

Abstract

The activity of light-activatable (“caged”) compounds can be temporally and spatially controlled, thereby providing a means to interrogate intracellular biochemical pathways as a function of time and space. Nearly all caged peptides contain photocleavable groups positioned on the side chains of key residues. We describe an alternative active site targeted strategy that disrupts the interaction between the protein target (SH2 domain, kinase, and proteinase) and a critical amide NH moiety of the peptide probe.
Living cells have been referred to as the test tubes of the 21st century.1 Indeed, a host of reagents have been described for inhibiting, manipulating, or visualizing a wide variety of intracellularly relevant processes. Nonetheless, key challenges remain before the cell-as-a-test-tube analogy can be fully realized. A particularly confounding attribute that differentiates living cell biochemistry from its counterpart in the test tube is that the cell, not the investigator, controls where and when a given transformation occurs. Light-activatable (“caged”) compounds allow the investigator to retain control over the activity of the bio-reagent, even after it has entered the cell.2 In this regard, a number of caged derivatives of small molecules, including ATP, glutamate, NO, and many others, has been described.2c Furthermore, recent interest in defining the temporal and spatial dynamics of signaling pathways, biochemical cascades largely driven by protein–protein interactions, has led to the construction of caged peptide derivatives.3 The latter compounds are designed to engage specific protein recognition motifs, but only upon photoactivation. Several examples of peptide-based protein kinase sensors3e,g and inhibitors3b as well as 14-3-33d and SH23c,f domain-targeting species have been reported. The general strategy for the preparation of these peptidic species is based on the side-chain modification (with photolabile groups) of key residues required for biorecognition, such as Ser or Tyr for protein kinases or phosphorylated Ser or Tyr for 14-3-3 and SH2 domains, respectively. Unfortunately, many amino acids lack side chain functionality necessary for modification whereas the preparation of caged derivatives of those that can be modified is typically a multistep, off-resin, process.
Protein–protein interactions are often dependent upon one or a few key amino acid residues. These residues must be able to achieve the requisite contacts with the protein-binding partner in order for recognition and/or catalysis to occur. In many instances, the amide NH of the essential and/or adjacent residue is crucial for proper orientation of the critical side chain. We reasoned that the incorporation of a photolabile moiety on this key amide nitrogen could significantly compromise recognition or catalysis, via loss of amide hydrogen bond donating ability and/or the presence of a sterically demanding light-cleavable substituent. This notion has been examined via the design, synthesis, and characterization of caged peptides for three different protein interaction domains.
To the best of our knowledge, there have only been two reports of backbone-caged peptides. Darszon, Yumoto, and their colleagues described a backbone-substituted glycine residue that was prepared off-resin as an Fmoc(N-o-nitrobenzyl) derivative and subsequently coupled to the growing peptide chain.4 These investigators reported difficulties in coupling the subsequent residue, a serine moiety, to the N-alkylated glycine. However, they found that acyl fluorides or diisopropylcarbodiimide furnished the desired material. More recently, Johnson and Kent examined photoremovable N-benzylated protecting groups as part of Boc chemistry solid-phase peptide synthesis.5 These investigators likewise found that the subsequent addition of a standard amino acid to the caged residue could be problematic, particularly if hindered amino acids were involved.
We examined whether the caged residue could be prepared directly on the resin, a strategy that could be immediately applied to conventional commercially available side chain protected amino acids. Solid-phase peptide synthesis was performed using Fmoc chemistry on the Rink resin. The Fmoc group was removed, via standard piperidine treatment, from the residue to be caged (Scheme 1). The free N-terminal amine of 1 was exposed twice to 4,5-dimethoxy-2-nitrobenzaldehyde to ensure complete conversion to the imine and then reduced with NaBH3CN to furnish the desired N-benzylated derivative 2 (HPLC and electrospray mass spectrometry).4,5 Unfortunately, as reported by Darszon, Yumoto, and Kent,6,7 we found that addition of the subsequent amino acid to the N-benzylated residue was problematic. For example, Darszon and Yumoto’s diisopropylcarbodiimide protocol was employed to couple Fmoc-Ser to (N-o-nitrobenzyl)Gly-peptide resin, but failed to furnish the desired product 3. This may be a consequence of the significantly more sterically demanding nature of the side chain phosphorylated Fmoc-Tyr residue to be coupled. Ultimately, we discovered that the activating agent PyBroP8 provided the desired pTyr-derivatized peptide 3 (however, see compound 10, vide infra). Subsequent solid-phase synthesis proceeded uneventfully to furnish 4.
