

Abstract
Exposure of yeast cells to an increase in external osmolarity induces a temporary growth arrest. Recovery from this stress is mediated by the accumulation of intracellular glycerol and the transcription of several stress response genes. Increased external osmolarity causes a transient accumulation of 1N and 2N cells and a concomitant depletion of S phase cells. Hypertonic stress triggers a cell cycle delay in G2 phase cells that appears distinct from the morphogenesis checkpoint, which operates in early S phase cells. Hypertonic stress causes a decrease in CLB2 mRNA, phosphorylation of Cdc28p, and inhibition of Clb2p-Cdc28p kinase activity, whereas Clb2 protein levels are unaffected. Like the morphogenesis checkpoint, the osmotic stress-induced G2 delay is dependent upon the kinase Swe1p, but is not tightly correlated with inhibition of Clb2p-Cdc28p kinase activity. Thus, deletion of SWE1 does not prevent the hypertonic stress-induced inhibition of Clb2p-Cdc28p kinase activity. Mutation of the Swe1p phosphorylation site on Cdc28p (Y19) does not fully eliminate the Swe1p-dependent cell cycle delay, suggesting that Swe1p may have functions independent of Cdc28p phosphorylation. Conversely, deletion of the mitogen-activated protein kinase HOG1 does prevent Clb2p-Cdc28p inhibition by hypertonic stress, but does not block Cdc28p phosphorylation or alleviate the cell cycle delay. However, Hog1p does contribute to proper nuclear segregation after hypertonic stress in cells that lack Swe1p. These results suggest a hypertonic stress-induced cell cycle delay in G2 phase that is mediated in a novel way by Swe1p in cooperation with Hog1p.
INTRODUCTION
The cell cycle is the orderly progression of events that allows a cell to replicate and segregate its genome. In yeast, progression of the cell cycle is driven by a single cyclin-dependent kinase, called Cdc2 in fission yeast and Cdc28p in budding yeast (reviewed in Hayles and Nurse, 1989; Nasmyth, 1993; Lew et al., 1997). Cdc28p and Cdc2 trigger cell cycle phase-specific events by differential association with obligatory-activating subunits called cyclins. During the cell cycle, cyclin levels are regulated by a complex system of transcriptional regulation and proteolysis (Deshaies, 1995; Nasmyth, 1996). In Saccharomyces cerevisiae, Cdc28p is activated in late G1 phase by the G1 cyclins Cln1p and Cln2p, and in G2/M phase by the B-type cyclins Clb1p and Clb2p.
Cell cycle progression is regulated by checkpoint mechanisms that monitor critical processes and delay the cell cycle to allow error-free completion of such processes before later cell cycle events are initiated. Some examples of checkpoint targets are DNA damage (Weinert and Hartwell, 1988), DNA replication (Weinert et al., 1994), kinetochore attachment to the mitotic spindle (Rudner and Murray, 1996), and bud morphogenesis (Lew and Reed, 1995). Inhibition of cyclin-dependent kinase activity (Cdc2 or Cdc28p) is the means by which some but not all checkpoint pathways enforce a delay in cell cycle progression. As an example, in fission yeast, DNA damage and defective DNA replication halt entry into mitosis by triggering the Wee1/Mik1-dependent phosphorylation of Cdc2 on tyrosine 15, which inhibits the kinase activity (Gould and Nurse, 1989; Lundgren et al., 1991; Rhind et al., 1997; Rhind and Russell, 1998). Cells that lack Wee1 are thus sensitive to DNA damage. Tyrosine phosphorylation of Cdc2 plays an important role in the normal progression of the cell cycle because wee1 mutants do not sense nutrient conditions properly and proceed through mitosis prematurely (reviewed in MacNeill and Nurse, 1997). Conversely, cells that lack Cdc25, a tyrosine phosphatase that removes the inhibitory phosphate from Y15 of Cdc2, arrest in G2 phase (Russell and Nurse, 1986). Regulation of Cdc25 phosphatase activity (Rhind et al., 1997) and regulation of Cdc25 localization (Lopez-Girona et al., 1999) are additional components of the response to DNA damage that affect Cdc2 phosphorylation.
Budding yeast contains an analogous regulatory circuit based on phosphorylation of the corresponding tyrosine (Y19) on Cdc28p by the Swe1p kinase, and removal of the inhibitory phosphate by the Mih1p phosphatase (Russell et al., 1989; Booher et al., 1993). However, deletion of SWE1 or a Y19F point mutation in Cdc28p has no obvious effect on growth or viability in S. cerevisiae (Sorger and Murray, 1992; Booher et al., 1993). Despite this, Cdc28p phosphorylation has been identified as a component of the bud morphogenesis checkpoint (Lew and Reed, 1995). Activation of this checkpoint by a cdc24-1 block in bud formation stimulates Swe1p-dependent tyrosine phosphorylation of Cdc28p, inhibition of Clb2p and Clb3p associated Cdc28p kinase activity, a delay in the accumulation of CLB2 mRNA and a consequent delay in G2 phase (Sia et al., 1996). Recent studies indicate that the morphogenesis checkpoint responds to disruption of the actin cytoskeleton (McMillan et al., 1998) and/or disruption of septin structures required for cytokinesis (Barral et al., 1999).
Yeast cells are normally exposed to a variety of environmental stresses. Some of these stresses, for example, oxidative stress (Lee et al., 1996; Wanke et al., 1999) and mild heat shock (Rowley et al., 1993; Raboy et al., 1999), cause arrest of the cell cycle in G1. Increases in extracellular osmolarity also induce a variety of cellular responses. Many of these responses are mediated by the HOG pathway (Banuett, 1998; Gustin et al., 1998) in one of five mitogen-activated protein (MAP) kinase signaling pathways in S. cerevisiae. Increasing the external osmolarity induces expression of osmoregulatory genes such as GPD1 and HOR2/GPP2 and stress response genes such as CTT1 and HSP12. Deletion of HOG pathway genes, such as the MAP kinase Hog1p, specifically blocks induction of these osmoregulation and stress response genes by osmotic stress, but has little or no effect on regulation of these genes by other stresses (Albertyn et al., 1994a; Schuller et al., 1994; Hirayama et al., 1995; Varela et al., 1995; Norbeck et al., 1996). Osmotic stress activation of Hog1p, measured as increases in Hog1p phosphorylation (Brewster et al., 1993) or Hog1p movement into the nucleus (Ferrigno et al., 1998; Reiser et al., 1999), occurs within minutes after increasing the osmolarity, correlating with the activation of gene expression. HOG pathway mutants have a complex phenotype, which includes cell morphological defects that suggest a lack of coordination between the cell cycle and cell growth (Brewster and Gustin, 1994). Not all responses to osmotic stress involve the HOG pathway. For example, the osmotic stress induced loss of actin cytoskeleton organization (Chowdhury et al., 1992) is unaffected by HOG pathway mutations (Brewster and Gustin, 1994).
