection competition assay (Urban et al., 1998), corresponds to the binding site of duCPD (Ishikawa et al., 1994; Breiner et al., 1998) Finally, duCPD is drastically downregulated in DHBV-infected hepatocytes (our unpublished data).
In this study, we used neutralizing anti-duCPD antibodies to demonstrate the fundamental role of CPD as a HBV receptor. We show that the C-domain of duCPD represents the virus binding domain. By using surface plasmon resonance (SPR) analysis and two-dimensional (2-D) NMR spectroscopy, we reveal the conformational requirements for receptor interaction. We provide evidence that the mechanism of receptor recognition of hepadnaviruses is exceptional and differs fundamentally from ligand–receptor interactions described so far. It offers an alternative route to solve the fundamental problem of how viral surface proteins maintain receptor affinity and simultaneously modulate their primary sequence in order to escape immune surveillance.
Results
The CPD-mediated entry of DHBV into hepatocytes constitutes the major route of infection
We recently provided evidence that duCPD functions as a receptor for DHBV (Breiner et al., 1998; Urban et al., 1998). To answer the question whether duCPD represents the key route of DHBV infection, we generated antibodies against sduCPD to test them for interference with a DHBV infection. As shown in Figure 1, these antibodies greatly inhibited infection of primary duck hepatocytes with DHBV, as visualized by a nucleocapsid-specific immunofluorescence. The absence of newly synthesized intracellular viral DNA in an equivalent experiment (Figure 1C) confirmed this result and demonstrated that an infection could be almost completely blocked (>97% compared with an infection in the presence of the pre-immune serum). It is important to note that the antiserum was generated against native sduCPD. Attempts to block the infection with antibodies recognizing only primary sequence elements of duCPD or denatured sduCPD were not successful or resulted in strongly reduced inhibition (our unpublished data). We therefore conclude that CPD plays a key role in the initial step of a DHBV infection and conformational epitopes are essential for mediating virus–receptor recognition.
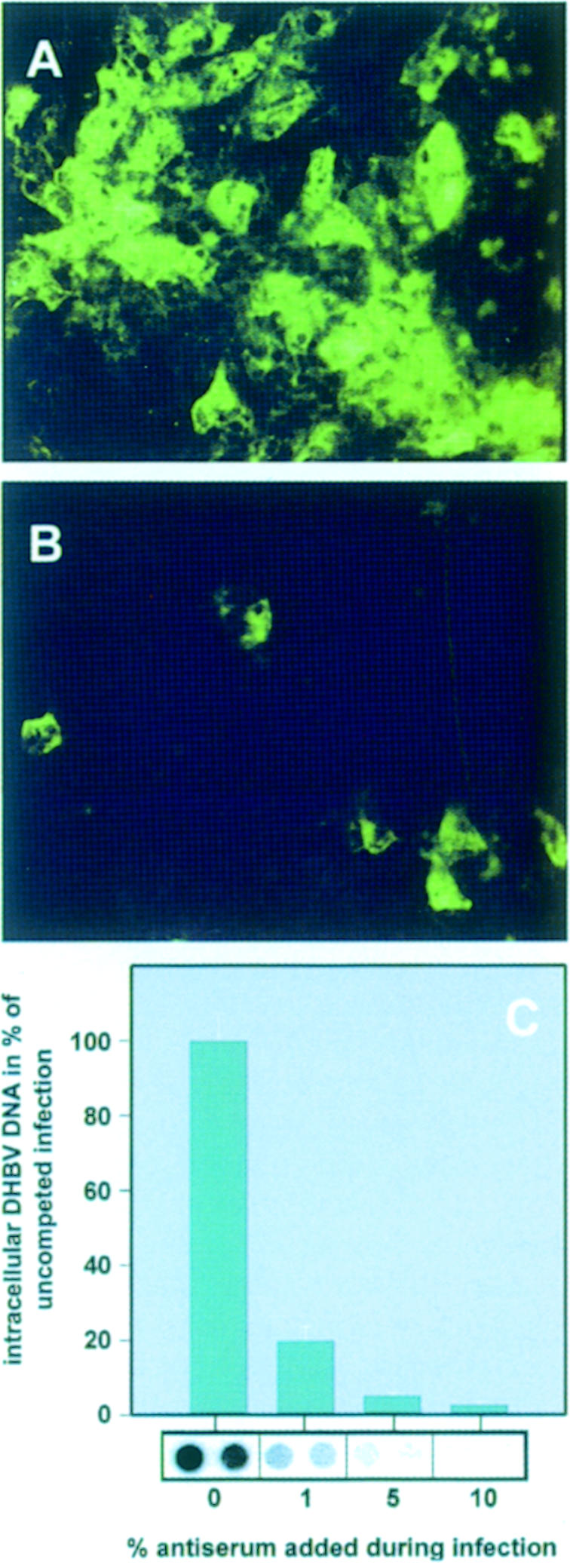
Fig. 1. CPD-mediated entry of DHBV into hepatocytes constitutes the key route of DHBV infection. Primary duck hepatocytes were infected with DHBV in the absence (A) or presence of 180 μg/ml α-sduCPDnat IgGs (B). Seven days post-infection, intracellular core protein was visualized by immunofluorescence. (C) Primary duck hepatocytes were infected with DHBV in the presence of the indicated concentrations of α-sduCPDnat antiserum. Seven days post-infection, intracellular viral DNA was quantified by dot-blot analysis. Mean values of two independent measurements are depicted as percentages of the uncompeted infections in the bar chart. The corresponding DNA dot-blot is shown below the bars. As a control, infection was performed with the respective pre-immune serum, leading to only insignificant reduction of viral DNA synthesis (our unpublished data).
Purification of a soluble C-domain of duCPD
Binding of duCPD to DpreS polypeptides has been shown to depend on the integrity of the C-domain of the receptor (Eng et al., 1998). To test whether the individually expressed domain can represent a functional virus attachment site and to perform binding studies with preS polypeptides we constructed a recombinant baculovirus encoding the secretory duCPD C-domain (duCPD–C). Since the standard protocol for the purification of carboxypeptidases failed, we purified duCPD–C from culture supernatants on a DpreS affinity column. Elution was performed by shifting the temperature from 4 to 37°C and the pH from 7.0 to 4.0, conditions that have been described to disrupt the sduCPD–preS complex (Urban et al., 1999). Silver staining (Figure 2A) and Western blot analysis (Figure 2B) confirmed the purity and identity of duCPD–C. The yield of purified duCPD–C was ∼1 mg/107 High Five cells. Analysis by size exclusion chromatography showed that the purified duCPD–C eluted in a single peak representing the monomeric protein (our unpublished data).

