

Abstract
We have reported that a population of chromaffin cell mitochondria takes up large amounts of Ca2+ during cell stimulation. The present study focuses on the pathways for mitochondrial Ca2+ efflux. Treatment with protonophores before cell stimulation abolished mitochondrial Ca2+ uptake and increased the cytosolic [Ca2+] ([Ca2+]c) peak induced by the stimulus. Instead, when protonophores were added after cell stimulation, they did not modify [Ca2+]c kinetics and inhibited Ca2+ release from Ca2+-loaded mitochondria. This effect was due to inhibition of mitochondrial Na+/Ca2+ exchange, because blocking this system with CGP37157 produced no further effect. Increasing extramitochondrial [Ca2+]c triggered fast Ca2+ release from these depolarized Ca2+-loaded mitochondria, both in intact or permeabilized cells. These effects of protonophores were mimicked by valinomycin, but not by nigericin. The observed mitochondrial Ca2+-induced Ca2+ release response was insensitive to cyclosporin A and CGP37157 but fully blocked by ruthenium red, suggesting that it may be mediated by reversal of the Ca2+ uniporter. This novel kind of mitochondrial Ca2+-induced Ca2+ release might contribute to Ca2+ clearance from mitochondria that become depolarized during Ca2+ overload.
INTRODUCTION
Mitochondrial Ca2+ fluxes are the subject of renewed attention because of their possible involvement on modulation of Ca2+ signals and secretion (Montero et al., 2000) and on triggering of apoptosis (Di Lisa and Bernardi, 1998; Green and Reed, 1998; Crompton, 1999). Mitocondrial [Ca2+] ([Ca2+]M) is low in resting cells but, during cell activation, mitochondria take up large amounts of Ca2+ from local cytosolic [Ca2+] ([Ca2+]c) microdomains generated in the vicinity of plasma membrane Ca2+ channels (Rizzuto et al., 1993, 1994; Brini et al., 1997; Montero et al., 2000). The increase in [Ca2+]M activates mitochondrial dehydrogenases and respiration (McCormack et al., 1990; Robb-Gaspers et al., 1998; Duchen, 1999), thus matching the rate of ATP production to the increasing energy demands. On the other hand, neurons and chromaffin cell mitochondria accumulate large amounts of Ca2+ during cell stimulation (Werth and Thayer, 1994; Herrington et al., 1996; Park et al., 1996; Babcock et al., 1997; White and Reynolds, 1997). This large mitochondrial Ca2+ uptake may damp the [Ca2+]c increase thus modulating secretion. In fact, using mitochondrially targeted aequorin, we have shown that chromaffin cell mitochondria can take up Ca2+ up to near the millimolar level during stimulation and that this Ca2+ uptake regulates catecholamine release (Montero et al., 2000).
Mitochondria take up Ca2+ through the Ca2+ uniporter, a specific Ca2+ pathway driven by the electrical potential difference across the mitochondrial membrane. The mitochondrial membrane potential, −150 to −180 mV, is enough to accumulate Ca2+ within the mitochondrial matrix up to 5–6 orders of magnitude above [Ca2+]c. However, the Ca2+ uniporter requires a high extramitochondrial [Ca2+]c to be activated (Kroner, 1986; Igbavboa and Pfeiffer, 1988; Gunter and Pfeiffer, 1990; Rizzuto et al., 1993; Xu et al., 1997; Bernardi, 1999; Csordás et al., 1999; Montero et al., 2000), a condition that is fulfilled only for a brief period of time during cell stimulation. Protonophores collapse mitochondrial membrane potential and prevent mitochondrial Ca2+ accumulation. Consistently, it has been shown that cell stimulation produces much larger [Ca2+]c increases in cells treated with protonophores (Friel and Tsien, 1994; Werth and Thayer, 1994; Hehl et al., 1996; Herrington et al., 1996; Park et al., 1996; Babcock et al., 1997; Tang and Zucker, 1997; Giovanucci et al., 1999; Pivovarova et al., 1999). Moreover, addition of protonophores immediately after cell stimulation produces in many cells a large increase in [Ca2+]c due to release of the Ca2+ accumulated into mitochondria (Werth and Thayer, 1994; Hehl et al., 1996; Herrington et al., 1996; Tang and Zucker, 1997; White and Reynolds, 1997).
The pathway for this protonophore-induced mitochondrial Ca2+ release is unclear. Although the Ca2+ uniporter shares most of the properties of a channel, flux through this system is not easily reverted after mitochondrial depolarization (Petronilli et al., 1993; Bernardi, 1999). Normally, Ca2+ exit from mitochondria occurs via Na+/Ca2+ exchange and an Na+-independent pathway that may include a H+/Ca2+ exchange. There is evidence that both kinds of transporters are electrogenic, and stoichiometries of 3Na+/1Ca2+ (Baysal et al., 1994; Jung et al., 1995) and more than 2H+/Ca2+ have been proposed (Gunter et al., 1991). If this is correct, Ca2+ release through the exchangers would be favored by the mitochondrial membrane potential and hence it should be inhibited on mitochondrial membrane depolarization by protonophores. In fact, it has been shown that mitochondrial Na+-independent Ca2+ exchange pathways are inhibited by mitochondrial depolarization (Bernardi and Azzone, 1982, 1983).
Another relevant mitochondrial transport mechanism, which could be activated by protonophores, is the so-called mitochondrial permeability transition pore (MPT, reviewed in Zoratti and Szabó, 1995; Di Lisa and Bernardi, 1998; Ichas and Mazat, 1998; Crompton, 1999). MPT allows passage of molecules with molecular weights below 1500 Da and is inhibited by cyclosporin A. A series of factors, including high mitochondrial Ca2+, mitochondrial depolarization, oxidized state of pyridine nucleotides and of critical dithiols, etc., favor pore opening, whereas the opposite conditions stabilize it in the closed conformation. Opening of MPT triggers a fast release of Ca2+ and mitochondrial metabolites, and prolonged opening of this pathway has been associated to the release of apoptotic factors and cell death. MPT may also be involved in the development of the so-called mitochondrial Ca2+-induced Ca2+ release (mCICR) responses (Ichas et al., 1997), and has been shown to be activated by protonophores after Ca2+-loading of mitochondria (Bernardi et al., 1984; Igbavboa and Pfeiffer, 1988; Bernardi, 1992).
Here we use a targeted aequorin to investigate the mechanisms for Ca2+ exit from mitochondria of intact chromaffin cells, previously loaded with Ca2+ by high-K+ depolarizing stimuli. Surprisingly, protonophores did not accelerate but inhibited Ca2+ exit from mitochondria, probably by interfering with electrogenic Na+/Ca2+ exchange. In these cells, with depolarized and Ca2+-loaded mitochondria, increasing the extramitochondrial [Ca2+]c triggered a massive mitochondrial Ca2+ release. This mCICR was sensitive to ruthenium red, but not to CGP37157 or cyclosporin A, revealing that the mitochondrial Ca2+ uniporter can behave, under certain conditions, as a [Ca2+]c-activated Ca2+ release channel functionally similar to the ryanodine receptor.
