

Abstract
Streptococcus pneumoniae hyaluronate lyase (spnHL) is a pathogenic bacterial spreading factor and cleaves hyaluronan, an important constituent of the extra– cellular matrix of connective tissues, through an enzymatic β–elimination process, different from the hyaluronan degradation by hydrolases in animals. The mechanism of hyaluronan binding and degradation was proposed based on the 1.56 Å resolution crystal structure, substrate modeling and mutagenesis studies on spnHL. Five mutants, R243V, N349A, H399A, Y408F and N580G, were constructed and their activities confirmed our mechanism hypothesis. The important roles of Tyr408, Asn349 and His399 in enzyme catalysis were proposed, explained and confirmed by mutant studies. The remaining weak enzymatic activity of the H399A mutant, the role of the free carboxylate group on the glucuronate residue, the enzymatic behavior on chondroitin and chondroitin sulfate, and the small activity increase in the N580G mutant were explained based on this mechanism. A possible function of the C–terminal β–sheet domain is to modulate enzyme activity through binding to calcium ions.
Keywords: enzyme catalytic mechanism/hyaluronan/hyaluronate lyase/protein crystallography/Streptococcus pneumoniae
Introduction
Streptococcus pneumoniae is a bacterial pathogen and an important cause of many diseases such as pneumonia, bacteremia, meningitis, otitis and sinusitis (Boulnois, 1992; Berry et al., 1994; Fiore et al., 1999). The bacterium produces several virulence factors to penetrate the physical defenses of the host, such as pneumolysin (PLY), pneumococcal surface protein A (PspA) and hyaluronate lyase (spnHL). Clinical studies have shown that the vast majority of isolates of S.pneumoniae produce spnHL, which catalyzes the degradation of hyaluronan (HA), chondroitin and chondroitin sulfates (Linker et al., 1955; Boulnois, 1992). In cultures of S.pneumoniae, spnHL is found both in the liquid media and in the cell-associated fractions, suggesting that circulating enzyme may be responsible for continuing bacterial invasion within the host. The HA degradation directly contributes to bacterial invasion by allowing greater microbial access to, or migration between, host tissues for colonization (Berry et al., 1994).
HA, the primary substrate of spnHL, is a highmolecular-weight negatively charged polysaccharide. Its average molecular weight is usually several million daltons (Laurent, 1970). It forms a strikingly viscous solution and functions primarily as a joint lubricant and shock absorber. It is widely distributed in various tissues such as the vitreous humor of the eye, the aorta, blood, brain, liver and skin (Lindahl, 1978). HA is a member of the glycosaminoglycans (GAGs) and is produced by almost all members of the animal kingdom as well as by certain members of streptococci (Laurent and Fraser, 1992). GAGs are a group of heterogeneous polysaccharides built of repeating disaccharide units that contribute to the architecture of the extracellular matrix. GAGs are classified into three structural groups according to the disaccharide units that are employed and their linkage pattern (Scott and Heatley, 1999): (i) cellobiose type (HA); (ii) polylactose type (chondroitin and keratan sulfates); and (iii) poly– maltose type (heparan sulfate). The diversification of GAGs was driven by evolutionary selection pressures and the exogenous selection pressures mediated by the microbial pathogens that recognize them (Gagneux and Varki, 1999). In addition to the structural role in connective tissues, many biologically important molecules are attached to HA. Binding of HA to the cell surface receptors CD44 and RHAMM has been implicated in various types of normal cell signaling pathways (Laurent and Fraser, 1992; Yang et al., 1994). By attachment to these and other surface receptors, HA plays an important role in various physiological processes such as cell proliferation, recognition, locomotion, interaction with leukocytes, etc. (Fraser and Laurent, 1989; Laurent and Fraser, 1992). To ensure proper cellular activity, HA levels in the body are finely regulated to control HA biosynthesis and degradation. A rapid turnover rate facilitates the use of HA and its products in many physiological regulatory mechanisms. Hyaluronidase is the enzyme responsible for HA degradation in animals and bacteria.
There are two main groups of hyaluronidases identified to date: (I) hydrolases, including testicular-type hyaluronidase and leech hyaluronidase; and (II) lyases, bacterial hyaluronidase (hyaluronate lyase) secreted by certain strains of Streptococcus, Propionibacterium, Peptostreptococcus, Staphylococcus and Streptomyces genera (Park et al., 1997). Considerable differences exist between these two groups of hyaluronidases. The bacterial enzymes are less stable and degrade HA at a faster rate and to a greater extent, and also produce unsaturated products. Conversely, the group I hyaluronidases act by hydrolysis of the same glycosidic linkage, giving oligosaccharide products composed of unchanged repeating disaccharide units (Linker et al., 1955).
Four bacterial hyaluronate lyase protein sequences from S.pneumoniae (Berry et al., 1994), Streptococcus agalactiae (Lin et al., 1994), Staphylococcus aureus (Farrell et al., 1995) and Propionibacterium acnes (Steiner et al., 1997) are known at present. Their sequence homologies range from 25 to 53% suggesting their functional, structural and evolutionary similarities. spnHL has 21.3% homology to chondroitin AC lyase, another member of the GAG-degrading enzymes from the bacterium Sphingobacterium heparinum (DDBJ/EMBL/GenBank accession No. U27583 or gi1002525) (Figure 4). The structural architecture of the chondroitin AC lyase (Fethiere et al., 1999) is similar to the spnHL structure presented in this paper. Residues concerning substrate binding and degradation are highly conserved among these sequences. Therefore, the spnHL structure and the HA binding and degradation mechanism may be similar to other GAG-degrading enzymes.

Fig. 4. Sequence alignment. spnHL sequence (S. pneu) was aligned to the other three known hyaluronate lyase sequences from Streptococcus agalactiae (S. agal; 53.3% homology), Staphylococcus aureus (S. aure; 37.4%) and Propionibacterium acnes (P. acne; 24.9%), and to one chondroitin lyase sequence from Sphingobacterium heparinum (Flavobacterium heparinum) (S. hepa; 21.3%). spnHL sequence was colored according to residue properties: red, positively charged; blue, negatively charged; pink, hydrophobic. Conserved residues in other sequences are colored in the same scheme. Residues marked with ‘*’ are active site residues in the spnHL. This figure was prepared using FASTA3 (Pearson, 1990).
The fully functional truncated form of spnHL was cloned, overexpressed in Escherichia coli, purified, and crystallized as described previously (Jedrzejas et al., 1998a, b). The current structure was determined by multiple isomorphous replacement (MIR) methods and refined to 1.56 Å. The mechanism of HA degradation is proposed, based on the spnHL crystal structure, substrate modeling, degradation product determination and site-directed mutagenesis studies.
Results and discussion
Overall structure of spnHL
The crystal structure of the spnHL molecule has approximate dimensions of 59 × 59 × 88 Å and is divided into two distinct structural domains connected by one short linker (Figure 1A). Both domains are spherical and are of approximately the same size. The N–terminal α–helical domain (α–domain) contains the first 361 residues (Lys171–Ser531) of the enzyme, and is composed of 13 α–helices connected by 12 loops (residues taking coil conformation, including various turns and 2–3 residues of short β–sheet and 310 helix). Only one 11–residue linker (Asp532–Ser542) is located in between the two structural domains. The C–terminal β–sheet domain (β–domain) contains the following 347 residues (Tyr543–Lys889) in 24 β-strands (packed into five antiparallel β–sheets), four short α–helices and 25 connecting loops. The topology diagram of the spnHL molecule secondary structure composition is shown in Figure 1B.