Fig. 2. Purification of duCPD–C by DpreS affinity chromatography. Culture supernatants of High Five insect cells, infected with a recombinant baculovirus encoding sduCPD–C, were purified on a DpreS affinity column. After washing, elution was performed at 37°C by shifting the pH to 4.0. (A) The applied culture supernatant (load), the flow through (FT), the wash fractions (wash) and the eluted protein (elution pH 4) were analyzed by SDS–PAGE and subsequent silver staining. (B) Western blot analysis of the pooled eluate using a duCPD-specific antibody. The arrow indicates the 63 kDa band, representing duCPD–C.
duCPD–C binds DpreS with high affinity and represents the functional virus binding domain
To determine the association and dissociation rates of preS–duCPD–C complex formation we performed a real-time SPR analysis (Figure 3). For this purpose a DpreS fragment (amino acid residues 30–115), representing the receptor binding site of the viral L-protein, was immobilized onto a CM5 sensor chip surface. Three different concentrations (5, 10 and 20 μg/ml) of duCPD–C (Figure 3A) and full-length sduCPD as a control (Figure 3B) were injected onto the surface at 37°C. The observed changes in the relative diffraction indices (response units; RU) were recorded as a function of time. The kinetic constants of association (150–400 s) and dissociation (400–600 s) were calculated as mean values from the slopes of the curves. The association rate ka for the binding of duCPD–C to DpreS30–115 was determined as 0.9 × 105 M–1 s–1, the dissociation rate kd as 1.7 × 10–4 s–1. The derived dissociation constant KD was calculated as 1.9 nM. This matches the rates determined for the interaction of sduCPD with DpreS30–115 (ka = 1.0 × 105 M–1 s–1, kd = 3.0 × 10–4 s–1, KD = 1.5 nM). Thus, the C-domain of duCPD and the full-length soluble receptor bind preS at similar rates with extraordinarily high affinity. Likewise, a high affinity interaction also resulted in the ability of duCPD–C to inhibit DHBV infection in vitro (Figure 3C). To visualize binding of duCPD–C to viral particles directly we performed immunogold electron microscopy. As shown in Figure 4, duCPD–C specifically localizes at the particle surface. No free duCPD–C was detectable, indicating tight interaction with viral particles.

Fig. 3. duCPD–C binds preS polypeptides with high affinity and represents the virus binding domain. Three different concentrations of duCPD–C (A) or sduCPD (B) were injected onto DpreS30–115, covalently immobilized onto a CM5 sensor chip. Binding was allowed to occur for 250 s. At 400 s, dissociation started in running buffer for 200 s. Association and dissociation rates were determined as mean values from the slope of the curves using the BIAevaluation program 2.1. The resulting dissociation constants were 1.5 nM for duCPD–C and 1.9 nM for sduCPD. Note that because of the differences in their molecular weight, identical concentrations of duCPD–C and sduCPD do not lead to identical sensorgrams. (C) sduCPD-C efficiently competes DHBV infection. Primary duck hepatocytes were infected with DHBV in the absence (–) or in the presence of 17, 50 and 120 nM of duCPD–C. Neither virus nor competitor was removed until 6 days post-infection when equal amounts of total cellular lysates were analyzed by Western blotting for the presence of viral L-protein.

Fig. 4. sduCPD–C binds DHBV particles with high affinity. Immuno- electron microscopy of duCPD–C bound to DHBV particles. Complexes of DHBV SVPs with duCPD–C were prepared and duCPD–C was stained with α-sduCPDnat and a gold-conjugated secondary antibody. Note that all gold granules are particle associated with a preferentially asymmetrical distribution. As a control, subviral particles were stained with an anti-DpreS specific antibody (inserted picture). The bar represents 100 nm.
Binding of DpreS to duCPD reflects a new mode of high affinity ligand–receptor interaction
Using an infection competition assay, we defined the receptor binding site of DHBV as an internal preS subdomain composed of amino acids 30–115 (Urban et al., 1998). This subdomain bound sduCPD with equal affinity compared with full-length preS (Urban et al., 1999). To evaluate the contribution of different preS elements to complex formation we developed a competition binding assay for SPR-based analysis. Increasing concentrations of free DpreS were incubated with duCPD–C and analyzed for binding sensor chip-bound preS (Figure 5). At a 5-fold molar excess of soluble preS, virtually all soluble receptor was blocked. At a ratio of 1:1, the apparent association rate was 50% retarded compared with the control, indicating a 1:1 stoichiometry of complex formation. This result particularly shows that almost all preS molecules were involved in high affinity complex formation. We used the same approach to investigate the activity of 18 different mutant preS polypeptides listed in Figure 6A. The molar ratios of the preS mutants to duCPD–C were altered in a way that even low affinity interactions could be monitored. Mutants could be divided into three classes. The first group of mutants, termed Δ85–96, Δ101–109, Δ107–125 (Figure 6C) and 86–115mut (Figure 6D), failed to inhibit complex formation of duCPD–C–DpreS, even exceeding a 20-fold molar excess of the competitor. These mutants had alterations within amino acids 85–115, indicating that this sequence is absolutely essential for complex formation. A second group, represented by 30–115 (Figure 6B), Δ128–139 (Figure 6C), 23–115, 23–130 (not shown), competed binding as efficiently as full-length preS (1–165). These mutants apparently contain an entirely functional receptor binding site, which is unimpaired by the introduced deletions. The third type, represented by 38–115, 43–115, 52–130 (Figure 6B), Δ52–61, Δ62–65, Δ67–70, Δ74–84 (Figure 6C), 86–115 and 88-SI-89 (Figure 6D), showed gradual effects on duCPD–C interaction. Surprisingly, competition activity increased progressively with an increasing number of amino acids located N–terminal to the essential sequence element. It was irrelevant whether internal or terminal deletions were applied. For example, binding of mutant Δ67–70, lacking only four amino acids, was more severely affected than that of mutant 52–130, suggesting that amino acids N–terminal of the gap were unable to contribute to complex stability. We observed this continuous sequential stabilization up to amino acid 30 (Figure 6B). Consistent with the delineation of the receptor binding site of DpreS to amino acids 30–115, mutations N–terminal to amino acid 30 had no further effect (not shown).

Fig. 5. An SPR-based binding competition assay. Binding of duCPD–C to immobilized DpreS30–115 in the presence of increasing amounts of the free ligand. duCPD–C was incubated with free DpreS30–115 at the indicated molar ratios and injected onto a DpreS30–115-coated CM5 sensor chip. Binding occurred between 100 and 400 s. Dissociation followed from 400 to 600 s. Note that the presence of an equimolar amount of free DpreS30–115 in the binding mixture reduces the apparent association rate to ∼50%, indicating that every free preS molecule is capable of binding duCPD–C in a 1:1 stoichiometry.

