

Abstract
Fibrates, the ligands of peroxisome proliferator-activated receptor-α, have been shown to have a renal protective action in diabetic models of renal disease, but the mechanisms underlying this effect are unknown. In the present study, we sought to investigate in greater detail the effect of fenofibrate and its mechanism of action on renal inflammation and tubulointerstitial fibrosis in an animal model of type 2 diabetes mellitus. Twelve-week-old non-diabetic Zucker lean (ZL) and Zucker diabetic fatty (ZD) rats were treated with vehicle or fenofibrate for 10 weeks. mRNA and protein analyses were performed by real-time polymerase chain reaction, Western blot and immunostaining. The diabetic condition of ZD rats was associated with an increase in collagen and α-smooth muscle actin accumulation in the kidney, which was significantly reduced by fenofibrate. Chronic treatment of ZD rats with fenofibrate attenuated renal inflammation and tubular injury as evidenced by a decrease in mRNA and protein expression of secreted phosphoprotein-1, monocyte chemotactic protein-1 and kidney injury molecule-1 in the kidneys. Renal interstitial macrophage infiltration was also significantly reduced in the kidneys of fenofibrate-treated diabetic animals. Moreover, renal nuclear factor (NF)-κB DNA-binding activity, transforming growth factor (TGF)-β1 and phospho-Smad3 proteins were significantly higher in ZD animals compared with ZL ones. This increase in NF-κB activity, TGF-β1 expression and Smad3 phosphorylation was greatly attenuated by fenofibrate in the diabetic kidneys. Taken together, fenofibrate suppressed NF-κB and TGF-β1/Smad3 signaling pathways and reduced renal inflammation and tubulointerstitial fibrosis in diabetic ZD animals.
Keywords: inflammation, kidney, peroxisome proliferator-activated receptor-α, renal injury, type 2 diabetes
Introduction
Peroxisome proliferator-activated receptor-α (PPARα) is a member of the nuclear hormone receptor superfamily and actively involved in lipid metabolism. Fenofibrate, a PPARα agonist, has been reported to improve the microcirculation and retard the angiographic progression of coronary atherosclerosis in patients with hyperlipidemia and/or diabetes.1–5 Studies performed in clinical and experimental models also suggest that PPARα may play an important role in the progression of kidney disease in both type 1 and type 2 diabetes.6–12 For example, PPARα deficiency aggravates the severity of diabetic nephropathy by increasing extracellular matrix (ECM) formation and circulating free fatty acid and triglyceride concentration in streptozotocin-induced diabetes in mice.11 Chronic treatment with fenofibrate improves renal structure and function by decreasing glomerular hypertrophy and mesangial matrix in animals with type 1 or type 2 diabetes.11–13 However, the exact mechanisms involved in the beneficial effect of fenofibrate in the diabetic kidneys have not been fully elucidated.
There is growing evidence that inflammatory processes play an important role in the pathogenesis of diabetic nephropathy, and that nuclear factor-kappaB (NF-κB) and transforming growth factor (TGF)-β have been identified as key mediators of these events. Increased macrophage infiltration has been reported in the glomeruli and in the interstitia of the kidneys of patients with diabetic nephropathy.14,15 The suppression of macrophage infiltration by radiation or the use of immunosuppressive agents could ameliorate the development and/or progression of diabetic glomerular injury.16,17 Lee et al.18 have recently reported that NF-κB inhibition with pyrrolidine dithiocarbamate also reduced monocyte chemoattractant protein-1 (MCP-1) and macrophage infiltration in the diabetic kidneys, supporting the implication of NF-κB-dependent pathways in the development and/or progression of diabetic nephropathy. In addition to the NF-κB pathway, the role of TGF-β/Smad pathway in inflammation and renal fibrosis has also been recognized.19–21 TGF-β promotes inflammation, stimulates fibroblast proliferation and the synthesis of a number of ECM proteins including collagens. Interestingly, PPARα ligands have been shown to inhibit TGF-β1 activation in cultured mesangial cells treated with H2O222 or high glucose.11 However, the in vivo function of PPARα activators on renal NF-κB and TGF-β1 signaling pathways still remains elusive.