Scheme 1.
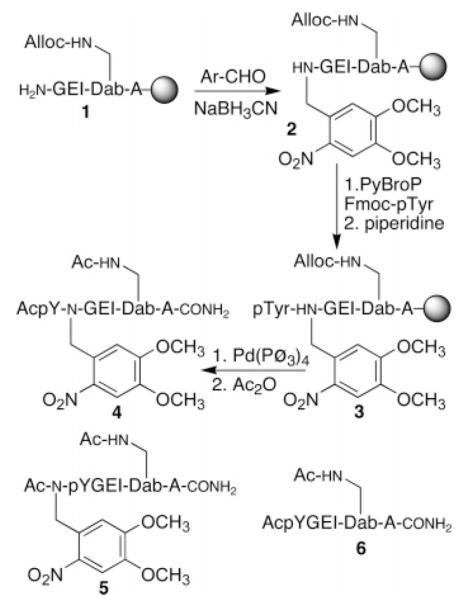
On-Resin Synthesis of Caged Peptide 4, Structure of the Alternatively Caged Peptide 5, and Structure of the Parent SH2 Domain-Directed Peptide 6
Src homology (SH) 2 domains play a key role in organizing, assembling, and activating signaling complexes.9 These domains bind to phosphorylated Tyr- (pTyr) containing residues positioned within an appropriate amino acid sequence context on a peptide or protein.8 For example, the Lck SH2 domain displays a moderate affinity (KD ≈ 1–5 μM) for peptides of the general form Ac-pTyr-Xaa-Xaa-Ile-amide (Figure 1). Previous structural studies have reported that the amide moiety linking the pTyr-Xaa dyad forms a key hydrogen bond with the SH2 domain.9b The caged derivative 4 was prepared via the Scheme 1 protocol. We also prepared the corresponding analogue 5, which contains the photolabile group on the pTyr-amide moiety. Kd values were acquired via a competition assay using a previously described dapoxyl-labeled peptide,10 which displays a 4-fold fluorescence enhancement upon binding to the Lck SH2 domain (Supporting Information). The caged derivative 4 has a significantly lower SH2 domain affinity (127 ± 6 μM) than the parent peptide 6 (2.6 ± 0.2 μM). By contrast, the difference in affinity between 5 (43 ± 10 μM) and 6 is somewhat more modest. The latent ligand 4 is converted to the high affinity form 6 upon photolysis. Furthermore, longer photolysis times generate larger yields of the active peptide 6 (Supporting Information). The latter property represents one of the distinct advantages associated with caged compounds, namely the ability to control both the timing and the quantity of the photoactivated species.
Figure 1.
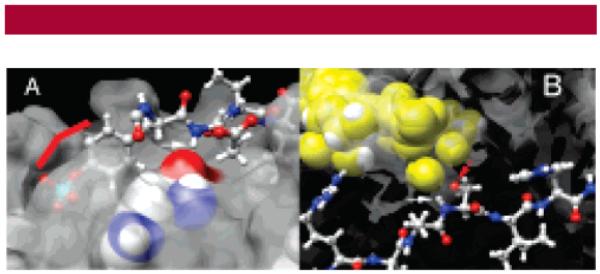
(A) pTyr peptide (ball and stick) bound to the SH2 domain of Lck. The key NH amide moiety linking the pTyr (red bar) and Glu residues is highlighted (white arrow) and shown within hydrogen-bonding distance of the His-180 carbonyl (space filling).9b (B) PKA substrate peptide (ball and stick) complexed with an ATP transition state mimic (ADP/AlF3; yellow space filling). The phosphoryl acceptor Ser (red arrow) and the key amide NH (white arrow) are highlighted.14
Proteinases participate in a diverse array of biochemical processes that range from the generation of biologically active proteins from their immature counterparts to the degradation of aged or otherwise defective proteins. Like many other proteins that act on protein substrates, proteinases are capable of utilizing short peptides of the appropriate amino acid sequence as substrates. Chymotrypsin’s distinct preference for Phe at the scissile bond is most noteworthy within the context of caged peptides ligands. The aromatic side chain lacks a modifiable substituent and is therefore not susceptible to the standard side chain caging strategy.