Although previous work has suggested that hypertonic shock early in the cell cycle can affect Swe1p stability (Sia et al., 1998), the effect of hypertonic shock on cell cycle components and cell cycle progression has not been thoroughly investigated. In this report, we show that an increase in extracellular osmolarity also triggers a G2 delay that is similar to, but distinct from the morphogenesis checkpoint. The delay caused by hypertonic shock involves changes in Cdc28p phosphorylation and changes in Cdc28p enzymatic activity that are dependent on Swe1p and the MAP kinase Hog1p, respectively. These observations suggest a complex interplay between the Swe1p and Hog1p pathways underlies the cell cycle response to hypertonic stress.
MATERIALS AND METHODS
Yeast Strains and Growth Conditions
Cells were grown in YEP medium (1% yeast extract, 2% peptone) supplemented with either 2% dextrose or 2% raffinose where indicated. Galactose induction was accomplished by initial growth in YEP + raffinose, followed by addition of galactose to 2%. Strains used are listed in Table 1 and were derivatives of W303. The strain used as a positive control for the antiphospho-Cdc2 antibody was created by deleting MIH1 in W303 as described in Booher et al. (1993). Wee1 under the control of a galactose-inducible promoter was subsequently integrated into the genome of the mih1Δ::LEU2 strain as described in Russell et al. (1989).
Table 1.
Yeast strains
| W303 | MATa ura3-1 ade2-1 trp1-1 his3-11 leu2-3,112 can1-100 |
| MT588 | MATa CLB2-3XHA |
| MT588 swe1Δ | MATa CLB2-3XHA swe1Δ∷LEU2 |
| MT588 hog1Δ | MATa CLB2-3XHA hog1Δ∷TRP1 |
| MT588 gpd1Δ gpd2Δ | MATa CLB2-3XHA gpd1Δ∷LEU2 gpd2Δ∷URA3 |
| MT588 swe1Δ gpd1Δ gpd2Δ | MATa CLB2-3XHA swe1Δ∷LEU2 gpd1Δ∷TRP1 gpd2Δ∷URA3 |
| MT588 hog1Δ swe1Δ | MATa CLB2-3XHA hog1Δ∷TRP1 swe1Δ LEU2 |
| MT588 hog1Δ gpd1Δ gpd2Δ | MATa CLB2-3XHA hog1Δ∷TRP1 gpd1Δ∷LEU2 gpd2Δ∷URA3 |
| CDC28-HA | MATa CDC28-HA |
| CDC28-HAY19F | MATa CDC28-HAYF |
Flow Cytometry, Cell Synchrony, and Determination of Mitotic Index
DNA content of cell cultures was determined as described in Tyers et al. (1993) by using a FACScan flow cytometer (Becton Dickinson, San Jose, CA). Synchronized G1 cells were obtained from a 1.5-liter mid-log phase YEP + raffinose culture by using centrifugal elutriation as described in Tyers et al. (1993). Fractions that contained >95% unbudded cells were incubated at 30°C with shaking until ∼80% budded at which point the culture was split in two and NaCl was added to 0.4 M to one half, whereas the other control culture was untreated. At each time point, aliquots were removed to determine the percentage of divided cells, the percentage of cells that had undergone nuclear division but not cytokinesis, and the mitotic index (Lew and Reed, 1995). Mitotic index was determined as the percentage of cells with two nuclei plus the percentage of cells that had completed cell division.
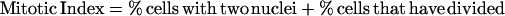 |
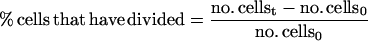 |
1 |
Nuclei were visualized by staining the DNA with 4,6-diamidino-2-phenylindole. The number of cells that had completed cell division was calculated by determining the cell density at each time point, by using a hemacytometer. For each parameter, a minimum of 100 cells was counted at each time point.
For alpha factor experiments cells were grown in YEPD to an A600 of 0.3. Alpha mating factor was added at 24 μg/ml and the cultures incubated at room temperature for 2.5 h. The cultures were released from arrest by two washes with fresh media.
Analysis of mRNA, Protein, and Kinase Activity
Total RNA was isolated and analyzed as described (Cross and Tinkelenberg, 1991). Clb2p-associated Cdc28p kinase activity was determined by using an in vitro immunoprecipitation kinase assay on strains with an unmarked genomic copy of a triple hemagglutinin (HA) epitope-tagged Clb2p. Cell extracts were prepared and histone H1 kinase assays performed on Clb2p immunoprecipitates prepared from 1 mg of total cellular protein as described in Tyers et al. (1993). 32P phosphorylated histone H1 was visualized by autoradiography and quantitated by using a MacBas Phosphorimager (Fuji, Stamford, CT) and a MacBas software package.
To analyze Cdc28p phosphorylation levels, cell extracts were prepared as described above and 1 mg of cell extract protein was incubated with p13suc1 agarose beads (Upstate Biotechnology, Lake Placid, NY). Alternatively, cell extracts were prepared as described above and HA-tagged Clb2p–Cdc28p complexes isolated by immunoprecipitation from 1 mg of cell extract protein by using the anti-HA 12CA5 monoclonal antibody plus protein A agarose beads. In each case, the beads were washed twice with lysis buffer, proteins extracted from the beads with SDS loading buffer, separated by SDS-PAGE, and transferred to nitrocellulose. Immunoblots were probed with an antibody specific for phospho-Tyr15 of Cdc2 (New England Biolabs, Beverly, MA). Antibody cross-reactivity with phospho-Cdc28p was initially determined by using samples containing hyperphosphorylated Cdc28p protein extracts from a GAL-wee1+ mih1Δ strain, and also samples containing nonphosphorylatable Cdc28p protein extracts from a cdc28Y19F mutant. Anti-phospho-Cdc2 antibodies showed significantly stronger specific immunoreactivity than anti-phosphotyrosine (our unpublished results). Antiphospho-Cdc2 antibody, anti-Cdc28p polyclonal antibody and 12CA5 monoclonal anti-HA antibody (Boehringer-Mannheim, Indianapolis, IN) were used at a 1:1000 dilution and detected with horseradish peroxidase secondary antibodies and enhanced chemiluminescence.
RESULTS
Hypertonic Shock Causes a Depletion of S Phase Cells
To determine the effect of mild osmotic stress conditions on cell cycle progression, flow cytometry was used to analyze the cellular DNA content of cultures that were exposed to 0.4 M NaCl (Figure 1A). We will subsequently refer to an increase in extracellular osmolarity as a hypertonic shock or stress. Within 30 min., addition of NaCl caused a reduction in the number of S phase cells with corresponding increases in the fraction of G1 and G2/M cells. Within 60 min, the S phase population had recovered and the histogram was very similar to that of an untreated control culture, indicating that the hypertonic stress response is transient. This result, along with an analysis of bud emergence and DNA content of synchronous cultures stressed in G1 (Figure 1B) suggested that hypertonic stress induces a cell cycle delay in both G2/M phase and G1 phase.
Figure 1.

(A) Hypertonic stress results in a depletion of S phase cells from an asynchronous culture. Log phase cultures of wild-type MT588 were stressed by the addition of NaCl to a final concentration of 0.4 M. Samples were taken at the indicated times, and processed as described (see MATERIALS AND METHODS). The cellular DNA was stained with propidium iodide and analyzed by flow cytometry. Histograms were generated by using WinMDI software for the PC. Similar results were obtained in three separate experiments. (B) Hypertonic stress delays exit from G1. Wild-type MT588 were arrested in alpha factor, released, and stressed at time zero by the addition of 0.4 M NaCl. Budding index and ratio of 1N to 2N cells was determined for no salt control (solid line) and 0.4 M NaCl treated (dashed line) and plotted versus time in minutes after addition of NaCl.
