

Abstract
Pancreatic cancer (PC) is one of the most lethal malignant diseases with the worst prognosis. It is ranked as the fourth leading cause of cancer-related deaths in the United States. Many risk factors have been associated with PC. Interestingly, large numbers of epidemiological studies suggest that obesity and diabetes, especially type-2 diabetes, are positively associated with increased risk of PC. Similarly, these chronic diseases (obesity, diabetes and cancer) are also a major public health concern. In the U.S. population, 50 percent are overweight, 30 percent are medically obese and 10 percent have diabetes mellitus (DM). Therefore, obesity and DM have been considered as potential risk factors for cancers; however, the focus of this article is restricted to PC. Although the mechanisms responsible for the development of these chronic diseases leading to the development of PC are not fully understood, the biological importance of the activation of insulin, insulin like growth factor-1 (IGF-1) and its receptor (IGF-1R) signaling pathways in insulin resistance mechanism and subsequent induction of compensatory hyperinsulinemia has been proposed. Therefore, targeting insulin/IGF-1 signaling with anti-diabetic drugs for lowering blood insulin levels and reversal of insulin-resistance could be useful strategy for the prevention and/or treatment of PC. A large number of studies have demonstrated that the administration of anti-diabetic drugs such as metformin and thiazolidinediones (TZD) class of PPAR-γ agonists decreases the risk of cancers, suggesting that these agents might be useful anti-tumor agents for the treatment of PC. In this review article, we will discuss the potential roles of metformin and TZD anti-diabetic drugs as anti-tumor agents in the context of PC, and will further discuss the complexities and the possible roles of microRNAs (miRNAs) in the pathogenesis of obesity, diabetes and PC.
Keywords: obesity, diabetes mellitus, pancreatic cancer, metformin, and TZD
Introduction
Pancreatic cancer (PC) is one of the most lethal malignant diseases with the worst prognosis. The incidence of PC is higher in American men compared with women and its occurrence is seen more frequently in African Americans compared to Caucasian Americans [1]. It has been estimated that 37,000 patients are newly diagnosed with PC, and 34,000 patients die of PC each year in the United States [2]. High cancer mortality confers this malignancy as the fourth leading cause of cancer-related deaths in the U.S. [2]. PC is also responsible for about 227,000 deaths each year in the world [3]. Due to the absence of specific symptoms and the lack of early detection tests, PC is usually diagnosed at an advanced incurable stage [1,4]. Thus, the median overall survival is 5–6 months after conventional therapy for locally advanced and metastatic disease. Consequently, the 5-year overall survival rate is less than five percent [1]. Therefore, identification of risk factors, especially those that are modifiable for PC is a very important task for preventing the development and progression of PC. Tobacco smoking, alcohol consumptions, chronic pancreatitis, and genetic risk factors, have been recognized as potential risk factors for the development and progression of PC [5–7]. However, the etiology of PC is poorly understood. A large number of epidemiological studies have suggested that obesity and diabetes are associated with increased risk of PC, and thus the focus of our article is restricted to the role of obesity, diabetes and PC.
1. Obesity and the risk of the development and progression of PC
Obesity and overweight conditions are prevalent in the world. A World Health Organization (WHO) report demonstrates that more than 400 million people are obese in the world, with a predicted increase to 700 million by 2015 [8,9]. In the United States, the current prevalence of obesity for adults is 30–35 percent [10,11], and thus the general population combining obesity and overweight is a major public health concern. According to WHO/Food and Agriculture Organization (FAO), obesity and overweight conditions can be diagnosed by measuring the body mass index (BMI) of the individual. The BMI is calculated by dividing body weight in kilograms by height squared in meters kg/m2). The current standard categories of BMI are as follows: underweight, <18.5; normal weight, 18.5–24.9; overweight, 25.0–29.9; obese, 30.0–34.9; and severely obese, > or = 35.0) [11–13].
It has long been recognized that being obese or overweight increases the risks of cardiovascular disease, hypertension, and type-2 DM. Similarly, numerous epidemiological studies have demonstrated that high BMI is positively associated with increased risk of many common cancers, such as liver, endometrium, breast, pancreas, and colorectal cancers [14–23]. Recently, high BMI has also been considered as an additional risk factor for PC [4,15,17,18]. Although early epidemiological studies showed inconsistent or inconclusive results concerning the association between BMI and the risk of PC, the majority of prospective epidemiological studies have shown that high BMI is indeed associated with an increased risk in the incidence or mortality of PC, independent of prior history of DM [24–33]. The inconsistencies in early findings are believed to be in part due to small sample size, study design bias, retrospective approach, residual or uncontrolled confounders such as smoking history and gender, as well as inadequate statistical power [4,33–36].
In 2003, a meta-analysis of six case-control and eight prospective studies including 6,391 PC cases were conducted to examine the association of high BMI with the risk of PC [37]. The results demonstrated a weak positive association between them (2% increase in risk per 1 kg/m2 increase in BMI). In 2007, a meta-analysis of 21 prospective studies was conducted including 3,495,981 individuals and 8,062 PC cases [38]. Of the 21 prospective studies, 10 were from the United States, 9 were from Europe, and 2 were from Asia. With the exception of one nested case-control prospective cohort study, all studies were controlled for cigarette smoking, and 13 studies also adjusted for DM. One cohort study including 63,000 individuals and 166 PC cases was excluded because BMI was classified by two categories only (high versus low BMI: >25.2 vs. ≤25.2 kg/m2 in men; >24.7 vs. ≤24.7 kg/m2 in women) [35]. The results show that the estimated summary risk ratio (RR) per 5 kg/m2 increase in BMI was 1.12 (95% CI, 1.06–1.17) in men and women combined. There was no significant difference of summary RR between men and women. Therefore, the authors have concluded that there was a positive association between BMI and risk of PC in both men and women. Overall, a 5 kg/m2 increase in BMI was associated with a 12% increased risk of PC. In 2008, another meta-analysis from 221 datasets, including 282,137 incidence of cancer cases [39]. For analysis of high BMI associated with the risk of PC, the total sample size was 3,338,001 subjects (2,390 PC cases in men and 2,053 PC cases in women). Like the 2003 findings, results also show a weak association of high BMI with the risk of PC (RR, 1.12; CI, 1.02–1.22).
Recently, one large prospective cohort study including 138,503 postmenopausal women in the Women’s Health Initiative study in the United States was conducted. This study examined obesity, especially central obesity, in relation to PC (n=251) for the duration of an average of 7.7 years of follow-up [40]. The results show that women in the highest quintile of waist-to-hip ratio have a 70 percent (95% CI, 10–160%) greater risk of PC compared with women in the lowest quintile. This suggests that obesity, especially central adiposity, is positively associated with the increased risk of PC. Another new case-control study included 841 PC patients and 754 healthy individuals and the purpose of this study was to examine the association of high BMI across an age cohort to determine at what age an individual became at risk, age of onset and overall survival of PC [41]. Individuals who were overweight from the ages of 14 to 39 years (highest odds ratio [OR], 1.67; 95% CI, 1.20–2.34) or obese from the ages of 20 to 49 years (highest OR, 2.58; 95% CI, 1.70–3.90) had an associated increased risk of PC, independent of DM status. This association was stronger in men (adjusted OR, 1.80; 95% CI, 1.45–2.23) from the ages of 14 to 59 years than in women (adjusted OR, 1.32; 95% CI, 1.02–1.70). Individuals with high BMI (= or > 25) from the ages of 20 to 49 years had an earlier onset of PC by 2 to 6 years. Individuals who were overweight or obese from the ages of 30 to 79 years or were overweight or obese in the year prior to recruitment had reduced overall survival of PC regardless of disease stage and tumor resection status (overweight patients: hazard ratio, 1.26, 95% CI, 0.94–1.69; obese patients: hazard ratio, 1.86, 95% CI, 1.35–2.56). Similarly, the authors concluded that being overweight or obese during early adulthood was associated with a greater risk of PC and a younger age of disease onset, whereas obesity at an older age was associated with a lower overall survival in patients diagnosed with PC.
More recently, several large prospective cohort studies with a long duration of follow-up has been conducted in the U.S. showing a positive association between high BMI and the risk of PC (adjusted RR 1.13–1.54) [42,43], suggesting the role of obesity and overweight with higher risk in the development and eventual death due to PC. Although the role of smoking and gender in the association of obesity and PC is not clear, the new evidence strongly supports a positive association of high BMI with increased risk of PC, consistent with the majority of early findings; however, all recent studies strongly suggest that obesity and overweight are independent risk factor of PC.
2. Obesity, insulin resistance, hyperinsulinemia, and the risk of PC
Obesity, especially central obesity, or central adiposity, has been well documented to be related to insulin resistance, hyperinsulinemia, glucose intolerance, and with subsequent development of type 2 DM [44–46]. A metabolic consequence of obesity, especially central adiposity, is the development of insulin resistance, which induces an increase in the secretion of insulin from the pancreas. Increased insulin production leads to compensatory hyperinsulinemia, which leads to insulin resistance [43,44,46]. The development of insulin resistance is one of the earliest negative effects of obesity, and is associated with the early altered glucose metabolism, chronic inflammation, oxidative stress and decreased levels of adipose hormone adiponectin and PPAR-γ, key regulators for adipogenesis [46–49]. Moreover, altered regulations in the expression of inflammatory cytokines, adipose hormones such as adipokines, and PPAR-γ also leads to enhance the development of insulin resistance in obese subjects [50]. Consequently, insulin resistance and abnormal glucose metabolism, even in the absence of diabetes, is associated with increased risk for the development of PC [27,51–53]. Several studies have demonstrated that the levels of fasting plasma glucose, insulin, and the degree of insulin resistance, are linearly associated with the risk for the development of PC after 10 or more years [27,54,55].
Insulin acts as a growth-promoting mitogen by binding to insulin receptor and causing activation of insulin signaling cascades, and its growth promoting effects in the pancreas have been shown by in vitro studies [56]. High levels of insulin can enhance insulin-like growth factor-1 (IGF-1) synthesis as well and down-regulate IGF-1 binding proteins in the liver, leading to an increase in the bioavailability of free or active IGF-1, which transducer it’s downstream signaling through binding to its receptor (IGF-R). IGF-1 has been shown to be associated with cell proliferation and the risk of PC [53,57]. Both insulin and IGF-1 stimulate anabolic processes as a function of available energy and elementary substrates (e.g. amino acids). The anabolic signals produced by insulin and IGF-1 can promote tumor development by inhibiting apoptosis, stimulating cell proliferation and enhancing angiogenesis [58–62]. Thus, one major biological contribution of obesity to the development of PC might be due to the development of insulin resistance, and its secondary impact on increased activity of IGF-1.
3. Alterations of adipocyte hormone in obesity and PC
Obesity, especially central adiposity, can alter the secretion and regulation of polypeptide hormones secreted by adipocytes in adipose tissues. Such hormones are known as adipokines, which plays important roles in the secretion, regulation and maintenance of normal metabolic and immune function [50,63,64]. Currently, there are more than 50 different types of adipokines such as adiponectin, leptin, resistin, and ghretin, etc. Emerging evidence suggests that adipokines plays a greater role in the regulation of insulin sensitivity, glucose and lipid metabolisms, immune response, and angiogenesis [50].
Adiponectin and leptin are the most abundantly expressed adipokines secreted by adipocytes. Adiponectin, a 244 amino acid polypeptide, is exclusively expressed and secreted by adipose tissue, which is present at high concentration in the blood (from 5 to 10 μg/mL) [50]. Moreover, it has been found to improve insulin sensitivity, decrease insulin resistance and possibly reduce the risk of type 2 DM [65,66], suggesting that adiponectin is an endogenous negative regulator of diabetes. Contrary to other adipokines that are increased in obesity, the level of adiponectin is decreased in obesity and DM patients along with those that are insulin resistant [67,68]. Therefore, it has been suggested that adiponectin is down-regulated in obesity, and thus higher levels of adiponectin is associated with decreased risk of DM in older men and women [66,69], suggesting that the replacement of adiponectin could be useful for reverting the severity of diabetes although it may not be curative. Interestingly, it has been documented that interventions that reduce adipose mass percentage and overall body weight leads to increased levels of adiponectin with a concomitant improvement in insulin sensitivity [65,70,71], further suggesting that low levels of adiponectin may lead to the development of insulin resistance in obesity, which is mechanistically associated with diabetes, resulting in the development of PC.
A number of studies demonstrate that adiponectin is similarly linked with the risk of cancers such as colon, breast, and PC [72–76]. Increased levels of adiponectin decrease the risk of cancer, while its low levels increases the risk of cancer [75]. Although the molecular mechanism of adiponectin action against tumor development is not clear, its potential role in the development of PC may be implicated by several direct and indirect mechanisms. First, adiponectin can directly increase insulin sensitivity and reduce insulin resistance through tyrosine phosphorylation of insulin receptors in muscle tissue [77], leading to the down-regulation of insulin/IGF-1 signaling. Secondly, adiponectin is also known as an anti-inflammatory cytokine, thus it can down-regulate the expression of inflammatory cytokines, such as TNF-α and IL-6, and inhibit NF-κB activation [78], which is likely to be the molecular mechanism involved in these chronic diseases. Therefore, the low levels of adiponectin will induce the expression of inflammatory cytokines in obese subjects, which will lead to subsequent development of diabetes and cancers. Moreover, the in vitro and in vivo studies have shown that adiponectin could affect cancer development more directly through the activation of the AMPK pathway at a local tissue level [79,80]. Adiponectin has also been shown to directly inhibit angiogenesis through the activation of PPAR-γ and by promoting apoptosis in vivo through the activation of the caspase cascade [81], which could be responsible for the anti-diabetes and anti-cancer effects of adiponectin. Collectively, these studies suggest that decreased levels of adiponectin in obesity may contribute to the development of PC by complex mechanisms.
Leptin is expressed and secreted by adipocytes, which is positively associated with adiposity and insulin [82]. Specifically, leptin binds to its receptors in the hypothalamus, thereby activating signals to suppress appetite and increasing energy expenditure. Insulin acts as a positive feedback on leptin gene expression and it has been shown that leptin and insulin levels are increased in obesity [82]. Interestingly, epidemiological and xenograft mouse model studies have shown that leptin is positively associated with increased risk of cancer, and in particular promotes PC growth [65,83–85]. Leptin has also been shown to act as a mitogen for various types of cells, including cancer cells [69,83]. In vitro studies show that leptin exerts its anti-apoptotic and pro-angiogenic activity mediated by the activation of vascular endothelial growth factor (VEGF), and thus may potentially function as a pro-inflammatory agent. In addition, leptin when binds to its receptor is known to activate PI3K/Akt/MAP kinase and STAT signaling pathways, and these signaling pathways are critical for cell survival, growth, proliferation and differentiation [82,86]. Therefore, leptin may play a key role in the development of PC; however, further in-depth studies are warranted in order to fully recognize the mechanistic role of leptin in obesity, diabetes and cancer.