Chymotrypsin substrate recognition and subsequent hydrolysis is dependent upon the amide moieties of the scissile bond and of adjacent residues. Indeed, N-methyl substitution at the scissile amide (and at the amide moieties at nearby residues) renders chymotrypsin substrates resistant to proteolysis.11 N-methylation not only results in the loss of a key hydrogen bond required for substrate recognition, but also introduces a steric factor that compromises active site incorporation. With these features in mind, we prepared a caged fluorescent substrate for chymotrypsin that contains a C-terminal coumarin at the scissile position. Previous studies have demonstrated that the free coumarin, generated upon proteolysis, is highly fluorescent.12 The peptide substrate 7 (Scheme 2) was synthesized using an Fmoc solid-phase protocol (Supporting Information). The corresponding caged derivative 8 was prepared by interrupting the standard protocol at the Phe-courmarin stage with the Scheme 1 caging strategy. Peptide 7, and its caged analogue 8, were examined as chymotrypsin substrates. Compound 7 is an efficient substrate (Km = 6.3 ± 1.3 μM; Vmax = 7.2 ± μmol/min-mg) and exhibits a 6-fold enhancement in fluorescence upon complete hydrolysis. By contrast, the caged derivative 8 is inactive as a chymotrypsin substrate (Figure 2). As in the case of the caged SH2 ligand 4, increasing photolysis times converts increasing amounts of caged analogue (8) to the active substrate (7). To the best of our knowledge, compound 8 is the first example of a caged proteinase sensor.
Scheme 2.
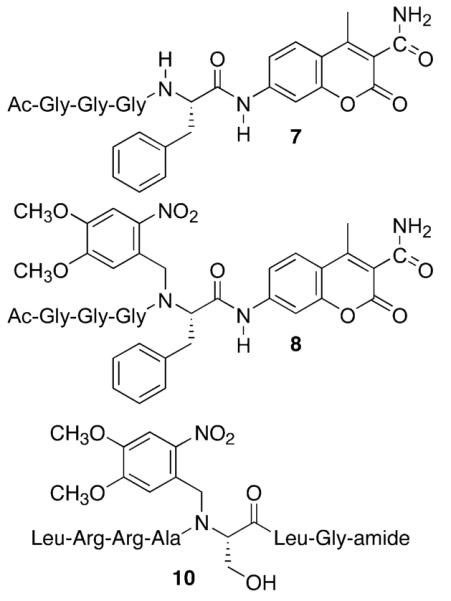
Chymotrypsin Substrate 7, its Caged Counterpart 8, and the Caged PKA Substrate 10
Figure 2.
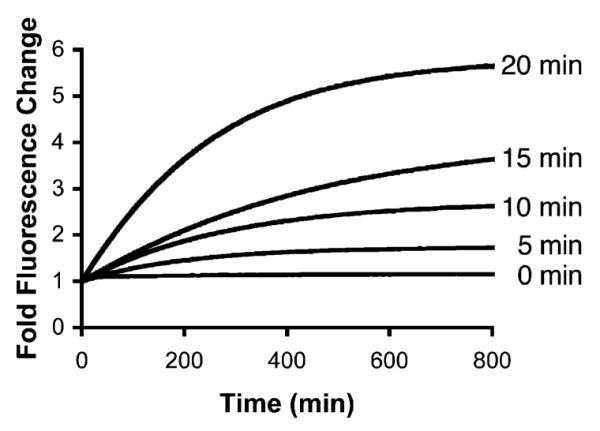
Caged peptide 8 (0 min) is not hydrolyzed by chymotrypsin as assessed by a fluorescent continuous assay (λex = 380 nm; λem = 460 nm). Longer photolysis times produce greater amounts of active substrate and therefore enhanced reaction rates.