MATERIALS AND METHODS
Cell Preparation and Culture
Bovine adrenal medulla chromaffin cells were isolated following standard methods (Livett, 1984) with some modifications (Moro et al., 1990). Cells were suspended in DMEM supplemented with 5% fetal calf serum, 10 μM cytosine arabinoside, 10 μM fluorodeoxyuridine, 50 IU ml−1 penicillin and 50 IU ml−1 streptomycin. Cells were plated in 12-mm glass polilysine-coated coverslips (0.25 × 106 cells/1 ml of DMEM). Cultures were maintained at 37°C in a humidified atmosphere of 5% CO2, and used within 1–3 d.
Expression of Aequorin with Defective Herpes Virus (HSV) as Vectors
The mitochondrial aequorin was a kind gift of Prof. Tullio Pozzan, Padova, Italy. The EcoRI-fragment containing the cDNA encoding for mitochondrially targeted aequorin (mitAEQ) was isolated from the original vector and inserted into the recipient vector pHSVpuc (Geller et al., 1993) to generate the pHSVmitAEQ. For construction of the mitochondrially targeted mutated (Asp119→Ala) aequorin cDNA we replaced in frame the wild-type aequorin in pHSVmitAEQ with the mutated aequorin (Montero et al., 1995), obtaining the pHSVmutmitAEQ. The expression was confirmed after transfection of 2-2 cells by inmunofluorescence with an antibody against the HA1 epitope. These two vectors were packaged into HSV particles by using a deletion mutant packaging system (Lim et al., 1997). In brief, 3 × 105 2-2 cells were seeded on 60-mm dishes and transfected with 6 μg of pHSVmitAEQ or pHSVmutmitAEQ by using lipofectamine. 24 h later the cells were infected with ∼2 × 106 plaque-forming units of 5dl1.2 helper virus, which contains a deletion in the IE2 gene of HSV-1. On the following day virus was harvested and subsequently passaged on fresh 2-2 cells twice to increase both the ratio of vector to helper and the total amount of virus. Titering has been previously described (Alonso et al., 1998). The titers of packaged pHSVmitAEQ and pHSVmutmitAEQ, as determined by immunocytochemistry in PC12 cells, were 1.5 × 105 infectious virus units (ivu)/ml and 2.4 × 105 ivu/ml, respectively. For expression in chromaffin cells, the coverslips containing 0.25 × 106 cells were suspended in 0.5 ml of DMEM and infected with 2 × 103 ivu 12–24 h before measurements. Immunofluorescence with the anti-HA1 antibody showed a typical mitochondrial pattern in chromaffin cells infected with HSV expressing mitochondrially targeted aequorin (our unpublished results).
[Ca2+]M Measurements
For aequorin reconstitution, the coverslip with the cells expressing mitochondrial mutated aequorin was incubated with 1 μM coelenterazine n in 0.2 ml of standard medium containing 145 mM NaCl, 5 mM KCl, 1 mM MgCl2, 1 mM CaCl2, 10 mM glucose, and 10 mM HEPES, pH 7.4. Reconstitution was carried out in the dark at room temperature for 1–2 h. Reconstitution of aequorin with coelenterazine n is much slower than with wild-type coelenterazine (the half-time in vitro is 5 h compared with 22 min for the wild-type; Shimomura et al., 1993). This means that longer times of reconstitution will give an almost linear increase in the total luminescence output. A very important factor is also to keep low the temperature of reconstitution. Increasing the temperature to 37°C during reconstitution decreases considerably the total luminescence of the sample, probably because of increased aequorin consumption. Cells were then placed in the perfusion chamber of a purpose-built luminometer thermostatized at 37°C. Perfusion of up to eight different solutions was controlled by a system of electrovalves. For the experiments with permeabilized cells, standard medium containing 0.5 mM EGTA instead of Ca2+ was perfused for 1min. Then, intracellular medium (130 mM KCl, 10 mM NaCl, 1 mM MgCl2, 1 mM K3PO4, 0,2 mM EGTA, 1 mM ATP, 20 μM ADP, 2 mM succinate, 20 mM HEPES, pH 7) containing 20 μM digitonin was perfused during 1 min, followed by intracellular medium without digitonin for 1–2 min before the experiment. Buffer containing 10 μM [Ca2+] in EGTA-free intracellular medium was prepared using an HEDTA/Ca2+/Mg2+ mixture (5 mM HEDTA, 0.84 mM Ca2+, 4.71 mM Mg2+).
To calibrate the data obtained in terms of [Ca2+]M, we need to know the total amount of luminescence that can be emitted by the sample (see below). For that, at the end of every experiment it is essential to perfuse lysis solution containing detergent (100 μM digitonin) and excess Ca2+ (10 mM) to measure all the remaining aequorin luminescence. To transform luminescence data in [Ca2+], a computer program subtracts the background and calculates the fractions L/Lmax at every point along the experiment. L is the luminescence value at every point (minus the background) and Lmax is the integral of luminescence (minus the background) from that point to the end of the experiment. L/Lmax values are then transformed into [Ca2+] values by using the following mathematical algorithm:
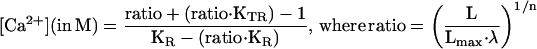 |
1 |
The values for the parameters of the algorithm used to calculate [Ca2+] in experiments at 37°C with mutated aequorin reconstituted with coelenterazine n have been obtained previously (Montero et al., 1997): KR = 8.47 · 107, KTR = 1.656 · 105, n = 1.2038, and λ = 0.138. This algorithm and the parameters KR, KTR, and n derive from a mathematical model proposed originally to explain from a molecular point of view the Ca2+ dependence of aequorin luminescence (Allen et al., 1971), but can be used as a simple mathematical transformation independent of the model. The parameter λ is the rate constant for aequorin consumption at saturating [Ca2+]. This parameter was not included in the original description of the algorithm (Brini et al., 1995), because the maximum rate constant of native aequorin reconstituted with wild-type coelenterazine is 1.0 s−1. Reconstitution with coelenterazine n reduces considerably the maximum rate constant, and this allows recording high [Ca2+] values with slower aequorin consumption. In some experiments, Lmax was reduced 50% to take into account that only ∼50% of mitochondria undergo large increases in [Ca2+]M after high-K+ stimulation (Montero et al., 2000).