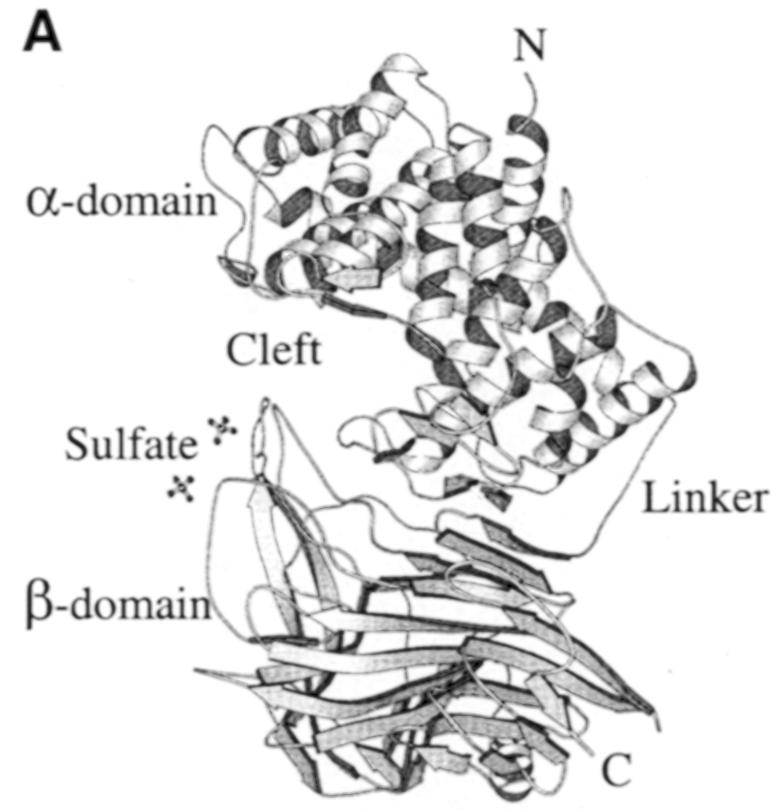

Fig. 1. (A) spnHL crystal structure overview. The figure shows the α–domain, the β–domain, the linker, the position of the two termini, the cleft and the sulfate ions. The figure was prepared using the program MOLSCRIPT (Kraulis, 1991). (B) Schematic diagram of spnHL secondary structure. Secondary structure segments are named as domain, type of secondary structure and sequential number. αH2 means the segment is in the α–domain, an α–helix, and the second one in the sequence. βS3s5 means that this segment belongs to the β–domain, β–sheet III and the fifth strand in the sheet.
Structure of the N–terminal α–domain
The α–domain is predominantly composed of 13 α–helices. Ten α–helices (αH3–αH12) are arranged into a twisted (α/α)–barrel structure (Figure 2A). The barrel is incomplete, leaving one side open. Every two adjacent helices of these 10, e.g. αH3 and αH4, form a basic hairpin structure. Five hairpins compose an (α5/α5)–barrel structure. Helices αH2 and αH13 are almost perpendicular to the barrel axis and block the barrel opening. The even-numbered helices are located at the outer layer of the barrel and the odd-numbered helices are located at the inner layer. The outer layer helices, therefore, are unidirectional and nearly parallel to each other, while the inner layer helices point to the opposite direction. The two helical layers are in a left-handed twist (∼30°) to the barrel axis, with αH9 in a near parallel position to the barrel axis. The outer layer helices twist more than their paired helices in the hairpin structure in the inner layer. The smaller numbered helices twist even more. In addition to the twist, these 10 helices are also tilted at a small angle, ∼5° to the barrel axis. The two sides of the barrel structure are, therefore, not the same size. The small end of the barrel is covered by helix αH1, while the large end faces the cleft and the β–domain. It is this large end of the barrel structure, together with the loops connecting adjacent hairpins at this end, that forms the majority of the prominent cleft in the middle of the spnHL molecule where spnHL is likely to bind to HA.


Fig. 2. (A) α5/α5-barrel structure in the α–domain. Five hairpins are αH3 and αH4, αH5 and αH6, αH7 and αH8, αH9 and αH10, and αH11 and αH12. αH2 and αH12 blocked the barrel opening. The end facing the reader is the large end and the further end is the small end. αH1 helix that blocked the small end was not shown. (B) The β–domain. Five β–sheets and the two loops participating in the formation of one edge of the cleft are shown. The figure was prepared using the program MOLSCRIPT (Kraulis, 1991).
At the small end of the barrel, loops connecting two helices in a hairpin are short and usually have 3–6 residues. At the large end of the barrel, loops connecting adjacent hairpins are significantly longer and form their own short secondary structures in several places (Figure 1B). Glu227–Phe229 in loop αL2 forms a short 310 helix. His334–Phe335 forms a small anti-parallel β–sheet with Phe343–Lys344 in helix αL6. In loop αL8, three segments Gly389–Phe390, Tyr396–Asp398 and Val402–Ala403, form an antiparallel β–sheet. In loop αL10, Ala447–Leu449, Asp457–Ser465 and Ala467–Ser469 form three 310 helices, and Leu450–Val451 and Glu454–Leu455 form an antiparallel β–sheet. In loop αL12, the Val513–Asn516 segment adopts a 310 helix conformation.
Similar barrel structures were seen in other polysaccharide-degrading enzymes such as glucoamylase, Protein Data Bank (PDB; Berstein et al., 1977) ID code 1ayx (Sevcik et al., 1998), and alginate lyase (PDB ID code 1qaz; Yoon et al., 1999). Glucoamylase uses a hydrolase mechanism to catalyze the degradation of starch. Alginate lyase uses the lyase mechanism and catalyzes the degradation of a polysaccharide alginate in seaweed and certain kinds of bacteria (Preiss and Ashwell, 1962). It is rather impressive that the two groups of polysaccharide-degrading enzymes, hydrolases and lyases, may have similar tertiary structure. This barrel infrastructure may be effective in holding the polysaccharide chain, while the detailed substrate-degrading mechanisms are determined by regional residue conformation, geometry and organization.
Structure of the C–terminal β–domain
In the β–domain, 24 β-strands are arranged into five β–sheets (Figure 2B). All β–sheets are antiparallel in the structure and the five sheets are parallel to each other. β–sheet I is composed of five successive strands in sequence. It is parallel to the small β–sheets in loops αL6 and αL8 in the α–domain. This arrangement composes the main interface between the two structural domains, the α–domain and the β–domain. β–sheet II is adjacent to sheet I and both are parallel to the largest nine-stranded β–sheet III. Sheets II and III are closely related. No break is found between strands βS2s5 and βS3s7 or βS3s6 and βS2s4. They are connected as two long continuous strands in the β–domain. Parallel to sheet III are β–sheets IV and V. The whole β–domain assumes the shape of a four-layered sandwich. Four small α–helix segments are present in the long loops, βL4, βL15, βL17 and βL25. Helix βH1 is a four-residue helix located between β–sheets I and II. Helix βH2 is a four-residue helix before loop βL15. The longest helix in the β–domain, helix βH3, is a 10–residue helix and is located between sheets III and IV. Helix βH4 is a four-residue helix in β–sheet V.
Two sulfate ion binding sites are observed in the β–domain (Figure 1A). These ions may originate from the crystallization solution since ammonium sulfate was used as the main precipitant in the spnHL crystallization condition. Their positions are in between β–sheets I and II, near to βL15 and βL3. βL15 and βL3 are the two loops from the β–domain that participate in the formation of one outer edge of the cleft.
Interface between the two structural domains
The interface between the α- and β–domains is 1776 Å2 in area and is composed of 37 residues from the α–domain and 34 residues from the β–domain forming 343 interactions (contacting atom pairs within a distance of 4.1 Å). Interface residues from the α–domain mainly come from the large end of the barrel and the inter-hairpin loops, αL8, αL10 and αL12. The interface at the β–domain side includes residues from the whole β–sheet I and several loops connecting strands in β–sheets I and II (Table I). Many aromatic residues, six from the α–domain and seven from the β–domain, were found in this interface, while the charged residues are arranged at, or point their charged side chain towards, the outer edge. This interface is hydrophobic in the middle and hydrophilic at the outer edge.