Hydrogen bonds play a critical role in orienting the phosphorylatable Ser of Leu-Arg–Arg-Ala-Ser-Leu-Gly-amide (9) into the active site of the PKA. Indeed, N-methylation of the Ala-Ser-amide moiety reduces the PKA-catalyzed efficiency (kcat/Km) of phosphorylation by 7 orders of magnitude.13 Crystallographic studies revealed that the amide NH of Ser is within hydrogen-bonding distance of the side chain hydroxyl and is also oriented toward the incoming phosphoryl group from ATP (Figure 1B).14 Consequently, we anticipated that steric bulk, positioned at this sensitive site (10), could have a deleterious effect on phosphoryl acceptor capability. Compound 10 was prepared in a fashion analogous to that outlined in Scheme 1. However, we discovered that Fmoc-Ala could not be coupled to the reductively alkylated Ser residue using the PyBroP protocol. Instead, coupling was achieved using the acid chloride of Fmoc-Ala.15 The efficiency of PKA-catalyzed phosphorylation of peptide 10 was assessed via a [γ-33P]-ATP assay. In the absence of photolysis, peptide 10 is not phosphorylated (Figure 3). Photolysis of 10 generates the active substrate 9 (mass spectrometry) that, upon addition of PKA, furnishes the phospho-product (scintillation counting). Furthermore, longer photolysis times generate larger quantities of radiolabeled product.
Figure 3.
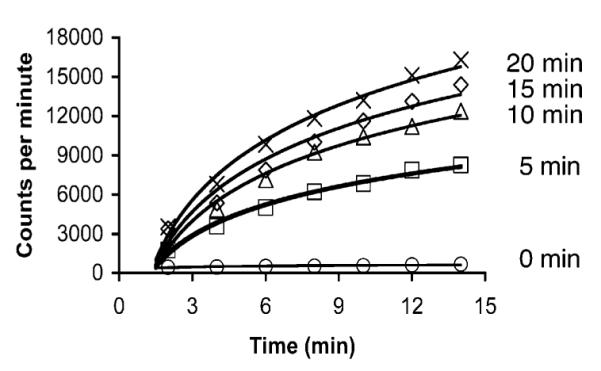
Caged peptide 10 (Y) is not phosphorylated by PKA as assessed by a fixed time point assay. Longer photolysis times produce larger amounts of active substrate and therefore enhanced reaction rates.
In summary, we have described an on-resin, solid-phase method that directly furnishes caged peptides in a straight-forward fashion. These derivatives, represented by an SH2 ligand and substrates for a protein kinase and a protease, provide a means to control both the timing of target protein interaction as well as the amount of active material unleashed even after the bio-agent has been delivered to the cell (e.g., via microinjection).
Supplementary Material
Acknowledgment
We thank the National Institutes of Health for financial support (GM067198 and CA79954).
Footnotes
Supporting Information Available: Experimental details of peptide synthesis and characterization and enzyme assays. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Hansen PM, Oddershede LB. Proc. SPIE. 2005;5930:5930031–9. [Google Scholar]
- (2) (a).Lawrence DS. Curr. Opin. Chem. Biol. 2005;9:570–575. doi: 10.1016/j.cbpa.2005.09.002. [DOI] [PubMed] [Google Scholar]; (b) Rothamn DM, Shults MD, Imperiali B. Trends Cell Biol. 2005;15:502–510. doi: 10.1016/j.tcb.2005.07.003. [DOI] [PubMed] [Google Scholar]; (c) Goeldner M, Givens R, editors. Dynamic Studies in Biology. Wiley; New York: 2005. [Google Scholar]
- (3) (a).Walker JW, Gilbert SH, Drummond RM, Yamada M, Sreekumar R, Carraway RE, Ikebe M, Fay FS. Proc. Natl. Acad. Sci. U.S.A. 1998;95:1568–1573. doi: 10.1073/pnas.95.4.1568. Wood JS, Koszelak M, Liu J, Lawrence DS. J. Am. Chem. Soc. 1998;120:7145–7146. Zou K, Miller WT, Givens RS, Bayley H. Angew. Chem., Int. Ed. 2001;40:3049–3051. doi: 10.1002/1521-3773(20010817)40:16<3049::AID-ANIE3049>3.0.CO;2-N. Nguyen A, Rothman DM, Stehn J, Imperiali B, Yaffe MB. Nat. Biotech. 2004;22:993–1000. doi: 10.1038/nbt997. Veldhuyzen WF, Nguyen Q, McMaster G, Lawrence DS. J. Am. Chem. Soc. 2003;125:13358–13359. doi: 10.1021/ja037801x. Humphrey D, Rajfur Z, Vazquez ME, Scheswohl D, Schaller MD, Jacobson K, Imperiali B. J. Biol. Chem. 2005;280:22091–22101. doi: 10.1074/jbc.M502726200. Wang Q, Dai Z, Cahill SM, Blumenstein M, Lawrence DS. J. Am. Chem. Soc. 2006;128:14016–14017. doi: 10.1021/ja065852z. For an alternative strategy to side chain modification, see Taniguchi A, Sohma Y, Kimura M, Okada T, Ikeda K, Hayashi Y, Kimura T, Hirota S, Matsuzaki K, Kiso Y. J. Am. Chem. Soc. 2006;128:696–697. doi: 10.1021/ja057100v.