4. Obesity, chronic inflammation, and PC
Chronic inflammation is well known to be involved in the initiation and progression of tumors including PC [18,87,88]. For example, chronic pancreatitis is directly linked with increased risk of PC [5,7,18]. Similarly, obesity has been found to be positively associated with low-grade chronic inflammation [89–91]. Consequently, obesity-induced chronic inflammation represents one of the key features of adipose tissue dysfunction. Local inflammatory responses include macrophage infiltration and resistance to hormones such as insulin and leptin. Systemic inflammatory responses such as increased production of TNF-α, IL-6, IL-1, CRP, plasminogen activator inhibitor-1 (PAI-1), fibrinogen and tissue specific hormone activity mediated through the activation of NF-κB and PPAR-γ pathways, appears to be mechanistically linked with chronic inflammation and obesity [90,92,93]. The loss of body weight leads to decreased levels of inflammatory markers whereas weight gain leads to an increase in these markers [94], suggesting that controlling body weight may reduce chronic inflammation, and thus will reduce the risk for the development of PC.
Although the causes of inflammation in obesity are not fully understood, existing evidence suggests that increased levels of inflammatory cytokines and proteins such as matrix metalloproteinases (MMPs), key mediators of cancer cell invasion and metastasis, are correlated with the high risk of cancers including PC [87,88,95–101]. Chronic inflammation also induces the development of insulin resistance [102], which may eventually contribute to the increased risk of PC mediated through insulin/IGF-1 signaling cascades. Therefore, obesity-induced chronic inflammation appears to contribute to the development of PC.
5. Obesity, oxidative stress, and PC
Oxidative stress is caused by an imbalance between the production of reactive oxygen species (ROS) such as, peroxides and free radicals, and the detoxification of reactive intermediates in a biological system. The maintenance of homeostasis is therefore important for maintaining normal health. Evidence suggests that obesity may induce systemic oxidative stress, which if unresolved, could lead to the development of PC. The oxidative stress created by chronic high exposure to glucose and lipids, and decreased activity of antioxidants in the accumulated adipose tissue leads to dysfunction of adipose tissues such as altered expression of adipokines and cytokines [103]. Several observations from human and animal models have suggested that obesity is involved in lipid peroxidation, which is an oxidative stress marker [50]. For example, one study showed that obese, non-diabetic subjects have increased levels of lipid peroxidation [103]. Obese mouse models show similar increase along with a decrease in the activity of antioxidants [103]. These studies suggest that obesity could induce oxidative stress, which in turn, could induce a series of alterations in the biological processes such as DNA damage, inflammation, apoptosis, and insulin resistance, resulting in the development of cancer [104,105]. ROS can also induce chronic inflammation by activation of NF-κB, and could alter the regulation of adiponectin in adipose tissue [50,106,107]. Thus, obesity-induced oxidative stress may contribute to the increased risk of PC; however, the exact mechanism requires further investigation.
6. Altered regulation of sex hormones in obesity and PC
Sex hormones are known to maintain homeostasis between cellular differentiation, proliferation and apoptosis, and deregulated functioning may promote cancer cell growth [15,108]. The altered regulation of sex hormones is also another key feature of obesity. Obesity-induced high levels of insulin along with IGF-1 can inhibit the synthesis of hormone binding globulin (SHBG) in the liver, the only source of SHBG synthesis [109]. SHBG is a major carrier protein for sex hormones such as testosterone and estradiol in the blood. Thus, a decreased level of SHBG gives rise to an increase in the levels of unbound sex hormones, resulting in increased bioavailability of these hormones to activate signaling cascades [110]. Furthermore, adipose tissue is a major site for the synthesis of estrogens from androgenic precursors [111].
Epidemiological studies have demonstrated that increased risk of breast, endometrial, colon, and prostate cancers are associated with high BMI or obesity, especially central adiposity, is probably mediated by the alterations in the free levels of sex hormones in obesity [65,112,113]. For example, increased risk of postmenopausal breast cancer is associated with a low plasma level of SHBG and an increased level of total and free androgens and estrogens [114]. Although the alterations in the levels of sex hormones have been observed in PC patients, there is no consistent data showing any significant difference in the risk for the development of PC between obese women and men. Thus, the role of sex hormones such as estrogen and testosterone in the development of PC can not be established based on existing evidence, and thus further rigorous studies are required for establishing the role of sex hormones in the context of obesity for the development of PC.
7. Adipose tissue hypoxia and PC
Hypoxia is one of the fundamental features associated with solid tumors including PC [115–118]. Hypoxia-inducible factor-(HIF)-1α, a key transcription factor for hypoxia responsive genes, plays a critical role in increased cell proliferation, survival, angiogenesis and metastasis by primarily regulating the VEGF expression. Tumor hypoxia and over-expression of HIF-1α have been reported to be associated with radiation and chemotherapy resistance, risk of invasion and metastasis, and poor prognosis of solid tumors, all of which lead to higher mortality [118–120]. Hypoxia has been observed in the white adipose tissue in obese mouse models [121–123]. Obesity-induced hypoxia, especially in adipose tissue, is involved in the development of local and systemic insulin resistance, the up-regulation of chronic inflammation, reduced adiponectin and increased leptin gene expressions [122–124]. Thus, adipose tissue hypoxia might play a key role in increased risk for the development and chronicity of cancers including PC.
Recent studies have shown that HIF-1a is involved in the up-regulation of leptin in colorectal and breast tumors [83,89,125]. Hyperinsulinemia might induce HIF-1α-mediated over-expression of leptin in breast cancer cells, contributing to disease progression [83,126]. Furthermore, obesity-induced hypoxia and HIF-1α increases the expression of MMPs and VEGF, suggesting that obesity-induced hypoxia and HIF-1α could be involved in the angiogenic and metastatic processes of tumors [124]. These findings are consistent with clinical data showing that the level of VEGF, regulated mainly by HIF-1α and NF-κB, increases with high BMI [127]. Furthermore, it is well known that VEGF plays an important role in tumor angiogenesis [128]. Thus, increased levels of VEGF, known as a key endothelial mitogen, in obesity may contribute to the development and progression of PC, suggesting that obesity-induced hypoxia and HIF-1α may play a key role in the development and progression of PC.
8. Diabetes Mellitus (DM) and the risk of PC
DM is a very common metabolic disorder with hyperglycemia, eventually affecting all systems in the body, and is it is highly prevalent in the world. There are two types of DM, type-1 and type-2. In adults, type-2 DM accounts for 90–95 percent of all diagnosed cases of DM. There are more than 1.5 million of people newly diagnosed with DM each year in the U.S. This number is expected to double in the next 15–20 years [129]. It is known that one of the main consequences attributed to DM is the impairment of multiple tissues and systems, including the cardiovascular and immune response systems. Moreover, the relationship between DM and cancer risk has been investigated for more than a decade as briefly stated under obesity and the risk of PC.
Emerging evidence indicates that DM, particularly type-2 DM, is positively associated with an increased risk of certain cancers including PC [130–134]. This association is complex because DM may be the cause of PC or often the precursor to the onset of its symptoms and signs. In addition, PC induced DM could also exist which is usually characterized by a short duration of course (several months to several years), a negative family history, the absence of obesity, and rapid progression to insulin dependence [135] whereas DM mediated development of PC has a longer duration (>4 years) before its onset [54,135–137]. Therefore, the examination of the duration of DM prior to the onset of PC is important to elucidate the cause and effect relationship between DM and PC so that appropriate customize management could be discovered.
A large number of epidemiological data supports the hypothesis that DM is positively associated with increased risk of PC as summarized earlier in this chapter. In 1995, a meta-analysis of 20 retrospective cohort studies demonstrates that longstanding DM (5 years or more) increased pancreatic cancer risk by two-fold (RR, 2.0; 95% CI: 1.2–3.2) [138]. In 2005, a meta-analysis of 17 case-control and 19 cohort or nested case-control studies including 9,220 cases of PC shows a modest association between DM and PC (the combined summary OR, 1.82; 95% CI, 1.66–1.89). Individuals with DM history less than four years had a 50 percent greater risk of PC compared with individuals who had DM history for more than 5 years (OR 2.1 vs 1.5) [139], suggesting a modest causal association between DM and PC. In 2007, another meta-analysis of three cohort and six case-control studies specifically conducted in type-1 and young-onset DM patients, confirmed that even in this population a two fold higher risk of PC was observed compared with individuals without DM (RR, 2.0; 95% CI 1.37–3.01) [140]. Recently, one large case-control cohort study including 278,761 DM and 836,283 control subjects was conducted to investigate the association of DM with PC and biliary cancer. The results demonstrate that DM has a three-fold increased risk for the development of PC and a two-fold risk for biliary cancer [141]. Other recent studies also provide supportive evidence documenting that DM is positively associated with increased risk of PC [54,142–144].
9. DM, insulin/IGF-1R signaling pathways and the risk of PC
Accumulating evidence clearly suggests that DM is positively associated with increased risk of PC (RR, 1.4–2.2). However, the exact molecular mechanism(s) has not been fully understood. Similar to the role of obesity in the development of PC, insulin resistance and induced compensatory hyperinsulinemia are widely considered to be mechanisms underlying the causal association of DM and cancers, including PC.
Insulin resistance and altered glucose metabolism are common features for both obesity and type-2 DM. Moreover, DM has a more extensive degree of insulin resistance, hyperinsulinemia and hyperglycemia, which may contribute to the development of PC. Emerging evidence has shown that high levels of insulin, increased fasting and postprandial blood glucose increases the risk of PC [27,145,146]. A high level of insulin stimulates the insulin/IGF-1R signaling pathway via insulin receptor substrates (IRS). In most cell types, these substrates mediate the PI3K/Akt/mTOR signaling pathway [147], which is known to play a pivotal role in the proliferation and survival of PC cells [148,149]. Human insulin users have been found to have an increased risk for the development of cancers including PC [150,151]. It is known that hyperinsulinemia increases IGF-1 by increasing its synthesis and inhibiting its binding protein. Thus, the increased level of insulin and consequently higher levels of IGF-1 promotes cell proliferation, inhibits apoptosis and enhances angiogenesis, all are involved in the development and progression of PC [60,60,152]. Insulin/IGF-1 could also activate PI3K/Akt/mTOR signaling pathway by activation of insulin receptor substrate 1–4 [153], which will contribute to the development of cancers including PC. Moreover, the activation of insulin/IGF-1 receptors by insulin can interplay positively with the hedgehog signaling pathway, which is involved in the regulation of cancer cell growth by regulating cell proliferation and differentiation [154]. It is well known that PC cells express insulin and IGF-1 receptors and IRS-1,2 in large amounts [153,155], suggesting that insulin/IGF-1R signaling pathway may play an important role in the development and progression of PC. Further evidence showed that intrapancreatic insulin/IGF-1 in combination with the activation of K-Ras, a known oncogene for the development of PC, can stimulate PI3K/Akt signaling pathway, which contributes to the development and conferring aggressiveness of PC. Likewise, the inhibition of IGF-1R has been shown to increase the sensitivity of colon cancer stem-like cells to chemotherapeutic drugs [156]. However, the signaling pathways activated by insulin/IGF-1 receptors in PC have not been fully elucidated, suggesting that further mechanistic studies are required to elucidate the role of this signaling pathway in the development and progression of PC.
10. The role of anti-diabetic drugs in PC
Anti-diabetic drugs are known to lower blood insulin levels and increases insulin sensitivity. Due to the positive association of DM with PC, and existing knowledge on the role of insulin/IGF-1R signaling pathways in the development and progression of PC, anti-diabetic drugs have been investigated for their use in prevention and treatment of cancers such as PC for more than a decade. Currently, extensive attention has been given to the anti-diabetic drugs metformin and thiazolidinediones (TZD).
10.1. The role of Metformin in PC
Metformin (1,1-dimethylbiguanide hydrochloride) is a biguanide class of oral hypoglycemic agents and it is the world’s most widely used anti-diabetic drug for the treatment of type-2 DM. Although it has been in clinical use for decades, its precise molecular mechanism of action is still not fully understood. The primary systemic effect of metformin is to decrease blood glucose levels through reduced hepatic gluconeogenesis and increased glucose uptake in peripheral tissues, including skeletal muscles and adipose tissue [157]. It also increases insulin sensitivity which results in decreased insulinemia. Metformin is a very safe drug, which is also well tolerated although it is associated with very low incidence of lactic acidosis (<1/10,000), which is seen predominantly in patients with poor renal function [158,159]. Numerous epidemiological studies have demonstrated that the administration of metformin in DM patients exhibits a protective effect by decreasing incidence of different tumors and improving prognosis of patients with cancer [160–164]. These protective roles of metformin in the development and progression of tumors prompted an investigation into its anti-tumor effect on site-specific tumors. Specifically, clinical studies in both diabetic and non-diabetic patients clearly suggest that metformin may decrease the risk of developing cancers such as breast cancer and PC. Furthermore, several randomized trials have demonstrated its protective role when used as adjuvant therapy and for the prevention of breast and colorectal cancers [165–168].
One recent study showed that women taking metformin had a pathologic complete response rate of 24 percent as compared to 8 percent of women who were not [169]. An additional new, large case-control clinical trial regarding the use of metformin for the treatment of PC was conducted from 2004 to 2008 involving 973 PC patients (including 259 DM patients) and 863 controls (including 109 DM patients) [170]. The results showed that DM patients who were administrated metformin had a significantly lower risk of PC compared to those who did not receive metformin. In contrast, DM patients administered insulin had a significantly higher risk of PC compared to those without insulin. These recent findings clearly suggest that the administration of metformin is positively associated with the decreased risk of PC in DM patients while the use of insulin or other agents is associated with an increased risk. Therefore, metformin appears to exert a protective role against the development and progression of PC.
Animal studies have also demonstrated that metformin can inhibit tumor growth in xenograft models [171–174]. The anti-tumor effect of metformin is probably related to its direct and indirect mechanisms although the detailed molecular mechanisms in support of these findings are not fully understood, several mechanisms have been proposed. It has been shown that increasing insulin sensitivity and decreasing insulin level by metformin could inhibit cancer cell growth by activation of AMP kinase (AMPK), which in turn inhibits mTOR signaling [175,176]. AMPK is usually activated when the ratio of AMP/ATP is increased and the activation of AMPK results in the down-regulation of the PI3K/Akt/mTOR signaling pathway by phosphorylation of mTOR. The mTOR is usually activated by mitogenic-responsive pathways such as Ras/ERK and PI3K/Akt as well as pathways that signal the availability of intracellular energy and nutrients including amino acids. However, the inhibition of mTOR pathway by metformin through activation of AMPK leads to a rapid inhibition of cellular protein synthesis and growth [165–167]. Moreover, metformin can directly inhibit cancer cell growth and proliferation through the regulation of cyclin D1-mediated cell cycle, p53 expression and phosphorylation in breast cancer and prostate cancer cells [177–182]. Metformin has also been found to inhibit the production of inflammatory cytokines such as TNF-α and IL-6 as well as VEGF, probably by inactivation of NF-κB and HIF-1α [183–185]. In vitro and in vivo studies show that metformin can block tumor growth by inactivation of breast cancer stem-like cells and epithelial-mesenchymal transition (EMT) phenotype, and these cells are believed to be the root cause of tumor recurrence and metastasis [186,187].