Table I. Interface residues between two structural domains.
| Residues from the α–domain | Contacting residues from the β–domaina |
|---|---|
| Asn290 | Asn580 |
| Tyr391 | Lys549 |
| Asp393 | Ala546, Phe547, Asn548, Lys549 |
| Gly394 | Phe547 |
| Ser395 | Phe547, Asn548, Lys549, Met550 |
| Tyr396 | Met550 |
| Ile397 | Lys549, Met550 |
| His399 | Glu577 |
| Asn401 | Arg572, Thr573, Leu574, Val635 |
| Val402 | Leu574, Glu577 |
| Ala403 | Met550, Lys552 |
| Tyr404 | Glu577 |
| Thr405 | Met550 |
| Lys444 | Phe547 |
| Ser445 | Phe547 |
| Ala447 | Tyr543 |
| Pro448 | Tyr543, Ser545, Phe547, Ala554 |
| Leu449 | Phe547 |
| Leu450 | Tyr543 |
| Val451 | Tyr543, Tyr597 |
| Asn452 | Leu601 |
| Glu454 | Tyr597, Leu601, Tyr604 |
| Met456 | Gly565, Tyr597 |
| Asp457 | Tyr595, Tyr597, Tyr604, Trp609 |
| Met458 | Lys552, Gly565, Leu566, Ser567, Tyr595 |
| Gly461 | Asn583 |
| Arg462 | Glu577, Met579, Asn580, Glu582, Asn583 |
| Ile464 | Asn583, Trp609, Trp763 |
| Ser465 | Glu582, Asn583, Trp763, Ile766, Asn767, Gln770 |
| Arg466 | Glu582, Trp763, Asn767, Gln770 |
| Ala467 | Trp763, Gln770 |
| Glu470 | Ser605, Asp606, Gly607, Trp763 |
| His472 | Leu601, Ser605 |
| Ser507 | Leu601 |
| Asp508 | Leu601 |
| Ser509 | Leu601 |
| Tyr510 | Leu601, Ser602, Ser605, Asp606 |
aTwo residues with atom pairs within a distance of 4.1 Å are considered to be in contact.
The cleft
The cleft in between the α- and β–domains on the spnHL molecule surface is ∼30 Å in length and 10 Å in width (Figure 3A). The cleft is the place where HA binds and is degraded (Fethiere et al., 1999). The cleft is wider at the two ends and narrower in the middle. The shortest distance across this cleft is 3.94 Å, which is between Asn290 in αL4 and Asn590 in βL3. Most of the cleft residues come from the large end of the barrel structure and the inter-hairpin loops in the α–domain. Two loops, βL3 in β–sheet I and βL15 in β–sheet II from the β–domain, form one edge of the cleft. Part of the interface between the two structural domains lies in the cleft. The widely examined key catalyzing residue, His399, is located inside the cleft and at the edge of this interface, and is in contact with Glu577 from loop βL3 in the β–domain. Most of the residues along the cleft are either charged or aromatic. The surface electronic potential distribution showed that the center of the cleft is highly positively charged (Figure 3). These positively charged residues include Lys200 and Lys280 in αH2; Arg243, Lys244 and Lys250 in αH3; Arg300 in αH5; Arg336 and Lys337 in αL6; Arg355 in αH7; Tyr408 in the end of αL8; Arg462 and Arg466 in αL10; Arg480 in αH11; Lys515 and Lys518 in αL12; Lys581 in βL3; Lys764 in βH2; and Lys773 in βL15.


Fig. 3. (A) spnHL surface electronic potential distribution. The figure shows the surface electronic potential distribution on the spnHL surface. The positively charged cleft, the negative patch, the aromatic patch and the active center are indicated. The figure was prepared using the program GRASP (Nicholls et al., 1991). (B) Stereo diagram of the aromatic patch. The geometry of the aromatic patch formed by Trp291, Trp292 and Phe343, and its relative position to the active center and the substrate are shown. Trp291 is right over the glycosidic linkage. At one side of Trp291, the Trp292 indole plane is in parallel to one disaccharide unit. At the other side, the Phe343 side chain phenyl plane is in parallel to another disaccharide unit. The active center is located at the opposite side of the cleft. The figure was prepared using the program MOLSCRIPT (Kraulis, 1991).
Four negatively charged residues, Glu388, Asp398, Thr400 and Glu577, accumulate at one end of the cleft and no positively charged residues are present in this region. This makes one side of this end of the cleft slightly negative, forming a negative patch (Figure 3A).
Three aromatic residues, Trp291, Trp292 and Phe343, are aligned at one side of the cleft, opposite to the above negative patch and His399. These three residues form a small aromatic patch (Figure 3B). The existence of this aromatic patch in the cleft provides the spnHL molecule with a way to select the cleavage sites on the substrate. A more detailed discussion can be seen in section on the structural basis for HA binding.
The spnHL sequence comparison with known hyaluronate lyase sequences from three other species, S.agalactiae, S.aureus and P.acnes, showed 25 conserved residues along the cleft (Figure 4): Thr239, Arg243, Asn290, Trp291, Trp292, Asp293, Glu295, Gly347, Asn349, Asp352, Phe390, His399, Tyr404, Tyr408, Val411, Leu412, Arg460, Arg462, Ser465, Arg466, Glu577, Asn580, Glu582, Trp763 and Asn767. Most of these residues are arranged along the middle narrow segment of the cleft. Most of these residues are also conserved in the chondroitin lyase sequence from S.heparinum (Flavobacterium heparinum).
Structural basis for HA binding and the spnHL catalytic active center
HA is a linear polysaccharide built of repeating units of β–d–glucuronic acid and 2–acetamino-2–deoxy-β–dglucose. Since the pKa of the glucuronic acid carboxylate group is ∼3.2, HA is negatively charged under physiological conditions (Laurent, 1970). The interaction between spnHL and its main substrate, HA, is dependent, in part, upon ionic interactions and involves basic amino acids (Yang et al., 1993, 1994). Therefore, salt bridges and hydrogen bonds play key roles in this interaction. The positive charges in the cleft facilitate the attachment of the spnHL molecule to the negatively charged HA, and these charges hold the substrate chain in position during catalysis.
When the hexasaccharide model of the HA structure was modeled and refined into the cleft as described in Materials and methods, 23 residues from spnHL were shown to form 78 contacts with the hexasaccharide (Table II). Of these 23 residues, 12 are conserved among four known hyaluronate lyase sequences (Figure 4). The sequential piece from Phe343 to Leu346 is conserved in group B Streptococcus hyaluronate lyase (gbsHL), suggesting that spnHL and gbsHL are more closely related in substrate-binding features. Two unconserved residues present in spnHL, Phe343 and Asn468, are intensively involved in the contact interface with HA. Also, most of these spnHL–substrate interface residues are charged.
Table II. spnHL–HA interface.
| (A) Residues in the spnHL–HA interface | |
|---|---|
| Hexasaccharide sugar ringsa | Residues in spnHL |
| Sugar 1 | Arg336, Phe343, Lys344, Ala345, Asn349, Glu388, Thr400 |
| Sugar 2 | Trp291, Arg336, Phe343, Asn349, Asp398, His399, Tyr408 |
| Sugar 3 | Asn290, Trp291, Trp292, Asn349, Tyr408, Val411, Arg462, Asn580 |
| Sugar 4 | Arg243, Trp292, Asp293, Arg466 |
| Sugar 5 | Thr239, Arg243, Arg466, Asn468, Ser469 |
| Sugar 6 | Phe229, Glu247 |
| (B) Selected distances in the spnHL–HA interface | ||
|---|---|---|
| Atoms in HAb | Atoms in spnHL | Distance (Å) |
| C25 | His399 NE2 | 2.46 |
| O22 | Arg336 NH1 | 2.64 |
| O2E | Asn349 OD1 | 2.06 |
| O2F | Asn349 OD1 | 2.73 |
| O23 | Trp291 NE1 | 2.38 |
| O24 | Tyr408 OH | 2.67 |
| O25 | Asn349 OD1 | 3.08 |
| O34 | Asn290 ND2 | 3.16 |
| O36 | Asn290 OD1 | 2.79 |
| O36 | Asn580 ND2 | 3.10 |
aSugars 1, 3 and 5 are 2–acetamino-2–deoxy-β–d–glucose residues. Sugars 2, 4 and 6 are β–d–glucuronic residues.
bThe first digit stands for the ring number and the second digit is the conventional atom numbering for sugars.
One of the residues at the spnHL–substrate interface, Asn349, forms several hydrogen bonds with the carboxylate group in glucuronic acid and one salt bridge with the oxygen in the glycosidic bond. Also, Tyr408, which is located at the bottom of the cleft, forms a hydrogen bond to the oxygen atom in this glycosidic bond. NE2 in His399 is pointing towards the C5 hydrogen in the glucuronate residue of the substrate. Thus, we propose that the active center is composed of Asn349, His399 and Tyr408. The position and geometry of these three residues are shown in Figure 3B. A detailed discussion of these functional residues in HA degradation can be found in the mechanism section.