- (4).Sasaki Y, Coy DH. Peptides. 1987;8:119–121. doi: 10.1016/0196-9781(87)90174-4. [DOI] [PubMed] [Google Scholar]
- (5).An alternative strategy, which we did not explore, is the direct alkylation of an o-nitrobenzenesulfonyl-activated amine: Miller SC, Scanlan TS. J. Am. Chem. Soc. 1997;119:2301–2302. Biron E, Chatierjee J, Kessler H. J. Peptide Sci. 2006;12:213–219. doi: 10.1002/psc.711. and references cited therein.
- (6).Tatsu Y, Nishigaki N, Darszon A, Yumoto N. FEBS Lett. 2002;525:20–24. doi: 10.1016/s0014-5793(02)03000-4. [DOI] [PubMed] [Google Scholar]
- (7).Johnson EC, Kent SB. Chem. Commun. 2006:1557–1559. doi: 10.1039/b600304d. [DOI] [PubMed] [Google Scholar]
- (8).Coste J, Dufour M-N, Panatloni A, Castro B. Tetrahedron Lett. 1990;31:669–672. [Google Scholar]
- (9) (a).Bradshaw JM, Waksman G. AdV. Protein Chem. 2002;61:161–210. doi: 10.1016/s0065-3233(02)61005-8. Coordinates obtained from the Protein Data Bank (1LCJ); Eck MJ, Shoelson SE, Harrison SC. Nature. 1993;362:87–91. doi: 10.1038/362087a0.
- (10).Wang Q, Lawrence DS. J. Am. Chem. Soc. 2005;127:7684–7685. doi: 10.1021/ja050789j. [DOI] [PubMed] [Google Scholar]
- (11).Haviv F, Fitzpatrik TD, Swenson RE, Nichols CJ, Mort NA, Bush EN, Diaz G, Bammert G, Nguyen A, Rhutasel NS, Nellans HN, Hoffman DJ, Johnson ES, Greer J. J. Med. Chem. 1993;36:363–369. doi: 10.1021/jm00055a007. [DOI] [PubMed] [Google Scholar]
- (12) (a).Zimmerman M, Ashe B, Yurewicz EC, Patel G. Anal. Biochem. 1977;78:47–51. doi: 10.1016/0003-2697(77)90006-9. [DOI] [PubMed] [Google Scholar]; (b) Backes BJ, Harris JL, Leonetti F, Craik CS, Ellman JA. Nat. Biotechnol. 2000;18:187–193. doi: 10.1038/72642. [DOI] [PubMed] [Google Scholar]
- (13).Bramson HN, Thomas NE, Kaiser ET. J. Biol. Chem. 1985;260:15452–15457. [PubMed] [Google Scholar]
- (14).Coordinates, obtained from the Protein Data Bank (1L3R); Akamine P. Madhusudan, Xuong N-H, Taylor SS. Nature Struc. Biol. 2002;9:273–277. doi: 10.1038/nsb780.
- (15).Carpino LA, Cohen BJ, Stephens KE, Jr., Sadat-Aalaee Y, Tien J-H, Langridge DC. J. Org. Chem. 1986;51:3732–3734. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.