Measurements of Single-Cell [Ca2+]c
Single cell measurements of [Ca2+]c were performed in cells loaded with the low-affinity Ca2+ dye fura-4F (4 μM; 60 min at 25°C). Other details were as described previously (Nuñez et al., 1995). Cells were epi-illuminated alternatively at 340 and 380 nm and light emitted above 520 nm was recorded by an extended ISIS-M camera (Photonic Science, Robertbridge, East Sussex, United Kingdom) and analyzed using an Applied Imaging Magical image processor (Sunderland, TyneandWear, United Kingdom). Eight frames excited at every wavelength were averaged by hardware, with a time resolution of 1.7 s for each pair of images. [Ca2+]c was estimated from the formula (Grynkiewicz et al., 1985):
 |
2 |
where R is the ratio between the fluorescence emissions measured at 340 and 380 nm excitation; Rmax and Rmin are the ratios obtained at saturation of the dye with Ca2+ and in the absence of Ca2+, respectively; Kd is the dissociation constant for the dye (0.77 μM); and β is the ratio of the maximal (in the absence of Ca2+) and minimum (at saturation with Ca2+) fluorescence emissions measured at 380-nm excitation. The values of Rmax, and Rmin and β were determined in cells permeabilized to Ca2+ with ionomycin and perfused with media containing either no Ca2+ (5 mM EGTA) or 10 mM Ca2+. These values were similar to the ones obtained with fura-2. All the experiments were performed at 37°C.
Materials
Fura-4F and coelenterazine n were obtained from Molecular Probes, Eugene, OR. Carbonyl cyanide m-chlorophenyl-hydrazone (CCCP) and cyclosporin A were from Sigma (St. Louis, MO). CGP37157 was from Tocris, Bristol, United Kingdom. Ruthenium red was from Calbiochem, La Jolla, CA. Other reagents were from Sigma or Merck (Darmstadt, Germany).
RESULTS
We have shown recently that stimulation of chromaffin cells with high-K+ medium triggers a fast Ca2+ uptake into ∼50% of the total mitochondrial pool. Mitochondrial depolarization by treatment with the protonophore CCCP prevented mitochondrial Ca2+ accumulation during stimulation with high K+ (Montero et al., 2000). We wanted now to investigate the effect of the protonophore on Ca2+ exit from mitochondria (Figure 1). In control cells [Ca2+]M increased to ∼300 μM during the 10-s stimulation period with high K+ and returned to the low resting levels with a half-time of <30 s (Figure 1A, first trace). Adding CCCP just after high-K+ stimulation slowed Ca2+ exit from mitochondria (Figure 1A, second trace). The inhibitory effect started a few seconds after addition of the protonophore, probably the time required to collapse the mitochondrial membrane potential. Figure 1, B and C, compare the effects of the inhibitor of the mitochondrial Na+/Ca2+ exchanger CGP37157 and of both CGP37157 and CCCP on Ca2+ exit from mitochondria after stimulation with high K+. The inhibition by CGP37157 was similar to the one effected by CCCP, except for the fact that inhibition by CGP37157 had no delay (it was preincubated for 2 min). In addition, the effects of both compounds were scarcely additive. As a representative parameter to compare the rate of mitochondrial Ca2+ efflux under the different conditions, we have measured the rate of [Ca2+]M decrease between 150 and 100 μM. The average values obtained (mean ± SEM) were 4.0 ± 0.4 μM/s (n = 7) in control cells, 1.1 ± 0.13 μM/s (n = 6) in the presence of CCCP, 1.22 ± 0.11 μM/s (n = 7) in the presence of CGP37157, and 0.95 ± 0.03 μM/s (n = 5) in the presence of both CGP37157 and CCCP. These results suggest that CCCP inhibits Ca2+ release by acting on the mitochondrial Na+/Ca2+ exchanger.
Figure 1.

Effects of CCCP and CGP37157 on Ca2+ release from mitochondria after high-K+ cell stimulation. Chromaffin cells expressing mitochondrial mutated aequorin reconstituted with coelenterazine n were stimulated by 10-s pulses of medium containing 70 mM K+ (indicated at the bottom). When indicated, cells were perfused with 2 μM CCCP added immediately after the high-K+ pulse (CCCP), with 10 μM CGP37157 (CGP, preincubated for 2 min before high-K+ stimulation), or with both together. For [Ca2+]M calculations, total luminescence was reduced 50%, assuming that [Ca2+]M changes take place in only 50% of the mitochondrial space.
Inhibition of mitochondrial Ca2+ exit by CCCP came as a surprise because protonophores have been reported to induce release of mitochondrial Ca2+ in several neuronal cells (see the Introduction). This interpretation was based mainly on measurements of cytosolic [Ca2+], nominally in the induction of an increase of [Ca2+]c by CCCP added just after mitochondrial Ca2+ loading. We then studied the effect of CCCP on the high-K+-induced [Ca2+]c transient. The low-Ca2+ affinity fluorescent probe fura-4F was used to avoid saturation of the dye at high-Ca2+ loads. Figure 2A shows that when CCCP was perfused before stimulation with high K+, the [Ca2+]c peak was considerably increased both in amplitude and duration, and this effect was reverted on washing CCCP. This outcome illustrates the action of mitochondria as a damper of the [Ca2+]c peak. In contrast, when CCCP was perfused just after high-K+ stimulation, the relaxation of the [Ca2+]c peak was indistinguishable from the control condition and we did not find a secondary [Ca2+]c peak (Figure 2B). This result indicates that, at least under the experimental conditions used in this work, CCCP did not release the Ca2+ accumulated in mitochondria during the stimulation period. This is consistent with the results seen with aequorin in Figure 1, where CCCP did not increase the rate of Ca2+ exit from mitochondria (as a matter of fact, it decreased it). The mitochondrial Na+/Ca2+ inhibitor CGP37157 had no significant effect on [Ca2+]c or on the kinetics of the high-K+-induced [Ca2+]c peak (our unpublished results).
Figure 2.

Effects of CCCP on [Ca2+]c. Cell were loaded with fura-4F and stimulated with 10-s pulses of medium containing 70 mM K+. When indicated, 2 μM CCCP was perfused. Traces correspond to the average of 25 (A) and 58 cells (B) present in the microscope field.
CCCP induced by itself a small increase in [Ca2+]c in resting cells (Figure 2A). This effect was still present in Ca2+-free medium, indicating that Ca2+ comes, at least partially, from intracellular stores. This kind of effect has been observed previously in different cell types, and it was attributed to Ca2+ release from mitochondria (Werth and Thayer, 1994; Babcock et al., 1997; White and Reynolds, 1997). However, [Ca2+]M has been shown to be in the same range as [Ca2+]c (100–200 nM) in resting cells (Rizzuto et al., 1993, 1994; Babcock et al., 1997). In these reports, protonophores produced a decrease in resting [Ca2+]M, but too small (≤100 nM) to justify the observed [Ca2+]c changes. Here we show that the initial resting [Ca2+]M values reported by the low-Ca2+-affinity aequorin were higher (5.8 ± 0.5 μM; mean ± SEM, n = 16), and that CCCP induced a reversible decrease in [Ca2+]M of several micromolar (Figure 3A). In contrast, using the mitochondrially targeted aequorin with high affinity for Ca2+ (native aequorin reconstituted with wild-type coelenterazine), the estimated resting [Ca2+]M value was 73 ± 7 nM, (mean ± SEM, n = 20). In this case, perfusion of the cells with CCCP induced a transient increase in [Ca2+]M to 300–400 nM (Figure 3B). The conflicting results obtained with both aequorins suggest that different mitochondria maintain different resting [Ca2+]M, which therefore cannot be considered as homogeneous among mitochondrial subpopulations. CCCP would be able to release Ca2+ from a subpopulation of mitochondria that contains micromolar levels of Ca2+ under resting conditions. Release of Ca2+ from these mitochondria may explain the increase in [Ca2+]c induced by CCCP. The endoplasmic reticulum could also be a possible source for that Ca2+, but using aequorin targeted to this organelle we were not able to detect any release of Ca2+ induced by CCCP (our unpublished results).