The Zucker diabetic fatty (ZD) rat strain has been well characterized as an animal model of metabolic syndrome and type 2 diabetes, and has been widely used to investigate vascular and renal function in diabetes. We recently reported that fenofibrate down-regulated cyclin D1 expression and attenuated glomerular hypertrophy in Zucker diabetic rats.12 This study was designed to more closely investigate the anti-inflammatory and antifibrotic effects of fenofibrate in the kidney of Zucker diabetic animals by (1) determining the expression of biomarkers of inflammation and renal injury; (2) evaluating macrophage infiltration in glomeruli as well as interstitial spaces; and (3) examining the expression and activity of NF-κB and TGF-β1/Smad3.
Materials and methods
Experimental animals
Twelve-week-old male Zucker lean (ZL) and ZD rats (Charles River Laboratories, Wilmington, MA, USA) were divided into four experimental groups: vehicle (0.5% carboxymethylcellulose, intragastric gavage)-treated ZL, fenofibrate (150 mg/kg/day, intragastric gavage)-treated ZL (F-ZL), vehicle-treated ZD and fenofibrate-treated ZD (F-ZD) rats. Animals were treated for 10 weeks and kidney tissues were harvested. Blood glucose was monitored using the Accu-chek glucometer by tail-vein blood sampling. Rats were housed in an animal care facility at the Morehouse School of Medicine. All animal protocols were approved by the Institutional Animal Care and Use Committee and were in accordance with the requirements stated in the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
RNA extraction and quantitative real-time polymerase chain reaction analysis
Total RNA was prepared from isolated kidney cortex by using ultra-pure TRIzol reagent according to the manufacturer’s instructions (GIBCO-BRL, Grand Island, NY, USA). Reverse transcription was performed on equal amounts of total RNA (2 μg) by using random hexanucleotide primers to produce a cDNA library for each sample. Real-time polymerase chain reaction (PCR) reactions were run on an iCycler iQ Real-Time PCR Detection System by using Taqman Universal PCR Master Mix (P/N 4304437, Applied Biosystems, Foster City, CA, USA).23 Kidney injury molecule-1 (KIM-1), secreted phosphoprotein-1 (SPP-1), monocyte chemotactic protein-1 (MCP-1), TGF-β1 and β-actin gene-specific Taqman probe and primer sets were obtained from Applied Biosystems as Assays-on-Demand gene expression products. The Assays-on-Demand identification numbers were Rn00597703_m1 for KIM-1, Rn00563571_m1 for SPP-1, Rn00580555_m1 for MCP-1 (chemokine ligand 2 [CCL2]), Rn00572010_m1 for TGF-β1 and 4331182 for rat β-actin endogenous control. Each sample was run in triplicate, and the comparative threshold cycle (Ct) method was used to quantify fold increase (2−ΔΔCt) compared with lean controls.
Immunoblot analysis
Kidney cortex was harvested and processed as described previously.24 Samples were separated by electrophoresis on a 10% stacking Tris-glycine gel, and proteins were transferred electrophoretically to a nitrocellulose membrane. The primary antibodies were mouse anti-α-smooth muscle actin (SMA; Santa Cruz Biotechnology, Inc, Santa Cruz, CA, USA), rabbit anti-TGF-β1 (Santa Cruz Biotechnology, Inc), rabbit anti-Smad3 and rabbit anti-phospho-Smad3 (Cell Signaling, Danvers, MA, USA). The blots were then washed in Tris-buffered saline (20 mmol/L Tris-HCl, 150 mmol/L NaCl, pH 7.5)–0.3% Tween-20 and incubated with the second antibody conjugated to horseradish peroxidase for 90 min at room temperature and washed. Detection was accomplished by enhanced chemiluminescence Western blotting (ECL, Amersham), and blots were exposed to X-ray film (Hyperfilm-ECL, Amersham). Band intensity was measured densitometrically, and the values were normalized to β-actin internal controls. Values are expressed as relative optical density.