Three clustered hydrophobic residues, Trp291, Trp292 and Phe343, in the protein–substrate interface form an aromatic patch, suggesting that hydrophobic interactions may also be involved in the binding of the substrate. The substrate HA molecule possesses a hydrophobic patch built of eight or nine C–H units stretching along three neighboring sugar rings and presented on alternating sides of the HA chain (Scott, 1992). The concept of the hydrophobic patch was used to explain the ability of HA to interact with lipids and membranes and also to explain its characteristic formation of a viscous high-macromolecular-mass solution (Scott and Heatley, 1999). It was also suspected that these hydrophobic patches in HA may be involved in interactions with proteins including hyaluronate lyase (Park et al., 1997). From the spnHL crystal structure and the substrate modeling studies, the structural basis for this protein–HA hydrophobic interaction is clear. Because Trp and Phe are two hydrophobic amino acids, the aromatic patch in spnHL is also hydrophobic. Therefore, except for the charged interactions, part of the substrate–enzyme interaction is likely to be due to the match of the hydrophobic patch in the HA molecule to the aromatic patch in the spnHL cleft.
The substrate modeling studies suggest that for each sugar ring there are at least two charged residues located on either side of the cleft, which hold the sugar rings in position (Table II; Figure 5). These residues are Glu388 and Arg336 for sugar 1, Asp398 and Asn349 for sugar 2, Asn580 and Asn290 for sugar 3, Arg293 and Arg466 for sugar 4, and Asn468 and Arg243 for sugar 5. The aromatic patch interacts well with the hydrophobic patch on the HA chain (Figure 3B). The geometry of the aromatic patch suggests that it may play a far more important role than simply holding the HA chain in position in the cleft. The side chains of Trp292 and Phe343 are positioned to face the cleft cavity and in parallel to the sugar rings across two disaccharide units over the glycosidic linkage to be cleaved. At the opposite side of the cleft is the active center. Thus, the hydrophobic matching features of Trp292 and Phe343 are most likely to facilitate the spnHL selection of the cleavage site on the HA chain and accurately anchor the selected site into position. This interaction may also be responsible for determining the size of the degradation product, a disaccharide unit for the spnHL enzyme.

Fig. 5. spnHL–HA interface. The figure shows the modeled HA-binding site in the spnHL structure. The hexasaccharide segment of HA was colored in lavender. The six sugar rings are labeled 1–6. Sugars 1 and 2, 3 and 4, and 5 and 6 are the three disaccharide units. Sugars 1, 3 and 5 are 2–acetamino-2–deoxy-β–d–glucose residues. Sugars 2, 4 and 6 are β–d–glucuronic residues. This figure was prepared using the program RIBBONS (Carson, 1997).
The enzymatic products of hyaluronate lyase differ between lyases of different origins. Studies of the Streptomyces hyalurolyticus hyaluronate lyase had shown that octasaccharides, either saturated or unsaturated, were the minimum size substrates and that tetra- and hexasaccharides were not degraded further by this enzyme and remained as final products (Shimada and Matsumura, 1980), while gbsHL produced only disaccharide as its final HA degradation product (Baker et al., 1997). The product of the spnHL studied in this paper was identi– fied as pure disaccharide only using mass spectrometry. The same experiment was also performed on the S.hyalurolyticus hyaluronate lyase. Results confirmed that it produces tetra- and hexasaccharides and no disaccharides.
Mechanism of HA degradation
There have been many previous efforts to elucidate the mechanism of hyaluronate lyase action. When the degradation reaction was carried out in H218O, no 18O was incorporated into the product. In contrast, the action of the testicular hyaluronidase incorporates 18O into the oligosaccharide products (Ludowieg et al., 1961). Markowitz et al. (1959) found that during lyase action, tritium was released into the medium from HA when the glucuronic acid moiety was 3H-labeled, suggesting that hyaluronate lyase attacks the bond between O and C4 of glucuronic acid and results in a β–elimination reaction. Since the HA methyl ester is not cleaved by hyaluronate lyase and, therefore, a free carboxylate group is essential for hyaluronate lyase degradation (Greiling et al., 1975), Greiling proposed that histidine, with a pKa value of 6.1 for its side chain, would act successively as proton donor and acceptor and could supply the energy required to change the glucuronate moiety into the tautomeric carbeniate structure by splitting the glucosaminidic bond and forming a double bond between C4 and C5. Using the site-directed mutagenesis method, the histidine residue involved in the gbsHL catalytic activity was identified (Lin et al., 1997). This histidine residue is conserved among all known hyaluronate lyases (Figure 4). Gacesa (1987) proposed a three-step mechanism of action for the lyases and epimerases based on biochemical studies on alginate-modifying enzymes. The first step consists of the neutralization of the carboxylate anion on the glucuronic acid, which provides resonance stabilization of the enolate anion intermidiate. Then a general base-catalyzed abstraction of the C5 proton takes place with the change of hybridization of C5. The final step breaks the O–C4 glycosidic linkage.
Based on the spnHL crystal structure, the substrate interaction modeling and the geometry of the active center, a clear picture of the possible mechanism for the spnHL enzymatic action emerges. The proposed mechanism of spnHL enzymatic action is shown in Figure 6. The positive charges in the center of the cleft guide the HA chain into the cleft. The aromatic patch in the cleft accurately binds the cleavage sites on the substrate chain, anchoring them into their correct positions (Figure 6A). Since Asn349 forms several hydrogen bonds with the carboxylate group in the glucuronate residue of HA and one salt bridge with the glycosidic oxygen, these interactions lead to a more acidic C5 proton on the glucuronate residue. This proton, therefore, can more easily be removed. The nucleophilic NE2 atom of the His399 residue accomplishes the removal of this acidic, axially positioned C5 proton, resulting in the formation of an unsaturated C4–C5 bond on glucuronic acid (Figure 6B). The glucuronate moiety changes into an unstable intermediate that requires a leaving group on the C4 atom. There are two possible choices for the leaving group: the axially positioned hydrogen atom on C4 or the equatorially positioned O–C4 glycosidic bond. In the spnHL structure the hydroxyl group of the Tyr408 side chain is H–bonded to the oxygen in the glycosidic bond (Table II). Tyr408 provides a proton to this oxygen atom and, therefore, satisfies the electron requirement for breaking down this glycosidic bond (Figure 6C). As a result, rather than choosing the axial hydrogen on C4 as the leaving group, the glycosidic bond is broken. This completes the β–elimination process of substrate degradation. The negative patch in the cleft provides an electrostatic force for the release of the unsaturated and negatively charged disaccharide product. His399 loses the extra proton and Tyr408 obtains a proton from its microenvironment, leaving these residues ready for the next round of catalysis (Figure 6D). In total, the spnHL molecule removes one proton by His399 from the β–d–glucuronic residue, and provides a proton by Tyr408 to the 2–acetamino-2–deoxy-β–d–glucose residue at the reducing end. The enzyme returns to its original state by proton exchange with its microenvironment, probably with water molecules.

Fig. 6. Schematic diagram of the spnHL mechanism. (A) Two disaccharide units of HA are shown. The His399 imidazole side chain is in a nucleophilic attacking position to C5 in glucuronic acid. Tyr408 is H-bonded to the glycosidic oxygen atom. (B) His399 extracts the acidic C5 proton on the glucuronate residue, resulting in the formation of a double bond between C4 and C5. (C) Tyr408 provides a proton to the glycosidic oxygen and the glycosidic linkage is broken. (D) Products are released, His399 and Tyr408 balance their proton with their microenvironment and are ready for the next round of catalysis.
It was assumed in the earlier studies that this glycosidic bond should be in diaxial conformation and His399 alone acting as proton donor and acceptor in the HA enzymatic degradation (Greiling et al., 1975). The currently proposed mechanism model suggests that spnHL uses one more residue, Tyr408, to solve this chemical challenge. Tyr408 provides a proton to the oxygen in the glycosidic bond and determines the leaving group on glucuronic acid C4. This tyrosine is conserved in all hyaluronate lyase sequences and in the chondroitin lyase sequence as well (Figure 4).