Figure 3.

Effects of CCCP on [Ca2+]M. (A) Cells expressing mitochondrially targeted low-Ca2+-affinity mutated aequorin were reconstituted with coelenterazine n, and 2 μM CCCP was added as indicated. (B) Cells expressing mitochondrially targeted wild-type aequorin were reconstituted with native coelenterazine, and 2 μM CCCP was added as indicated.
Protonophores collapse the electrical potential difference that drives Ca2+ accumulation into the mitochondria. This abolishes mitochondrial Ca2+ uptake (Rizzuto et al., 1994; Montero et al., 2000). However, stimulation of Ca2+ release requires, in addition, the opening of a pathway for Ca2+ exit. The possible implication of MPT was examined first. Figure 4 compares the [Ca2+]M peaks induced by stimulation with high K+ in the control condition and in the presence of cyclosporin A, an inhibitor of MPT. Except for the small differences in the lower part of the traces, cyclosporin A had little effect on the rate of Ca2+ efflux from mitochondria, suggesting no participation of MPT. On the other hand, inhibition of mitochondrial Ca2+ exit by CCCP (Figure 1) is inconsistent with MPT opening. Therefore, under the conditions used in our experiments, neither the large mitochondrial Ca2+ accumulation observed nor the mitochondrial depolarization induced by CCCP was enough to induce MPT opening.
Figure 4.

Effect of cyclosporin A on the [Ca2+]M transient induced by high-K+ stimulation. Either control cells or cells preincubated for 5 min with 10 μM cyclosporin A were stimulated with a 10-s pulse of medium containing 70 mM K+. The figure shows the mean of 11 experiments of each type (bars indicate the SEM of each point). Other details as in Figure 1. For [Ca2+]M calculations, total luminescence was reduced 50%.
Another possible pathway for Ca2+ exit from mitochondria would be the reversal of the Ca2+ uniporter. After treatment with the protonophore, the mitochondrial membrane potential collapses and the electrochemical gradient for Ca2+ becomes directed outwards. However, the uniporter might be inactive because of lack of cytosolic Ca2+. High local [Ca2+]c is required to activate mitochondrial Ca2+ uptake through the uniporter and half-maximum rates of transport are only reached at [Ca2+]c of 15–50 μM (Xu et al., 1997; Csordás et al., 1999; Montero et al., 2000). Then the failure of CCCP to induce mitochondrial Ca2+ release through the uniporter may be due to inactivation of this system following the rapid decrease of local [Ca2+]c after high-K+ stimulation. If that was the case, increasing [Ca2+]c at that time should, paradoxically, promote mitochondrial Ca2+ release. This rationale was tested by performing a second stimulation with high K+ in the presence of CCCP at the time [Ca2+]M was returning toward the resting level. The first trace in Figure 5 shows the slow decline of [Ca2+]M obtained in the presence of CCCP (compare to Figure 1). In the second trace a new stimulation with high K+ was applied during the decline of [Ca2+]M, ∼90 s after the first stimulus. This maneuver induced a very fast release of mitochondrial Ca2+. Of course, in the absence of CCCP, repetitive cell stimulation even at short periods (every 15–30 s) always induced repetitive increases in [Ca2+]M (our unpublished results; Montero et al., 2000). The third trace in Figure 5 shows that caffeine was also able to accelerate the release of mitochondrial Ca2+ in the presence of CCCP. We have shown before that caffeine induces a large local increase of [Ca2+]c by releasing Ca2+ from the endoplasmic reticulum (Alonso et al., 1999; Montero et al., 2000). Thus, the increase of [Ca2+]c, either due to Ca2+ entry through the plasma membrane or to release from the endoplasmic reticulum, was able to induce fast release of Ca2+ from depolarized mitochondria. The rate constants for efflux increased similarly on stimulation with either high K+ or caffeine from 0.7–0.9 to 3–5 μM/s. The same effects of CCCP on mitochondrial Ca2+ release were obtained in the presence of 2 μM oligomycin, to prevent reversal of the ATP synthase, or 10 μM cyclosporin A (preincubated for 2 min before high-K+ stimulation) to inhibit MPT. In addition, the effects of CCCP on Ca2+ exit from mitochondria were tested also at 22°C because, in many previous studies, release of Ca2+ from mitochondria by CCCP was studied at room temperature (Friel and Tsien, 1994; Herrington et al., 1996; Park et al., 1996; Babcock et al., 1997; Tang and Zucker, 1997; White and Reynolds, 1997). The results were similar to those shown in Figure 5. CCCP strongly inhibited mitochondrial Ca2+ exit at 22°C, and a second stimulation with high K+ produced a massive Ca2+ release.
Figure 5.

Cell stimulation induces Ca2+ release from depolarized Ca2+-loaded mitochondria. Cells were stimulated with a 10-s pulse of medium containing 70 mM K+ (indicated at the bottom), and 2 μM CCCP was perfused immediately afterward (A). Addition of a later 10-s high-K+ pulse (B) or a 30-s pulse with 50 mM caffeine (C) triggered Ca2+ release from mitochondria. Other details as in Figure 1. For [Ca2+]M calculations, total luminescence was reduced 50%.
The results shown in Figure 5 strongly suggest that mitochondrial Ca2+ release induced by cell stimulation in the presence of CCCP is due to reversal of the mitochondrial Ca2+ uniporter, which may require relatively high [Ca2+]c to open. To obtain further evidence on this point, we studied the effect of CCCP in permeabilized cells, where extramitocondrial [Ca2+] can be tightly controlled. Cells were permeabilized by a 1-min treatment with 20 μM digitonin (Montero et al., 2000). Cells were then perfused with Ca2+-free intracellular medium, and a mitochondrial [Ca2+]M transient was triggered by brief (3-s) perfusion with a 10 μM Ca2+ buffer (Figure 6A). The rates of Ca2+ efflux from mitochondria were very similar to the ones measured in intact cells (compare to Figure 1). Figure 6B shows that the effect of CCCP in the permeabilized cells was nearly the same as in the intact cells. After a short lag, Ca2+ exit was strongly inhibited. Figure 6,C and D, show the effects of CGP37157 and both CGP37157 and CCCP on Ca2+ exit. As observed in the intact cells, the effects of these inhibitors were scarcely additive. The rate of Ca2+ release between 150 and 100 μM [Ca2+]M (mean ± SEM) was 4.4 ± 0.3 μM/s (n = 10) in control cells, 1.42 ± 0.16 μM/s (n = 6) in the presence of CCCP, 1.56 ± 0.03 μM/s in the presence of CGP37157 (n = 3), and 1.4 ± 0.3 μM/s (n = 3) in the presence of both CGP37157 and CCCP.