The first animal study using metformin on pancreatic tumors showed that metformin can prevent the growth of carcinogen-induced pancreatic tumors in hamsters maintained on a high-fat diet [188]. It has also been found that metformin could induce apoptosis in PC cells, and also inhibited tumor growth in xenograft model of tumors induced by PANC-1 and MIAPaCa-2 cell lines [189,190]. Additional evidence showed that metformin can inhibit PC cell growth and proliferation by disruption of insulin/IGF-1R and GPCR crosstalk through the activation of AMPK [189,191]. These results suggest that metformin could be useful anti-tumor agent for the treatment of PC by targeting AMPK/mTOR, insulin/IGF-1R, GPCR, and other signaling pathways.
10.2. PPAR-γ ligands, thiazolidinediones, and PC treatment
Peroxisome proliferator-activated receptor-γ (PPAR-γ), expressed predominantly in adipose tissue and in the immune system [161,192,193], is a ligand-activated transcription factor involved in the regulation of lipid and glucose homeostasis, and also control of inflammation [194]. Upon the binding of the ligand, PPAR-γ translocates to the nucleus where it heterodimerizes with RXR [161,195,196] and activate genes with a peroxisome proliferator response element (PPRE) in their promoter regions initiating their transcription [197,198]. Altered regulations of PPAR-γ have been implicated in a number of disease processes, including obesity, type-2 DM, and carcinogenesis [161,199–201]. Thiazolidinediones (TZD), an agonist of PPAR-γ have been used effectively to mediate insulin sensitivity and decrease high insulin levels through the activation PPAR-γ however, the molecular mechanism has not fully established. Currently, two TZD classes of synthetic PPAR-γ agonist (ligand), pioglitazone and rosiglitazone, have been approved by the United States Food and Drug Administration (FDA) as anti-diabetic drugs for the treatment of type-2 DM although the use of pioglitazone in more safer than rosiglitazone.
Because TZD has the ability to modulate insulin signaling, the use of TZD has been proposed as a potential anti-tumor agent for the treatment of cancers [202,203]. A number of clinical studies have been conducted for assessing the efficacy of the TZD class of PPAR-γ agonists. The majority of the studies showed inconsistent or inconclusive results [164,204]. This is probably due to the short duration of treatment, study design bias, residual confounders, or cell- or tissue-specific targeting [203]. In recent years, several clinical studies have suggested that the treatment of DM patients with PPAR-γ agonist may have preventive effects on the development of cancer. As a result, PPAR-γ agonist could be useful as adjuvant therapy with conventional chemotherapeutics [205–207]. There has been no clinical report regarding the use of PPAR-γ ligands in PC patients. However, the over-expression of PPAR-γ activation has been found to be positively correlated with higher tumor stage and grade, and associated with shorter patient survival, suggesting its potential role in PC progression [131,208]. Although the biological role of PPAR-γ in the development of PC is not clear, the TZD class of PPAR-γ ligands have been shown to have activity in pre-clinical cell culture and xenograft mouse models [209–212]. One animal study, using Syrian golden hamsters, demonstrated that dietary intake of 800 ppm pioglitazone for 22 weeks correlated with a decreased incidence and multiplicity of carcinogen-induced pancreatic tumors, implicating the potential chemopreventive role of TZD in the development of PC [213].
The inhibitory effects of the TZD class of PPAR-γ ligands on the development and progression of cancer have been proposed, which involves PPAR-γ-dependent and independent mechanisms. First, PPAR-γ ligands have been shown to increase insulin sensitivity and decrease the insulin level through the activation of PPAR-γ, leading to the inhibition of insulin/IGF-1R signaling pathways [209,210]. Secondly, the activation of PPAR-γ can inhibit TNF-α, IL-6, IL-8, PAI-1, VEGF, COX2, MMPs, uPA, NF-κB, AP-1, STAT, and leptin [212,214–223]. PPAR-γ ligands can directly inhibit inflammatory response by the activation of AMPK through the regulation of mitochondrial function and ROS production in a PPAR-γ-independent pathways [224]. There is also evidence showing that PPAR-γ ligands can induce apoptosis and inhibit proliferation of brain cancer stem cells [225]. Furthermore, PPAR-γ ligands are also shown to increase the expression of PTEN, an oncogene suppressor, and could inhibit the activation of β-catenin, a positive modulator of cell growth through PPAR-γ-independent mechanism [211,222,226,227]. Based on these experimental evidences, it is important to note that the anti-tumor effects of TZD class of PPAR-γ ligand, which are anti-diabetic drugs, could be useful for the prevention and/or treatment of human malignancies especially PC.
11. The regulatory role of microRNAs (miRNAs) in obesity, DM and PC
MicroRNAs (miRNAs) are small non-protein-coding RNAs (around 21–24 nucleotides) that function as post-transcriptional gene regulators by specific binding to the 3′ untranslated region (3′UTR) of target mRNAs to control protein synthesis or degradation of the mRNA. They are currently recognized as regulator of expression of most genes, and consequently they play critical roles in a wide array of biological processes, including cell differentiation, proliferation, death, metabolism and energy homeostasis [228,229]. A large number of miRNAs have been reported to be associated with chronic diseases such as obesity, DM and cancers, and are suspected to be mechanistically associated with these chronic diseases. However, their exact role in the pathogenesis of obesity, DM and PC are not fully understood.
A number of studies have demonstrated that miR-21 is a potential oncogene and it is up-regulated in PC cells [230–237]. Over-expression of miR-21 results in decreased expression of PTEN, a known tumor suppressor in PC [237]. The miR-21 has also been reported to show anti-apoptotic, proliferative, invasive and angiogenic properties in cancer cells [230,236,238]. Moreover, decreased expression of miR-200 has been observed in many cancer cells [230,236,238], suggesting that altered expression of these miRNAs may contribute to the invasiveness and metastatic characteristics of PC. The possible roles of these two miRNAs in obesity and DM associated with the development of PC require further studies.
Let7 family, especially let7b, is abundantly expressed in pancreatic islet cells and postulated to be important ribo-regulators of blood glucose [239–241]. Let7 has been found to regulate the expression of PPAR-γ2 in adipocytes. In the pathologenesis of cancer, let7 has been found to be down-regulated thereby increasing the expression of Ras and c-Myc in malignant cells [234–236]. In addition, the altered regulation of let7 has been observed in DM and PC [234,235], suggesting that let7 may play a key role in the pathologenesis of obesity, DM and PC.
Another class of miRNA, miR-34a and miR-146a have been found to be highly expressed in obesity and DM [241–243], and over-expressions of miR-34a and miR-146a are known to cause alterations in glucose-stimulated insulin secretion and the induction apoptosis in β-cells [241,242]. In contrast, miR-34a has been shown to be induced by p53 and exhibit potent anti-proliferative and pro-apoptotic activities in cancer cells including PC cells [234]. Studies have also shown that miR-146a have angiogenic activity [241,242], and altered expression of miR-34a and miR-146a are typically found in PC cells [234,235,244] although the exact role of these miRNAs in obesity, DM and PC has not been fully elucidated.
The up-regulation of miR-143 has been found in obesity, DM and PC [232,235,240,241]. One identified target gene of miR-143a is MAPK7, which is involved in the regulation of MAPK signaling pathway. While, over-expression of miR-143 is related to altered regulation of PPAR-γ in adipose tissue of obese mice [240,241], the potential role of miR-143 in the development of PC associated with obesity and DM is not clear, suggesting that further in-depth mechanistic studies are warranted for elucidating the possible role of miR-143 in the development of PC in obese and DM patients.
The expression of miR-29 has been found to be elevated in the insulin targeted tissues [239]. Over-expression of miR-29 represses insulin-stimulated glucose uptake, leading to the development of insulin resistance [239,241,242]. Moreover, high level of miR-29 has been observed in obesity and DM animals and DM patients [239–241,245]. Moreover, miR-29 was also found to be up-regulated in PC cells [235], suggesting the potential role of miR-29 in obesity, DM, and PC and thus, novel strategies by which specific miRNA could be down-regulated or up-regulated would become a novel approach for the treatment of chronic diseases including obesity, diabetes and PC. Further studies have shown that miR-375 is highly expressed in β-cells in pancreatic islet. It down-regulates glucose-stimulated insulin secretion by controlling the expressions of 3′-phosphoinositide-dependent protein kinase-1 (PDK1), a key regulator of β-cell function, and myotrophin, a regulator of insulin secretion [241,242]. An increased level of miR-375 has been found in obesity and type-2 DM patients [241,246] where it represses insulin secretion. Inhibition of miR-375 leads to an increase in insulin secretion [241,246]; however, miR-375 knockout mice display marked hyperglycemia, suggesting that miR-375 is essential to maintain normal pancreatic function [246]. Interestingly, miR-375 has been found to down-regulated in PC cells [235], suggesting the potential role of miR-375 in the development of PC associated with obesity and DM although mechanistic studies are lacking. The above sections underscore the importance of miRNA in chronic diseases; however, the field is still in its infancy and clearly more in-depth mechanistic studies are warranted in order to fully appreciate the roles and therapeutic regulations of miRNAs in obesity, diabetes and cancer especially PC.
12. Conclusions and Perspectives
A large number of epidemiological studies have documented a clear association between obesity and diabetes, which is also positively associated with increased risk of PC, suggesting that both obesity and diabetes are additional risk factors for PC. Insulin resistance and the induction of compensatory high levels of insulin, leading to the alteration of glucose and lipid metabolism, are common characteristics of obesity and type-2 DM. They appear to be primarily contributes to the development of PC by activation of insulin/IGF-1R signaling pathways. Increased levels of insulin, fasting and post-prandial blood glucose have also been reported to be related to the high risk of PC. Thus, targeting insulin/IGF-1 signaling by anti-diabetic drugs which will cause lowering of blood insulin level, could be useful for the prevention and/or treatment of PC although further novel well thought-out clinical trial design is warranted. A large number of studies have demonstrated that the administration of anti-diabetic drugs, such as metformin and the TZD class of PPAR-γ agonist decreases the risk of cancers, suggesting that these drugs could be useful anti-tumor agents for PC especially because metformin could effectively eliminate cancer stem cells which is the root cause of tumor recurrence and metastasis. In vitro and in vivo studies have also shown that these drugs can inhibit PC growth by different mechanisms including alterations in the expression of genes mediated by the regulation of miRNAs. Although the exact roles of miRNAs in the pathogenesis of obesity, DM and PC are not fully understood, selective up-regulation and down-regulation of important miRNAs such as miR-21, let7, miR-29, miR-34a, miR-134, miR-146a, and miR-375 could become novel and newer strategies for the treatment of obesity, DM and PC in the future. Overall, the future looks brighter than ever before for exploration of targeting miRNAs by novel approaches for the management of chronic diseases such as obesity, diabetes and cancer especially in subjects who are at high risk for the development of PC.
Figure 1.
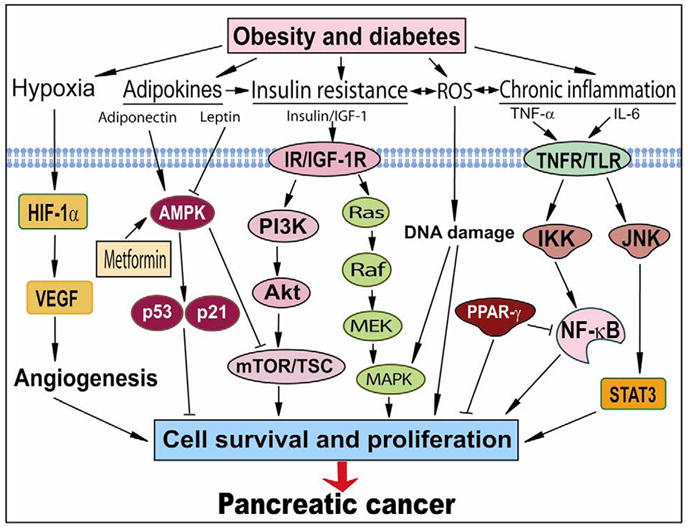
Potential pathways directly linking obesity and diabetes to pancreatic cancer. Obesity and diabetes cause mutiple alterations in glucose and lipid hemastasis, microenvironments, and immune responses, which result in the activation of several oncogenic signaling pathways. These deregulations increase cell survival and proliferation, eventually leading to the development and progression of pancreatic cancer. ROS, reactive oxygen species; IGF-1, insulin-like growth factor-1; IR, insulin receptors; IGF-1R, insulin-like growth factor-1 receptors; TNFR, tumor necrosis factor receptors; TLR, Toll-like receptors; HIF-1α, hypoxia-inducible factor-α1; AMPK, AMP kinase; IKK, IκB kinase; PPAR-γ, peroxisome proliferator-activated receptor-γ; VEGF, vascular endothelial growth factor; MAPK, MAP kinase; mTOR, mammalian target of rapamycin; TSC, tuberous sclerosis complex; Akt, protein kinase B. PI3K, phosphoinositide-3-kinase; STAT3, activator of transcription-3; JNK, c-Jun NH2-terminal kinase.
Acknowledgments
We thank Puschelberg and Guido foundations for their generous contribution. We also thank Ms. Amanda C. Sehmer for editing the manuscript.
Grant Support: National Cancer Institute, NIH grants 5R01CA131151, 3R01CA131151-02S1, and 5R01CA132794 (F.H. Sarkar).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Edwards BK, Brown ML, Wingo PA, Howe HL, Ward E, Ries LA, Schrag D, Jamison PM, Jemal A, Wu XC, Friedman C, Harlan L, Warren J, Anderson RN, Pickle LW. Annual report to the nation on the status of cancer, 1975–2002, featuring population-based trends in cancer treatment. J Natl Cancer Inst. 2005;97:1407–1427. doi: 10.1093/jnci/dji289. [DOI] [PubMed] [Google Scholar]
- 2.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 3.Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55:74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- 4.Arslan AA, Helzlsouer KJ, Kooperberg C, Shu XO, Steplowski E, Bueno-de-Mesquita HB, Fuchs CS, Gross MD, Jacobs EJ, Lacroix AZ, Petersen GM, Stolzenberg-Solomon RZ, Zheng W, Albanes D, Amundadottir L, Bamlet WR, Barricarte A, Bingham SA, Boeing H, Boutron-Ruault MC, Buring JE, Chanock SJ, Clipp S, Gaziano JM, Giovannucci EL, Hankinson SE, Hartge P, Hoover RN, Hunter DJ, Hutchinson A, Jacobs KB, Kraft P, Lynch SM, Manjer J, Manson JE, McTiernan A, McWilliams RR, Mendelsohn JB, Michaud DS, Palli D, Rohan TE, Slimani N, Thomas G, Tjonneland A, Tobias GS, Trichopoulos D, Virtamo J, Wolpin BM, Yu K, Zeleniuch-Jacquotte A, Patel AV. Anthropometric measures, body mass index, and pancreatic cancer: a pooled analysis from the Pancreatic Cancer Cohort Consortium (PanScan) Arch Intern Med. 2010;170:791–802. doi: 10.1001/archinternmed.2010.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lowenfels AB, Maisonneuve P. Epidemiologic and etiologic factors of pancreatic cancer. Hematol Oncol Clin North Am. 2002;16:1–16. doi: 10.1016/s0889-8588(01)00003-x. [DOI] [PubMed] [Google Scholar]
- 6.Lowenfels AB, Sullivan T, Fiorianti J, Maisonneuve P. The epidemiology and impact of pancreatic diseases in the United States. Curr Gastroenterol Rep. 2005;7:90–95. doi: 10.1007/s11894-005-0045-6. [DOI] [PubMed] [Google Scholar]
- 7.Michaud DS. Epidemiology of pancreatic cancer. Minerva Chir. 2004;59:99–111. [PubMed] [Google Scholar]
- 8.Schuster DP. Obesity and the Development of Type 2 Diabetes: the Effects of Fatty Tissue Inflamation. Dovepress; 2010. pp. 253–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.WHO. World Health Organization Fact Sheet for World Wide Prevalence of Obesity. 2006 http://www.who.int/mediacentre/factsheets/fs311/en/index.html.