The observation that a free carboxylate group on the glucuronate moiety of the substrate is essential for hyaluronate lyase degradation (Greiling et al., 1975) can also be explained by our proposed mechanism. A free carboxylate group is needed to interact with Asn349. This interaction will make the C5 proton more acidic and stabilize the intermediate. This also explains the enzymatic behavior of hyaluronate lyases on chondroitin and chondroitin sulfate (Baker et al., 1997). Chondroitin and chondroitin sulfate are usually sulfated at C4 or C6. Hence, only non-sulfated disaccharide units can be degraded since the sulfate group will prevent the interaction of Asn349 with the carboxylate group and hence the C5 proton can not be acidic.
The role of the β–domain in spnHL catalysis
In structurally related enzymes, glucoamylase (1ayx; Sevcik et al., 1998) and alginate lyase (1qaz; Yoon et al., 1999), only the α–domain is present. In the spnHL structure, there is only one residue from the β–domain, Asn580 in βL3, that is in direct contact with the substrate in our substrate modeling studies. The βL3 segment participates in the formation of one edge of the cleft and makes the shortest cross distance of the cleft with Asn290. Such an arrangement provides a clue for the possible function of the β–domain in spnHL enzymatic activity. Two sulfate ions were observed near to βL3 and βL15, and between β–sheet I and β–sheet II. These are the only two loops from the β–domain that form one edge of the cleft. The movement of βL3 and βL15 may act as a control of the entrance into the cleft, although a detailed mechanism for this movement is not clear at present. It is not known whether the sulfate ions are present in the native spnHL or if these two sulfate positions could be taken by other ions. But it is well known that spnHL enzymatic activity depends upon the presence of calcium ions, which are most likely to bind to the β–domain (Fethiere et al., 1999). Our spnHL activity experiment also confirmed that calcium ions are essential for the normal enzymatic activity of spnHL (Jedrzejas et al., 1998a). However, no calcium density was observed in the spnHL crystal structure. This is a result of EDTA added in the spnHL crystallization conditions to prevent the autodegradation of the spnHL sample (Berry et al., 1994). We suspect that the functional significance of the existence of the β–domain might be to modulate spnHL activity through controlling the substrate entrance, accomplished by the binding of small molecules such as calcium ions. A spnHL crystal structure with calcium ions is currently in preparation.
Mechanism evaluation by site-directed mutagenesis studies
To confirm our proposed catalytic mechanism of HA degradation by spnHL and to evaluate the role of the residues implicated in this mechanism, five single mutations, R243V, N349A, H399A, Y408F and N580G, were generated using site-directed mutagenesis as described in Materials and methods. Their enzymatic activities were 67, 6, 12, 0 and 115% relative to the spnHL enzyme activity, respectively (Figure 7). Asn349, His399 and Tyr408 are the three key residues for the spnHL catalytic activity in the proposed mechanism. Asn580 is the only residue from the β–domain in contact with the substrate in our substrate modeling study. Arg243 is believed to stabilize sugar 5 in the cleft by interacting with the third disaccharide unit of the substrate (Table II). All these five residues are strictly conserved among all known bacterial hyaluronate lyases (Figure 4). The enzymatic properties of the above mutants strongly support the proposed HA degradation mechanism.

Fig. 7. Enzymatic activities of spnHL mutants. Absorption changes at 232 nm were measured. An absorption coefficient of 5.5 × 103 mol–1cm–1 for the product was used. The enzymatic activity of mutants is scaled to the native spnHL activity (100%) and standard deviations are shown (n ≥2).
In the H399A mutant, Asn349 is still available and in contact with the glucuronic moiety carboxylate group to make the C5 proton more acidic. The 12% remaining activity of the H399A mutant indicates that this acidic proton on the C5 carbon atom may be lost slowly and steadily to its environment even without the proton acceptor from the imidazole group of His399. The small remaining activity for the N349A mutant (6% activity) is attributed to a compensatory effect from residue Asp398, which is also in contact with the carboxylate group on the glucuronate residue in the leaving disaccharide unit of the substrate. In the native spnHL, Asp398 may give a small contribution to make the C5 proton acidic too. In mutant N349A, when Asn349 is eliminated, the existence of Asp398 affords the enzyme weak but not omissible activity. The lethal mutation Y408F emphasizes the functional importance of this residue and provides strong support for our mechanism. The donation of a proton from Tyr408 to the glycosidic oxygen atom is essential for the breaking down of this glycosidic bond. The enzymatic activity result indicates that its function can not be compensated for by any other residues nearby or water molecules in the environment. The hydrogen bond formed between Tyr408 and the glycosidic oxygen atom is absolutely necessary in passing this proton. Therefore, Tyr408 is acting as a proton pump, pumping a proton from environmental water molecules to degrade the glyco– sidic bond in HA. This is the first time that the important role of Tyr408 and Asn349 in HA degradation has been proposed, identified, and further confirmed by site-directed mutagenesis.
Mutant R243V was prepared to test the relative roles of residues in contact with the substrate but outside of the active center. The substrate modeling studies revealed that Arg243 is one of the 23 residues that were implicated in substrate binding and is far from the active center (Figure 5). Arg243 also provides positive charges to the center of the cleft and forms several hydrogen bonds or salt bridges with the sugar rings of the third disaccharide unit of the modeled substrate (Figure 5). The R243V mutant retains 67% of the activity of the wild-type spnHL. This result indicates that the positive charge and proper substrate binding are important for the normal function of spnHL.
In our proposed mechanism, the β–domain is more important in modulating spnHL activity rather than directly involved in HA degradation. Asn580 plays a key role in controlling the substrate entrance into the cleft. Mutating Asn580 to Gly creates a wider entrance into the cleft, allowing the substrate to reach the active residues more easily. The observed increase in the activity of this mutant supported this hypothesis.
Conclusions and general implications
The spnHL crystal structure was determined at 1.56 Å resolution. It consists of two distinct domains, an α–helical and a β–sheet domain, connected by a short linker. A deep cleft transverses the middle of the molecule and is responsible for HA binding and degradation. Using the spnHL crystal structure, substrate modeling and site-directed mutagenesis, the mechanism of action of the enzyme becomes evident. The amino acids along the cleft are highly conserved among the known hyaluronate lyases and can be classified into four groups: (i) the positively charged residues; (ii) the aromatic patch; (iii) the active center; and (iv) the negative patch. Substrate modeling studies suggest that the substrate is bound to the cleft by charged residues aligned on two sides of each sugar ring. The aromatic patch selects the cleavage sites on the HA chain and anchors these sites accurately into the degradation position in the cleft. In the active center, Asn349 attracts electrons on the carboxylate group of glucuronic acid, making the C5 proton more acidic. This is followed by the removal of a relatively acidic C5 proton by the His399 imidazole side chain. This results in the formation of an unsaturated C4–C5 bond. The OH group of Tyr408 provides a proton to the glycosidic bond oxygen to break this linkage. The negative patch is related to the release of the HA enzymatic degradation products. The function of the β–domain is to modulate the activity of spnHL through binding to small ions such as calcium.
Compared with animals, bacteria use a different chemical solution for HA degradation. spnHL cleaves HA through β–elimination in which unsaturation of the glucuronate moiety in the leaving disaccharide product occurs. Hyaluronidases of animal origin hydrolyze HA by adding a water between the disaccharide unit, or by a process of transglycosylation (Highsmith et al., 1975). The activity of hyaluronidase from higher organisms, testicular-type hyaluronidase for example, is usually modulated by various activators (adrenalin, histamine and acid phosphatases) and inhibitors (antihistaminica, salicylate, heparin, dicumarene, vitamin C and flavonoids) (Menzel and Farr, 1998). Conversely, spnHL function is modulated by small, simple molecules such as calcium. Such a dramatic difference in the HA degradation mechanism between bacteria and animals may be the result of living competition in the course of evolution, in which the emergence of an alternative HA degradation pathway would provide animals with certain means of resistance to bacterial invasion. This evolutionary force drives the branching of hyaluronidases into different catalytic mechanisms in bacteria and animals.