Figure 6.

Effects of CCCP and CGP37157 on Ca2+ release from mitochondria in permeabilized cells. Cells expressing mutated aequorin reconstituted with coelenterazine n were permeabilized on line as described in MATERIALS AND METHODS. Then, transient Ca2+ uptake was induced by a 3-s perfusion of intracellular medium containing a 10 μM [Ca2+] buffer (arrows). When indicated, cells were perfused with 2 μM CCCP immediately after the Ca2+ pulse (CCCP), with 10 μM CGP37157 (CGP) or with both together.
The question was now whether we could induce Ca2+ release from depolarized Ca2+-loaded mitochondria by increasing extramitochondrial [Ca2+], as in the experiments with intact cells (Figure 5). Figure 7 shows that this was the case. In Figure 7A short (3-s) pulses with a 10 μM Ca2+ buffer induced a fast Ca2+ release from Ca2+-loaded mitochondria. Once CCCP was washed away, mitochondria recovered the ability to accumulate Ca2+ and to release it slowly. The [Ca2+]c-induced mCICR was not prevented by the Na+/Ca2+ blocker CGP37157 (Figure 7B) but it was fully prevented by ruthenium red (Figure 7C), an inhibitor of the mitochondrial Ca2+ uniporter. This confirms that mCICR takes place though reversal of the mitochondrial Ca2+ uniporter.
Figure 7.

Ca2+-induced Ca2+ release from depolarized Ca2+-loaded mitochondria. Cells expressing mutated aequorin reconstituted with coelenterazine n were permeabilized on line as described in MATERIALS AND METHODS. The arrows indicate 3-s pulses with intracellular medium containing a 10 μM [Ca2+] buffer. When indicated, either 2 μM CCCP, 10 μM CGP37157 (CGP), or 4 μM ruthenium red (RR) was perfused.
An important point was to show that the observed mCICR develops after mitochondrial depolarization induced by CCCP but is independent of other effects of CCCP such as matrix acidification. We have tested the effects of other ionophores such as valinomycin and nigericin. Figure 8A shows the effect of nigericin. This ionophore carries out an electroneutral H+/K+ exchange that should acidify the mitochondrial matrix similarly to CCCP but without the associated depolarization. Consistently, it produced little effects on the kinetics of Ca2+ release. Figure 8B shows the effect of valinomycin. This ionophore carries out an electrogenic K+ transport, and should depolarize mitochondria and increase the matrix pH. In spite of its opposite effect on matrix pH, valinomycin produced the same effects as CCCP: Ca2+ exit from mitochondria was slowed and mCICR could be induced by a second Ca2+ pulse. Finally, Figure 8C shows the effects of a combination of valinomycin and nigericin. This combination should be equivalent to CCCP, and produced in fact the same effects.
Figure 8.

Effects of nigericin and valinomycin on Ca2+ release from mitochondria in permeabilized cells. Cells expressing mutated aequorin reconstituted with coelenterazine n were permeabilized on line as described in MATERIALS AND METHODS. The arrows indicate 3-s pulses with intracellular medium containing a 10 μM [Ca2+] buffer. When indicated, either 0.5 μM nigericin, 0.5 μM valinomycin, or both were perfused. In B and C, the ionophores were perfused just after the Ca2+ pulse.
DISCUSSION
In this article we have studied the dynamics of [Ca2+]M after high mitochondrial Ca2+ loads and the effects of collapsing the mitochondrial membrane potential on the rate of Ca2+ exit from mitochondria. We have reported recently that Ca2+ entry through voltage-gated Ca2+ channels of plasma membrane induces, in chromaffin cells, a fast Ca2+ uptake to near millimolar levels into a population of mitochondria, probably with subplasmalemmal location (Montero et al., 2000). Then, Ca2+ exits from mitochondria within 1–2 min, mainly through the mitochondrial Na+/Ca2+ exchanger. The large increase in [Ca2+]M was apparently not enough to activate the MPT, which should have produced an abrupt drop in [Ca2+]M. In fact, a series of such millimolar mitochondrial transients could be elicited with no sign of activation of MPT (Montero et al., 2000). In addition, we show here that cyclosporin A, an inhibitor of MPT, had almost no effect on this mitochondrial Ca2+ release (Figure 4), suggesting that MPT did not open under our experimental conditions. However, some contribution of MPT to Ca2+ release cannot be completely excluded, because the degree of inhibition of MPT by cyclosporin A may be variable or transient depending on a series of factors (Bernardi, 1999).
Mitochondrial depolarization induces Ca2+ release from Ca2+-loaded mitochondria under several conditions (Bernardi et al., 1984; Igbavboa et al., 1988; Bernardi, 1992). In resting cells, measurements of [Ca2+]M by using either high-affinity aequorin (Rizzuto et al., 1993, 1994) or the fluorescent Ca2+ indicator rhod-2 (Babcock et al., 1997) have provided very low values, close to the resting [Ca2+]c. In both cases, however, protonophores still produced a small but significant reduction in [Ca2+]M. We obtain here conflicting results on this matter by using either the high- or the low-Ca2+-affinity aequorin. In the first case we estimate resting [Ca2+]M levels of 70 nM, which increase transiently to 300–400 nM on addition of CCCP. In contrast, with the low-Ca2+-affinity aequorin we estimate resting [Ca2+]M values around 5 μM, which decrease on addition of CCCP. Low-Ca2+-affinity aequorin has little sensitivity at these [Ca2+], and the level of luminescence produced is very low compared with the peaks induced by cell stimulation. However, because of the large dynamic range of aequorin, the luminescence observed at resting [Ca2+]M is still severalfold the background. In addition, CCCP produced a clear and reversible decrease in the resting [Ca2+]M, demonstrating the presence of mitochondria with micromolar resting [Ca2+] that can release their Ca2+ upon CCCP addition. The discrepancies among the results obtained with both types of aequorin may be due to the presence of heterogeneity in the resting [Ca2+]M levels. Although some mitochondria keep a low resting [Ca2+]M around 100 nM, others may have values in the micromolar range or perhaps oscillating [Ca2+]M values due to spontaneous opening of nearby Ca2+ channels. When using high-Ca2+-affinity aequorin, the photoprotein present in these last ones would be consumed during the reconstitution period before measurements, so that they are never observed. The remaining aequorin would be present in low-Ca2+ mitochondria, which may take up transiently small amounts of Ca2+ from the cytosol after CCCP addition (up to 300–400 nM, undetectable for low-Ca2+-affinity aequorin).