Fig. 6. Binding of DpreS to duCPD proceeds by a potential two-step mechanism. (A) Schematic drawing of DpreS deletion mutants. Numbers on the right indicate amino acids that define the positions of the deletions. Mutant 88-SI-89 had two amino acid exchanges at the indicated positions 88 and 89 (88-WT-89 → 88-SI-89); the peptide 86–115mut carries a point mutation at position 93 (D → Q). Bars in light gray symbolize polypeptides that competed DpreS binding to duCPD–C like full-length DpreS (1–165, 23–130, 23–115, 30–115, Δ128–139), whereas bars in black represent polypeptides that showed no detectable competition activity even at a 20-fold molar excess (Δ85–96, 86–115mut, Δ101–109, Δ107–125). Competition activities of mutants are illustrated by the gray scale of the bars (38–115, 43–115, 52–130, Δ52–61, Δ62–65, Δ67–70, Δ74–84, 88-SI-89, 86–115). (B) Sensorgram overlays of binding competition assays using mutants 30–115, 38–115, 43–115 and 52–130. duCPD–C was injected onto a DpreS sensor chip in the presence of a 20-fold molar excess of the indicated preS mutants. Binding occurred between 100 and 400 s; dissociation followed between 400 and 700 s. The scale of the ordinate is extended compared with (C) and (D) in order to resolve the relatively high competition activity. (C) Binding competition assays using a 10-fold molar excess of mutants 52–130, Δ52–61, Δ62–65, Δ67–70, Δ74–84, Δ85–96, Δ101–109, Δ107–125, Δ128–139, DpreS (1–165) and buffer. (D) Binding competition assays using a 100-fold molar excess over duCPD–C of the synthetic DpreS peptides 86–115, 86–115mut or DpreS 88-SI-89. As a control, full-length DpreS (1–165) in a 2.5-fold molar excess and buffer was used. (E) Schematic representation of the primary binding site (86–115) and the stabilizing part (30–85) of the receptor binding site of DHBV. The two α–helical regions are boxed. The sequential character of complex stabilization is indicated by the grayscale N–terminal of the primary binding site.
Evidence that allows the dissection of primary attachment and complex stabilization as a potential two-step process was derived from a binding competition analysis using the synthetic peptide 86–115 and the double point preS mutant 88-SI-89 (Figure 6D). Mutations of those two amino acids have been shown to render the respective virus non-infective (Sunyach et al., 1999). Using a 100-fold molar excess of both molecules over duCPD–C revealed similar, although weak, competition activities. The inhibition profile of the peptide demonstrated that interaction of duCPD–C with a site restricted to the essential sequence of the receptor binding domain occurs independently of the stabilizing N–terminal element. The specificity of competition became most evident when a mutant peptide with a point mutation at position 93 (D to Q) failed to show inhibition. In addition, binding of the 88-SI-89 mutant with similar affinity suggests that despite the presence of 56 potentially stabilizing amino acids N–terminal to amino acid 86, no high affinity complex is formed. The replacement of W88 and T89 with S and I, respectively, apparently allows primary complex formation, but prevents the subsequent stabilization reaction to occur.
Formation of a stable ligand–receptor complex induces significant conformational alterations
To investigate whether receptor binding induces conformational changes, we recorded fluorescence spectra of duCPD–C (Figure 7A) and of duCPD–C bound to DpreS30–115 (Figure 7B). We found a significant shift of λmax from 326 to 344 nm and a reduction of fluorescence intensity upon preS binding. As, due to the much lower number of Trp and Tyr residues, the contribution of DpreS30–115 to the emission signal of the complex was minor, we conclude that the observed shifts cannot be explained by alterations in the ligand alone, but mirror conformational changes of the whole complex.

Fig. 7. Formation of a high affinity preS–duCPD–C complex induces conformational changes. Fluorescence spectra of duCPD–C and a duCPD–C–DpreS30–115 complex. Trp and Tyr residues of an aqueous duCPD–C solution (38 nM) were excited at 280 nm and the emission spectrum was recorded between 280 and 450 nm, showing a maximum at 326 nm (A). Addition of a 1.5-fold excess of DpreS30–115 led to a shift of λmax to 344 nm and a change in the fluorescence intensity (B).
Conformational analysis of the DHBV receptor binding domain
Secondary structure predictions using several algorithms suggest an overall random structure of DpreS30–115 with two helical parts from L43 to Q60 and E91 to Q104 (Figure 8A) with the exception of method 8, predicting only the C–terminal α–helix. These predictions were to a certain extent confirmed by CD and NMR spectroscopy: evaluation of the CD spectrum of DpreS30–115 indicates the presence of both random coil and ∼19% of α–helix (Figure 8B) (Greenfield and Fasman, 1969; Morrisett et al., 1973; Schmid, 1997). The homonuclear two-dimensional 1H 1H NMR spectrum (Figure 8C) showed a dispersion of the amide proton resonances of <1 p.p.m., and no upfield-shifted methyl proton resonances, indicating the absence of defined tertiary structure. Additionally, the dispersion of side chain proton resonances was within the range of random coil structure (Wüthrich, 1986). The lack of downfield-shifted Cα proton resonances points to the absence of β-sheet structure. The narrow dispersion of α–proton resonances and their tendency to be upfield shifted (Wishart et al., 1992), as well as the appearance of NH NH NOESY cross-peaks, suggest a partial α–helical character of the polypeptide.

Fig. 8. Secondary structure prediction, CD and NMR spectroscopic analysis of the receptor binding domain of DHBV. (A) Predicted secondary structure of DpreS30–115 using the methods of GORI (Garnier et al., 1978) (1), GORIII (Gibrat et al., 1987) (2), GORIV (Garnier et al., 1996) (3), PHD (Rost and Sander, 1993, 1994; Rost et al., 1994) (4), Predator (Frishman and Argos, 1996, 1997) (5), PSIpred (Jones, 1999) (6), SOPM (Geourjon and Deléage, 1994) (7) and AGADIR (Muñoz and Serrano, 1994a,b, 1997; Lacroix et al., 1998) (8). The amino acid sequence of DpreS30–115 is shown above the secondary structure prediction. H, α–helix; E, extended; C, random coil; T, turn. A consensus was assumed when five of the eight methods indicate the same structure prediction for a given amino acid. (B) Far-UV CD spectrum of His-tagged DpreS30–115 in 25 mM phosphate buffer pH 6.2 at 15°C. The protein concentration was 20 μM. (C) 200 ms NOESY spectrum of His-tagged DpreS30–115 under the same solution conditions as in (B). The protein concentration was 1.6 mM.
Sequence-specific resonance assignment of DpreS30–115 was carried out for 53 amino acid spin systems: P30–P41 (except T35), H45 and V46, V50–T58, G63–T89 [with gaps of single amino acids (67, 69 and 72)], K95, A99–Y103 (except 102) and R107 and P108. Owing to frequency degeneration, further spin systems, albeit identified, remained unassigned. Thus, a complete structure determination using NOESY distance information and molecular dynamics simulation was impossible. Owing to frequency degeneration and the likely simultaneous existence of sequential and (i,i+3) NOESY cross-peaks of comparable intensities between backbone protons, the two regions of potential helical character were also only partially assigned. Nevertheless, within the predicted C–terminal helix (91–104) the Cα proton resonances of all allocated spin systems (K95 and A99–Y103) were upfield shifted by >0.1 p.p.m. compared with the corresponding random coil values. This indicates the propensity to form an α–helix (Wishart et al., 1992, 1995). For the second predicted helix between amino acids 43 and 60, only residues T52–I56 showed upfield shifts of >0.1 p.p.m., the remainder were within the range of random coil values. dαβ(i,i+3) NOE assignments for V50–N57 indicate helical structure within this region.