Hypertonic Shock Causes a Decrease in CLB2 mRNA, and Inhibits Clb2p-Cdc28p Kinase Activity, but Does Not Affect Clb2p Levels
The mRNA levels of different cyclins were examined before and after hypertonic shock. Treatment with 0.4 M NaCl caused a rapid but temporary decrease in the level of CLB2 mRNA (Figure 2A). Levels of other cyclin transcripts were also reduced, but not to the same degree as CLB2 (our unpublished results). Because Clb2p-Cdc28p kinase activity stimulates CLB2 transcription in a positive feedback loop (Amon et al., 1993), the decrease in CLB2 mRNA following hypertonic shock might be a consequence of reduced Clb2p-Cdc28p kinase activity. To test this idea, the Cdc28p kinase activity associated with wild-type levels of Clb2p was assayed in Clb2p immune complexes isolated from cells before and after exposure to hypertonic shock. These in vitro kinase assays used exogenously added histone H1 as a substrate. A decrease in Clb2p-Cdc28p kinase activity was observed following addition of 0.4 M NaCl, but with a substantial lag compared with repression of CLB2 mRNA (Figures 2B and 5A). Thus, it appears that hypertonic stress affects CLB2 transcription before its effects on Clb2p-Cdc28p kinase activity. Inhibition of Clb2p-Cdc28p activity was also observed when cells were stressed by 1 M sorbitol (our unpublished results).
Figure 2.

CLB2 mRNA levels and Clb2p-Cdc28p activity decrease in response to hypertonic shock. (A) Wild-type MT588 cells were grown to log phase and stressed by the addition of NaCl to 0.4 M. Samples were taken at the indicated times. Total RNA was isolated and probed for CLB2. The same membrane was reprobed for ACT1 as a loading control. (B) Clb2p-Cdc28p kinase activity was assessed by in vitro kinase assays by using histone H1 (HH1) as a substrate. Kinase assays were performed on immunoprecipitated Clb2p-HA Cdc28p complexes isolated from wild-type MT588 cells treated the same as in A. (C) Clb2-HA and Cdc28p levels in total cell lysate used for B were determined by Western blot analysis. (D) Stability of Clb2–HA/Cdc28p complexes was determined by immunoprecipitating Clb2-HAp from the samples used for B and C. Western blots were then probed with anti-Cdc28 and anti-HA antibodies. The band marked with ∗ is IgG. W303 cells lacking the HA tag on Clb2p were used as a control. Similar results were obtained in at least three separate experiments.
Figure 5.

Inhibition of Clb2p-Cdc28p kinase activity by exposure to 0.4 M NaCl. Wild-type MT588 (A), swe1Δ (B), hog1Δ (C), and hog1Δ swe1Δ (D) strains were grown to log phase and stressed by the addition of NaCl to 0.4 M. Clb2p–Cdc28p complexes were immunoprecipitated and assayed for activity before and after the addition of NaCl. A strain lacking the HA tag on Clb2p was used as a control. Similar results were observed in at least three independent experiments.
To determine whether hypertonic stress had an effect on the level of Clb2 or Cdc28 protein, the amount of Clb2-HAp and Cdc28p present in total cell lysate was determined by Western blot analysis. The addition of 0.4 M NaCl did not affect the abundance of either protein (Figure 2C). Although the abundance of Clb2p and Cdc28p was unaffected by hypertonic stress, we were interested in determining whether the Clb2p/Cdc28p complex remained intact following treatment with 0.4 M NaCl. Anti-HA antibodies were used to immunoprecipitate Clb2-HAp. Western blots were probed with anti-HA and anti-Cdc28 antibodies. The amount of Cdc28p that coprecipitated with Clb2-HAp was not affected by hypertonic stress, indicating that the Clb2–HA/Cdc28p complex remained intact (Figure 2D).
Hypertonic Stress Causes a G2 Delay
The accumulation of 2N cells, the decrease in CLB2 mRNA, and the inhibition of Clb2p-Cdc28p kinase activity all suggest that hypertonic stress causes a cell cycle delay in G2/M. To examine this possibility more directly, we studied the effect of osmotic stress on the timing of mitosis in synchronized cultures. Cultures enriched in G2 phase cells were obtained by first isolating small unbudded cells by centrifugal elutriation and then allowing the cells to grow synchronously until the population was >80% budded with only 10–20% of the cells having completed mitosis. The fraction of cells that had completed mitosis was scored as the total number of cells that had either two separated nuclei or had completed cell division (Lew and Reed, 1995). The culture was split in two and NaCl added to a final concentration of 0.4 M to half of the culture. Note that this regimen is in contrast to previous studies on the morphogenesis checkpoint, in which cells were perturbed well before bud emergence and completion of S phase (Lew and Reed, 1995; Sia et al., 1996, 1998). Initial experiments indicated that NaCl-treated cells showed an ∼30-min delay in the onset of mitosis compared with the control culture (our unpublished results).
To examine the effects of different regulatory protein mutations on the kinetics of cell cycle delay after osmotic stress, we used mutants that were defective in the osmoregulation response. The assumption behind this approach is that, like DNA damage-signaling pathways, osmotic stress-signaling pathways control two different responses, one delaying the cell cycle, and the other allowing adaptation to the stress. Thus, to determine the effects of different mutations on the kinetics of cell cycle delay per se, experiments are best done in a genetic background that prevents the adaptation to osmotic stress. Exposure of yeast to increases in the external osmolarity induces the synthesis and accumulation of glycerol, leading to a restoration of the osmotic gradient and resumption of cell growth (Brown, 1990; Ansell et al., 1997). This glycerol-based osmoregulation is eliminated by deletion of GPD1 and GPD2, enzymes responsible for catalyzing glycerol production, or deletion of HOG pathway genes that regulate expression of glycerol synthesis genes (Albertyn et al., 1994a,b; Eriksson et al., 1995). When a gpd1Δ gpd2Δ mutant was tested for cell cycle progression after osmotic stress, the mitotic index did not rise (Figure 3A), showing a sustained cell cycle arrest. It could be argued that the slow growth rate of these cells (Figure 3, A–E) in raffinose leads to reduced synchrony. However, the low number of cells that have completed mitosis (10–20%) coupled with the immediate rise in the mitotic index of no salt control samples to 100% within 120 min shows that these cultures are enriched for G2/M cells. Note also that S phase cells under osmotic stress appear to complete DNA synthesis before arresting at G2/M (Figure 1A), suggesting that any delay in cell cycle progression is therefore likely to be explained by a G2/M delay.
Figure 3.