- 10.Chang S, Masse LC, Moser RP, Dodd KW, Arganaraz F, Fuemmler BF, Jemal A. State ranks of incident cancer burden due to overweight and obesity in the United States, 2003. Obesity (Silver Spring) 2008;16:1636–1650. doi: 10.1038/oby.2008.228. [DOI] [PubMed] [Google Scholar]
- 11.Flegal KM, Carroll MD, Kuczmarski RJ, Johnson CL. Overweight and obesity in the United States: prevalence and trends, 1960–1994. Int J Obes Relat Metab Disord. 1998;22:39–47. doi: 10.1038/sj.ijo.0800541. [DOI] [PubMed] [Google Scholar]
- 12.FAO/WHO/UN. Energy and Protein Requirements: Report of a Joint Expert Consultation. WHO; Geneva: 1985. [PubMed] [Google Scholar]
- 13.Flegal KM, Carroll MD, Ogden CL, Curtin LR. Prevalence and trends in obesity among US adults, 1999–2008. JAMA. 2010;303:235–241. doi: 10.1001/jama.2009.2014. [DOI] [PubMed] [Google Scholar]
- 14.Anderson AS, Caswell S. Obesity management--an opportunity for cancer prevention. Surgeon. 2009;7:282–285. doi: 10.1016/s1479-666x(09)80005-x. [DOI] [PubMed] [Google Scholar]
- 15.Bianchini F, Kaaks R, Vainio H. Overweight, obesity, and cancer risk. Lancet Oncol. 2002;3:565–574. doi: 10.1016/s1470-2045(02)00849-5. [DOI] [PubMed] [Google Scholar]
- 16.bu-Abid S, Szold A, Klausner J. Obesity and cancer. J Med. 2002;33:73–86. [PubMed] [Google Scholar]
- 17.Calle EE, Thun MJ. Obesity and cancer. Oncogene. 2004;23:6365–6378. doi: 10.1038/sj.onc.1207751. [DOI] [PubMed] [Google Scholar]
- 18.Gumbs AA. Obesity, pancreatitis, and pancreatic cancer. Obes Surg. 2008;18:1183–1187. doi: 10.1007/s11695-008-9599-3. [DOI] [PubMed] [Google Scholar]
- 19.Hsing AW, Sakoda LC, Chua S., Jr Obesity, metabolic syndrome, and prostate cancer. Am J Clin Nutr. 2007;86:s843–s857. doi: 10.1093/ajcn/86.3.843S. [DOI] [PubMed] [Google Scholar]
- 20.Kuriyama S, Tsubono Y, Hozawa A, Shimazu T, Suzuki Y, Koizumi Y, Suzuki Y, Ohmori K, Nishino Y, Tsuji I. Obesity and risk of cancer in Japan. Int J Cancer. 2005;113:148–157. doi: 10.1002/ijc.20529. [DOI] [PubMed] [Google Scholar]
- 21.Percik R, Stumvoll M. Obesity and cancer. Exp Clin Endocrinol Diabetes. 2009;117:563–566. doi: 10.1055/s-0029-1241870. [DOI] [PubMed] [Google Scholar]
- 22.Pischon T, Nothlings U, Boeing H. Obesity and cancer. Proc Nutr Soc. 2008;67:128–145. doi: 10.1017/S0029665108006976. [DOI] [PubMed] [Google Scholar]
- 23.Teucher B, Rohrmann S, Kaaks R. Obesity: focus on all-cause mortality and cancer. Maturitas. 2010;65:112–116. doi: 10.1016/j.maturitas.2009.11.018. [DOI] [PubMed] [Google Scholar]
- 24.Berrington de GA, Spencer EA, Bueno-de-Mesquita HB, Roddam A, Stolzenberg-Solomon R, Halkjaer J, Tjonneland A, Overvad K, Clavel-Chapelon F, Boutron-Ruault MC, Boeing H, Pischon T, Linseisen J, Rohrmann S, Trichopoulou A, Benetou V, Papadimitriou A, Pala V, Palli D, Panico S, Tumino R, Vineis P, Boshuizen HC, Ocke MC, Peeters PH, Lund E, Gonzalez CA, Larranaga N, Martinez-Garcia C, Mendez M, Navarro C, Quiros JR, Tormo MJ, Hallmans G, Ye W, Bingham SA, Khaw KT, Allen N, Key TJ, Jenab M, Norat T, Ferrari P, Riboli E. Anthropometry, physical activity, and the risk of pancreatic cancer in the European prospective investigation into cancer and nutrition. Cancer Epidemiol Biomarkers Prev. 2006;15:879–885. doi: 10.1158/1055-9965.EPI-05-0800. [DOI] [PubMed] [Google Scholar]
- 25.Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med. 2003;348:1625–1638. doi: 10.1056/NEJMoa021423. [DOI] [PubMed] [Google Scholar]
- 26.Friedman GD, van den Eeden SK. Risk factors for pancreatic cancer: an exploratory study. Int J Epidemiol. 1993;22:30–37. doi: 10.1093/ije/22.1.30. [DOI] [PubMed] [Google Scholar]
- 27.Gapstur SM, Gann PH, Lowe W, Liu K, Colangelo L, Dyer A. Abnormal glucose metabolism and pancreatic cancer mortality. JAMA. 2000;283:2552–2558. doi: 10.1001/jama.283.19.2552. [DOI] [PubMed] [Google Scholar]
- 28.Isaksson B, Jonsson F, Pedersen NL, Larsson J, Feychting M, Permert J. Lifestyle factors and pancreatic cancer risk: a cohort study from the Swedish Twin Registry. Int J Cancer. 2002;98:480–482. doi: 10.1002/ijc.10256. [DOI] [PubMed] [Google Scholar]
- 29.Larsson SC, Permert J, Hakansson N, Naslund I, Bergkvist L, Wolk A. Overall obesity, abdominal adiposity, diabetes and cigarette smoking in relation to the risk of pancreatic cancer in two Swedish population-based cohorts. Br J Cancer. 2005;93:1310–1315. doi: 10.1038/sj.bjc.6602868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Michaud DS, Giovannucci E, Willett WC, Colditz GA, Stampfer MJ, Fuchs CS. Physical activity, obesity, height, and the risk of pancreatic cancer. JAMA. 2001;286:921–929. doi: 10.1001/jama.286.8.921. [DOI] [PubMed] [Google Scholar]
- 31.Patel AV, Rodriguez C, Bernstein L, Chao A, Thun MJ, Calle EE. Obesity, recreational physical activity, and risk of pancreatic cancer in a large U.S. Cohort. Cancer Epidemiol Biomarkers Prev. 2005;14:459–466. doi: 10.1158/1055-9965.EPI-04-0583. [DOI] [PubMed] [Google Scholar]
- 32.Rapp K, Schroeder J, Klenk J, Stoehr S, Ulmer H, Concin H, Diem G, Oberaigner W, Weiland SK. Obesity and incidence of cancer: a large cohort study of over 145,000 adults in Austria. Br J Cancer. 2005;93:1062–1067. doi: 10.1038/sj.bjc.6602819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shibata A, Mack TM, Paganini-Hill A, Ross RK, Henderson BE. A prospective study of pancreatic cancer in the elderly. Int J Cancer. 1994;58:46–49. doi: 10.1002/ijc.2910580109. [DOI] [PubMed] [Google Scholar]
- 34.Howe GR, Jain M, Miller AB. Dietary factors and risk of pancreatic cancer: results of a Canadian population-based case-control study. Int J Cancer. 1990;45:604–608. doi: 10.1002/ijc.2910450405. [DOI] [PubMed] [Google Scholar]
- 35.Nilsen TI, Vatten LJ. A prospective study of lifestyle factors and the risk of pancreatic cancer in Nord-Trondelag, Norway. Cancer Causes Control. 2000;11:645–652. doi: 10.1023/a:1008916123357. [DOI] [PubMed] [Google Scholar]
- 36.Zatonski W, Przewozniak K, Howe GR, Maisonneuve P, Walker AM, Boyle P. Nutritional factors and pancreatic cancer: a case-control study from south-west Poland. Int J Cancer. 1991;48:390–394. doi: 10.1002/ijc.2910480314. [DOI] [PubMed] [Google Scholar]
- 37.Berrington de GA, Sweetland S, Spencer E. A meta-analysis of obesity and the risk of pancreatic cancer. Br J Cancer. 2003;89:519–523. doi: 10.1038/sj.bjc.6601140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Larsson SC, Orsini N, Wolk A. Body mass index and pancreatic cancer risk: A meta-analysis of prospective studies. Int J Cancer. 2007;120:1993–1998. doi: 10.1002/ijc.22535. [DOI] [PubMed] [Google Scholar]
- 39.Renehan AG, Tyson M, Egger M, Heller RF, Zwahlen M. Body-mass index and incidence of cancer: a systematic review and meta-analysis of prospective observational studies. Lancet. 2008;371:569–578. doi: 10.1016/S0140-6736(08)60269-X. [DOI] [PubMed] [Google Scholar]
- 40.Luo J, Margolis KL, Adami HO, LaCroix A, Ye W. Obesity and risk of pancreatic cancer among postmenopausal women: the Women’s Health Initiative (United States) Br J Cancer. 2008;99:527–531. doi: 10.1038/sj.bjc.6604487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li D, Morris JS, Liu J, Hassan MM, Day RS, Bondy ML, Abbruzzese JL. Body mass index and risk, age of onset, and survival in patients with pancreatic cancer. JAMA. 2009;301:2553–2562. doi: 10.1001/jama.2009.886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jiao L, Berrington de GA, Hartge P, Pfeiffer RM, Park Y, Freedman DM, Gail MH, Alavanja MC, Albanes D, Beane Freeman LE, Chow WH, Huang WY, Hayes RB, Hoppin JA, Ji BT, Leitzmann MF, Linet MS, Meinhold CL, Schairer C, Schatzkin A, Virtamo J, Weinstein SJ, Zheng W, Stolzenberg-Solomon RZ. Body mass index, effect modifiers, and risk of pancreatic cancer: a pooled study of seven prospective cohorts. Cancer Causes Control. 2010;21:1305–1314. doi: 10.1007/s10552-010-9558-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Johansen D, Stocks T, Jonsson H, Lindkvist B, Bjorge T, Concin H, Almquist M, Haggstrom C, Engeland A, Ulmer H, Hallmans G, Selmer R, Nagel G, Tretli S, Stattin P, Manjer J. Metabolic factors and the risk of pancreatic cancer: a prospective analysis of almost 580,000 men and women in the Metabolic Syndrome and Cancer Project. Cancer Epidemiol Biomarkers Prev. 2010;19:2307–2317. doi: 10.1158/1055-9965.EPI-10-0234. [DOI] [PubMed] [Google Scholar]
- 44.Godsland IF. Insulin resistance and hyperinsulinaemia in the development and progression of cancer. Clin Sci (Lond) 2010;118:315–332. doi: 10.1042/CS20090399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kahn BB, Flier JS. Obesity and insulin resistance. J Clin Invest. 2000;106:473–481. doi: 10.1172/JCI10842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pisani P. Hyper-insulinaemia and cancer, meta-analyses of epidemiological studies. Arch Physiol Biochem. 2008;114:63–70. doi: 10.1080/13813450801954451. [DOI] [PubMed] [Google Scholar]
- 47.Jazet IM, Pijl H, Meinders AE. Adipose tissue as an endocrine organ: impact on insulin resistance. Neth J Med. 2003;61:194–212. [PubMed] [Google Scholar]
- 48.Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2006;444:840–846. doi: 10.1038/nature05482. [DOI] [PubMed] [Google Scholar]
- 49.Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest. 2006;116:1793–1801. doi: 10.1172/JCI29069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Becker S, Dossus L, Kaaks R. Obesity related hyperinsulinaemia and hyperglycaemia and cancer development. Arch Physiol Biochem. 2009;115:86–96. doi: 10.1080/13813450902878054. [DOI] [PubMed] [Google Scholar]
- 51.Boyd DB. Insulin and cancer. Integr Cancer Ther. 2003;2:315–329. doi: 10.1177/1534735403259152. [DOI] [PubMed] [Google Scholar]
- 52.Fisher WE, Boros LG, Schirmer WJ. Insulin promotes pancreatic cancer: evidence for endocrine influence on exocrine pancreatic tumors. J Surg Res. 1996;63:310–313. doi: 10.1006/jsre.1996.0266. [DOI] [PubMed] [Google Scholar]
- 53.McCarty MF. Insulin secretion as a determinant of pancreatic cancer risk. Med Hypotheses. 2001;57:146–150. doi: 10.1054/mehy.2001.1316. [DOI] [PubMed] [Google Scholar]
- 54.Pannala R, Basu A, Petersen GM, Chari ST. New-onset diabetes: a potential clue to the early diagnosis of pancreatic cancer. Lancet Oncol. 2009;10:88–95. doi: 10.1016/S1470-2045(08)70337-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stolzenberg-Solomon RZ, Graubard BI, Chari S, Limburg P, Taylor PR, Virtamo J, Albanes D. Insulin, glucose, insulin resistance, and pancreatic cancer in male smokers. JAMA. 2005;294:2872–2878. doi: 10.1001/jama.294.22.2872. [DOI] [PubMed] [Google Scholar]
- 56.Mossner J, Logsdon CD, Goldfine ID, Williams JA. Do insulin and the insulin like growth factors (IGFs) stimulate growth of the exocrine pancreas? Gut. 1987;28(Suppl):51–55. doi: 10.1136/gut.28.suppl.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Conover CA, Lee PD, Kanaley JA, Clarkson JT, Jensen MD. Insulin regulation of insulin-like growth factor binding protein-1 in obese and nonobese humans. J Clin Endocrinol Metab. 1992;74:1355–1360. doi: 10.1210/jcem.74.6.1375600. [DOI] [PubMed] [Google Scholar]
- 58.Kaaks R, Lukanova A. Energy balance and cancer: the role of insulin and insulin-like growth factor-I. Proc Nutr Soc. 2001;60:91–106. doi: 10.1079/pns200070. [DOI] [PubMed] [Google Scholar]
- 59.Kaaks R. Nutrition, insulin, IGF-1 metabolism and cancer risk: a summary of epidemiological evidence. Novartis Found Symp. 2004;262:247–260. [PubMed] [Google Scholar]
- 60.Khandwala HM, McCutcheon IE, Flyvbjerg A, Friend KE. The effects of insulin-like growth factors on tumorigenesis and neoplastic growth. Endocr Rev. 2000;21:215–244. doi: 10.1210/edrv.21.3.0399. [DOI] [PubMed] [Google Scholar]
- 61.Verheus M, Peeters PH, Rinaldi S, Dossus L, Biessy C, Olsen A, Tjonneland A, Overvad K, Jeppesen M, Clavel-Chapelon F, Tehard B, Nagel G, Linseisen J, Boeing H, Lahmann PH, Arvaniti A, Psaltopoulou T, Trichopoulou A, Palli D, Tumino R, Panico S, Sacerdote C, Sieri S, van Gils CH, Bueno-de-Mesquita BH, Gonzalez CA, Ardanaz E, Larranaga N, Garcia CM, Navarro C, Quiros JR, Key T, Allen N, Bingham S, Khaw KT, Slimani N, Riboli E, Kaaks R. Serum C-peptide levels and breast cancer risk: results from the European Prospective Investigation into Cancer and Nutrition (EPIC) Int J Cancer. 2006;119:659–667. doi: 10.1002/ijc.21861. [DOI] [PubMed] [Google Scholar]