The structure and sequence similarities of spnHL to chondroitin lyase, glucoamylase and alginate lyase suggest that the substrate binding and degradation mechanism proposed for spnHL may be significant for the understanding of other polysaccharide-degrading enzymes.
Materials and methods
Crystallization and data collection
The full sequence spnHL sample was not stable in vitro (Berry et al., 1994). Therefore, a fully functional truncated spnHL enzyme comprising residues Ala168–Ala893 of the native enzyme has been cloned, overexpressed in E.coli, and purified to homogeneity as described previously (Jedrzejas et al., 1998a,b). The spnHL crystal structure was solved by MIR methods. The crystals belong to the orthorhombic space group P212121 with cell dimensions a = 84.04, b= 104.28 and c = 101.76 Å.
A native data set at 1.56 Å resolution was collected at 100 K using cryocooling techniques and synchrotron radiation on the beamline X12C at Brookhaven National Laboratory. The crystal was treated with a cryoprotecting solution obtained by adding 30% xylitol to the crystal harvest solution (3.5 M ammonium sulfate, 200 mM sodium cacodylate pH 6.0). Diffraction data were collected using a Brandeis4K CCD detector at a wavelength of 0.950 Å and processed using the HKL package (Otwinowski and Minor, 1997) (Table III).
Table III. Statistics of diffraction data collection and structure refinement.
| Crystal system | orthorhombic |
| Space group | P212121 |
| Unit cell dimensions (Å) | a = 84.04, b = 104.28, c = 101.76 |
| Molecules/asymmetric unit | 1 |
| Matthews coefficient Vm (Å3/Da) | 2.81 |
| Data collection | |
| X-ray source | X12C synchrotron beamline at BNL |
| wavelength (Å) | 0.90 |
| detector | CCD |
| resolution limit (Å) | 1.56 |
| oscillation (°) | 0.6 |
| oscillation range (°) | 0–105 |
| measured reflections | 411 697 |
| unique reflections | 124 955 |
| completeness (last shell) (%) | 98.6 (96.0) |
| Rmerge (last shell) (%) | 4.1 (20.4) |
| final model | 719 aa, 529 water and 2 sulfate |
| resolution range for refinement (Å) | 40.0–1.56 |
| completeness (%) | 98.6 |
| R-factor (%) | 20.0 |
| Rfree (%) | 23.6 |
| Root-mean-square deviation | |
| bond (Å) | 0.011 |
| angle (°) | 2.38 |
| Average B-factor (Å2) | |
| protein | 23.88 |
| α–domain | 26.75 |
| β–domain | 19.22 |
| water | 31.19 |
Heavy atom derivatives were prepared by soaking the spnHL crystals in heavy atom solutions with 3.5 M ammonium sulfate or 1.9 M lithium sulfate and 200 mM sodium cacodylate at pH 6.0. Diffraction data for heavy atom derivatives were collected at room temperature on an R–axis IV detector using a Rigaku RU–300 rotating anode generator with CuKα radiation at a wavelength of 1.5418 Å. Reflections were reduced and scaled by the HKL package (Otwinowski and Minor, 1997). Parameters of diffraction data collection are summarized in Table IV.
Table IV. Summary of the heavy atom crystallographic analysis.
| MHAa | Se-Met | PIP1b | PIP2 | HgI2 | KAu(CN)2 | |
|---|---|---|---|---|---|---|
| Concentration (mM)c | 10 | 0.5 | saturated | saturated | 10 | |
| Soaking time | 3 days | 12 h | 1 h | 18 h | 30 h | |
| Cell dimensions | ||||||
| a (Å) | 84.15 | 84.36 | 84.22 | 84.24 | 84.27 | 84.16 |
| b (Å) | 104.95 | 104.24 | 103.31 | 104.29 | 103.38 | 103.21 |
| c (Å) | 104.36 | 104.98 | 104.41 | 105.00 | 105.40 | 104.73 |
| Resolution (Å) | 3.0 | 2.5 | 2.5 | 2.7 | 2.3 | 2.5 |
| Redundancy | 3.1 | 5.6 | 2.5 | 3.6 | 5.8 | 3.0 |
| Completeness (%) (last shell) | 97.3 (97.3) | 96.9 (96.0) | 99.9 (97.3) | 97.4 (98.2) | 99.8 (100.0) | 90. (90.4) |
| Rmerge (%) (last shell) | 12.8 (27.1) | 10.6 (20.3) | 6.5 (14.2) | 10.7 (24.7) | 9.3 (26.0) | 11.6 (31.7) |
| Riso (%) | 23.9 | 14.7 | 21.3 | 10.7 | 20.5 | 14.9 |
| MIR analysis | ||||||
| resolution (Å) | 20.0–3.2 | 20.0–3.0 | 20.0–3.0 | 20.0–3.0 | 20.0–3.0 | 20.0–3.0 |
| no. of sites | 3 | 22 | 4 | 2 | 1 | 3 |
| Rcullis | 0.80 | 0.72 | 0.82 | 0.88 | 0.92 | 0.88 |
| phasing power | 1.03 | 1.27 | 1.05 | 0.84 | 0.67 | 0.70 |
aMHA, methylmercury acetate.
bPIP, di-m-iodobi(ethylenediamine)-diplatinum.
cDerivatives were prepared in a solution of the heavy atoms in 3.5 M ammonium sulfate or 1.9 M lithium sulfate and 200 mM sodium cacodylate pH 6.0.
The spnHL Se-Met derivative was also produced according to the standard methodology (Doublie, 1997). Escherichia coli containing the spnHL clone was grown at 37°C in LB medium containing 100 μg/ml ampicillin to an OD600 of ∼0.8. The cells were harvested, washed three times with 37°C M9 medium (Sambrook et al., 1989) and resuspended in 1 l of M9 medium. After incubation at 37°C until an OD600 of ∼0.8, spnHL overexpression was induced by the addition of isopropyl-β-d-thiogalactopyranoside (IPTG) to 1 mM. High concentrations of lysine (100 mg/l), phenylalanine (100 mg/l), threonine (100 mg/l), isoleucine (50 mg/l), leucine (50 mg/l) and valine (50 mg/l) were added to the E.coli culture to inhibit the bacterial methionine biosynthesis. High concentrations of l-selenomethionine (60 mg/l) were added to incorporate Se–Met into the protein. The sample was purified with DEAE–Sepharose (Sigma), Superdex–200 and Mono–Q (Amersham Pharmacia Biotech) columns successively. The enzyme was concentrated to 5 mg/ml. Se–Met-substituted spnHL crystals were obtained at similar conditions to the native ones with the presence of an additional 2 mM EDTA and 10 mM dithiothreitol. The diffraction data were collected on an R-axis IV detector at a wavelength of 1.5418 Å. Reflections were reduced and scaled by the HKL package (Otwinowski and Minor, 1997). The presence of Se–Met residues in the spnHL sample was confirmed by matrix assisted laser desorption ionization (MALDI) time of flight (TOF) spectrometry experiments.
MIR phasing and primary structure model building
Heavy atom sites were determined from difference Patterson maps and difference Fourier maps using routines from the Xtalview package (McRee, 1995). The selenium sites were found from difference Fourier maps using the phases from the methylmercury acetate derivative. All heavy atom sites were refined with MLPHARE (CCP4, 1994). Initial phases from conventional MIRAS methods (Hendrickson, 1991) with a figure of merit of 0.61 (at 20.0–3.0 Å resolution range) were improved by solvent flattening (40% of content solvent), and the resolution was extended to 2.0 Å with the program DM (CCP4, 1994). Phases (50.0–2.0 Å resolution) after density modification were then combined with the Fobs derived from the native data.
The resulting electron density map was of good quality. Most β–sheets and some α–helices were clearly identified. A primary polyALA structure model was built based on the 2.0 Å MIRAS Fourier map and on maps calculated with MIRAS phases combined with phases from the partial polyALA model using SIGMAA (CCP4, 1994). The sequence was identified gradually and was built on graphics using O (Jones et al., 1991).