Addition of protonophores after cell stimulation induces, in several cell types, a large increase in [Ca2+]c, due to release of Ca2+ from mitochondria (Werth and Thayer, 1994; Hehl et al., 1996; Herrington et al., 1996; Tang and Zucker, 1997; White and Reynolds, 1997). At variance with these observations, we find that CCCP inhibited Ca2+ exit from Ca2+-loaded mitochondria both in intact and in permeabilized chromaffin cells. Because both the Na+/Ca2+ and Ca2+/H+ antiporters are electrogenic (see the Introduction), inhibition of Ca2+ exit through these systems by depolarization should be expected and is consistent with the results obtained here. On the contrary, opening of MPT would favor Ca2+ release because, once mitochondria are depolarized, the Ca2+ electrochemical gradient is directed outwards. Depolarization of Ca2+-loaded mitochondria is known to favor opening of MPT (Igbavboa and Pfeiffer, 1988; Bernardi, 1992; Di Lisa and Bernardi, 1998). However, we could not evidence such behavior in intact or permeabilized chromaffin cells under our experimental conditions, in spite of the large accumulation of Ca2+ into mitochondria. This may be due to the presence of other factors promoting MPT closure, such as nucleotides or reduced pyridine nucleotides (Di Lisa and Bernardi, 1998; Bernardi, 1999; Crompton, 1999).
Another possible pathway for Ca2+ exit from depolarized mitochondria would be reversal of the mitochondrial uniporter. However, Ca2+ release through this system has been difficult to observe (Petronilli et al., 1993; Bernardi, 1999), this leading to the suggestion that flux through the uniporter may be scarcely reversible. However, a high [Ca2+]c is necessary to activate Ca2+ entry through this system (Rizzuto et al., 1993; Xu et al., 1997; Csordás et al., 1999; Montero et al., 2000) and probably a threshold concentration of Ca2+ is also required at the extramitochondrial side to allow Ca2+ exit (Kroner, 1986; Gunter and Pfeiffer, 1990, Igbavboa and Pfeiffer, 1988). Should this condition apply, then an increase of [Ca2+]c would, paradoxically, release Ca2+ from depolarized Ca2+-loaded mitochondria. Cell stimulation with high K+ generates high local [Ca2+]c microdomains that trigger mitochondrial Ca2+ uptake (Montero et al., 2000), but they dissipate rapidly after closure of the Ca2+ channels. Measurements with fura-4F show that [Ca2+]c decreases to <1 μM within ∼10 s of stimulation (Figure 2). Thus, when CCCP is added after stimulation, local [Ca2+]c microdomains may return below the threshold for opening of the uniporter before mitochondrial depolarization is achieved. This rationale predicts that increasing [Ca2+]c at this time would trigger mitochondrial Ca2+ release through the uniporter. This prediction was confirmed by further stimulating the Ca2+-loaded cells with either high K+ or caffeine (Figure 4). The massive Ca2+ release induced by these maneuvers was insensitive to cyclosporin A. In addition, Ca2+ release from depolarized Ca2+-loaded mitochondria could be induced in permeabilized cells by brief perfusion with 10 μM Ca2+, and this Ca2+ release was blocked by ruthenium red. These results demonstrate that the Ca2+ uniporter is readily reversible after mitochondrial depolarization in the presence of micromolar extramitochondrial Ca2+ and can mediate mCICR.
It could be argued that the effects of CCCP on mitochondrial Ca2+ release could be due to acidification of the mitochondrial matrix rather than to depolarization. Aequorin is scarcely sensitive to pH, and acidification should, in any case, decrease the luminescence (Moisescu and Ashley, 1977) and thus the apparent [Ca2+]M levels. Instead, we observe that CCCP slows down the [Ca2+]M decrease after cell stimulation. To exclude other possible effects of matrix acidification, we used nigericin and valinomycin. Nigericin, which decreases matrix pH and does not depolarize the mitochondrial membrane (it may actually hyperpolarize it), had no effect on Ca2+ release. In contrast, valinomycin, which depolarizes mitochondria but does not acidify the mitochondrial matrix (in fact, it should probably increase matrix pH), produced effects similar to CCCP. Finally, the combination of valinomycin + nigericin also reproduced the effects of CCCP on Ca2+ release. This demonstrates that the effect of CCCP on Ca2+ exit from mitochondria is due to mitochondrial depolarization.
It is not clear whether this mCICR may occur under physiological conditions. A mCICR has been described previously (Ichas et al., 1997), but mediated by opening of the MPT after mitochondrial Ca2+ overload. For the mCICR described here, mediated by the Ca2+ uniporter, mitochondrial depolarization after the Ca2+ loading would be required. Both, Ca2+ uptake through the uniporter and Ca2+ release through the exchangers tend to depolarize mitochondria. It would then be conceivable that mitochondrial depolarization after a large Ca2+ uptake, particularly if respiration is somewhat compromised, could render mitochondria sensitive to mCICR through the Ca2+ uniporter. Opening of other mitochondrial membrane channels, such as uncoupling proteins or KATP channels, could also contribute or trigger mitochondrial depolarization. In addition, opening of any of these channels would be essential to allow net Ca2+ exit from mitochondria through the uniporter by providing charge compensation. At this stage, further increase in local [Ca2+]c would trigger opening of the uniporter and Ca2+ release from mitochondria. mCICR through the Ca2+ uniporter could be advantageous over MPT for clearing of mitochondrial Ca2+ overload. Because the uniporter is a pathway specific for Ca2+, loss of metabolites would not happen in this case, and quick recovery of the ability to produce ATP would be easier.
Our results point out the subtlety of the Ca2+-handling mechanisms of mitochondria and how slight differences can lead to very different outcomes. Mitochondria take up large amounts of Ca2+ though the uniporter when a high-[Ca2+]c microdomain is generated close enough after cell stimulation (Montero et al., 2000). Exit of Ca2+ from the loaded mitochondria can then follow several alternative pathways, with very different consequences for the cell. If the mitochondrial membrane potential remains high, Ca2+ exits mitochondria through the Ca2+/Na+ and the Ca2+/H+ exchangers, as it happens in most of the experiments shown here. If the mitochondrial membrane potential collapses, Ca2+ can exit mitochondria by reversal of the uniporter, provided that mitochondrial H+ or K+ pathways allow charge compensation and local [Ca2+]c is high enough at that time (mCICR). Finally, if added circumstances favoring opening of MPT exist, Ca2+ can exit mitochondria through this pathway, but now accompanied by other low-molecular weight mitochondrial constituents. Transition from one alternative to the other may depend on subtle differences in kinetics of the [Ca2+]c changes, oxygen supply, metabolic state, oxidative stress, etc.
The discrepancies of our results with previous reports showing CCCP-induced Ca2+-release from mitochondria may be explained on the basis of the above-mentioned complexity. It seems clear that the pathway responsible for the CCCP-induced Ca2+ release observed in previous studies should not be the Ca2+ uniporter acting in reverse mode, because the local [Ca2+]c returns rapidly to resting values once stimulation has ceased, leading to inactivation of this system. A more probable alternative is the MPT, given that MPT open-closed transitions are highly regulated by multiple effectors (Di Lisa and Bernardi, 1998; Bernardi, 1999; Crompton, 1999), including [Ca2+]M and mitochondrial membrane potential, so that opening may critically depend on the particular experimental conditions. Our data suggest that, in intact cells and under our conditions, mitochondrial Ca2+ accumulation and depolarization are not enough to promote opening of MPT. Use of uncouplers to study mitochondrial Ca2+ homeostasis should therefore take into account that, even though uncouplers certainly abolish mitochondrial Ca2+ uptake, they do not necessarily induce Ca2+ release from mitochondria.