- 62.Wei EK, Ma J, Pollak MN, Rifai N, Fuchs CS, Hankinson SE, Giovannucci E. A prospective study of C-peptide, insulin-like growth factor-I, insulin-like growth factor binding protein-1, and the risk of colorectal cancer in women. Cancer Epidemiol Biomarkers Prev. 2005;14:850–855. doi: 10.1158/1055-9965.EPI-04-0661. [DOI] [PubMed] [Google Scholar]
- 63.Engeli S, Feldpausch M, Gorzelniak K, Hartwig F, Heintze U, Janke J, Mohlig M, Pfeiffer AF, Luft FC, Sharma AM. Association between adiponectin and mediators of inflammation in obese women. Diabetes. 2003;52:942–947. doi: 10.2337/diabetes.52.4.942. [DOI] [PubMed] [Google Scholar]
- 64.Straczkowski M, Kowalska I, Stepien A, Dzienis-Straczkowska S, Szelachowska M, Kinalska I. Increased plasma-soluble tumor necrosis factor-alpha receptor 2 level in lean nondiabetic offspring of type 2 diabetic subjects. Diabetes Care. 2002;25:1824–1828. doi: 10.2337/diacare.25.10.1824. [DOI] [PubMed] [Google Scholar]
- 65.Khalili P, Flyvbjerg A, Frystyk J, Lundin F, Jendle J, Engstrom G, Nilsson PM. Total adiponectin does not predict cardiovascular events in middle-aged men in a prospective, long-term follow-up study. Diabetes Metab. 2010;36:137–143. doi: 10.1016/j.diabet.2009.10.004. [DOI] [PubMed] [Google Scholar]
- 66.Spranger J, Kroke A, Mohlig M, Bergmann MM, Ristow M, Boeing H, Pfeiffer AF. Adiponectin and protection against type 2 diabetes mellitus. Lancet. 2003;361:226–228. doi: 10.1016/S0140-6736(03)12255-6. [DOI] [PubMed] [Google Scholar]
- 67.Chandran M, Phillips SA, Ciaraldi T, Henry RR. Adiponectin: more than just another fat cell hormone? Diabetes Care. 2003;26:2442–2450. doi: 10.2337/diacare.26.8.2442. [DOI] [PubMed] [Google Scholar]
- 68.Swarbrick MM, Havel PJ. Physiological, pharmacological, and nutritional regulation of circulating adiponectin concentrations in humans. Metab Syndr Relat Disord. 2008;6:87–102. doi: 10.1089/met.2007.0029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Krakoff J, Funahashi T, Stehouwer CD, Schalkwijk CG, Tanaka S, Matsuzawa Y, Kobes S, Tataranni PA, Hanson RL, Knowler WC, Lindsay RS. Inflammatory markers, adiponectin, and risk of type 2 diabetes in the Pima Indian. Diabetes Care. 2003;26:1745–1751. doi: 10.2337/diacare.26.6.1745. [DOI] [PubMed] [Google Scholar]
- 70.Samaras K, Botelho NK, Chisholm DJ, Lord RV. Subcutaneous and visceral adipose tissue gene expression of serum adipokines that predict type 2 diabetes. Obesity (Silver Spring) 2010;18:884–889. doi: 10.1038/oby.2009.443. [DOI] [PubMed] [Google Scholar]
- 71.Viljanen AP, Lautamaki R, Jarvisalo M, Parkkola R, Huupponen R, Lehtimaki T, Ronnemaa T, Raitakari OT, Iozzo P, Nuutila P. Effects of weight loss on visceral and abdominal subcutaneous adipose tissue blood-flow and insulin-mediated glucose uptake in healthy obese subjects. Ann Med. 2009;41:152–160. doi: 10.1080/07853890802446754. [DOI] [PubMed] [Google Scholar]
- 72.Dalamaga M, Migdalis I, Fargnoli JL, Papadavid E, Bloom E, Mitsiades N, Karmaniolas K, Pelecanos N, Tseleni-Balafouta S, onyssiou-Asteriou A, Mantzoros CS. Pancreatic cancer expresses adiponectin receptors and is associated with hypoleptinemia and hyperadiponectinemia: a case-control study. Cancer Causes Control. 2009;20:625–633. doi: 10.1007/s10552-008-9273-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Stolzenberg-Solomon RZ, Weinstein S, Pollak M, Tao Y, Taylor PR, Virtamo J, Albanes D. Prediagnostic adiponectin concentrations and pancreatic cancer risk in male smokers. Am J Epidemiol. 2008;168:1047–1055. doi: 10.1093/aje/kwn221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chang MC, Chang YT, Su TC, Yang WS, Chen CL, Tien YW, Liang PC, Wei SC, Wong JM. Adiponectin as a potential differential marker to distinguish pancreatic cancer and chronic pancreatitis. Pancreas. 2007;35:16–21. doi: 10.1097/MPA.0b013e3180547709. [DOI] [PubMed] [Google Scholar]
- 75.Tworoger SS, Eliassen AH, Kelesidis T, Colditz GA, Willett WC, Mantzoros CS, Hankinson SE. Plasma adiponectin concentrations and risk of incident breast cancer. J Clin Endocrinol Metab. 2007;92:1510–1516. doi: 10.1210/jc.2006-1975. [DOI] [PubMed] [Google Scholar]
- 76.Grossmann ME, Ray A, Nkhata KJ, Malakhov DA, Rogozina OP, Dogan S, Cleary MP. Obesity and breast cancer: status of leptin and adiponectin in pathological processes. Cancer Metastasis Rev. 2010;29:641–653. doi: 10.1007/s10555-010-9252-1. [DOI] [PubMed] [Google Scholar]
- 77.Stefan N, Vozarova B, Funahashi T, Matsuzawa Y, Weyer C, Lindsay RS, Youngren JF, Havel PJ, Pratley RE, Bogardus C, Tataranni PA. Plasma adiponectin concentration is associated with skeletal muscle insulin receptor tyrosine phosphorylation, and low plasma concentration precedes a decrease in whole-body insulin sensitivity in humans. Diabetes. 2002;51:1884–1888. doi: 10.2337/diabetes.51.6.1884. [DOI] [PubMed] [Google Scholar]
- 78.Ouchi N, Walsh K. Adiponectin as an anti-inflammatory factor. Clin Chim Acta. 2007;380:24–30. doi: 10.1016/j.cca.2007.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yamauchi T, Kamon J, Minokoshi Y, Ito Y, Waki H, Uchida S, Yamashita S, Noda M, Kita S, Ueki K, Eto K, Akanuma Y, Froguel P, Foufelle F, Ferre P, Carling D, Kimura S, Nagai R, Kahn BB, Kadowaki T. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med. 2002;8:1288–1295. doi: 10.1038/nm788. [DOI] [PubMed] [Google Scholar]
- 80.Zakikhani M, Dowling RJ, Sonenberg N, Pollak MN. The effects of adiponectin and metformin on prostate and colon neoplasia involve activation of AMP-activated protein kinase. Cancer Prev Res (Phila) 2008;1:369–375. doi: 10.1158/1940-6207.CAPR-08-0081. [DOI] [PubMed] [Google Scholar]
- 81.Brakenhielm E, Veitonmaki N, Cao R, Kihara S, Matsuzawa Y, Zhivotovsky B, Funahashi T, Cao Y. Adiponectin-induced antiangiogenesis and antitumor activity involve caspase-mediated endothelial cell apoptosis. Proc Natl Acad Sci U S A. 2004;101:2476–2481. doi: 10.1073/pnas.0308671100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Margetic S, Gazzola C, Pegg GG, Hill RA. Leptin: a review of its peripheral actions and interactions. Int J Obes Relat Metab Disord. 2002;26:1407–1433. doi: 10.1038/sj.ijo.0802142. [DOI] [PubMed] [Google Scholar]
- 83.Cascio S, Bartella V, Auriemma A, Johannes GJ, Russo A, Giordano A, Surmacz E. Mechanism of leptin expression in breast cancer cells: role of hypoxia-inducible factor-1alpha. Oncogene. 2008;27:540–547. doi: 10.1038/sj.onc.1210660. [DOI] [PubMed] [Google Scholar]
- 84.White PB, True EM, Ziegler KM, Wang SS, Swartz-Basile DA, Pitt HA, Zyromski NJ. Insulin, Leptin, and Tumoral Adipocytes Promote Murine Pancreatic Cancer Growth. J Gastrointest Surg. 2010 doi: 10.1007/s11605-010-1349-x. [DOI] [PubMed] [Google Scholar]
- 85.Grossmann ME, Ray A, Nkhata KJ, Malakhov DA, Rogozina OP, Dogan S, Cleary MP. Obesity and breast cancer: status of leptin and adiponectin in pathological processes. Cancer Metastasis Rev. 2010;29:641–653. doi: 10.1007/s10555-010-9252-1. [DOI] [PubMed] [Google Scholar]
- 86.Considine RV, Sinha MK, Heiman ML, Kriauciunas A, Stephens TW, Nyce MR, Ohannesian JP, Marco CC, McKee LJ, Bauer TL. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N Engl J Med. 1996;334:292–295. doi: 10.1056/NEJM199602013340503. [DOI] [PubMed] [Google Scholar]
- 87.Aggarwal BB, Shishodia S, Sandur SK, Pandey MK, Sethi G. Inflammation and cancer: how hot is the link? Biochem Pharmacol. 2006;72:1605–1621. doi: 10.1016/j.bcp.2006.06.029. [DOI] [PubMed] [Google Scholar]
- 88.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.van Kruijsdijk RC, Evan der W, Visseren FL. Obesity and cancer: the role of dysfunctional adipose tissue. Cancer Epidemiol Biomarkers Prev. 2009;18:2569–2578. doi: 10.1158/1055-9965.EPI-09-0372. [DOI] [PubMed] [Google Scholar]
- 90.Ramos EJ, Xu Y, Romanova I, Middleton F, Chen C, Quinn R, Inui A, Das U, Meguid MM. Is obesity an inflammatory disease? Surgery. 2003;134:329–335. doi: 10.1067/msy.2003.267. [DOI] [PubMed] [Google Scholar]
- 91.Wellen KE, Hotamisligil GS. Obesity-induced inflammatory changes in adipose tissue. J Clin Invest. 2003;112:1785–1788. doi: 10.1172/JCI20514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Visser M, Bouter LM, McQuillan GM, Wener MH, Harris TB. Elevated C-reactive protein levels in overweight and obese adults. JAMA. 1999;282:2131–2135. doi: 10.1001/jama.282.22.2131. [DOI] [PubMed] [Google Scholar]
- 93.Visser M, Bouter LM, McQuillan GM, Wener MH, Harris TB. Low-grade systemic inflammation in overweight children. Pediatrics. 2001;107:E13. doi: 10.1542/peds.107.1.e13. [DOI] [PubMed] [Google Scholar]
- 94.Fogarty AW, Glancy C, Jones S, Lewis SA, McKeever TM, Britton JR. A prospective study of weight change and systemic inflammation over 9 y. Am J Clin Nutr. 2008;87:30–35. doi: 10.1093/ajcn/87.1.30. [DOI] [PubMed] [Google Scholar]
- 95.Naugler WE, Karin M. The wolf in sheep’s clothing: the role of interleukin-6 in immunity, inflammation and cancer. Trends Mol Med. 2008;14:109–119. doi: 10.1016/j.molmed.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 96.Kim S, Keku TO, Martin C, Galanko J, Woosley JT, Schroeder JC, Satia JA, Halabi S, Sandler RS. Circulating levels of inflammatory cytokines and risk of colorectal adenomas. Cancer Res. 2008;68:323–328. doi: 10.1158/0008-5472.CAN-07-2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2:161–174. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- 98.Chavey C, Mari B, Monthouel MN, Bonnafous S, Anglard P, Van OE, Tartare-Deckert S. Matrix metalloproteinases are differentially expressed in adipose tissue during obesity and modulate adipocyte differentiation. J Biol Chem. 2003;278:11888–11896. doi: 10.1074/jbc.M209196200. [DOI] [PubMed] [Google Scholar]
- 99.Kulbe H, Thompson R, Wilson JL, Robinson S, Hagemann T, Fatah R, Gould D, Ayhan A, Balkwill F. The inflammatory cytokine tumor necrosis factor-alpha generates an autocrine tumor-promoting network in epithelial ovarian cancer cells. Cancer Res. 2007;67:585–592. doi: 10.1158/0008-5472.CAN-06-2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Davies FE, Rollinson SJ, Rawstron AC, Roman E, Richards S, Drayson M, Child JA, Morgan GJ. High-producer haplotypes of tumor necrosis factor alpha and lymphotoxin alpha are associated with an increased risk of myeloma and have an improved progression-free survival after treatment. J Clin Oncol. 2000;18:2843–2851. doi: 10.1200/JCO.2000.18.15.2843. [DOI] [PubMed] [Google Scholar]
- 101.Il’yasova D, Colbert LH, Harris TB, Newman AB, Bauer DC, Satterfield S, Kritchevsky SB. Circulating levels of inflammatory markers and cancer risk in the health aging and body composition cohort. Cancer Epidemiol Biomarkers Prev. 2005;14:2413–2418. doi: 10.1158/1055-9965.EPI-05-0316. [DOI] [PubMed] [Google Scholar]
- 102.Bruce WR, Wolever TM, Giacca A. Mechanisms linking diet and colorectal cancer: the possible role of insulin resistance. Nutr Cancer. 2000;37:19–26. doi: 10.1207/S15327914NC3701_2. [DOI] [PubMed] [Google Scholar]
- 103.Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y, Nakayama O, Makishima M, Matsuda M, Shimomura I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest. 2004;114:1752–1761. doi: 10.1172/JCI21625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gago-Dominguez M, Castelao JE, Pike MC, Sevanian A, Haile RW. Role of lipid peroxidation in the epidemiology and prevention of breast cancer. Cancer Epidemiol Biomarkers Prev. 2005;14:2829–2839. doi: 10.1158/1055-9965.EPI-05-0015. [DOI] [PubMed] [Google Scholar]
- 105.Gago-Dominguez M, Jiang X, Castelao JE. Lipid peroxidation, oxidative stress genes and dietary factors in breast cancer protection: a hypothesis. Breast Cancer Res. 2007;9:201. doi: 10.1186/bcr1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Robertson RP, Harmon J, Tran PO, Poitout V. Beta-cell glucose toxicity, lipotoxicity, and chronic oxidative stress in type 2 diabetes. Diabetes. 2004;53(Suppl 1):S119–S124. doi: 10.2337/diabetes.53.2007.s119. [DOI] [PubMed] [Google Scholar]