Histology and immunohistochemical staining
Perfusion-fixed paraffin sections (5 μm) were stained with Masson’s trichrome method to identify collagen fibers.25 For immunohistochemical analysis, paraffin sections (5 μm) were dewaxed and washed in phosphate-buffered saline (PBS). Sections were then incubated in preheated 10 mmol/L sodium citrate buffer at 95°C for 20 min. Slides were washed in PBS (137 mmol/L NaCl, 2.68 mmol/L KCl, 10 mmol/L Na2HPO4, 1.76 mmol/L KH2PO4, pH 7.4) and blocked with protein-blocking solution (10% normal horse serum) for one hour. Slides were then incubated with mouse SMA antibody (1:100, Santa Cruz Biotechnology, Inc), mouse anti-MPIIIB10 (SPP-1 antibody, 1:200, Developmental Studies Hybridoma Bank, Iowa City, IA, USA) or mouse anti-CD68 (ED-1, 1:100, AbD Serotec, Raleigh, NC, USA) in protein-blocking solution for 30–60 min. The primary antibody was removed, washed in PBS and incubated for 30 min with the secondary antibody. Slides were then incubated with ABC reagents for 30 min, and then stained with diaminobenzidine for 1–3 min.
For immunofluorescent staining, 5-μm-thick cryostat sections of optical coherence tomography-embedded kidney samples were fixed in precooled methanol for five minutes. Sections were washed in PBS and blocked with protein blocking solution (10% normal donkey serum) for one hour. Slides were incubated overnight with goat anti-rat TIM-1/KIM-1/HAVCR antibody (1:100, R&D Systems, Minneapolis, MN, USA) in protein-blocking solution. The primary antibody was removed, and sections were washed in PBS. Tissues were then incubated for 30 min with the secondary antibody (Rhodamine Red-conjugated donkey anti-rabbit IgG, Jackson ImmunoResearch, West Grove, PA, USA). This and subsequent steps were conducted in the dark. Sections were washed in PBS, counterstained with 4′,6-diamidino-2-phenylindole and mounted with Prolong antifade mounting agent (Invitrogen, Camarillo, CA, USA), and a cover slide was added to each.
Electrophoretic mobility shift assay
Nuclear protein was extracted from the kidney cortex with lysis buffer (50 mmol/L KCl, 0.5% NP-40, 25 mmol/L 4--(2-hydroxyethyl)-1-piperazineethanesulfonic acid, 100 mmol/L dithiothreitol [DTT], proteinase inhibitor [1 mmol/L phenyl-methanesulfonylfluoride, 10 mg/mL leupeptin, 20 mg/mL aprotinin], pH 7.8) and then nuclear protein extraction buffer (500 mmol/L KCl, 10% glycerol, 100 mmol/L DTT, proteinase inhibitor [same as lysis buffer]). The binding reactions were performed with a biotin end-labeled NF-κB oligonucleotide (5′-AGT TGA GGG GAC TTT CCC AGG C-3′) (Research Genetics Inc, Hunstville, AL, USA) using LightShift™ chemiluminescent electrophoretic mobility shift assay (EMSA) kit (Pierce Chemical Co, Rockford, IL, USA) as described previously.26 Biotin-labeled NF-κB oligonucleotide was then incubated with the nuclear extract of the cells for 20 min at room temperature. These DNA–protein complexes were resolved on a 6% polyacrylamide gel electrophoresis and transferred to a Hybound-N+ membrane (Amersham and Pharmacia Biotech, Piscataway, NJ, USA). The biotin end-labeled DNA was detected using a streptavidin–horseradish peroxidase conjugate and a chemiluminescent substrate according to the manufacturer’s instructions.
Statistical analysis
Data are expressed as mean ± standard error of mean. Statistical analysis was performed with analysis of variance followed by the Student’s unpaired two-tailed test. Statistical significance was set at P < 0.05.