More detailed information about the C–terminal helix was obtained by the homonuclear 1H 1H NMR spectroscopic analysis of peptide DpreS86–115. The chemical shift data were used to perform a secondary structure estimation based on the chemical shift index strategy (Wishart et al., 1992, 1995). This procedure suggests a helical conformation for amino acids 89/90 to 104 (Figure 9A). The beginning of the helix could not be determined precisely as the Cα chemical shift of T89 is only within the range typical for a helix after using proline correction. However, threonine has a propensity to function as an N-cap and proline is often found to follow the N-cap residue of an α–helix (Richardson and Richardson, 1988). Owing to frequency degeneration helix typical (i,i+3) NOEs could not be unambiguously identified in this region, preventing structure calculation. A helix wheel presentation of residues T89–E104 shows that this helix is amphipathic with alternating charges along the helix axis (K95 + R102, E91 + E98, R97 + R101) at the hydrophilic side (Figure 9B).

Fig. 9. Chemical shifts and helical wheel representation of DpreS sequences. Difference values of the observed Cα proton chemical shifts relative to the ‘random coil’ values according to Wishart et al. (1995). The thresholds of ±0.1 p.p.m. are indicated by dashed lines, values below –0.1 p.p.m. are shaded in gray. (A) DpreS86–115 in aqueous solution. (B) Helix wheel presentation of residues T89–E104. Hydrophobic residues are boxed, basic residues are underlined, acidic residues are in italic.
Discussion
We have studied the interaction of the virus binding domain of the DHBV receptor duCPD with its ligand, the preS moiety of the large viral envelope protein. Combining SPR spectroscopy, CD spectroscopy and an NMR-based structure analysis we demonstrate that avian HBVs evolved a hitherto unknown mode to achieve a specific and high affinity interaction with their receptor. In particular, we provide evidence for a potential two-step process in which a mostly unstructured stretch of amino acid sequence reaches the potential to interact strongly with its receptor. This mechanism explains how the virus can greatly modulate its receptor binding site in order to escape immune surveillance and cope with the evolutionary restrictions of the overlapping polymerase and envelope reading frames characteristic for hepadnaviral genomes.
Recently, we provided evidence that CPD serves as an avian HBV receptor (Breiner et al., 1998; Urban et al., 1998). The almost complete competition of DHBV infection by antibodies directed against duCPD described here substantiates this hypothesis by blocking cellular duCPD during infection. It furthermore demonstrates that DHBV entry into hepatocytes predominantly proceeds via the CPD-dependent pathway. Since competition was only efficient with antibodies raised against native duCPD, we assume that tertiary or quaternary structural elements of the virus binding site of the receptor play important roles in virus recognition.
By SPR analysis we show that the purified C-domain of duCPD binds DpreS with a binding constant similar to that of the full-length soluble receptor (KD = 1–2 nM at 37°C). This affinity is, for unknown reasons, 20-fold higher than that reported previously (Urban et al., 1999). This finding further strengthens our conclusion that avian hepadnaviruses bind their receptor with very high affinity. Furthermore, it shows that the enzymatically active duCPD A- and B-domains do not contribute to preS interaction. The ability of duCPD–C to compete with the membrane-bound receptor for viral particles strongly suggests that this domain represents the functional virus binding domain. Compared with receptor interactions described for other viruses, e.g. HIV, poliovirus or measles virus (Wimmer, 1994; Manchester et al., 1997; Casasnovas et al., 1999), DHBV binds to a receptor domain located in close proximity to the cellular membrane. Owing to the lack of a reconstitutable infection system we do not know whether membrane vicinity is required for productive infection. However, considering that DHBV infection is supposed to depend on a yet unidentified co-factor, which, similar to CD4 and the chemokine receptors in HIV infection, might cross-talk to each other (for a review, see Berger et al., 1999), domain shuffling within duCPD would be expected to destroy receptor functionality. Another surprising aspect is the exceptionally high affinity determined for the interaction between DpreS and duCPD–C (KD = 1–2 nM at 37°C). Although the typical affinities for interactions of viruses with cells were found in this range, most dissociation constants for the bare ligand–receptor interactions generally lay ∼20- to 250-fold above this value. For example, the KD of the interaction of poliovirus with its purified receptor was determined to be 45 nM at 4°C (Arita et al., 1998), CD55 binds human echovirus 11 with a KD of 3 μM (Lea et al., 1998), the cellular receptor for herpes simplex virus type 1, HveAt, binds various viral glycoprotein D variants with KD values between 33 nM and 3.2 μM (Willis et al., 1998), the murine polyomavirus binds sialyloligosaccharide chains with a KD of 5–10 mM (Stehle and Harrison, 1996), and the influenza hemagglutinin binds sialic acid residues with a KD of 100 nM (Takemoto et al., 1996). The only dissociation constant that is similar to the one determined by us was reported for the interaction of a subgroup A avian sarcoma and leukosis virus with a soluble form of its receptor Tva (KD = 0.3 nM) (Balliet et al., 1999). It has been speculated that strong binding of a viral ligand to its receptor indicates that only a single or very few molecules interact with a virus particle during infection, either because of a low receptor density on the cell surface, or the possibility that the virus is hindered in binding for steric reasons (Wickham et al., 1990). Supporting evidence for these assumptions came from experiments showing that the number of high affinity virus binding sites on the surface of cultured hepatocytes is <100 (our unpublished data).
The receptor binding domain of DHBV has been functionally defined as an internal preS sequence including amino acids 30–115 (Urban et al., 1998). Consistently, this fragment competed binding of duCPD to DpreS efficiently. Evidence that binding of preS to duCPD–C can be divided into two discrete steps came from the binding inhibition analysis using peptide 86–115, which represents the essential part of the receptor binding domain and the 88-SI-89 mutant encompassing the whole sequence with two amino acids mutated. As both molecules display similar low binding competition activities, we conclude that they both form a low affinity primary complex. Thus, the 58 amino acids N–terminal of the mutations in DpreS 88-SI-89 cannot participate in receptor interaction, probably because of steric reasons. Interestingly, the increase in complex stability depends on the sequence uninterruptedly foregoing the primary attachment site. Thus, potential stabilizing sequence elements that have been shifted by a gap lost their ability to participate in receptor binding. From these observations we propose the following model for DHBV binding to its receptor. Initially, the primary attachment site of the viral L-protein located within amino acids 86–115 forms a low affinity complex with duCPD. This initial complex sequentially proceeds to form a tight complex, including ∼60 amino acids. Both steps are required for productive infection (Sunyach et al., 1999).