- 107.Katiyar SK, Meeran SM. Obesity increases the risk of UV radiation-induced oxidative stress and activation of MAPK and NF-kappaB signaling. Free Radic Biol Med. 2007;42:299–310. doi: 10.1016/j.freeradbiomed.2006.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Dickson RB, Thompson EW, Lippman ME. Regulation of proliferation, invasion and growth factor synthesis in breast cancer by steroids. J Steroid Biochem Mol Biol. 1990;37:305–316. doi: 10.1016/0960-0760(90)90479-5. [DOI] [PubMed] [Google Scholar]
- 109.Jones JI, Clemmons DR. Insulin-like growth factors and their binding proteins: biological actions. Endocr Rev. 1995;16:3–34. doi: 10.1210/edrv-16-1-3. [DOI] [PubMed] [Google Scholar]
- 110.Pugeat M, Crave JC, Elmidani M, Nicolas MH, Garoscio-Cholet M, Lejeune H, Dechaud H, Tourniaire J. Pathophysiology of sex hormone binding globulin (SHBG): relation to insulin. J Steroid Biochem Mol Biol. 1991;40:841–849. doi: 10.1016/0960-0760(91)90310-2. [DOI] [PubMed] [Google Scholar]
- 111.Siiteri PK. Adipose tissue as a source of hormones. Am J Clin Nutr. 1987;45:277–282. doi: 10.1093/ajcn/45.1.277. [DOI] [PubMed] [Google Scholar]
- 112.Kaaks R, Lukanova A, Kurzer MS. Obesity, endogenous hormones, and endometrial cancer risk: a synthetic review. Cancer Epidemiol Biomarkers Prev. 2002;11:1531–1543. [PubMed] [Google Scholar]
- 113.Kaaks R, Berrino F, Key T, Rinaldi S, Dossus L, Biessy C, Secreto G, Amiano P, Bingham S, Boeing H, Bueno de Mesquita HB, Chang-Claude J, Clavel-Chapelon F, Fournier A, van Gils CH, Gonzalez CA, Gurrea AB, Critselis E, Khaw KT, Krogh V, Lahmann PH, Nagel G, Olsen A, Onland-Moret NC, Overvad K, Palli D, Panico S, Peeters P, Quiros JR, Roddam A, Thiebaut A, Tjonneland A, Chirlaque MD, Trichopoulou A, Trichopoulos D, Tumino R, Vineis P, Norat T, Ferrari P, Slimani N, Riboli E. Serum sex steroids in premenopausal women and breast cancer risk within the European Prospective Investigation into Cancer and Nutrition (EPIC) J Natl Cancer Inst. 2005;97:755–765. doi: 10.1093/jnci/dji132. [DOI] [PubMed] [Google Scholar]
- 114.Key TJ. Serum oestradiol and breast cancer risk. Endocr Relat Cancer. 1999;6:175–180. doi: 10.1677/erc.0.0060175. [DOI] [PubMed] [Google Scholar]
- 115.Gort EH, Groot AJ, van der WE, van Diest PJ, Vooijs MA. Hypoxic regulation of metastasis via hypoxia-inducible factors. Curr Mol Med. 2008;8:60–67. doi: 10.2174/156652408783565568. [DOI] [PubMed] [Google Scholar]
- 116.Jiang BH, Agani F, Passaniti A, Semenza GL. V-SRC induces expression of hypoxia-inducible factor 1 (HIF-1) and transcription of genes encoding vascular endothelial growth factor and enolase 1: involvement of HIF-1 in tumor progression. Cancer Res. 1997;57:5328–5335. [PubMed] [Google Scholar]
- 117.Zhong H, De Marzo AM, Laughner E, Lim M, Hilton DA, Zagzag D, Buechler P, Isaacs WB, Semenza GL, Simons JW. Overexpression of hypoxia-inducible factor 1alpha in common human cancers and their metastases. Cancer Res. 1999;59:5830–5835. [PubMed] [Google Scholar]
- 118.Marignol L, Coffey M, Lawler M, Hollywood D. Hypoxia in prostate cancer: a powerful shield against tumour destruction? Cancer Treat Rev. 2008;34:313–327. doi: 10.1016/j.ctrv.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 119.Vaupel P, Hoeckel M. Predictive power of the tumor oxygenation status. Adv Exp Med Biol. 1999;471:533–539. doi: 10.1007/978-1-4615-4717-4_63. [DOI] [PubMed] [Google Scholar]
- 120.Feldmann HJ, Molls M, Vaupel P. Blood flow and oxygenation status of human tumors. Clinical investigations. Strahlenther Onkol. 1999;175:1–9. doi: 10.1007/BF02743452. [DOI] [PubMed] [Google Scholar]
- 121.Roberts DL, Dive C, Renehan AG. Biological mechanisms linking obesity and cancer risk: new perspectives. Annu Rev Med. 2010;61:301–316. doi: 10.1146/annurev.med.080708.082713. [DOI] [PubMed] [Google Scholar]
- 122.Trayhurn P, Wang B, Wood IS. Hypoxia in adipose tissue: a basis for the dysregulation of tissue function in obesity? Br J Nutr. 2008;100:227–235. doi: 10.1017/S0007114508971282. [DOI] [PubMed] [Google Scholar]
- 123.Ye J, Gao Z, Yin J, He Q. Hypoxia is a potential risk factor for chronic inflammation and adiponectin reduction in adipose tissue of ob/ob and dietary obese mice. Am J Physiol Endocrinol Metab. 2007;293:E1118–E1128. doi: 10.1152/ajpendo.00435.2007. [DOI] [PubMed] [Google Scholar]
- 124.Lolmede K, Durand de SFV, Galitzky J, Lafontan M, Bouloumie A. Effects of hypoxia on the expression of proangiogenic factors in differentiated 3T3-F442A adipocytes. Int J Obes Relat Metab Disord. 2003;27:1187–1195. doi: 10.1038/sj.ijo.0802407. [DOI] [PubMed] [Google Scholar]
- 125.Koda M, Sulkowska M, Kanczuga-Koda L, Cascio S, Colucci G, Russo A, Surmacz E, Sulkowski S. Expression of the obesity hormone leptin and its receptor correlates with hypoxia-inducible factor-1 alpha in human colorectal cancer. Ann Oncol. 2007;18(Suppl 6):vi116–vi119. doi: 10.1093/annonc/mdm238. [DOI] [PubMed] [Google Scholar]
- 126.Bartella V, Cascio S, Fiorio E, Auriemma A, Russo A, Surmacz E. Insulin-dependent leptin expression in breast cancer cells. Cancer Res. 2008;68:4919–4927. doi: 10.1158/0008-5472.CAN-08-0642. [DOI] [PubMed] [Google Scholar]
- 127.Silha JV, Krsek M, Sucharda P, Murphy LJ. Angiogenic factors are elevated in overweight and obese individuals. Int J Obes (Lond) 2005;29:1308–1314. doi: 10.1038/sj.ijo.0802987. [DOI] [PubMed] [Google Scholar]
- 128.Kerbel RS. Tumor angiogenesis. N Engl J Med. 2008;358:2039–2049. doi: 10.1056/NEJMra0706596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.CDC. National Diabetes Fact Sheet. Atlanta, GA: 2007. http://www.cdc.gov/diabetes/pubs/factsheet07.htm. [Google Scholar]
- 130.Chowdhury TA. QJM. 2010. Diabetes and Cancer. [DOI] [PubMed] [Google Scholar]
- 131.Giaginis C, Katsamangou E, Tsourouflis G, Zizi-Serbetzoglou D, Kouraklis G, Theocharis S. Peroxisome proliferator-activated receptor-gamma and retinoid X receptor-alpha expression in pancreatic ductal adenocarcinoma: association with clinicopathological parameters, tumor proliferative capacity, and patients’ survival. Med Sci Monit. 2009;15:BR148–BR156. [PubMed] [Google Scholar]
- 132.Giovannucci E, Harlan DM, Archer MC, Bergenstal RM, Gapstur SM, Habel LA, Pollak M, Regensteiner JG, Yee D. Diabetes and cancer: a consensus report. Diabetes Care. 2010;33:1674–1685. doi: 10.2337/dc10-0666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Grote VA, Becker S, Kaaks R. Diabetes mellitus type 2 - an independent risk factor for cancer? Exp Clin Endocrinol Diabetes. 2010;118:4–8. doi: 10.1055/s-0029-1243193. [DOI] [PubMed] [Google Scholar]
- 134.Schott S, Schneeweiss A, Sohn C. Breast Cancer and Diabetes Mellitus. Exp Clin Endocrinol Diabetes. 2010 doi: 10.1055/s-0030-1254116. [DOI] [PubMed] [Google Scholar]
- 135.Czyzyk A, Szczepanik Z. Diabetes mellitus and cancer. Eur J Intern Med. 2000;11:245–252. doi: 10.1016/s0953-6205(00)00106-0. [DOI] [PubMed] [Google Scholar]
- 136.Gullo L, Pezzilli R, Morselli-Labate AM. Diabetes and the risk of pancreatic cancer. N Engl J Med. 1994;331:81–84. doi: 10.1056/NEJM199407143310203. [DOI] [PubMed] [Google Scholar]
- 137.Pezzilli R, Casadei R, Morselli-Labate AM. Is type 2 diabetes a risk factor for pancreatic cancer? JOP. 2009;10:705–706. [PubMed] [Google Scholar]
- 138.Everhart J, Wright D. Diabetes mellitus as a risk factor for pancreatic cancer. A meta-analysis. JAMA. 1995;273:1605–1609. [PubMed] [Google Scholar]
- 139.Huxley R, nsary-Moghaddam A, Berrington de GA, Barzi F, Woodward M. Type-II diabetes and pancreatic cancer: a meta-analysis of 36 studies. Br J Cancer. 2005;92:2076–2083. doi: 10.1038/sj.bjc.6602619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Stevens RJ, Roddam AW, Beral V. Pancreatic cancer in type 1 and young-onset diabetes: systematic review and meta-analysis. Br J Cancer. 2007;96:507–509. doi: 10.1038/sj.bjc.6603571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Jamal MM, Yoon EJ, Vega KJ, Hashemzadeh M, Chang KJ. Diabetes mellitus as a risk factor for gastrointestinal cancer among American veterans. World J Gastroenterol. 2009;15:5274–5278. doi: 10.3748/wjg.15.5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Chari ST, Leibson CL, Rabe KG, Timmons LJ, Ransom J, AMde, Petersen GM. Pancreatic cancer-associated diabetes mellitus: prevalence and temporal association with diagnosis of cancer. Gastroenterology. 2008;134:95–101. doi: 10.1053/j.gastro.2007.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Pannala R, Leirness JB, Bamlet WR, Basu A, Petersen GM, Chari ST. Prevalence and clinical profile of pancreatic cancer-associated diabetes mellitus. Gastroenterology. 2008;134:981–987. doi: 10.1053/j.gastro.2008.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Chodick G, Heymann AD, Rosenmann L, Green MS, Flash S, Porath A, Kokia E, Shalev V. Diabetes and risk of incident cancer: a large population-based cohort study in Israel. Cancer Causes Control. 2010;21:879–887. doi: 10.1007/s10552-010-9515-8. [DOI] [PubMed] [Google Scholar]
- 145.Butler AE, Galasso R, Matveyenko A, Rizza RA, Dry S, Butler PC. Pancreatic duct replication is increased with obesity and type 2 diabetes in humans. Diabetologia. 2010;53:21–26. doi: 10.1007/s00125-009-1556-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Renehan AG, Berster JM. Pediatr Endocrinol Rev; Insulin and Cancer: Report of the Proceedings of the First International Workshop; October 27–28, 2007; Dusseldorf, Germany. 2008. pp. 810–816. [PubMed] [Google Scholar]
- 147.Taniguchi CM, Emanuelli B, Kahn CR. Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol. 2006;7:85–96. doi: 10.1038/nrm1837. [DOI] [PubMed] [Google Scholar]
- 148.Asano T, Yao Y, Shin S, McCubrey J, Abbruzzese JL, Reddy SA. Insulin receptor substrate is a mediator of phosphoinositide 3-kinase activation in quiescent pancreatic cancer cells. Cancer Res. 2005;65:9164–9168. doi: 10.1158/0008-5472.CAN-05-0779. [DOI] [PubMed] [Google Scholar]
- 149.Asano T, Yao Y, Zhu J, Li D, Abbruzzese JL, Reddy SA. The rapamycin analog CCI-779 is a potent inhibitor of pancreatic cancer cell proliferation. Biochem Biophys Res Commun. 2005;331:295–302. doi: 10.1016/j.bbrc.2005.03.166. [DOI] [PubMed] [Google Scholar]
- 150.Hemkens LG, Grouven U, Bender R, Gunster C, Gutschmidt S, Selke GW, Sawicki PT. Risk of malignancies in patients with diabetes treated with human insulin or insulin analogues: a cohort study. Diabetologia. 2009;52:1732–1744. doi: 10.1007/s00125-009-1418-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Campbell PT, Deka A, Jacobs EJ, Newton CC, Hildebrand JS, McCullough ML, Limburg PJ, Gapstur SM. Prospective study reveals associations between colorectal cancer and type 2 diabetes mellitus or insulin use in men. Gastroenterology. 2010;139:1138–1146. doi: 10.1053/j.gastro.2010.06.072. [DOI] [PubMed] [Google Scholar]
- 152.Simon D, Balkau B. Diabetes mellitus, hyperglycaemia and cancer. Diabetes Metab. 2010;36:182–191. doi: 10.1016/j.diabet.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 153.Kornmann M, Maruyama H, Bergmann U, Tangvoranuntakul P, Beger HG, White MF, Korc M. Enhanced expression of the insulin receptor substrate-2 docking protein in human pancreatic cancer. Cancer Res. 1998;58:4250–4254. [PubMed] [Google Scholar]
- 154.Nakamura K, Sasajima J, Mizukami Y, Sugiyama Y, Yamazaki M, Fujii R, Kawamoto T, Koizumi K, Sato K, Fujiya M, Sasaki K, Tanno S, Okumura T, Shimizu N, Kawabe J, Karasaki H, Kono T, Ii M, Bardeesy N, Chung DC, Kohgo Y. Hedgehog promotes neovascularization in pancreatic cancers by regulating Ang-1 and IGF-1 expression in bone-marrow derived pro-angiogenic cells. PLoS One. 2010;5:e8824. doi: 10.1371/journal.pone.0008824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Kolb S, Fritsch R, Saur D, Reichert M, Schmid RM, Schneider G. HMGA1 controls transcription of insulin receptor to regulate cyclin D1 translation in pancreatic cancer cells. Cancer Res. 2007;67:4679–4686. doi: 10.1158/0008-5472.CAN-06-3308. [DOI] [PubMed] [Google Scholar]