Hypertonic shock causes a G2 delay. For each experiment, cells were grown and eluted in YEP + raffinose to reduce the incidence of clumping and disruption of media flow. The synchronized culture was grown until ∼80% budded. The culture was split and NaCl was added to one flask to a final concentration of 0.4 M (dashed line). The remaining flask served as a no-salt control (solid line). The mitotic index, an indication of the percentage of cells that have completed nuclear division, was determined as described (see MATERIALS AND METHODS). Time corresponds to minutes after addition of NaCl. (A) Cell cycle progression is halted when MT588 gpd1Δ gpd2Δ cells are stressed by 0.4 M NaCl. (B) swe1Δ gpd1Δ gpd2Δ cells do not halt cell cycle progression in response to osmotic stress, as indicated by the immediate increase in the mitotic index after the addition of 0.4 M NaCl. (C) Hypertonic stress causes a brief transient delay in cdc28Y19F-HA gpd1Δ gpd2Δ cells. (D) Deletion of HOG1 has a limited effect on the hypertonic stress-induced delay. (E) Response of swe1Δ hog1Δ cells to hypertonic stress was comparable to the response of swe1Δ gpd1Δ gpd2Δ cells. Similar results were obtained in at least three separate experiments.
Swe1p Is Required for the Hypertonic Stress-induced Cell Cycle Delay
The results from the mitotic index experiments with gpd1Δ gpd2Δ cells suggest a cell cycle delay in G2/M in response to hypertonic shock. To determine what pathway(s) might signal the cell cycle delay, we examined the effects of hypertonic shock by using various mutant strains. Swe1p phosphorylates and inhibits Cdc28p (Booher et al., 1993) and is required for a cell cycle delay in response to disruption of the normal organization of the actin cytoskeleton or the septin ring (Sia et al., 1996; McMillan et al., 1998; Barral et al., 1999). Because hypertonic stress also disrupts the actin cytoskeleton (Chowdhury et al., 1992; Brewster and Gustin, 1994) and apparently stabilizes Swe1p (Sia et al., 1998), the role of Swe1p in the response to hypertonic shock was examined. To determine the effect of hypertonic stress on cell cycle progression in a cell lacking Swe1p, the timing of mitosis was determined in synchronous cultures of swe1Δ gpd1Δ gpd2Δ mutants following addition of 0.4 M NaCl. In the absence of Swe1p, the mitotic index increased immediately following hypertonic shock, suggesting a defective delay mechanism (Figure 3B). This result contrasts with the results from gpd1Δ gpd2Δ cells, where the mitotic index did not rise after the addition of NaCl (Figure 3A). Hypertonic stress did however, slow the rate of the increase in the mitotic index compared with the control.
The finding that deletion of SWE1 caused a loss of the hypertonic stress-induced cell cycle delay suggests that Swe1p-mediated phosphorylation of Cdc28p is involved in this process. To further examine the role of Cdc28p phosphorylation, the timing of mitosis following addition of 0.4 M NaCl was determined using cells expressing a mutant Cdc28p that cannot be phosphorylated by Swe1p. In these cdc28Y19F-HA gpd1Δ gpd2Δ cells, hypertonic stress caused a transient cell cycle delay, where the mitotic index did not rise for a period of ∼30 min (Figure 3C). This response is different from that of gpd1Δ gpd2Δ and swe1Δ gpd1Δ gpd2Δ, suggesting that phosphorylation of Cdc28p may not be the only factor contributing to this stress response. Consistent with this hypothesis is the finding that Swe1p has a phosphorylation-independent role in triggering the morphogenesis checkpoint (McMillan et al., 1999).
Because hypertonic stress causes the activation of the HOG pathway (Brewster et al., 1993), we tested the role of the MAP kinase (MAPK) Hog1p in the hypertonic stress-induced G2/M delay. Hog1p is required for optimal recovery from hypertonic stress and activation of Hog1p induces glycerol accumulation through induction of GPD1. Thus, deletion of HOG1, like deletion of GPD1 and GPD2 inhibits the ability of cells to accumulate glycerol and restore the osmotic gradient (Brewster et al., 1993). After addition of 0.4 M NaCl to hog1Δ mutants (Figure 3D), there was only a small increase in mitotic index over that seen in gpd1Δ gpd2Δ cells (Figure 3A). Analysis of cell cycle progression after hypertonic stress in hog1Δ gpd1Δ gpd2Δ triple mutants gave similar results to hog1Δ single mutants (our unpublished results). Thus, the addition of 0.4 M NaCl did not completely prevent an increase in the mitotic index in hog1Δ cells, but the effect of deleting HOG1 was not nearly as strong as the effect of deleting SWE1. Deletion of SWE1 in the hog1Δ background resulted in a mitotic index similar to swe1Δ mutants (Figure 3E). Thus, Swe1p appears to be the main effector of the mitotic delay.
Swe1-dependent Phosphorylation of Cdc28 and Hog1-dependent Inhibition of Cdc28
The results from Figure 3 suggest that Swe1p is an important component of a hypertonic stress-induced cell cycle delay. As mentioned previously, Swe1p has been shown to phosphorylate Cdc28p (Booher et al., 1993). To examine whether hypertonic shock induces Cdc28p phosphorylation by Swe1p, p13Suc1-agarose beads were used to precipitate Cdc28p from cell lysates and the phosphorylation state of the coprecipitated Cdc28p determined by Western blot with anti-phospho-Cdc2 antibody. Hypertonic shock caused an increase in Cdc28p phosphorylation within 30 min, which was prevented by deletion of SWE1 (Figure 4A). In contrast, deletion of HOG1 did not block the hypertonic stress-induced tyrosine phosphorylation of Cdc28p. To examine more specifically the phosphorylation of Cdc28p in complex with Clb2p, Clb2p–HA/Cdc28p complexes were isolated by anti-HA immunoprecipitation. Immunoblots were probed with anti-phospho-Cdc2 antibodies and reprobed with anti-Cdc28 and anti-HA antibodies (Figure 4B). Consistent with the results from p13Suc1 coprecipitation experiments, Clb2p associated Cdc28p was also phosphorylated in response to hypertonic shock. In hog1Δ mutants, the degree of Cdc28p phosphorylation in Clb2p–HA/Cdc28p complexes after hypertonic stress was similar to that of wild type.
Figure 4.

Hypertonic stress induces the phosphorylation of Cdc28p in a Swe1p-dependent manner. Asynchronous cultures of MT588 were stressed by the addition of 0.4 M NaCl. (A) Cdc28p was precipitated with p13Suc1-agarose beads and analyzed by Western blot. Anti-phospho-Cdc2 was used to determine the phosphorylation state of Cdc28p (see MATERIALS AND METHODS). Membranes were stripped and reprobed with anti-Cdc28. A GAL: wee1+ mih1Δ strain and a Cdc28Y19F-HA strain were used as positive and negative controls, respectively. The decrease in mobility of the Y19F control is the result of the HA epitope tag. (B) Phosphorylation of Clb2-Hap-associated Cdc28p was examined by immunoprecipitating Clb2–HAp/Cdc28p complexes with anti-HA antibodies and protein A/G agarose beads. As a positive control, hyperphosphorylated Cdc28p from GAL: wee1+ mih1Δ cells was coprecipitated with p13Suc1-agarose beads. Western blots were probed with anti-phospho-Cdc2 antibodies and then stripped and reprobed with anti-Cdc28 and anti-HA antibodies. Results were consistent in three separate experiments.