With respect to the ability of virtually every preS molecule to bind duCPD–C tightly, it was surprising that all attempts to determine its tertiary structure by 2-D NMR analysis failed. Except for two regions between amino acids 51–57 and 89–104, tending to form short α–helical segments (Figure 6E), we found for most amino acids of the preS receptor binding domain a random conformation without tertiary structure. This was particularly true for the stabilizing sequence preceding the essential C–terminal element. As this part contributes to complex stabilization in a sequential manner, we hypothesize that its predominantly random conformation is a characteristic feature that represents the structural basis for tight receptor binding. We propose that this mode of interaction allows avian hepadnaviruses to modulate the primary sequences of their receptor binding domains without losing the potential to interact with their receptor productively. Support for this assumption comes from the observation that the heron HBV preS polypeptide, although showing only 50% homology, still binds duCPD–C with only slightly reduced affinity (our unpublished data). Facing the same problem, other viruses have evolved different strategies. For rhinovirus-14, a representative of the picornavirus family, it has been shown that the receptor ICAM-1 binds to a site at the bottom of a canyon (Bella et al., 1998). For steric reasons, molecules of the immune system are excluded from this site, thus enabling the virus to escape an immune attack and at the same time to conserve the primary sequence constituting its receptor binding domain (Rossmann, 1989). A different strategy has been described for HIV. The 3D structure of the viral envelope protein gp120 in complex with a soluble form of its receptor CD4 showed a small array of the outer domain of CD4 contacting gp120 at different positions in the molecule. Interestingly, the majority of receptor contacts are established by the gp120 backbone, consequently allowing the side chains to vary, provided that the overall tertiary structure remains preserved (Kwong et al., 1998). Here we propose an alternative strategy, realized by avian HBVs. Distinct from HIV and picornaviruses, the primary sequence variation of the receptor binding site and simultaneous conservation of high affinity receptor interaction do not involve either hiding of the receptor attachment site or preservation of positional contact sites within a defined 3D structure. The interaction is characterized by a sequential, zipper-like participation of an extended unstructured sequence after initial contact with a short helix. This apparently enables the virus to expose and change the primary sequence of its receptor binding domain as long as it conserves some as yet undefined key elements involved in the binding process. In addition to the problem of escaping the host immune response, hepadnaviruses show extensive overlapping of open reading frames and are therefore subjected to a dual evolutionary pressure within these parts of their genome. This holds especially true for the preS domain of the viral L-protein, which completely overlaps with the polymerase gene. We therefore assume that in addition to the immunological aspect, the described receptor binding mode, providing preS with a higher degree of sequence variation, also enables the overlapping polymerase gene to vary its sequence evolutionarily.
Interestingly, binding of the cellular protein stathmin/OP18 to tubulin resembles the mode of interaction described here, in the way that stathmin/OP18 is unstructured in the unbound state (Wallon et al., 2000). This indicates that the mechanism of DpreS binding to duCPD–C might exemplify a general theme of protein–protein interactions.
Beside this unconventional method of receptor interaction, the predominantly random conformation might be responsible for some other unique features, described for the preS domain as part of the viral L-protein. One is the ability to cross cellular membranes, leading to a complex topology of the L-protein (Swameye and Schaller, 1997; Grgacic et al., 2000). A second aspect is the association of cellular Hsc-70 with the preS part of the DHBV L–protein (Hild, 1997). One could speculate that the preS moiety traps Hsc-70 by exposing sequence elements that serve as recognition sites for the chaperone and is thereby prevented from degradation. Interestingly, post-translational membrane translocation and Hsc-70 binding have also been described for the preS domain of the HBV L–protein (Bruss et al., 1994; Löffler-Mary et al., 1997). As both properties have been conserved between avian and mammalian hepadnaviruses, although sequence homologies between their preS domains are lacking, we consider structural similarities to be responsible for these unique preS features.
Despite major efforts that have been undertaken during the last decades, little is known about the initial steps in HBV infection, particularly the nature of the HBV receptor. With respect to the viral envelope proteins required for productive infection, it has recently been shown that an extended sequence of ∼75 amino acids in the preS1 domain of the viral L-protein is required for productive HBV infection of primary human hepatocytes (Le Seyec et al., 1999). We have investigated a recombinant HBV preS1 polypeptide by 2-D NMR analysis and found a similar predominantly random conformation (our unpublished data). For both reasons, the requirement for an extended sequence and its predominant random structure, it might well be that the mode of interaction described here for the avian HBV model system also holds true for the human HBV.
Materials and methods
Plasmid construction and generation of recombinant baculoviruses
The baculovirus transfer vector pVL-duCPD–C was constructed by ligating the NcoI–StuI fragment of plasmid pBCgp180-C-myc (provided by K.M.Breiner) into the NcoI–StuI-digested plasmid pVL-sduCPD (Urban et al., 1999). The resulting plasmid pVL-duCPD–C thus contains a unique NcoI site that is part of the start codon, the original signal sequence of full-length duCPD, a myc tag, the open reading frame of the duCPD C-domain, and a poly linker at the 3′ end containing an artificial stop codon. The recombinant baculovirus (AcNPV-duCPD–C) was obtained by the standard protocol (O'Reilly et al., 1992), amplified in Sf9 cells and used for infection of High Five insect cells (Invitrogen).
SDS–PAGE, silver staining and immunological techniques
SDS–PAGE, silver staining and Western blot analysis were performed by standard procedures. The monoclonal antibody 4F8 recognized the epitope 95-KAREAFRRYQE-105 of the preS part of the DHBV L–protein (provided by C.Kuhn). The duCPD-specific antibody used in Western blots was generated against a fragment of duCPD expressed in E.coli. For developing we used enhanced chemoluminescence (ECL; Amersham-Pharmacia). DHBV core protein was detected by immunofluorescence microscopy after fixation of hepatocytes with methanol, incubation with the DHBV core-specific antiserum D-084 and detection using fluorescein isothiocyanate-conjugated goat anti-rabbit immunoglobulin G (Dianova).
Generation and purification of neutralizing duCPD-specific antibodies
Antibody α-sduCPDnat recognizing conformational epitopes of duCPD was raised in rabbits by mixing 200 μg of purified soluble duCPD (Urban et al., 1999) in 250 μl of 25 mM NaPi pH 7.0 with 250 μl of complete Freund's adjuvant and injecting it subcutaneously and intramuscularly without prior denaturation. Ten and 16 weeks later immunization was repeated with 300 μg of sduCPD, respectively. Serum was prepared 18 weeks post-immunization. Immunoglobulins were purified using HiTrap Protein A–Sepharose columns according to the manufacturer's protocol (Amersham-Pharmacia). Concentrations were determined by measuring the extinction at 280 nm (Gill and von Hippel, 1989).
Protein purification and handling
High Five insect cells (5.4 × 107, 3 T175 flasks) grown in 100 ml of Express Five insect medium (Gibco) were infected with AcNPV-duCPD–C at a multiplicity of infection (m.o.i.) of 10. Secretion of the recombinant protein was allowed to proceed at 27°C for 70 h. The culture supernatant was applied to a DpreS 30-115 affinity column equilibrated with 50 mM Tris–HCl, 150 mM NaCl pH 7.4 (1 ml bed volume, flow rate of 0.25 ml/min, 4°C). After washing, duCPD–C was eluted with 100 mM NaAc pH 4.0 at 37°C. duCPD–C-containing fractions were immediately dialyzed against 25 mM NaPi pH 7.0. DpreS polypeptides were purified as His6 fusions from E.coli (Urban et al., 1998). DpreS30–115 was coupled to activated CH Sepharose 4B (Amersham-Pharmacia) according to the manufacturer's protocol. For NMR spectroscopy, DpreS30–115 was concentrated to 1.6 mM using a Centricon 3 concentrator (Amicon). The protein concentrations were determined by measuring the extinction at 280 nm, based on the molar extinction coefficient ɛ = 43.960 for duCPD–C and the respective ɛ values for the preS mutants calculated from the primary sequence using the Protean software (DNA-Star, Lasergene).