ACKNOWLEDGMENTS
We thank J. Fernández for technical assistance. Financial support from Dirección General de Enseñanza Superior to J.A. (PM-98/0142) and J.G.-S. (PB-97/0474), and from Junta de Castilla y León to J.A. (VA-19/99) and M.T.A. (VA-62/99) are gratefully acknowledged. A.A. holds a fellowship from Ministerio de Educación y Ciencia.
Abbreviations used:
- CCCP
carbonyl cyanide m-chlorophenyl-hydrazone
- HSV
herpes virus
- mCICR
mitochondrial Ca2+-induced Ca2+ release
- MPT
mitochondrial permeability transition
REFERENCES
- Alonso MT, Barrero MJ, Carnicero E, Montero M, García-Sancho J, Alvarez J. Functional measurements of [Ca2+] in the endoplasmic reticulum using a herpes virus to deliver targeted aequorin. Cell Calcium. 1998;24:87–96. doi: 10.1016/s0143-4160(98)90076-8. [DOI] [PubMed] [Google Scholar]
- Alonso MT, Barrero MJ, Michelena P, Carnicero E, Cuchillo I, García AG, García-Sancho J, Montero M, Alvarez J. Ca2+-induced Ca2+ release in chromaffin cells seen from inside the ER with targeted aequorin. J Cell Biol. 1999;144:241–254. doi: 10.1083/jcb.144.2.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen DG, Blinks JR, Prendergast FG. Aequorin luminescence. Relation of light emission to calcium concentration – a calcium-independent component. Science. 1971;195:996–998. doi: 10.1126/science.841325. [DOI] [PubMed] [Google Scholar]
- Babcock DF, Herrington J, Goodwin PC, Park Y-B, Hille B. Mitochondrial participation in the intracellular Ca2+ network. J Cell Biol. 1997;136:833–843. doi: 10.1083/jcb.136.4.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baysal K, Jung DW, Gunther KK, Gunter TE, Brierley GP. Na+-dependent Ca2+ efflux mechanism of heart mitochondria is not a passive Ca2+/2Na+ exchanger. Am J Physiol. 1994;266:C800–C808. doi: 10.1152/ajpcell.1994.266.3.C800. [DOI] [PubMed] [Google Scholar]
- Bernardi P. Modulation of the mitochondrial cyclosporin A-sensitive permeability transition pore by the proton electrochemical gradient. Evidence that the pore can be opened by membrane depolarization. J Biol Chem. 1992;267:8834–8839. [PubMed] [Google Scholar]
- Bernardi P. Mitochondrial transport of cations: channels, exchangers and permeability transition. Physiol Rev. 1999;79:1127–1155. doi: 10.1152/physrev.1999.79.4.1127. [DOI] [PubMed] [Google Scholar]
- Bernardi P, Azzone GF. A membrane potential-modulated pathway for Ca2+ efflux in rat liver mitochondria. FEBS Lett. 1982;139:13–16. doi: 10.1016/0014-5793(82)80476-6. [DOI] [PubMed] [Google Scholar]
- Bernardi P, Azzone GF. Regulation of Ca2+ efflux in rat liver mitochondria. Role of membrane potential. Eur J Biochem. 1983;134:377–383. doi: 10.1111/j.1432-1033.1983.tb07578.x. [DOI] [PubMed] [Google Scholar]
- Bernardi P, Paradisi V, Pozzan T, Azzone GF. Pathway for uncoupler-induced calcium efflux en rat liver mitochondria: inhibition by ruthenium red. Biochemistry. 1984;23:1645–1651. doi: 10.1021/bi00303a010. [DOI] [PubMed] [Google Scholar]
- Brini M, De Giorgi F, Murgia M, Marsault R, Massimino ML, Cantini M, Rizutto R, Pozzan T. Subcellular analysis of Ca2+ homeostasis in primary cultures of skeletal muscle myotubes. Mol Biol Cell. 1997;8:129–143. doi: 10.1091/mbc.8.1.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brini M, Marsault R, Bastianutto C, Alvarez J, Pozzan T, Rizzuto R. Transfected aequorin in the measurement of cytosolic Ca2+ concentration ([Ca2+]c). A critical evaluation. J Biol Chem. 1995;270:9896–9903. doi: 10.1074/jbc.270.17.9896. [DOI] [PubMed] [Google Scholar]
- Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochem J. 1999;341:233–249. [PMC free article] [PubMed] [Google Scholar]
- Csordás G, Thomas AP, Hajnóczky G. Quasi-synaptic calcium signal transmission between endoplasmic reticulum and mitochondria. EMBO J. 1999;18:96–108. doi: 10.1093/emboj/18.1.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Lisa F, Bernardi P. Mitochondrial function as a determinant of recovery or death in cell response to injury. Mol Cell Biochem. 1998;184:379–391. [PubMed] [Google Scholar]
- Duchen MR. Contributions of mitochondria to animal physiology: from homeostatic sensor to calcium signaling and cell death. J Physiol. 1999;516:1–17. doi: 10.1111/j.1469-7793.1999.001aa.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friel DD, Tsien RW. An FCCP-sensitive Ca2+ store in bullfrog sympathetic neurons and its participation in stimulus-evoked changes in [Ca2+]i. J Neurosci. 1994;14:4007–4024. doi: 10.1523/JNEUROSCI.14-07-04007.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geller AI, During MJ, Haycock A, Freese A, Neve R. Long-term increases in neurotransmitter release from neuronal cells expressing a constitutively active adenylate cyclase from a herpes simplex virus type 1 vector. Proc Natl Acad Sci USA. 1993;90:7603–7607. doi: 10.1073/pnas.90.16.7603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giovanucci DR, Hlubek MD, Stuenkel EL. Mitochondria regulate the Ca2+-exocytosis relationship of bovine adrenal chromaffin cells. J Neurosci. 1999;19:9261–9270. doi: 10.1523/JNEUROSCI.19-21-09261.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Gunter TE, Pfeiffer DR. Mechanisms by which mitochondria transport calcium. Am J Physiol. 1990;258:C755–C786. doi: 10.1152/ajpcell.1990.258.5.C755. [DOI] [PubMed] [Google Scholar]
- Gunter KK, Zuscik MJ, Gunter TE. The Na+-independent Ca2+ efflux mechanism of liver mitochondria is not a passive Ca2+/2H+ exchanger. J Biol Chem. 1991;266:21640–21648. [PubMed] [Google Scholar]
- Hehl S, Golard A, Hille B. Involvement of mitochondria in intracellular calcium sequestration by rat gonadotropes. Cell Calcium. 1996;20:515–524. doi: 10.1016/s0143-4160(96)90094-9. [DOI] [PubMed] [Google Scholar]
- Herrington J, Park YB, Babcock DF, Hille B. Dominant role of mitochondria in clearance of large Ca2+ loads from rat adrenal chromaffin cells. Neuron. 1996;16:219–228. doi: 10.1016/s0896-6273(00)80038-0. [DOI] [PubMed] [Google Scholar]