The hypertonic shock-induced, Swe1p-dependent phosphorylation of Cdc28p, and the Swe1p-dependent cell cycle delay suggested that Swe1p might also be required for the observed inhibition of Clb2p-Cdc28p kinase activity after hypertonic stress (Figure 2B). To test this idea, the activity of Clb2p–Cdc28p complexes from swe1Δ cultures was determined after addition of 0.4 M NaCl. Surprisingly, the activity of Clb2p–Cdc28p complexes isolated from swe1Δ cells was inhibited by 0.4 M NaCl to an extent similar to that observed for wild type (Figure 5B).
We next determined whether the inhibition of Clb2p-HA-associated Cdc28p kinase activity under hypertonic stress would be affected in a HOG1 mutant. Deletion of HOG1 blocked the inhibition of kinase activity following hypertonic shock (Figure 5C). Swe1p did not significantly contribute to inhibition of Clb2p-Cdc28p because there was no additional elevation of Clb2-Cdc28 kinase activity in hog1Δ swe1Δ double mutants (Figure 5D). Taken together, the above results suggest that following hypertonic shock, the inhibition of Clb2p-Cdc28p activity does not strongly correlate with mitotic delay, and that the inhibition of Clb2-Cdc28p does not solely depend upon tyrosine phosphorylation.
Swe1 and Hog1 Prevent Mislocalization of Mitosis under Hypertonic Stress
In S. cerevisiae, mitosis takes place at the bud neck, resulting in the segregation of a single nucleus to the mother cell, and a single nucleus to the daughter cell. Wild-type cells exposed to hypertonic stress show no defects in segregation of nuclei to mother and daughter cells. However, as shown previously (Lew and Reed, 1995), hypertonic stress under conditions where the Swe1p checkpoint is overridden in wild-type cells, by the overexpression of Clb2p, results in the accumulation of binucleated mother cells under conditions of hypertonic stress. We found that deletion of SWE1 also caused cultures to accumulate binuclear mother cells under conditions of hypertonic stress (Figure 6A). Consistent with our finding that elimination of Cdc28p tyrosine phosphorylation does not completely abrogate the mitotic delay in response to hypertonic stress, we found that nuclear mislocalization occurred only rarely in cdc28Y19F cells under hypertonic stress conditions (Figure 6A). Although deletion of HOG1 has little effect on proper nuclear segregation in elutriation synchronized salt stressed cells, a hog1Δ swe1Δ double mutant accumulates more cells with mislocalized nuclei than a swe1Δ culture (Figure 6A). A role for Hog1p can be more easily seen when hog1Δ cells are exposed to hypertonic stress following release from a mating pheromone-induced G1 arrest. In these cultures ∼70% of large budded cells had two nuclei localized to the mother cell (Figure 6B). The increase in mislocalized nuclei in a swe1Δ hog1Δ double mutant could be the result of the inability to restore the osmotic gradient caused by a disrupted HOG pathway. However, a swe1Δ gpd1Δ gpd2Δ culture treated in the same manner did not give rise to the same increase in mislocalized nuclei (Figure 6A). Thus, Hog1p appears to share overlapping functions with Swe1p in enforcing proper nuclear segregation under conditions of hypertonic stress.
Figure 6.

(A) Mislocalization of nuclei in cells that are unable to delay in G2. Cells were treated as described in Figure 3. 4,6-Diamidino-2-phenylindole was used to stain the nuclei (swe1Δ hog1Δ cells shown). The same samples were also used to determine mitotic indices (Figure 3). The percentage of cells with separated nuclei that had not properly segregated (both nuclei in the mother cell) was determined ±SD from a minimum of three independent experiments. (B) Nuclear segregation is abnormal in hog1Δ cells following salt stress after release from alpha mating factor. Wild-type and hog1Δ cultures were arrested in G1 with alpha factor and released into fresh media. After 100 min, the culture was stressed by the addition of 0.4 M NaCl. The appearance of mislocalized nuclei in budded cells following the addition of NaCl was plotted versus time. A representative experiment with wild-type cells with or without salt (○ and ●, respectively) and hog1Δ cells with and without salt (▵ and ▴, respectively) is shown here.
DISCUSSION
Our study shows that hypertonic stress induces a cell cycle delay in both G1 phase and G2 phase. The G2 delay is correlated with both a Swe1p-dependent increase in tyrosine phosphorylation of Cdc28p and a Hog1p-dependent decrease in kinase activity of the Clb2p–Cdc28p complex. Comparison of the phenotypes of swe1Δ strains to that of hog1Δ strains suggests that the tyrosine phosphorylation state of Cdc28p rather than its actual kinase activity appears to be more important in regulating cell cycle progression. However, Hog1p may also have effects on cell cycle progression as revealed by the increase in nuclear mis-segregation in hog1Δ swe1Δ double mutants.
Comparison of the Hypertonic Stress Response and Activation of the Morphogenesis Checkpoint
Mutational or chemical disruption of the actin cytoskeleton in cells with small buds triggers a G2 arrest termed the morphogenesis checkpoint (Lew and Reed, 1995; Sia et al., 1996; McMillan et al., 1998; Barral et al., 1999). In this pathway, a family of Swe1p inhibitory kinases monitors proper septin ring assembly, and in the absence of a septin ring is held in an inactive state, thereby allowing Swe1p to phosphorylate and inhibit Clb–Cdc28 complexes (Barral et al., 1999). Once septin assembly occurs, bud morphogenesis is initiated, and Swe1 is inactivated. Deletion of SWE1 abrogates the checkpoint response (Lew and Reed, 1995). Hypertonic stress also disrupts the actin cytoskeleton (Chowdhury et al., 1992), and consequently activates the morphogenesis checkpoint (Lew and Reed, 1995; Sia et al., 1998).
Through the use of synchronized cultures, we have found that Swe1p may also impose a hypertonic stress-induced cell cycle delay. This mechanism appears to be sensitive to stresses that occur later in the cell cycle than those that can trigger the morphogenesis checkpoint (McMillan et al., 1998). swe1Δ cells exposed to hypertonic stress appear to enter mitosis with little or no initial delay (Figure 3), although the loss of Swe1p does not fully restore cell cycle kinetics, suggesting that other factors may also contribute to the response. The effects of hypertonic shock on cell cycle progression are similar to those caused by activation of the morphogenesis checkpoint: Clb2p-Cdc28p kinase activity is inhibited (Lew and Reed, 1995), the accumulation of CLB2 mRNA is delayed (Sia et al., 1996), and tyrosine phosphorylation of Cdc28p is increased (Lew and Reed, 1995). However, the osmotic stress-induced G2 delay and the morphogenesis checkpoint are not identical. In the latter case, a delayed induction of CLB2 mRNA and protein correlates with a delay in entry into mitosis. In contrast, increased osmolarity induces a large drop in CLB2 mRNA, but has little effect on Clb2p levels (Figure 2). This difference is likely to derive in part from the timing of the actin cytoskeleton-disrupting stress relative to the state of a Clb1/2p-Cdc28p positive feedback loop that regulates entry of cells into mitosis (Amon et al., 1993). Activation of the morphogenesis checkpoint early in the cell cycle prevents the onset of a CLB2 positive feedback loop. In these experiments, however, the hypertonic stress is occurring later in the cell cycle after the feedback loop has already been established. Because Clb2p is stable in post-G1 phase cells, Clb2p persists even though CLB2 expression and Clb2p-Cdc28p kinase activity are diminished by hypertonic stress (Amon et al., 1993). In addition, hypertonic stress induces tyrosine phosphorylation of Cdc28p within 30 min (Figure 4), whereas a significant increase in Cdc28p phosphorylation in a cdc24-1 strain is not detected until 2–3 h after a shift to the nonpermissive temperature (Lew and Reed, 1995).