- 156.Dallas NA, Xia L, Fan F, Gray MJ, Gaur P, van BG, Samuel S, Kim MP, Lim SJ, Ellis LM. Chemoresistant colorectal cancer cells, the cancer stem cell phenotype, and increased sensitivity to insulin-like growth factor-I receptor inhibition. Cancer Res. 2009;69:1951–1957. doi: 10.1158/0008-5472.CAN-08-2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, DePinho RA, Montminy M, Cantley LC. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. 2005;310:1642–1646. doi: 10.1126/science.1120781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Lalau JD, Race JM. Lactic acidosis in metformin therapy. Drugs 58 Suppl. 1999;1:55–60. doi: 10.2165/00003495-199958001-00013. [DOI] [PubMed] [Google Scholar]
- 159.Lalau JD, Race JM. Lactic acidosis in metformin-treated patients. Prognostic value of arterial lactate levels and plasma metformin concentrations. Drug Saf. 1999;20:377–384. doi: 10.2165/00002018-199920040-00006. [DOI] [PubMed] [Google Scholar]
- 160.Bowker SL, Majumdar SR, Veugelers P, Johnson JA. Increased cancer-related mortality for patients with type 2 diabetes who use sulfonylureas or insulin. Diabetes Care. 2006;29:254–258. doi: 10.2337/diacare.29.02.06.dc05-1558. [DOI] [PubMed] [Google Scholar]
- 161.Heikkinen S, Auwerx J, Argmann CA. PPARgamma in human and mouse physiology. Biochim Biophys Acta. 2007;1771:999–1013. doi: 10.1016/j.bbalip.2007.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Landman GW, Kleefstra N, van Hateren KJ, Groenier KH, Gans RO, Bilo HJ. Metformin associated with lower cancer mortality in type 2 diabetes: ZODIAC-16. Diabetes Care. 2010;33:322–326. doi: 10.2337/dc09-1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Libby G, Donnelly LA, Donnan PT, Alessi DR, Morris AD, Evans JM. New users of metformin are at low risk of incident cancer: a cohort study among people with type 2 diabetes. Diabetes Care. 2009;32:1620–1625. doi: 10.2337/dc08-2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Monami M, Lamanna C, Balzi D, Marchionni N, Mannucci E. Sulphonylureas and cancer: a case-control study. Acta Diabetol. 2009;46:279–284. doi: 10.1007/s00592-008-0083-2. [DOI] [PubMed] [Google Scholar]
- 165.Cazzaniga M, Bonanni B, Guerrieri-Gonzaga A, Decensi A. Is it time to test metformin in breast cancer clinical trials? Cancer Epidemiol Biomarkers Prev. 2009;18:701–705. doi: 10.1158/1055-9965.EPI-08-0871. [DOI] [PubMed] [Google Scholar]
- 166.Goodwin PJ, Ligibel JA, Stambolic V. Metformin in breast cancer: time for action. J Clin Oncol. 2009;27:3271–3273. doi: 10.1200/JCO.2009.22.1630. [DOI] [PubMed] [Google Scholar]
- 167.Martin-Castillo B, Vazquez-Martin A, Oliveras-Ferraros C, Menendez JA. Metformin and cancer: Doses, mechanisms and the dandelion and hormetic phenomena. Cell Cycle. 2010;9 doi: 10.4161/cc.9.6.10994. [DOI] [PubMed] [Google Scholar]
- 168.Hosono K, Endo H, Takahashi H, Sugiyama M, Sakai E, Uchiyama T, Suzuki K, Iida H, Sakamoto Y, Yoneda K, Koide T, Tokoro C, Abe Y, Inamori M, Nakagama H, Nakajima A. Metformin suppresses colorectal aberrant crypt foci in a short-term clinical trial. Cancer Prev Res (Phila) 2010;3:1077–1083. doi: 10.1158/1940-6207.CAPR-10-0186. [DOI] [PubMed] [Google Scholar]
- 169.Jiralerspong S, Palla SL, Giordano SH, Meric-Bernstam F, Liedtke C, Barnett CM, Hsu L, Hung MC, Hortobagyi GN, Gonzalez-Angulo AM. Metformin and pathologic complete responses to neoadjuvant chemotherapy in diabetic patients with breast cancer. J Clin Oncol. 2009;27:3297–3302. doi: 10.1200/JCO.2009.19.6410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Li D, Yeung SC, Hassan MM, Konopleva M, Abbruzzese JL. Antidiabetic therapies affect risk of pancreatic cancer. Gastroenterology. 2009;137:482–488. doi: 10.1053/j.gastro.2009.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Anisimov VN, Egormin PA, Bershtein LM, Zabezhinskii MA, Piskunova TS, Popovich IG, Semenchenko AV. Metformin decelerates aging and development of mammary tumors in HER-2/neu transgenic mice. Bull Exp Biol Med. 2005;139:721–723. doi: 10.1007/s10517-005-0389-9. [DOI] [PubMed] [Google Scholar]
- 172.Anisimov VN, Berstein LM, Egormin PA, Piskunova TS, Popovich IG, Zabezhinski MA, Kovalenko IG, Poroshina TE, Semenchenko AV, Provinciali M, Re F, Franceschi C. Effect of metformin on life span and on the development of spontaneous mammary tumors in HER-2/neu transgenic mice. Exp Gerontol. 2005;40:685–693. doi: 10.1016/j.exger.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 173.Liu B, Fan Z, Edgerton SM, Deng XS, Alimova IN, Lind SE, Thor AD. Metformin induces unique biological and molecular responses in triple negative breast cancer cells. Cell Cycle. 2009;8:2031–2040. doi: 10.4161/cc.8.13.8814. [DOI] [PubMed] [Google Scholar]
- 174.Tomimoto A, Endo H, Sugiyama M, Fujisawa T, Hosono K, Takahashi H, Nakajima N, Nagashima Y, Wada K, Nakagama H, Nakajima A. Metformin suppresses intestinal polyp growth in ApcMin/+ mice. Cancer Sci. 2008;99:2136–2141. doi: 10.1111/j.1349-7006.2008.00933.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Dowling RJ, Zakikhani M, Fantus IG, Pollak M, Sonenberg N. Metformin inhibits mammalian target of rapamycin-dependent translation initiation in breast cancer cells. Cancer Res. 2007;67:10804–10812. doi: 10.1158/0008-5472.CAN-07-2310. [DOI] [PubMed] [Google Scholar]
- 176.Zakikhani M, Dowling R, Fantus IG, Sonenberg N, Pollak M. Metformin is an AMP kinase-dependent growth inhibitor for breast cancer cells. Cancer Res. 2006;66:10269–10273. doi: 10.1158/0008-5472.CAN-06-1500. [DOI] [PubMed] [Google Scholar]
- 177.Ben SI, Laurent K, Loubat A, Giorgetti-Peraldi S, Colosetti P, Auberger P, Tanti JF, Le Marchand-Brustel Y, Bost F. The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene. 2008;27:3576–3586. doi: 10.1038/sj.onc.1211024. [DOI] [PubMed] [Google Scholar]
- 178.Ben SI, Le Marchand-Brustel Y, Tanti JF, Bost F. Metformin in cancer therapy: a new perspective for an old antidiabetic drug? Mol Cancer Ther. 2010;9:1092–1099. doi: 10.1158/1535-7163.MCT-09-1186. [DOI] [PubMed] [Google Scholar]
- 179.Feng Z, Hu W, de SE, Teresky AK, Jin S, Lowe S, Levine AJ. The regulation of AMPK beta1, TSC2, and PTEN expression by p53: stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res. 2007;67:3043–3053. doi: 10.1158/0008-5472.CAN-06-4149. [DOI] [PubMed] [Google Scholar]
- 180.Guigas B, Bertrand L, Taleux N, Foretz M, Wiernsperger N, Vertommen D, Andreelli F, Viollet B, Hue L. 5-Aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside and metformin inhibit hepatic glucose phosphorylation by an AMP-activated protein kinase-independent effect on glucokinase translocation. Diabetes. 2006;55:865–874. doi: 10.2337/diabetes.55.04.06.db05-1178. [DOI] [PubMed] [Google Scholar]
- 181.Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, Birnbaum MJ, Thompson CB. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell. 2005;18:283–293. doi: 10.1016/j.molcel.2005.03.027. [DOI] [PubMed] [Google Scholar]
- 182.Okoshi R, Ozaki T, Yamamoto H, Ando K, Koida N, Ono S, Koda T, Kamijo T, Nakagawara A, Kizaki H. Activation of AMP-activated protein kinase induces p53-dependent apoptotic cell death in response to energetic stress. J Biol Chem. 2008;283:3979–3987. doi: 10.1074/jbc.M705232200. [DOI] [PubMed] [Google Scholar]
- 183.Ersoy C, Kiyici S, Budak F, Oral B, Guclu M, Duran C, Selimoglu H, Erturk E, Tuncel E, Imamoglu S. The effect of metformin treatment on VEGF and PAI-1 levels in obese type 2 diabetic patients. Diabetes Res Clin Pract. 2008;81:56–60. doi: 10.1016/j.diabres.2008.02.006. [DOI] [PubMed] [Google Scholar]
- 184.Lund SS, Tarnow L, Stehouwer CD, Schalkwijk CG, Teerlink T, Gram J, Winther K, Frandsen M, Smidt UM, Pedersen O, Parving HH, Vaag AA. Impact of metformin versus repaglinide on non-glycaemic cardiovascular risk markers related to inflammation and endothelial dysfunction in non-obese patients with type 2 diabetes. Eur J Endocrinol. 2008;158:631–641. doi: 10.1530/EJE-07-0815. [DOI] [PubMed] [Google Scholar]
- 185.Huang NL, Chiang SH, Hsueh CH, Liang YJ, Chen YJ, Lai LP. Metformin inhibits TNF-alpha-induced IkappaB kinase phosphorylation, IkappaB-alpha degradation and IL-6 production in endothelial cells through PI3K-dependent AMPK phosphorylation. Int J Cardiol. 2009;134:169–175. doi: 10.1016/j.ijcard.2008.04.010. [DOI] [PubMed] [Google Scholar]
- 186.Hirsch HA, Iliopoulos D, Tsichlis PN, Struhl K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. 2009;69:7507–7511. doi: 10.1158/0008-5472.CAN-09-2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Vazquez-Martin A, Oliveras-Ferraros C, Barco SD, Martin-Castillo B, Menendez JA. The anti-diabetic drug metformin suppresses self-renewal and proliferation of trastuzumab-resistant tumor-initiating breast cancer stem cells. Breast Cancer Res Treat. 2010 doi: 10.1007/s10549-010-0924-x. [DOI] [PubMed] [Google Scholar]
- 188.Schneider MB, Matsuzaki H, Haorah J, Ulrich A, Standop J, Ding XZ, Adrian TE, Pour PM. Prevention of pancreatic cancer induction in hamsters by metformin. Gastroenterology. 2001;120:1263–1270. doi: 10.1053/gast.2001.23258. [DOI] [PubMed] [Google Scholar]
- 189.Kisfalvi K, Eibl G, Sinnett-Smith J, Rozengurt E. Metformin disrupts crosstalk between G protein-coupled receptor and insulin receptor signaling systems and inhibits pancreatic cancer growth. Cancer Res. 2009;69:6539–6545. doi: 10.1158/0008-5472.CAN-09-0418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Wang LW, Li ZS, Zou DW, Jin ZD, Gao J, Xu GM. Metformin induces apoptosis of pancreatic cancer cells. World J Gastroenterol. 2008;14:7192–7198. doi: 10.3748/wjg.14.7192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Rozengurt E, Sinnett-Smith J, Kisfalvi K. Crosstalk between insulin/insulin-like growth factor-1 receptors and G protein-coupled receptor signaling systems: a novel target for the antidiabetic drug metformin in pancreatic cancer. Clin Cancer Res. 2010;16:2505–2511. doi: 10.1158/1078-0432.CCR-09-2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Barak Y, Nelson MC, Ong ES, Jones YZ, Ruiz-Lozano P, Chien KR, Koder A, Evans RM. PPAR gamma is required for placental, cardiac, and adipose tissue development. Mol Cell. 1999;4:585–595. doi: 10.1016/s1097-2765(00)80209-9. [DOI] [PubMed] [Google Scholar]
- 193.Burns KA, Vanden Heuvel JP. Modulation of PPAR activity via phosphorylation. Biochim Biophys Acta. 2007;1771:952–960. doi: 10.1016/j.bbalip.2007.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Zingarelli B, Sheehan M, Hake PW, O’Connor M, Denenberg A, Cook JA. Peroxisome proliferator activator receptor-gamma ligands, 15-deoxy-Delta(12,14)-prostaglandin J2 and ciglitazone, reduce systemic inflammation in polymicrobial sepsis by modulation of signal transduction pathways. J Immunol. 2003;171:6827–6837. doi: 10.4049/jimmunol.171.12.6827. [DOI] [PubMed] [Google Scholar]
- 195.Auwerx J, Martin G, Guerre-Millo M, Staels B. Transcription, adipocyte differentiation, and obesity. J Mol Med. 1996;74:347–352. doi: 10.1007/BF00210629. [DOI] [PubMed] [Google Scholar]
- 196.Papi A, Rocchi P, Ferreri AM, Orlandi M. RXRgamma and PPARgamma ligands in combination to inhibit proliferation and invasiveness in colon cancer cells. Cancer Lett. 2010 doi: 10.1016/j.canlet.2010.04.026. [DOI] [PubMed] [Google Scholar]
- 197.Crosby MB, Svenson J, Gilkeson GS, Nowling TK. A novel PPAR response element in the murine iNOS promoter. Mol Immunol. 2005;42:1303–1310. doi: 10.1016/j.molimm.2004.12.009. [DOI] [PubMed] [Google Scholar]
- 198.Tontonoz P, Hu E, Spiegelman BM. Regulation of adipocyte gene expression and differentiation by peroxisome proliferator activated receptor gamma. Curr Opin Genet Dev. 1995;5:571–576. doi: 10.1016/0959-437x(95)80025-5. [DOI] [PubMed] [Google Scholar]
- 199.Brun RP, Spiegelman BM. PPAR gamma and the molecular control of adipogenesis. J Endocrinol. 1997;155:217–218. doi: 10.1677/joe.0.1550217. [DOI] [PubMed] [Google Scholar]