Infection competition assays
Primary duck hepatocytes were prepared (Rigg and Schaller, 1992) and infection competition assays (Urban et al., 1998) were performed in the presence of increasing concentrations of duCPD–C, the duCPD-specific antiserum α-sduCPDnat or the purified immunoglobulin fraction thereof. Six days post-infection, cells were analyzed for the presence of L-protein (Western blot), DHBV core protein (immunofluorescence) or intracellular viral DNA (DNA dot-blot).
Immunoelectron microscopy of viral particles
Subviral particles of DHBV were prepared from serum of infected ducklings using a Sephacryl-S1000 column and sucrose density gradient centrifugation. Fractions containing subviral particles were collected and incubated with an excess of duCPD–C. The complex was isolated on a sucrose gradient. Aliquots thereof (20 μl) were placed onto a glow-discharged carbon-coated 300 mesh copper grid for 1 min. After rinsing with water the grid was placed face down onto a 50–100 μl drop of the rabbit antiserum α-sduCPDnat (0.2 mg/ml) diluted 1:400 in phosphate-buffered saline (PBS), 1% bovine serum albumin (BSA) (Sigma). After three washing steps with 100 μl of PBS, 1% BSA the grids were incubated on a 50 μl drop of a 1–10 dilution of 5 nm gold-labeled goat anti-rabbit IgG (H+L) (Amersham-Pharmacia) in PBS, 1% BSA for 1 h in the dark. The grids were rinsed and stained for 1 min with 20 μl of 2% aqueous uranyl acetate. Micrographs were recorded with a Zeiss 10 Å electron microscope at an acceleration voltage of 80 kV.
SPR spectroscopy
Binding DpreS and DpreS mutants to duCPD–C was investigated by real-time SPR spectroscopy (BIACore-Upgrade, BIAcore-System). Measurements were performed at 37°C in 1× HBS buffer (10 mM HEPES pH 7.4, 150 mM NaCl, 0.005% Surfactant P-20; Amersham-Pharmacia) at flow rates of 10 μl/min. DpreS polypeptides were coupled to a CM5 sensor chip via standard NHS/EDC chemistry in amounts that yielded 750–1500 RU. duCPD–C was injected for 300 s followed by 200–300 s elution in HBS buffer. The sensor chip surface was regenerated with 30 μl of 20 mM HCl to remove residual duCPD–C from immobilized DpreS. Association and dissociation constants of the duCPD–C–DpreS complex were calculated using the BIAevaluation program version 2.1 (Amersham-Pharmacia). KD was calculated by fitting the data to the equation R = R0e–KD(t – t0). The binding competition assays were performed by pre-incubating DpreS mutants with duCPD–C at the respective molar ratios prior to injection.
Fluorescence spectroscopy
Emission fluorescence spectra of duCPD–C and its preS complexes were recorded in the range of 280–450 nm using a Shimadzu RF 5000 spectrofluorophotometer. The excitation wavelength was 280 nm, addressing Trp and Tyr. Solutions of duCPD–C (38 nM in 25 mM NaPi pH 6.8) were adjusted to 20°C and spectra were recorded in the absence or presence of DpreS30–115. Spectra were corrected for the Raman signal of the buffer.
CD spectroscopy
CD spectra were recorded at 5, 15 and 30°C in 1 mM cells from 250 to 190 nm at 20 nm/min on a Jasco J. 600A CD spectropolarimeter. The concentration of DpreS30–115 (containing an N–terminal His tag of 12 amino acids) was 20 μM in 25 mM phosphate buffer pH 6.2. The reference sample contained buffer without protein. Four scans were accumulated at each temperature.
NMR spectroscopy
2-D NMR spectra of the recombinant DpreS30–115 polypeptide and the synthetic peptide DpreS86–115 were recorded on a Bruker AMX600 spectrometer at 15 and 30°C (DpreS30–115) and 25°C (DpreS86–115). The protein concentration was 1.6 mM for DpreS30–115 in 25 mM phosphate buffer pH 6.2 in H2O/D2O (9:1, v/v, 500 ml) and 4.1 mM for DpreS86–115 in aqueous solution pH 3.9 in H2O/D2O (9:1, v/v, 500 ml). The H2O resonance was pre-saturated by continuous coherent irradiation at the H2O resonance frequency prior to the reading pulse. The spectra were recorded with a spectral width of 7246.4 Hz in both dimensions and 4 K × 0.5 K data points in the time domain. Quadrature detection was used in both dimensions with the time proportional phase incrementation technique in ω1. Spectra were multiplied with a squared sinebell function phase shifted by π/4, π/3 or π/2 for the NOESY (mixing time 200 ms), the Clean-TOCSY (mixing time 80 ms) and the DQF-COSY spectra, respectively, prior to Fourier transformation. Application of zero filling resulted in 4 K × 1 K data points in the frequency domain. Sixth-order baseline and phase correction were used. Data were evaluated on X-window workstations with the NDee program package (Software Symbiose, Bayreuth).
Secondary structure estimation was performed by the chemical shift index strategy using random coil values (Wishart et al., 1992, 1995). This procedure depends on a direct correlation between the chemical shifts of Cα proton resonances of consecutive amino acids and the local secondary structure (Wishart et al., 1991). As the presence of proline causes significant chemical shift differences, for residues preceding a proline the corresponding corrected values from Wishart et al. (1995) were used. Indication for a helix is found if the Cα proton chemical shifts of an amino acid sequence are upfield shifted by >0.1 p.p.m. compared with the corresponding random coil values and if this stretch is not interrupted by an amino acid with a Cα proton resonance downfield shifted by >0.1 p.p.m.
Acknowledgments
Acknowledgements
Part of this work was performed in the laboratory of Paul Rösch, Universität Bayreuth, whom we thank for excellent support. S.U. is particularly obliged to Hans Ulrich Schairer for many discussions. We acknowledge Kazuyuki Kuroki for duCPD cDNA and Lucyna Cova for the DHBV mutant 88-SI-89. We thank Hüsseyin Sirma for contributing work to immunoelectron microscopy, Bärbel Glass for preparation of primary duck hepatocytes, Christa Kuhn for the gift of antibodies, Klaus M.Breiner for discussions and Sandra Reuter for contribution to the construction of recombinant baculoviruses. We also thank Thilo Stehle for critical reading of the manuscript. This work was supported by the grants from the Deutsche Forschungsgemeinschaft (UR 72/1-1 to S.U. and SFB 317 to G.M.) and by the Fonds der Chemischen Industrie H.S. and Konrad Beyreuther.