The morphogenesis checkpoint monitors the septin ring/actin cytoskeleton only in G1 cells and early S phase cells with a very small bud, but not later in the cell cycle (Lew and Reed, 1995; McMillan et al., 1998). In part, this is because the abundance of Swe1p is controlled by cell cycle-regulated transcription and ubiquitin-dependent proteolysis, which results in higher expression in G1 and early S phase cells (McMillan et al., 1998; Sia et al., 1998). Although this finding appears at odds with the Swe1p dependence of the hypertonic delay in G2 phase cells, residual Swe1 protein persists in cells that are refractory to perturbation of the actin cytoskeleton (McMillan et al. 1998). The lower level of Swe1p in G2 phase may be sufficient to mediate the hypertonic stress-induced delay but not the morphogenesis checkpoint delay. Consistent with this hypothesis is the recent finding that Swe1p is present and important at later phases (G2/M) of the cell cycle (Sreenivasan and Kellogg, 1999). Thus, although the morphogenesis checkpoint and the cell cycle delay caused by hypertonic shock share Swe1p as a common component, the signals that regulate Swe1 may be different in each response. The dependence of the hypertonic stress response on components upstream of Swe1 remains to be determined.
Finally, we note that hypertonic stress delays the initiation of DNA synthesis in G1 phase cells, as shown by the depletion of S phase cells in asynchronous cultures and by the delayed onset of DNA replication in cultures released from mating pheromone arrest. In contrast, perturbation of septin/actin function does not affect the onset of DNA replication (Lew and Reed, 1995). Taken together, the above-mentioned results suggest that hypertonic stress does disrupt the actin cytoskeleton and, like activation of the morphogenesis checkpoint, does trigger a Swe1p-dependent cell cycle delay in response to osmotic stress early in the cell cycle. However, unlike the morphogenesis checkpoint, hypertonic shock also delays cell cycle progression at later points in the cell cycle.
Role of the HOG Pathway
Our data show that Hog1p is required for the decrease in Clb2p-Cdc28p kinase activity following hypertonic stress. Furthermore, deletion of HOG1 increases the fraction of swe1Δ cells that accumulate two nuclei in the mother cell after hypertonic stress. Taken together, these data suggest that Hog1p might work together with Swe1p to impose a cell cycle delay in G2 phase, which prevents aberrant nuclear segregation. However, our data are also consistent with Hog1p playing a role in the proper orientation of the mitotic spindle. In hog1Δ mutants, Swe1p might therefore be important to halt cell cycle progression until the spindle is properly aligned. This would account for the increase in mislocalized nuclei in elutriation synchronized hog1Δ swe1Δ double mutants exposed to a hypertonic stress (Figure 6A). Surprisingly, when hog1Δ cells are exposed to hypertonic stress following release from mating pheromone, ∼70% of large-budded cells had two nuclei localized within the mother cell even though Swe1p should be present in these cells (Figure 6B). However, exposure to mating pheromone might be affecting Swe1p levels or activity in these experiments.
The mechanisms whereby Swe1p and Hog1p affect cell cycle progression are unclear and somewhat contrary to expectations. First, the Hog1p-dependent inhibition of Clb2p-Cdc28p kinase activity (Figure 5) is puzzling in light of the finding that cell cycle progression is significantly slowed, if not completely delayed, even though Clb2p-Cdc28p activity remains high following hypertonic stress. In cells with an intact Swe1p pathway, Hog1p-dependent effects may simply be too subtle to detect by the mitotic progression assay (see above). Conversely, although Swe1p does stimulate tyrosine phosphorylation of Cdc28p, and is necessary for the hypertonic stress-induced delay, these events do not correlate with inhibition of Clb2p-Cdc28p activity. Thus, tyrosine phosphorylation of Cdc28p, and not inhibition of Clb2p-Cdc28p activity, appears to be a more critical factor for the cell cycle delay. However, there does appear to be phosphorylation-independent effects of Swe1p as seen by the partial cell cycle delay observed in a CDC28y19F strain.
Previous work has shown that Clb2p-Cdc28p kinase activity is also depressed in cells arrested by the morphogenesis checkpoint, but in this instance decreased amount of Clb2 protein and not phosphorylation of Cdc28p seems to account for most of the reduction in kinase activity (Lew and Reed, 1995). Because it appears that tyrosine phosphorylation does not dramatically alter Cdc28p activity, it may be that Swe1-dependent phosphorylation alters the localization and/or assembly of Cdc28p complexes with other factors under conditions of hypertonic stress. The mechanism whereby the Hog1p pathway contributes to Cdc28p inhibition is also not understood at this time.
Regulation of the Cell Cycle by MAPK Pathways
Numerous examples suggest that MAPK signaling is a common means to regulate cell cycle progression. In budding yeast, the MAPK Fus3p activates the CDK inhibitor Far1p, which eliminates Cln-Cdc28p activity and causes G1 arrest in preparation for mating (Peter et al., 1993; Tyers and Futcher, 1993; Peter and Herskowitz, 1994). In fission yeast, the stress-activated MAPK pathway based on the Hog1p homolog StyI (also called Spc1 or Phh1) positively regulates cell cycle progression by an unknown mechanism (Shiozaki and Russell, 1995). In animal cells, the embryonic cell cycle is controlled by MAPK pathways that mediate hormone-dependent stimulation of the cell cycle. Finally, the Ras-Erk MAPK pathway helps couple growth factor stimulation to G1 progression in mammalian tissue cultures cells (Weber et al., 1997). The mechanisms that couple MAPK activity to the cell cycle machinery, as in the Hog1p-mediated inhibition of Cdc28p activity, represent an important means by which cell division is controlled by environmental cues.
ACKNOWLEDGMENTS
We thank the members of the Gustin lab for their help and advice. Anti-phospho Cdc2 antibody graciously provided by A. Nelsbach of New England Biolabs. We also thank Drs. Adler, Lew, Futcher, Reed, and Mendenhall for supplying strains and plasmids. This work was supported by a grant from the National Science Foundation (MCB-9506987) and a grant from the National Cancer Institute of Canada.