Structure refinement
X-PLOR protocols (Brunger and Krukowski, 1990) were used in spnHL structural refinement with 124 955 reflections (98% completeness) at 40.0–1.56 Å resolution range. The least sigma cut-off (0.001) was performed to include weak reflections in the calculation. One percent of reflections (1232 reflections) were assigned Rfree flags for the validation of the refinement process. Temperature factors for all atoms in the primary model were set to 15.00 Å2. Positional refinement was performed in the first few rounds. Side chains were manually fitted into a 2∣Fo∣ – 1∣Fc∣ map at the 1σ level on an INDIGO2 Silicon Graphics workstation using the O program (Jones et al., 1991) between each round of refinement calculations. Bulk solvent correction was applied. After most of the model was built, simulated annealing refinement and group B factor refinement were performed. Water molecules were picked using the standard protocol in X–PLOR (Brunger and Krukowski, 1990), where the electron densities were above the 4.0σ level in the difference map and were incorporated into the structure model only if it was clearly shown on the 2∣Fo∣ – 1∣Fc∣ map at the 1.0σ level and satisfied standard acceptance criteria. At the final stage of the refinement, torsion angle simulated annealing refinement and individual B factor refinement were performed.
Quality of the spnHL structural model
The final overall R-factor for the refined model was 0.200 and Rfree was 0.236. The final root-mean-square deviations from the standard bond geometry were 0.011 Å for bond lengths and 2.38° for bond angles. The average temperature factor for the whole model was 23.88 Å2, for α–domain 26.75 Å2, for β–domain 19.22 Å2 and for waters 31.19 Å2. In the Ramachandran plot (PROCHECK; Laskowski et al., 1993), 99.7% of residues fall in or near the energetically favored regions. However, two non-glycine residues, Thr400 and Lys674, are located in bad regions. They have well defined electron density. Both residues are exposed to solvent and are located at two sharp bends of the structure.
Out of all 731 residues in the expressed fully functional spnHL enzyme, 718 amino acid residues were modeled. Three residues at the N–terminus (Ala168, Ser169 and Val170), nine residues at the C–terminus (Leu890, Glu891, Gln892 and His6 tag), and one residue in the middle (Ser219) were not identified in electron density maps. The two termini protrude out of the molecular main body and the connected residues have high temperature factors. These 13 residues are not included in the present spnHL structure model. A total of 529 water molecules were incorporated.
spnHL product determination
One milliliter of 10 mg/ml solution of human umbilical cord HA (Sigma) in 50 mM sodium acetate and 10 mM CaCl2 buffered to pH 6.0 was incubated with 5 μg of spnHL sample at 37°C for 1.5 h. The mixture was then separated on a Superdex peptide HR 10/30 column (Amersham Pharmacia Biotech) using a 10 mM Tris–HCl pH 7.4 buffer, and monitored at 232 nm. The molecular weights of collected peak fractions were identified by electron-spray mass spectrometry using a Perkin Elmer-Sciex API3. The experiment was also performed with S.hyalurolyticus hyaluronate lyase under the same conditions.
Hexasaccharide model building
A hexasaccharide (a three disaccharide unit of HA) structure model was built based on the published X–ray coordinates (Winter and Arnott, 1977). The fractional coordinates were transformed into the standard PDB format. The coordinates of waters and calcium ions were not used. Necessary modifications were made. One sugar ring was moved manually from one end of the structure to the other to make the three disaccharide unit, the final hexasaccharide model. The hexasaccharide structure was then manually modeled into the cleft on graphics. These six sugar rings were easily fitted into the cleft space. The reducing end was positioned towards the His399 side of the cleft. The carboxylate group in the glucuronate residue of the first disaccharide unit and the acetyl group in sugar 3 were faced towards the bottom of the cleft. The substrate position was then adjusted in such a way to allow for interactions of C5 in the glucuronate residue in the first disaccharide unit with the His399 residue imidazole side chain in spnHL. Any other way of modeling the hexasaccharide into the cleft would cause more conflicts and less favorable hydrogen bonds and salt bridges. The model was then refined by molecular dynamic simulation using the program X-PLOR (Brunger and Krukowski, 1990) with the X–ray terms switched off and all protein atoms initially fixed and only hexasaccharide atoms allowed to move, followed by successive least-square refinement with the cleft residues initially restrained and then totally lifted. No large distortion or significant positional changes in the hexasaccharide structure occurred after refinement.
spnHL mutation
Single amino acid mutations of spnHL were created using the QuickChange™ Site-directed Mutagenesis Kit from Stratagene (La Jolla, CA). Reactions were carried out in a Perkin Elmer GeneAmp PCR system 2400 using synthetic oligonucleotide primers (sequences can be provided upon request). Mutant plasmids were purified using the Qiagen Plasmid Mini Kit (Valencia, CA) and were then sequenced to confirm the mutagenesis. The prepared plasmids were transformed into E.coli BL21 (DE3). Stocks of BL21 (DE3) cells harboring the mutant plasmids were frozen in 10% glycerol at –80°C. The mutants were then overexpressed and purified to homogeneity using methods described previously (Jedrzejas et al., 1998a, b).
Enzyme activity was measured using a Beckman DU 640 spectrometer based on the characteristic absorption at 232 nm of the double bond in the unsaturated product as described previously (Jedrzejas et al., 1998a, b). A 33 ng sample of enzyme was added to 1.0 ml of a 0.2 mg/ml solution of HA from human umbilical cord (Sigma) in 50 mM sodium acetate and 10 mM CaCl2 buffered to pH 6.0. Immediately following the addition of enzyme, the absorbance at 232 nm was measured at room temperature at 15 s intervals for 360 s. Enzyme activities were calculated as previously described (Jedrzejas et al., 1998a).
Acknowledgments
Acknowledgements
The authors would like to thank Dr Laurent Chantalat and R.Brandon Mewbourne for their assistance. Diffraction data of native crystals were collected at Brookhaven National Laboratory at the National Synchrotron Light Source on the X12C beamline. Mass spectrometry analysis was performed at the UAB Comprehensive Cancer Center shared facilities. The oligonucleotide primers were synthesized using the Oligonucleotide Synthesis Core Facility, UAB Comprehensive Cancer Center, and sequenced using the DNA Sequence Core Facility, Department of Microbiology, UAB. This work was supported by NIH grant No. R01 AI 44079 (M.J.J.).