- 200.Carter AB, Misyak SA, Hontecillas R, Bassaganya-Riera J. Dietary Modulation of Inflammation-Induced Colorectal Cancer through PPARgamma. PPAR Res. 2009;2009:498352. doi: 10.1155/2009/498352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Carter JC, Church FC. Obesity and Breast Cancer: The Roles of Peroxisome Proliferator-Activated Receptor-gamma and Plasminogen Activator Inhibitor-1. PPAR Res. 2009;2009:345320. doi: 10.1155/2009/345320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Aiello A, Pandini G, Frasca F, Conte E, Murabito A, Sacco A, Genua M, Vigneri R, Belfiore A. Peroxisomal proliferator-activated receptor-gamma agonists induce partial reversion of epithelial-mesenchymal transition in anaplastic thyroid cancer cells. Endocrinology. 2006;147:4463–4475. doi: 10.1210/en.2005-1610. [DOI] [PubMed] [Google Scholar]
- 203.Ramos-Nino ME, MacLean CD, Littenberg B. Association between cancer prevalence and use of thiazolidinediones: results from the Vermont Diabetes Information System. BMC Med. 2007;5:17. doi: 10.1186/1741-7015-5-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Koro C, Barrett S, Qizilbash N. Cancer risks in thiazolidinedione users compared to other anti-diabetic agents. Pharmacoepidemiol Drug Saf. 2007;16:485–492. doi: 10.1002/pds.1352. [DOI] [PubMed] [Google Scholar]
- 205.Nicolucci A. Epidemiological aspects of neoplasms in diabetes. Acta Diabetol. 2010;47:87–95. doi: 10.1007/s00592-010-0187-3. [DOI] [PubMed] [Google Scholar]
- 206.Roman J. Peroxisome proliferator-activated receptor gamma and lung cancer biology: implications for therapy. J Investig Med. 2008;56:528–533. doi: 10.2310/JIM.0b013e3181659932. [DOI] [PubMed] [Google Scholar]
- 207.Takahashi H, Hosono K, Uchiyama T, Sugiyama M, Sakai E, Endo H, Maeda S, Schaefer KL, Nakagama H, Nakajima A. PPARgamma Ligand as a Promising Candidate for Colorectal Cancer Chemoprevention: A Pilot Study. PPAR Res. 2010;2010 doi: 10.1155/2010/257835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Kristiansen G, Jacob J, Buckendahl AC, Grutzmann R, Alldinger I, Sipos B, Kloppel G, Bahra M, Langrehr JM, Neuhaus P, Dietel M, Pilarsky C. Peroxisome proliferator-activated receptor gamma is highly expressed in pancreatic cancer and is associated with shorter overall survival times. Clin Cancer Res. 2006;12:6444–6451. doi: 10.1158/1078-0432.CCR-06-0834. [DOI] [PubMed] [Google Scholar]
- 209.Eibl G, Wente MN, Reber HA, Hines OJ. Peroxisome proliferator-activated receptor gamma induces pancreatic cancer cell apoptosis. Biochem Biophys Res Commun. 2001;287:522–529. doi: 10.1006/bbrc.2001.5619. [DOI] [PubMed] [Google Scholar]
- 210.Eibl G. The Role of PPAR-gamma and Its Interaction with COX-2 in Pancreatic Cancer. PPAR Res. 2008;2008:326915. doi: 10.1155/2008/326915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Knauer SK. Prognostic and therapeutic potential of nuclear receptors in head and neck squamous cell carcinomas. J Oncol. 2009;2009:349205. doi: 10.1155/2009/349205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Panigrahy D, Shen LQ, Kieran MW, Kaipainen A. Therapeutic potential of thiazolidinediones as anticancer agents. Expert Opin Investig Drugs. 2003;12:1925–1937. doi: 10.1517/13543784.12.12.1925. [DOI] [PubMed] [Google Scholar]
- 213.Takeuchi Y, Takahashi M, Sakano K, Mutoh M, Niho N, Yamamoto M, Sato H, Sugimura T, Wakabayashi K. Suppression of N-nitrosobis(2-oxopropyl)amine-induced pancreatic carcinogenesis in hamsters by pioglitazone, a ligand of peroxisome proliferator-activated receptor gamma. Carcinogenesis. 2007;28:1692–1696. doi: 10.1093/carcin/bgm095. [DOI] [PubMed] [Google Scholar]
- 214.Dolezalova R, Haluzik MM, Bosanska L, Lacinova Z, Kasalova Z, Stulc T, Haluzik M. Effect of PPAR-gamma agonist treatment on markers of endothelial dysfunction in patients with type 2 diabetes mellitus. Physiol Res. 2007;56:741–748. doi: 10.33549/physiolres.931060. [DOI] [PubMed] [Google Scholar]
- 215.Quinn CE, Hamilton PK, Lockhart CJ, McVeigh GE. Thiazolidinediones: effects on insulin resistance and the cardiovascular system. Br J Pharmacol. 2008;153:636–645. doi: 10.1038/sj.bjp.0707452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Rumi MA, Ishihara S, Kazumori H, Kadowaki Y, Kinoshita Y. Can PPAR gamma ligands be used in cancer therapy? Curr Med Chem Anticancer Agents. 2004;4:465–477. doi: 10.2174/1568011043352678. [DOI] [PubMed] [Google Scholar]
- 217.Liu J, Lu H, Huang R, Lin D, Wu X, Lin Q, Wu X, Zheng J, Pan X, Peng J, Song Y, Zhang M, Hou M, Chen F. Peroxisome proliferator activated receptor-gamma ligands induced cell growth inhibition and its influence on matrix metalloproteinase activity in human myeloid leukemia cells. Cancer Chemother Pharmacol. 2005;56:400–408. doi: 10.1007/s00280-005-1029-9. [DOI] [PubMed] [Google Scholar]
- 218.Nagata D, Yoshihiro H, Nakanishi M, Naruyama H, Okada S, Ando R, Tozawa K, Kohri K. Peroxisome proliferator-activated receptor-gamma and growth inhibition by its ligands in prostate cancer. Cancer Detect Prev. 2008;32:259–266. doi: 10.1016/j.cdp.2008.05.008. [DOI] [PubMed] [Google Scholar]
- 219.Nunez NP, Liu H, Meadows GG. PPAR-gamma ligands and amino acid deprivation promote apoptosis of melanoma, prostate, and breast cancer cells. Cancer Lett. 2006;236:133–141. doi: 10.1016/j.canlet.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 220.Takashima T, Fujiwara Y, Higuchi K, Arakawa T, Yano Y, Hasuma T, Otani S. PPAR-gamma ligands inhibit growth of human esophageal adenocarcinoma cells through induction of apoptosis, cell cycle arrest and reduction of ornithine decarboxylase activity. Int J Oncol. 2001;19:465–471. [PubMed] [Google Scholar]
- 221.Zand H, Rhimipour A, Bakhshayesh M, Shafiee M, Nour MI, Salimi S. Involvement of PPAR-gamma and p53 in DHA-induced apoptosis in Reh cells. Mol Cell Biochem. 2007;304:71–77. doi: 10.1007/s11010-007-9487-5. [DOI] [PubMed] [Google Scholar]
- 222.Mansure JJ, Nassim R, Kassouf W. Peroxisome proliferator-activated receptor gamma in bladder cancer: a promising therapeutic target. Cancer Biol Ther. 2009;8:6–15. doi: 10.4161/cbt.8.7.7853. [DOI] [PubMed] [Google Scholar]
- 223.Panigrahy D, Huang S, Kieran MW, Kaipainen A. PPARgamma as a therapeutic target for tumor angiogenesis and metastasis. Cancer Biol Ther. 2005;4:687–693. doi: 10.4161/cbt.4.7.2014. [DOI] [PubMed] [Google Scholar]
- 224.Feinstein DL, Spagnolo A, Akar C, Weinberg G, Murphy P, Gavrilyuk V, Dello RC. Receptor-independent actions of PPAR thiazolidinedione agonists: is mitochondrial function the key? Biochem Pharmacol. 2005;70:177–188. doi: 10.1016/j.bcp.2005.03.033. [DOI] [PubMed] [Google Scholar]
- 225.Simpson-Haidaris PJ, Pollock SJ, Ramon S, Guo N, Woeller CF, Feldon SE, Phipps RP. Anticancer Role of PPARgamma Agonists in Hematological Malignancies Found in the Vasculature, Marrow, and Eyes. PPAR Res. 2010;2010:814609. doi: 10.1155/2010/814609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Feng YH, Velazquez-Torres G, Gully C, Chen J, Lee MH, Yeung SC. The impact of type 2 diabetes and antidiabetic drugs on cancer cell growth. J Cell Mol Med. 2010 doi: 10.1111/j.1582-4934.2010.01083.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Jin T, George FI, Sun J. Wnt and beyond Wnt: multiple mechanisms control the transcriptional property of beta-catenin. Cell Signal. 2008;20:1697–1704. doi: 10.1016/j.cellsig.2008.04.014. [DOI] [PubMed] [Google Scholar]
- 228.DeSano JT, Xu L. MicroRNA regulation of cancer stem cells and therapeutic implications. AAPS J. 2009;11:682–692. doi: 10.1208/s12248-009-9147-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Perera RJ, Ray A. MicroRNAs in the search for understanding human diseases. BioDrugs. 2007;21:97–104. doi: 10.2165/00063030-200721020-00004. [DOI] [PubMed] [Google Scholar]
- 230.Moriyama T, Ohuchida K, Mizumoto K, Yu J, Sato N, Nabae T, Takahata S, Toma H, Nagai E, Tanaka M. MicroRNA-21 modulates biological functions of pancreatic cancer cells including their proliferation, invasion, and chemoresistance. Mol Cancer Ther. 2009 doi: 10.1158/1535-7163.MCT-08-0592. [DOI] [PubMed] [Google Scholar]
- 231.Dillhoff M, Liu J, Frankel W, Croce C, Bloomston M. MicroRNA-21 is overexpressed in pancreatic cancer and a potential predictor of survival. J Gastrointest Surg. 2008;12:2171–2176. doi: 10.1007/s11605-008-0584-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Li Y, Kong D, Wang Z, Sarkar FH. Regulation of microRNAs by natural agents: an emerging field in chemoprevention and chemotherapy research. Pharm Res. 2010;27:1027–1041. doi: 10.1007/s11095-010-0105-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Sarkar FH, Li Y, Wang Z, Kong D, Ali S. Implication of microRNAs in drug resistance for designing novel cancer therapy. Drug Resist Updat. 2010;13:57–66. doi: 10.1016/j.drup.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Kent OA, Mullendore M, Wentzel EA, Lopez-Romero P, Tan AC, Alvarez H, West K, Ochs MF, Hidalgo M, Arking DE, Maitra A, Mendell JT. A resource for analysis of microRNA expression and function in pancreatic ductal adenocarcinoma cells. Cancer Biol Ther. 2009;8:2013–2024. doi: 10.4161/cbt.8.21.9685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Rachagani S, Kumar S, Batra SK. MicroRNA in pancreatic cancer: pathological, diagnostic and therapeutic implications. Cancer Lett. 2010;292:8–16. doi: 10.1016/j.canlet.2009.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Zhang B, Pan X, Cobb GP, Anderson TA. microRNAs as oncogenes and tumor suppressors. Dev Biol. 2007;302:1–12. doi: 10.1016/j.ydbio.2006.08.028. [DOI] [PubMed] [Google Scholar]
- 237.Ali S, Ahmad A, Banerjee S, Padhye S, Dominiak K, Schaffert JM, Wang Z, Philip PA, Sarkar FH. Gemcitabine sensitivity can be induced in pancreatic cancer cells through modulation of miR-200 and miR-21 expression by curcumin or its analogue CDF. Cancer Res. 2010;70:3606–3617. doi: 10.1158/0008-5472.CAN-09-4598. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 238.Olson P, Lu J, Zhang H, Shai A, Chun MG, Wang Y, Libutti SK, Nakakura EK, Golub TR, Hanahan D. MicroRNA dynamics in the stages of tumorigenesis correlate with hallmark capabilities of cancer. Genes Dev. 2009;23:2152–2165. doi: 10.1101/gad.1820109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.He A, Zhu L, Gupta N, Chang Y, Fang F. Overexpression of micro ribonucleic acid 29, highly up-regulated in diabetic rats, leads to insulin resistance in 3T3-L1 adipocytes. Mol Endocrinol. 2007;21:2785–2794. doi: 10.1210/me.2007-0167. [DOI] [PubMed] [Google Scholar]
- 240.Heneghan HM, Miller N, Kerin MJ. Role of microRNAs in obesity and the metabolic syndrome. Obes Rev. 2010;11:354–361. doi: 10.1111/j.1467-789X.2009.00659.x. [DOI] [PubMed] [Google Scholar]
- 241.Kong L, Zhu J, Han W, Jiang X, Xu M, Zhao Y, Dong Q, Pang Z, Guan Q, Gao L, Zhao J, Zhao L. Significance of serum microRNAs in pre-diabetes and newly diagnosed type 2 diabetes: a clinical study. Acta Diabetol. 2010 doi: 10.1007/s00592-010-0226-0. [DOI] [PubMed] [Google Scholar]
- 242.Kolfschoten IG, Roggli E, Nesca V, Regazzi R. Role and therapeutic potential of microRNAs in diabetes. Diabetes Obes Metab. 2009;11(Suppl 4):118–129. doi: 10.1111/j.1463-1326.2009.01118.x. [DOI] [PubMed] [Google Scholar]
- 243.Lovis P, Roggli E, Laybutt DR, Gattesco S, Yang JY, Widmann C, Abderrahmani A, Regazzi R. Alterations in microRNA expression contribute to fatty acid-induced pancreatic beta-cell dysfunction. Diabetes. 2008;57:2728–2736. doi: 10.2337/db07-1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Li Y, Vandenboom TG, Wang Z, Kong D, Ali S, Philip PA, Sarkar FH. miR-146a suppresses invasion of pancreatic cancer cells. Cancer Res. 2010;70:1486–1495. doi: 10.1158/0008-5472.CAN-09-2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Herrera BM, Lockstone HE, Taylor JM, Ria M, Barrett A, Collins S, Kaisaki P, Argoud K, Fernandez C, Travers ME, Grew JP, Randall JC, Gloyn AL, Gauguier D, McCarthy MI, Lindgren CM. Global microRNA expression profiles in insulin target tissues in a spontaneous rat model of type 2 diabetes. Diabetologia. 2010;53:1099–1109. doi: 10.1007/s00125-010-1667-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Lynn FC. Meta-regulation: microRNA regulation of glucose and lipid metabolism. Trends Endocrinol Metab. 2009;20:452–459. doi: 10.1016/j.tem.2009.05.007. [DOI] [PubMed] [Google Scholar]