REFERENCES
- Albertyn J, Hohmann S, Prior BA. Characterization of the osmotic-stress response in Saccharomyces cerevisiae: osmotic stress and glucose repression regulate glycerol-3-phosphate dehydrogenase independently. Curr Genet. 1994a;25:12–18. doi: 10.1007/BF00712960. [DOI] [PubMed] [Google Scholar]
- Albertyn J, Hohmann S, Thevelein JM, Prior BA. GPD1, which encodes glycerol-3-phosphate dehydrogenase, is essential for growth under osmotic stress in Saccharomyces cerevisiae, and its expression is regulated by the high-osmolarity glycerol response pathway. Mol Cell Biol. 1994b;14:4135–4144. doi: 10.1128/mcb.14.6.4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amon A, Tyers M, Futcher B, Nasmyth K. Mechanisms that help the yeast cell cycle clock tick: G2 cyclins transcriptionally activate G2 cyclins and repress G1 cyclins. Cell. 1993;74:993–1007. doi: 10.1016/0092-8674(93)90722-3. [DOI] [PubMed] [Google Scholar]
- Ansell R, Granath K, Hohmann S, Thevelein JM, Adler L. The two isoenzymes for yeast NAD+-dependent glycerol 3-phosphate dehydrogenase encoded by GPD1 and GPD2 have distinct roles in osmoadaptation and redox regulation. EMBO J. 1997;16:2179–2187. doi: 10.1093/emboj/16.9.2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banuett F. Signaling in the yeasts: an informational cascade with links to the filamentous fungi. Microbiol Mol Biol Rev. 1998;62:249–274. doi: 10.1128/mmbr.62.2.249-274.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barral Y, Parra M, Bidlingmaier S, Snyder M. Nim1-related kinases coordinate cell cycle progression with the organization of the peripheral cytoskeleton in yeast. Genes Dev. 1999;13:176–187. doi: 10.1101/gad.13.2.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booher RN, Deshaies RJ, Kirschner MW. Properties of S. cerevisiae wee1 and its differential regulation of p34CDC28 in response to G1 and G2 cyclins. EMBO J. 1993;12:3417–3426. doi: 10.1002/j.1460-2075.1993.tb06016.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewster JL, de Valoir T, Dwyer ND, Winter E, Gustin MC. An osmosensing signal transduction pathway in yeast. Science. 1993;259:1760–1763. doi: 10.1126/science.7681220. [DOI] [PubMed] [Google Scholar]
- Brewster JL, Gustin MC. Positioning of cell growth and division after osmotic stress requires a MAP kinase pathway. Yeast. 1994;10:425–439. doi: 10.1002/yea.320100402. [DOI] [PubMed] [Google Scholar]
- Brown AD. Microbial Water Stress Physiology. Principles, and Perspectives. New York: John Wiley & Sons; 1990. [Google Scholar]
- Chowdhury S, Smith KW, Gustin MC. Osmotic stress and the yeast cytoskeleton: phenotype-specific suppression of an actin mutation. J Cell Biol. 1992;118:561–571. doi: 10.1083/jcb.118.3.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross FR, Tinkelenberg AH. A potential positive feedback loop controlling CLN1 and CLN2 gene expression at the start of the yeast cell cycle. Cell. 1991;65:875–883. doi: 10.1016/0092-8674(91)90394-e. [DOI] [PubMed] [Google Scholar]
- Deshaies RJ. The self-destructive personality of a cell cycle in transition. Curr Opin Cell Biol. 1995;7:781–789. doi: 10.1016/0955-0674(95)80061-1. [DOI] [PubMed] [Google Scholar]
- Eriksson P, Andre L, Ansell R, Blomberg A, Adler L. Cloning and characterization of GPD2, a second gene encoding sn-glycerol 3-phosphate dehydrogenase (NAD+) in Saccharomyces cerevisiae, and its comparison with GPD1. Mol Microbiol. 1995;17:95–107. doi: 10.1111/j.1365-2958.1995.mmi_17010095.x. [DOI] [PubMed] [Google Scholar]
- Ferrigno P, Posas F, Koepp D, Saito H, Silver PA. Regulated nucleo/cytoplasmic exchange of HOG1 MAPK requires the importin beta homologs NMD5 and XPO1. EMBO J. 1998;17:5606–5614. doi: 10.1093/emboj/17.19.5606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould KL, Nurse P. Tyrosine phosphorylation of the fission yeast cdc2 protein kinase regulates entry into mitosis. Nature. 1989;32:39–45. doi: 10.1038/342039a0. [DOI] [PubMed] [Google Scholar]
- Gustin MC, Albertyn J, Alexander M, Davenport K. MAP kinase pathways in the yeast Saccharomyces cerevisiae. Microbiol Mol Biol Rev. 1998;62:1264–1300. doi: 10.1128/mmbr.62.4.1264-1300.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayles J, Nurse P. A review of mitosis in the fission yeast Schizosaccharomyces pombe. Exp Cell Res. 1989;184:273–286. doi: 10.1016/0014-4827(89)90327-3. [DOI] [PubMed] [Google Scholar]
- Hirayama T, Maeda T, Saito H, Shinozaki K. Cloning and characterization of seven cDNAs for hyperosmolarity-responsive (HOR) genes of Saccharomyces cerevisiae. Mol Gen Genet. 1995;249:127–138. doi: 10.1007/BF00290358. [DOI] [PubMed] [Google Scholar]
- Lee J, Romeo A, Kosman DJ. Transcriptional remodeling and G1 arrest in dioxygen stress in Saccharomyces cerevisiae. J Biol Chem. 1996;271:24885–24893. doi: 10.1074/jbc.271.40.24885. [DOI] [PubMed] [Google Scholar]
- Lew DJ, Reed SI. A cell cycle checkpoint monitors cell morphogenesis in budding yeast. J Cell Biol. 1995;129:739–749. doi: 10.1083/jcb.129.3.739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lew DJ, Weinert T, Pringle J. Cell cycle control in Saccharomyces cerevisiae. In: Pringle J, Broach J, Jones E, editors. The Molecular and Cellular Biology of the Yeast Saccharomyces: Cell Cycle and Cell Biology. Plainview, NY: Cold Spring Harbor Laboratory Press; 1997. pp. 607–695. [Google Scholar]
- Lopez-Girona A, Furnari B, Mondesert O, Russell P. Nuclear localization of Cdc25 is regulated by DNA damage and a 14-3-3 protein. Nature. 1999;397:172–175. doi: 10.1038/16488. [DOI] [PubMed] [Google Scholar]
- Lundgren K, Walworth N, Booher R, Dembski M, Kisrchner M, Beach D. mik1 and wee1 cooperate in the inhibitory tyrosine phosphorylation of cdc2. Cell. 1991;64:1111–1122. doi: 10.1016/0092-8674(91)90266-2. [DOI] [PubMed] [Google Scholar]
- MacNeill SA, Nurse P. Cell cycle control in fission yeast. In: Pringle JR, Broach JR, Jones WW, editors. The Molecular, and Cellular. Biology of the Yeast Saccharomyces. Plainview, NY: Cold Spring Harbor Laboratory Press; 1997. pp. 697–763. [Google Scholar]
- McMillan JN, Sia RA, Bardes ES, Lew DJ. Phosphorylation-independent inhibition of Cdc28p by the tyrosine kinase Swe1p in the morphogenesis checkpoint. Mol Cell Biol. 1999;19:5981–5990. doi: 10.1128/mcb.19.9.5981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan JN, Sia RAL, Lew DJ. A morphogenesis checkpoint monitors the actin cytoskeleton in yeast. J Cell Biol. 1998;142:1487–1499. doi: 10.1083/jcb.142.6.1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasmyth K. Control of the yeast cell cycle by the Cdc28 protein kinase. Curr Opin Cell Biol. 1993;5:166–179. doi: 10.1016/0955-0674(93)90099-c. [DOI] [PubMed] [Google Scholar]