References
- Arita M., Koike, S., Aoki, J., Horie, H. and Nomoto, A. (1998) Interaction of poliovirus with its purified receptor and conformational alteration in the virion. J. Virol., 72, 3578–3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balliet J.W., Berson, J., D'Cruz, C.M., Huang, J., Crane, J., Gilbert, J.M. and Bates, P. (1999) Production and characterization of a soluble, active form of Tva, the subgroup A avian sarcoma and leucosis virus receptor. J. Virol., 73, 3054–3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bella J., Prasanna, R.K., Christopher, W.M., Jeffrey, M.G. and Rossmann, M.G. (1998) The structure of the two amino–terminal domains of human ICAM-1 suggests how it functions as a rhinovirus receptor and as an LFA-1 integrin ligand. Proc. Natl Acad. Sci. USA, 95, 4140–4145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger E.A., Murphy, P.M. and Farber, J.M. (1999) Chemokine receptors as HIV-1 coreceptors: roles in viral entry, tropism and disease. Annu. Rev. Immunol., 17, 657–700. [DOI] [PubMed] [Google Scholar]
- Breiner K.M. and Schaller, H. (2000) Cellular receptor traffic is essential for productive duck hepatitis B virus infection. J. Virol., 74, 2203–2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breiner K.M., Urban, S. and Schaller, H. (1998) Carboxypeptidase D (gp180), a Golgi-resident protein, functions in the attachment and entry of avian hepatitis B viruses. J. Virol., 72, 8098–8104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruss V., Lu, X., Thomssen, R. and Gerlich, W.H. (1994) Post-translational alterations in transmembrane topology of the hepatitis B virus large envelope protein. EMBO J., 13, 2273–2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casasnovas J.M., Larvie, M. and Stehle, T. (1999) Crystal structure of two CD46 domains reveals an extended measles virus-binding surface. EMBO J., 18, 2911–2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng F.J., Novikova, E.G., Kuroki, K., Ganem, D. and Fricker, L.D. (1998) gp180, a protein that binds duck hepatitis B virus particles, has metallocarboxypeptidase D-like enzymatic activity. J. Biol. Chem., 273, 8382–8388. [DOI] [PubMed] [Google Scholar]
- Eng F.J., Varlamov, O. and Fricker, L.D. (1999) Sequences within the cytoplasmic domain of gp180/carboxypeptidase D mediate localization to the trans-Golgi network. Mol. Biol. Cell, 10, 35–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frishman D. and Argos, P. (1996) Incorporation of non-local interactions in protein secondary structure prediction from the amino acid sequence. Protein Eng., 2, 133–142. [DOI] [PubMed] [Google Scholar]
- Frishman D. and Argos, P. (1997) Seventy-five percent accuracy in protein secondary structure prediction. Proteins, 3, 329–335. [DOI] [PubMed] [Google Scholar]
- Ganem D. and Varmus, H.E. (1987) The molecular biology of the hepatitis B viruses. Annu. Rev. Biochem., 56, 651–693. [DOI] [PubMed] [Google Scholar]
- Garnier J., Osguthorpe, D.J. and Robson, B.A. (1978) Analysis of the accuracy and implications of simple methods for predicting the secondary structure of globular proteins. J. Mol. Biol., 120, 97–120. [DOI] [PubMed] [Google Scholar]
- Garnier J., Gibrat, J.F. and Robson, B. (1996) GOR secondary structure prediction method version IV. Methods Enzymol., 266, 540–553. [DOI] [PubMed] [Google Scholar]
- Geourjon C. and Deléage, G. (1994) SOPM: a self-optimized method for protein secondary structure prediction. Protein Eng., 7, 157–164. [DOI] [PubMed] [Google Scholar]
- Gibrat J.F., Garnier, J. and Robson, B. (1987) Further developments of protein secondary structure prediction using information theory. New parameters and consideration of residue pairs. J. Mol. Biol., 198, 425–443. [DOI] [PubMed] [Google Scholar]
- Gill S.C. and von Hippel, P.H. (1989) Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem., 182, 319–326. [DOI] [PubMed] [Google Scholar]
- Greenfield N. and Fasman, G. (1969) Computed circular dichroism spectra for the evaluation of protein conformation. Biochemistry, 8, 4108–4116. [DOI] [PubMed] [Google Scholar]
- Grgacic E.V.L., Kuhn, C. and Schaller, H. (2000) Hepatitis B virus envelope topology: membrane insertion of a loop region and role of S in L protein translocation. J. Virol., 74, 2455–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gripon P., Diot, C. and Guguen-Guillouzo, C. (1993) Reproducible high level infection of cultured adult human hepatocytes by hepatitis B virus: effect of polyethylene glycol on adsorption and penetration. Virology, 192, 534–540. [DOI] [PubMed] [Google Scholar]
- Hild M. (1997) Zelluläre Funktionen während der frühen und späten Schritte im Infektionszyklus des Enten-Hepatitis B Virus. PhD thesis, Ruprecht-Karls-Universität, Heidelberg, Germany.
- Hildt E., Hofschneider, P.H. and Urban, S. (1996) The role of hepatitis B virus in the development of hepatocellular carcinoma. Semin. Virol., 7, 333–347. [Google Scholar]
- Ishikawa T., Kuroki, K., Lenhoff, R., Summers, J. and Ganem, D. (1994) Analysis of the binding of a host cell surface glycoprotein to the pre-S protein of duck hepatitis B virus. Virology, 202, 1061–1064. [DOI] [PubMed] [Google Scholar]
- Jones D.T. (1999) Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol., 292, 195–202. [DOI] [PubMed] [Google Scholar]
- Klingmüller U. and Schaller, H. (1993) Hepadnavirus infection requires interaction between the viral pre-S domain and a specific hepatocellular receptor. J. Virol., 67, 7414–7422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroki K., Cheung, R., Marion, P.L. and Ganem, D. (1994) A cell surface protein that binds avian hepatitis B virus particles. J. Virol., 68, 2091–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroki K., Eng, F., Ishikawa, T., Turck, C., Harada, F. and Ganem, D. (1995) gp180, a host cell glycoprotein that binds duck hepatitis B virus particles, is encoded by a member of the carboxypeptidase gene family. J. Biol. Chem., 270, 15022–15028. [DOI] [PubMed] [Google Scholar]
- Kwong P.D., Wyatt, R., Robinson, J., Sweet, R.W., Sodroski, J. and Hendrickson, W.A. (1998) Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature, 18, 648–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacroix E., Viguera, A.R. and Serrano, L. (1998) Elucidating the folding problem of α–helices: Local motifs, long-range electrostatics, ionic strength dependence and prediction of NMR parameters. J. Mol. Biol., 284, 173–191. [DOI] [PubMed] [Google Scholar]
- Lea S.M., Powell, R.M., McKee, T., Evans, D.J., Brown, D., Stuart, D.I. and van der Merwe, P.A. (1998) Determination of the affinity and kinetic constants for the interaction between the human virus echovirus 11 and its cellular receptor, CD55. J. Biol. Chem., 273, 30443–30447. [DOI] [PubMed] [Google Scholar]