- Ichas F, Jouaville LS, Mazat J-P. Mitochondria are excitable organelles capable of generating and conveying electrical and calcium signals. Cell. 1997;89:1145–1153. doi: 10.1016/s0092-8674(00)80301-3. [DOI] [PubMed] [Google Scholar]
- Ichas F, Mazat J-P. From calcium signaling to cell death: two conformations for the mitochondrial permeability transition pore. Biochim Biophys Acta. 1998;1366:33–50. doi: 10.1016/s0005-2728(98)00119-4. [DOI] [PubMed] [Google Scholar]
- Igbavboa U, Pfeiffer DR. EGTA inhibits reverse uniport-dependent Ca2+ release from uncoupled mitochondria. J Biol Chem. 1988;263:1405–1412. [PubMed] [Google Scholar]
- Jung DW, Baysal K, Brierley GP. The sodium-calcium antiport of heart mitochondria is not electroneutral. J Biol Chem. 1995;270:672–678. doi: 10.1074/jbc.270.2.672. [DOI] [PubMed] [Google Scholar]
- Kroner H. Ca2+. ions, an allosteric activator of calcium uptake in rat liver mitochondria. Arch Biochem Biophys. 1986;251:525–535. doi: 10.1016/0003-9861(86)90360-7. [DOI] [PubMed] [Google Scholar]
- Lim F, Hartley D, Starr P, Song S, Yu L, Wang Y, Geller AI. Use of defective herpes-derived plasmid vectors. Methods Mol Biol. 1997;62:223–232. doi: 10.1385/0-89603-480-1:223. [DOI] [PubMed] [Google Scholar]
- Livett BG. Adrenal medullary chromaffin cells in vitro. Physiol Rev. 1984;64:1103–1161. doi: 10.1152/physrev.1984.64.4.1103. [DOI] [PubMed] [Google Scholar]
- McCormack JG, Halestrap AP, Denton RM. Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol Rev. 1990;70:391–425. doi: 10.1152/physrev.1990.70.2.391. [DOI] [PubMed] [Google Scholar]
- Moisescu DG, Ashley CC. The effect of physiologically occurring cations upon aequorin light emission. Determination of the binding constants. Biochim Biophys Acta. 1977;460:189–205. doi: 10.1016/0005-2728(77)90206-7. [DOI] [PubMed] [Google Scholar]
- Montero M, Barrero MJ, Alvarez J. [Ca2+]. microdomains control agonist-induced Ca2+ release in intact HeLa cells. FASEB J. 1997;11:881–885. doi: 10.1096/fasebj.11.11.9285486. [DOI] [PubMed] [Google Scholar]
- Montero M, Alonso MT, Carnicero E, Cuchillo I, Albillos A, García AG, García-Sancho J, Alvarez J. Physiological stimuli trigger fast millimolar [Ca2+] transients in chromaffin cell mitochondria that modulate secretion. Nat Cell Biol. 2000;2:57–61. doi: 10.1038/35000001. [DOI] [PubMed] [Google Scholar]
- Montero M, Brini M, Marsault R, Alvarez J, Sitia R, Pozzan T, Rizzuto R. Monitoring dynamic changes in free Ca2+ concentration in the endoplasmic reticulum of intact cells. EMBO J. 1995;14:5467–5475. doi: 10.1002/j.1460-2075.1995.tb00233.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moro MA, Lopez MG, Gandia L, Michelena P, Garcia AG. Separation and culture of living adrenaline- and noradrenaline-containing cells from bovine adrenal medullae. Anal Biochem. 1990;185:243–248. doi: 10.1016/0003-2697(90)90287-j. [DOI] [PubMed] [Google Scholar]
- Nuñez L, De La Fuente MT, Garcia AG, Garcia-Sancho J. Differential Ca2+ responses of adrenergic and noradrenergic chromaffin cells to various secretagogues. Am J Physiol. 1995;269:C1540–C1546. doi: 10.1152/ajpcell.1995.269.6.C1540. [DOI] [PubMed] [Google Scholar]
- Park YB, Herrington J, Babcock DF, Hille B. Ca2+ clearance mechanisms in isolated rat adrenal chromaffin cells. J Physiol. 1996;492:329–346. doi: 10.1113/jphysiol.1996.sp021312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petronilli V, Cola C, Bernardi P. Modulation of the mitochondrial cyclosporin A-sensitive permeability transition pore. II. The minimal requirements for pore induction underscore a key role for transmembrane electrical potential, matrix pH, and matrix Ca2+ J Biol Chem. 1993;268:1011–1016. [PubMed] [Google Scholar]
- Pivovarova NB, Hongpaisan J, Andrews SB, Friel DD. Depolarization-induced mitochondrial Ca accumulation in sympathetic neurons: spatial and temporal characteristics. J Neurosci. 1999;19:6372–6384. doi: 10.1523/JNEUROSCI.19-15-06372.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzuto R, Bastianutto C, Brini M, Murgia M, Pozzan T. Mitochondrial Ca2+ homeostasis in intact cells. J Cell Biol. 1994;126:1183–1194. doi: 10.1083/jcb.126.5.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzuto R, Brini M, Murgia M, Pozzan T. Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science. 1993;262:744–747. doi: 10.1126/science.8235595. [DOI] [PubMed] [Google Scholar]
- Robb-Gaspers LD, Burnett P, Rutter GA, Denton RM, Rizzuto R, Thomas AP. Integrating cytosolic calcium signals into mitochondrial metabolic responses. EMBO J. 1998;17:4987–5000. doi: 10.1093/emboj/17.17.4987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimomura O, Kishi Y, Inouye S. The relative rate of aequorin regeneration from apoaequorin and coelenterazine analogues. Biochem J. 1993;296:549–551. doi: 10.1042/bj2960549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Zucker RS. Mitochondrial involvement in post-tetanic potentiation of synaptic transmission. Neuron. 1997;18:483–491. doi: 10.1016/s0896-6273(00)81248-9. [DOI] [PubMed] [Google Scholar]
- Werth JL, Thayer SA. Mitochondria buffer physiological calcium loads in cultured rat dorsal root ganglion neurons. J Neurosci. 1994;14:346–356. doi: 10.1523/JNEUROSCI.14-01-00348.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White RJ, Reynolds IJ. Mitochondria accumulate Ca2+ following intense glutamate stimulation of cultured rat forebrain neurons. J Physiol. 1997;498:31–47. doi: 10.1113/jphysiol.1997.sp021839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T, Naraghi M, Kang H, Neher E. Kinetic studies of Ca2+ binding and Ca2+ clearance in the cytosol of adrenal chromaffin cells. Biophys J. 1997;73:532–545. doi: 10.1016/S0006-3495(97)78091-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoratti M, Szabó I. The mitochondrial permeability transition. Biochim Biophys Acta. 1995;1241:139–176. doi: 10.1016/0304-4157(95)00003-a. [DOI] [PubMed] [Google Scholar]