- Nasmyth K. At the heart of the budding yeast cell cycle. Trends Genet. 1996;12:405–412. doi: 10.1016/0168-9525(96)10041-x. [DOI] [PubMed] [Google Scholar]
- Norbeck J, Pahlman AK, Akhtar N, Blomberg A, Adler L. Purification and characterization of two isoenzymes of DL-glycerol-3-phosphatase from Saccharomyces cerevisiae. Identification of the corresponding GPP1 and GPP2 genes and evidence for osmotic regulation of Gpp2p expression by the osmosensing mitogen-activated protein kinase signal transduction pathway. J Biol Chem. 1996;271:13875–13881. doi: 10.1074/jbc.271.23.13875. [DOI] [PubMed] [Google Scholar]
- Peter M, Gartner A, Horecka J, Ammerer G, Herskowitz I. FAR1 links the signal transduction pathway to the cell cycle machinery in yeast. Cell. 1993;73:747–760. doi: 10.1016/0092-8674(93)90254-n. [DOI] [PubMed] [Google Scholar]
- Peter M, Herskowitz I. Direct inhibition of the yeast cyclin-dependent kinase Cdc28-Cln by Far1. Science. 1994;265:1228–1231. doi: 10.1126/science.8066461. [DOI] [PubMed] [Google Scholar]
- Raboy B, Marom A, Dor Y, Kulka RG. Heat-induced cell cycle arrest of Saccharomyces cerevisiae: involvement of the RAD6/UBC2 and WSC2 genes in its reversal. Mol Microbiol. 1999;32:729–739. doi: 10.1046/j.1365-2958.1999.01389.x. [DOI] [PubMed] [Google Scholar]
- Reiser V, Ruis H, Ammerer G. Kinase activity-dependent nuclear export opposes stress-induced nuclear accumulation and retention of Hog1 mitogen-activated protein kinase in the budding yeast Saccharomyces cerevisiae. Mol Biol Cell. 1999;10:1147–1161. doi: 10.1091/mbc.10.4.1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhind N, Furnari B, Russell P. Cdc2 tyrosine phosphorylation is required for the DNA damage checkpoint in fission yeast. Genes Dev. 1997;11:504–511. doi: 10.1101/gad.11.4.504. [DOI] [PubMed] [Google Scholar]
- Rhind N, Russell P. Tyrosine phosphorylation of cdc2 is required for the replication checkpoint in Schizosaccharomyces pombe. Mol Cell Biol. 1998;18:3782–3787. doi: 10.1128/mcb.18.7.3782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowley A, Johnston GC, Butler B, Werner-Washburne M, Singer RA. Heat shock-mediated cell cycle blockage and G1 cyclin expression in the yeast Saccharomyces cerevisiae. Mol Cell Biol. 1993;13:1034–1041. doi: 10.1128/mcb.13.2.1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudner AD, Murray AW. The spindle assembly checkpoint. Curr Opin Cell Biol. 1996;8:773–780. doi: 10.1016/s0955-0674(96)80077-9. [DOI] [PubMed] [Google Scholar]
- Russell P, Moreno S, Reed SI. Conservation of mitotic controls in fission and budding yeasts. Cell. 1989;57:295–303. doi: 10.1016/0092-8674(89)90967-7. [DOI] [PubMed] [Google Scholar]
- Russell P, Nurse P. cdc25+ functions as an inducer in the mitotic control of fission yeast. Cell. 1986;45:145–153. doi: 10.1016/0092-8674(86)90546-5. [DOI] [PubMed] [Google Scholar]
- Schuller C, Brewster JL, Alexander MR, Gustin MC, Ruis H. The HOG pathway controls osmotic regulation of transcription via the stress response element (STRE) of the Saccharomyces cerevisiae CTT1 gene. EMBO J. 1994;13:4382–4389. doi: 10.1002/j.1460-2075.1994.tb06758.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiozaki K, Russell P. Cell-cycle control linked to extracellular environment by MAP kinase pathway in fission yeast. Nature. 1995;378:739–743. doi: 10.1038/378739a0. [DOI] [PubMed] [Google Scholar]
- Sia RA, Bardes ES, Lew DJ. Control of Swe1p degradation by the morphogenesis checkpoint. EMBO J. 1998;17:6678–6688. doi: 10.1093/emboj/17.22.6678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sia RA, Herald HA, Lew DJ. Cdc28 tyrosine phosphorylation and the morphogenesis checkpoint in budding yeast. Mol Biol Cell. 1996;7:1657–1666. doi: 10.1091/mbc.7.11.1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorger PK, Murray WA. S-phase feedback control in budding yeast independent of tyrosine phosphorylation of p34cdc28. Nature. 1992;355:365–371. doi: 10.1038/355365a0. [DOI] [PubMed] [Google Scholar]
- Sreenivasan A, Kellogg D. The elm1 kinase functions in a mitotic signaling network in budding yeast. Mol Cell Biol. 1999;12:7983–7994. doi: 10.1128/mcb.19.12.7983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyers M, Futcher B. Far1 and Fus3 link the mating pheromone signal transduction pathway to three G1-phase Cdc28 kinase complexes. Mol Cell Biol. 1993;13:5659–5669. doi: 10.1128/mcb.13.9.5659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyers M, Tokiwa G, Futcher B. Comparison of the Saccharomyces cerevisiae G1 cyclins: Cln3 may be an upstream activator of Cln1, Cln2 and other cyclins. EMBO J. 1993;12:1955–1968. doi: 10.1002/j.1460-2075.1993.tb05845.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varela JC, Praekelt UM, Meacock PA, Planta RJ, Mager WH. The Saccharomyces cerevisiae HSP12 gene is activated by the high-osmolarity glycerol pathway and negatively regulated by protein kinase A. Mol Cell Biol. 1995;15:6232–6245. doi: 10.1128/mcb.15.11.6232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanke V, Accorsi K, Porro D, Esposito F, Russo T, Vanoni M. In budding yeast, reactive oxygen species induce both RAS-dependent and RAS-independent cell cycle-specific arrest. Mol Microbiol. 1999;32:753–764. doi: 10.1046/j.1365-2958.1999.01391.x. [DOI] [PubMed] [Google Scholar]
- Weber JD, Hu W, Jefcoat SC, Jr, Raben DM, Baldassare JJ. Ras-stimulated extracellular signal-related kinase 1 and RhoA activities coordinate platelet-derived growth factor-induced G1 progression through the independent regulation of cyclin D1 and p27. J Biol Chem. 1997;272:32966–32971. doi: 10.1074/jbc.272.52.32966. [DOI] [PubMed] [Google Scholar]
- Weinert TA, Hartwell LH. The RAD9 gene controls the cell cycle response to DNA damage in Saccharomyces cerevisiae. Science. 1988;241:317–322. doi: 10.1126/science.3291120. [DOI] [PubMed] [Google Scholar]
- Weinert TA, Kiser GL, Hartwell LH. Mitotic checkpoint genes in budding yeast and the dependence of mitosis on DNA replication and repair. Genes Dev. 1994;8:652–665. doi: 10.1101/gad.8.6.652. [DOI] [PubMed] [Google Scholar]