References
- Baker J.R., Yu, H., Morrison, K., Averett, W.F. and Pritchard, D.G. (1997) Specificity of the hyaluronate lyase of group-B streptococcus toward unsulphated regions of chondroitin sulphate. Biochem. J., 327, 65–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry A.M., Lock, R.A., Thomas, S.M., Rajan, D.P., Hansman, D. and Paton, J.C. (1994) Cloning and nucleotide sequence of the Streptococcus pneumoniae hyaluronidase gene and purification of the enzyme from recombinant Escherichia coli.Infect. Immun., 62, 1101–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berstein F.C., Koetzle, T.F., Williams, G.J.B., Meyer, E.F., Jr, Brice, M.D., Rodgers, J.R., Kennard, O., Shimanouchi, T. and Tasumi, M. (1977) The Protein Data Bank: a computer-based archival file for macromolecular structures. J. Mol. Biol., 112, 535–542. [DOI] [PubMed] [Google Scholar]
- Boulnois G.J. (1992) Pneumococcal proteins and the pathogenesis of disease caused by Streptococcus pneumoniae.J. Gen. Microbiol., 138, 249–259. [DOI] [PubMed] [Google Scholar]
- Brunger A.T. and Krukowski, A. (1990) Slow-cooling protocols for crystallographic refinement by simulated annealing. Acta Crystallogr. A, 46, 585–593. [DOI] [PubMed] [Google Scholar]
- Carson M. (1997) RIBBONS. Methods Enzymol., 277, 493–505. [PubMed] [Google Scholar]
- Collaborative Computational Project Number 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr D., 50, 760–763. [DOI] [PubMed] [Google Scholar]
- Doublie S. (1997) Preparation of selenomethionyl proteins for phase determination. Methods Enzymol., 276, 523–527. [PubMed] [Google Scholar]
- Farrell A.M., Taylor, D. and Holland, K.T. (1995) Cloning, nucleotide sequence determination and expression of the Staphylococcus aureus hyaluronate lyase gene. FEMS Microbiol. Lett., 130, 81–85. [DOI] [PubMed] [Google Scholar]
- Fethiere J., Eggimann, B. and Cygler, M. (1999) Crystal structure of chondroitin AC lyase, a representative of a family of glycosaminoglycan degrading enzymes. J. Mol. Biol., 288, 635–641. [DOI] [PubMed] [Google Scholar]
- Fiore A.E., Levine, O.S., Elliott, J.A., Facklam, R.R. and Butler, J.C. (1999) Effectiveness of pneumococcal polysaccharide vaccine for preschool-age children with chronic disease. Emerg. Infect. Dis., 5, 828–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser J.R.E. and Laurent, T.C. (1989) Turnover and metabolism of hyaluronan. The biology of hyaluronan. Ciba Found. Symp., 143, 41–49. [PubMed] [Google Scholar]
- Gacesa P. (1987) Alginate-modifying enzymes. A proposed unified mechanism of action for the lyases and epimerases. FEBS Lett., 212, 199–202. [Google Scholar]
- Gagneux P. and Varki, A. (1999) Evolutionary considerations in relating oligosaccharide diversity to biological function. Glycobiology, 9, 747–755. [DOI] [PubMed] [Google Scholar]
- Greiling H., Stuhlsatz, H.W., Eberhard, T. and Eberhard, A. (1975) Studies on the mechanism of hyaluronate lyase action. Connect. Tissue Res., 3, 135–139. [DOI] [PubMed] [Google Scholar]
- Hendrickson W.A. (1991) Determination of macromolecular structures from anomalous diffraction of synchrotron radiation. Science, 254, 51–58. [DOI] [PubMed] [Google Scholar]
- Highsmith S., Garvin, J.H. and Chipman, D.M. (1975) Mechanism of action of bovine testicular hyaluronidase, mapping of the active site. J. Biol. Chem., 18, 7473–7480. [PubMed] [Google Scholar]
- Jedrzejas M.J., Mewbourne, R.B., Chantalat, L. and McPherson, D.T. (1998a) Expression and purification of Streptococcus pneumoniae hyaluronate lyase from Escherichia coli.Protein Expr. Purif., 13, 83–89. [DOI] [PubMed] [Google Scholar]
- Jedrzejas M.J., Chantalat, L. and Mewbourne, R.B. (1998b) Crystallization and preliminary X-ray analysis of Streptococcus pneumoniae hyaluronate lyase. J. Struct. Biol., 121, 73–75. [DOI] [PubMed] [Google Scholar]
- Jones T.A., Zou, J.Y., Cowan, S.W. and Kjeldgaard, M. (1991) Improved methods for the building of protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A, 47, 110–119. [DOI] [PubMed] [Google Scholar]
- Kraulis P.J. (1991) MOLSCRIPT: a program to produce both detailed and schematic plots of protein structure. J. Appl. Crystallogr., 24, 946–950. [Google Scholar]
- Laskowski R.A., MacArthur, M.W., Moss, D.S. and Thornton, J.M. (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr., 26, 283–291. [Google Scholar]
- Laurent T.C. (1970) Structure of hyaluronic acid. In Balazs,E.A. (ed.), Chemistry and Molecular Biology of the Intercellular Matrix 2. Academic Press, London, UK, pp. 703–732. [Google Scholar]
- Laurent T.C. and Fraser, R.E. (1992) Hyaluronan. FASEB J., 6, 2397–2404. [PubMed] [Google Scholar]
- Lin B., Hollingshead, S.K., Coligan, J.E., Egan, M.L., Baker, J.R. and Pritchard, D.G. (1994) Cloning and expression of the gene for group B streptococcal hyaluronate lyase. J. Biol. Chem., 269, 30113–30116. [PubMed] [Google Scholar]
- Lin B., Averett, W.F. and Pritchard, D.G. (1997) Identification of a histidine residue essential for enzymatic activity of group B streptococcal hyaluronate lyase. Biochem. Biophys. Res. Commun., 231, 379–382. [DOI] [PubMed] [Google Scholar]
- Lindahl U. (1978) Glycosaminoglycans and their binding to biological macromolecules. Annu. Rev. Biochem., 47, 385–417. [DOI] [PubMed] [Google Scholar]
- Linker A., Meyer, K. and Hoffman, P. (1955) The production of unsaturated uronices by bacterial hyaluronidases. J. Biol. Chem., 219, 13–25. [PubMed] [Google Scholar]
- Ludowieg J., Vennesland, B. and Dorfman, A. (1961) The mechanism of action of hyaluronidases. J. Biol. Chem., 236, 333–337. [PubMed] [Google Scholar]
- Markowitz A., Cifonelli, J.A. and Dorfman, A. (1959) The biosynthesis of hyaluronic acid by group A Streptococcus.J. Biol. Chem., 234, 2343–2247. [PubMed] [Google Scholar]
- McRee D.E. (1995) Xtalview Version 3.0. Computational Center for Macromolecular Structure, San Diego Supercomputer Center, San Diego, CA. [Google Scholar]
- Menzel E.J. and Farr, C. (1998) Hyaluronidase and its substrate hyaluronan: biochemistry, biological activities and therapeutic uses. Cancer Lett., 131, 3–11. [DOI] [PubMed] [Google Scholar]
- Nicholls A., Sharp, K. and Honig, B. (1991) Protein folding and association: insights from the interfacial thermodynamic properties of hydrocarbons. Proteins, 11, 281–293. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z. and Minor, W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol., 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Park Y., Cho, S. and Linhardt, R.J. (1997) Exploration of the action pattern of Streptomyces hyaluronate lyase using high-resolution capillary electrophoresis. Biochim. Biophys. Acta, 1337, 217–226. [DOI] [PubMed] [Google Scholar]
- Pearson W.R. (1990) Rapid and sensitive sequence comparison with FASTP and FASTA. Methods Enzymol., 183, 63–98. [DOI] [PubMed] [Google Scholar]
- Preiss J. and Ashwell, G. (1962) Alginate acid metabolism in bacteria. J. Biol. Chem., 237, 309–316. [PubMed] [Google Scholar]
- Sambrook J., Fritsch,E.T. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Scott J.E. (1992) Supramolecular organization of extracellular matrix glycosaminoglycans, in vitro and in the tissues. FASEB J., 6, 2639–2645. [PubMed] [Google Scholar]
- Scott J.E. and Heatley, F. (1999) Hyaluronan forms specific stable tertiary structures in aqueous solution: a 13C NMR study. Proc. Natl Acad. Sci. USA, 96, 4850–4855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevcik J., Hostinova, E., Gasperik, J., Solovicova, A., Wilson, K.S. and Dauter, Z. (1998) The crystal structure of glucoamylase from Saccharomycopsis fibruligera at 1.7 Å resolution. Acta Crystallogr. D, 54, 854–866. [DOI] [PubMed] [Google Scholar]
- Shimada E. and Matsumura, G. (1980) Degradation process of hyaluronic acid by Streptomyces hyaluronidase. J. Biochem., 88, 1015–1023. [DOI] [PubMed] [Google Scholar]
- Steiner B.M., Romero-Steiner, S., Cruce, D. and George, R. (1997) Cloning and sequencing of the hyaluronate lyase gene from Propionibacterium acnes.Can. J. Microbiol., 43, 315–321. [DOI] [PubMed] [Google Scholar]
- Winter W.T. and Arnott, S. (1977) Hyaluronic acid: the role of divalent cations in conformation and packing. J. Mol. Biol., 117, 761–784. [DOI] [PubMed] [Google Scholar]
- Yang B., Zhang, L. and Turley, E.A. (1993) Identification of two hyaluronan-binding domains in the hyaluronan receptor RHAMM. J. Biol. Chem., 268, 8617–8623. [PubMed] [Google Scholar]
- Yang B., Yang, B.L., Savani, R.C. and Turley, E.A. (1994) Identification of a common hyaluronan binding motif in the hyaluronan binding proteins RHAMM, CD44 and link protein. EMBO J., 13, 288–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon H.J., Mikami, B., Hashimoto, W. and Murata, K. (1999) Crystal structure of alginate lyase A1-III from Sphingomonas species A1 at 1.78 Å resolution. J. Mol. Biol., 290, 505–514. [DOI] [PubMed] [Google Scholar]