- Le Seyec J., Chouteau, P., Cannie, I., Guguen-Guillouzo, C. and Gripon, P. (1999) Infection process of the hepatitis B virus depends on the presence of a defined sequence in the pre-S1 domain. J. Virol., 73, 2052–2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löffler-Mary H., Werr, M. and Prange, R. (1997) Sequence-specific repression of cotranslational translocation of the hepatitis B virus envelope proteins coincides with binding of heat shock protein Hsc70. Virology, 235, 144–152. [DOI] [PubMed] [Google Scholar]
- Manchester M., Gairin, J.E., Patterson, J.B., Alvarez, J., Liszewski, M.K., Eto, D.S., Atkinson, J.P. and Oldstone, M.B. (1997) Measles virus recognizes its receptor, CD46, via two distinct binding domains within SCR1–2. Virology, 233, 174–184. [DOI] [PubMed] [Google Scholar]
- Morrisett J.D., David, J.S.K., Pownall, H.J. and Gotto, A.M. (1973) Interaction of an apolipoprotein (apoLP-alanine) with phosphatidylcholine. Biochemistry, 12, 1290–1299. [DOI] [PubMed] [Google Scholar]
- Muñoz V. and Serrano, L. (1994a) Elucidating the folding problem of α–helical peptides using empirical parameters, II. Helix macrodipole effects and rational modification of the helical content of natural peptides. J. Mol. Biol., 245, 275–296. [DOI] [PubMed] [Google Scholar]
- Muñoz V. and Serrano, L. (1994b) Elucidating the folding problem of α–helical peptides using empirical parameters III: Temperature and pH dependence. J. Mol. Biol., 245, 297–308. [DOI] [PubMed] [Google Scholar]
- Muñoz V. and Serrano, L. (1997) Development of the multiple sequence approximation within the agadir model of α–helix formation. Comparison with Zimm–Bragg and Lifson–Roig formalisms. Biopolymers, 41, 495–509. [DOI] [PubMed] [Google Scholar]
- Nassal M. and Schaller, H. (1993) Hepatitis B virus replication. Trends Microbiol., 1, 221–228. [DOI] [PubMed] [Google Scholar]
- Nassal M. and Schaller, H. (1996) Hepatitis B virus replication, an update. J. Viral Hepatitis, 3, 217–226. [DOI] [PubMed] [Google Scholar]
- O'Reilly D., Miller,L.K. and Luckow,V.A. (1992) Baculovirus Expression Vectors: A Laboratory Manual. W.H.Freeman, New York, NY. [Google Scholar]
- Richardson J.S. and Richardson, D.C. (1988) Amino acid preferences for specific locations at the ends of α helices. Science, 240, 1648–1652. [DOI] [PubMed] [Google Scholar]
- Rigg R.J. and Schaller, H. (1992) Duck hepatitis B virus infection of hepatocytes is not dependent on low pH. J. Virol., 66, 2829–2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossmann M.G. (1989) The canyon hypothesis. Hiding the host cell receptor attachment site on a viral surface from immune surveillance. J. Biol. Chem., 264, 14587–14590. [PubMed] [Google Scholar]
- Rost B. and Sander, C. (1993) Prediction of protein structure at better than 70% accuracy. J. Mol. Biol., 232, 584–599. [DOI] [PubMed] [Google Scholar]
- Rost B. and Sander, C. (1994) Combining evolutionary information and neural networks to predict protein secondary structure. Proteins, 19, 55–72. [DOI] [PubMed] [Google Scholar]
- Rost B., Sander, C. and Schneider, R. (1994) PHD—an automatic mail server for protein secondary structure prediction. Comput. Appl. Biosci., 10, 53–60. [DOI] [PubMed] [Google Scholar]
- Schmid F.-X. (1997) Optical spectroscopy to characterize protein conformation and conformational changes. In Creighton,T.E. (ed.), Protein Structure: A Practical Approach. IRL Press at Oxford University Press, Oxford, UK, pp. 261–297. [Google Scholar]
- Stehle T. and Harrison, S. (1996) Crystal structures of murine polyoma virus in complex with straight-chain and branched-chain sialyloligosaccharide receptor fragments. Structure, 4, 183–194. [DOI] [PubMed] [Google Scholar]
- Sunyach C., Rollier, C., Robaczewska, M., Borel, C., Barraud, L., Kay, A., Trepo, C., Will, H. and Cova, L. (1999) Residues critical for duck hepatitis B virus neutralization are involved in host cell interaction. J. Virol., 73, 2569–2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swameye I. and Schaller, H. (1997) Dual topology of the large envelope protein of duck hepatitis B virus: determinants preventing pre-S translocation and glycosylation. J. Virol., 71, 9434–9441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemoto D.K., Skehel, J.J. and Wiley, D.C. (1996) A surface plasmon resonance assay for the binding of influenza virus hemagglutinin to its sialic acid receptor. Virology, 217, 452–458. [DOI] [PubMed] [Google Scholar]
- Tong S., Li, J. and Wands, J.R. (1995) Interaction between duck hepatitis B virus and a 170-kilodalton cellular protein is mediated through a neutralizing epitope of the pre-S region and occurs during viral infection. J. Virol., 69, 7106–7112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuttleman J.S., Pugh, J.C. and Summers, J.W. (1986) In vitro experimental infection of primary duck hepatocyte cultures with duck hepatitis B virus. J. Virol., 58, 17–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban S., Breiner, K.M., Fehler, F., Klingmüller, U. and Schaller, H. (1998) Avian hepatitis B virus infection is initiated by the interaction of a distinct pre-S subdomain with the cellular receptor gp180. J. Virol., 72, 8089–8097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban S., Kruse, C. and Multhaup, G. (1999) A soluble form of the avian hepatitis B virus receptor. Biochemical characterization and functional analysis of the receptor ligand complex. J. Biol. Chem., 274, 5707–5715. [DOI] [PubMed] [Google Scholar]
- Wallon G., Rappsilber, J., Mann, M. and Serrano, L. (2000) Model for stathmin/OP18 binding to tubulin. EMBO J., 19, 213–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham T.J., Grenados, R.R., Wood, H.A., Hammer, D.A. and Shuler, M.L. (1990) General analysis of receptor-mediated viral attachment to cell surfaces. Biophys J., 58, 1501–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis S.H., et al. (1998)Examination of the kinetics of herpes simplex virus glycoprotein D binding to the herpesvirus entry mediator, using surface plasmon resonance. J. Virol., 72, 5937–5947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimmer E. (ed.) (1994) Cellular Receptors for Animal Viruses. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Wishart D.S., Sykes, B.D. and Richards, F.M. (1991) Relationship between nuclear magnetic resonance chemical shift and protein secondary structure. J. Mol. Biol., 20, 311–333. [DOI] [PubMed] [Google Scholar]
- Wishart D.S., Sykes, B.D. and Richards, F.M. (1992) The chemical shift index: a fast and simple method for the assignment of protein secondary structure through NMR spectroscopy. Biochemistry, 31, 1647–1651. [DOI] [PubMed] [Google Scholar]
- Wishart D.S., Bigam, C.G., Holm, A., Hodges, R.S. and Sykes, B.D. (1995) 1H, 13C and 15N random coil NMR chemical shifts of the common amino acids. I. Investigations of nearest-neighbor effects. J. Biomol. NMR, 5, 67–81. [DOI] [PubMed] [Google Scholar]
- Wüthrich K. (1986) NMR of Proteins and Nucleic Acids. John Wiley, New York, NY. [Google Scholar]