Results
Fenofibrate reduced tubulointerstitial fibrosis
Collagen accumulation and SMA deposition were evaluated to assess the intensity of fibrogenic response in the diabetic kidneys. Compared with the lean controls, collagen deposition was significantly increased in the kidneys of 22-week-old ZD rats, which was greatly attenuated by fenofibrate in F-ZD group (Figure 1a). Consistent with our previous findings in Zucker diabetic rats at 32 weeks,12 SMA-positive staining was significantly increased in the renal interstitia of 22-week-old ZD rats (Figure 1b). Diabetes-associated interstitial SMA accumulation was markedly reduced by treatment with fenofibrate (Figure 1b). Western blot analysis further indicated that SMA protein was increased by 3.5-fold in the kidney cortex of ZD animals compared with the ZL controls. This increase in renal cortical SMA protein was partially but significantly blunted by fenofibrate in F-ZD animals (Figure 1c). As expected, fasting blood glucose significantly increased in ZD animals (425±35 mg/dL) compared with ZL controls (108±8 mg/dL). However, fenofibrate did not alter fasting blood glucose levels in F-ZD animals (392±56 mg/dL).
Figure 1.

Effects of fenofibrate on collagen and α-smooth muscle actin (SMA) deposition in the diabetic kidneys. (a) Masson staining of collagen. (b) Immunostaining of SMA in paraffin-embedded rat kidney sections. Collagen fibers (blue) and SMA (dark brown) are normally confined in the wall of the arterioles in the lean rats (ZL), and pathologically appear in the intraglomerular, periglomerular, interstitial and peritubular areas in the diabetic rats (ZD). Collagen and SMA deposition was decreased in fenofibrate-treated Zucker diabetic rats (F-ZD). Original magnifications, ×400. (c) Representative Western blots showing SMA and β-actin protein levels in the kidney cortex of Zucker rats. Densitometric evaluations of protein levels (20 μg/lane) were obtained from four different animals per group. Quantitative data are expressed as mean ± standard error of mean. *P < 0.05 versus ZL control; #P < 0.05 versus vehicle-treated ZD animals
Fenofibrate inhibited KIM-1 renal expression
KIM-1 is a sensitive biomarker of tubular injury in several different kidney diseases. We found that KIM-1 messenger RNA was present at a low level in normal lean kidneys but was up-regulated by 26-fold in the ZD kidneys (Figure 2). Immunostaining further revealed that KIM-1 protein was strongly induced in the tubules of ZD rats, whereas KIM-1 staining was undetectable in ZL controls (Figure 3). This up-regulation of KIM-1 mRNA and protein in the diabetic kidneys was greatly attenuated by fenofibrate administration (Figures 2 and 3). Ten-week treatment with fenofibrate decreased renal KIM-1 mRNA by 63%.
Figure 2.

Fenofibrate inhibited mRNA expression of kidney injury molecule-1 (KIM-1), secreted phosphoprotein-1 (SPP-1) and monocyte chemoattractant protein-1 (MCP-1) in the kidney cortex of diabetic rats. Renal cortical mRNA level was determined by real-time polymerase chain reaction. mRNA fold changes of KIM-1, SPP-1 and MCP-1 were calculated using β-actin as an internal control. Values are mean ± standard error of mean. n = 5–6 animals/group. *P < 0.05 versus Zucker lean controls; #P < 0.05 versus vehicle-treated Zucker diabetic fatty group
Figure 3.

Representative images of kidney injury molecule-1 (KIM-1) and secreted phosphoprotein-1 (SPP-1) protein in the kidney sections of Zucker lean (ZL), Zucker diabetic fatty (ZD) and fenofibrate-treated Zucker diabetic rats (F-ZD). KIM-1 and SPP-1 protein were almost undetectable in the lean kidneys. KIM-1 staining (red) was dramatically increased in the tubules of vehicle-treated ZD rats, whereas SPP-1 (brown) was increased in both glomeruli and tubules of ZD kidneys. Fenofibrate treatment decreased KIM-1 and SPP-1 staining in the diabetic kidneys. Original magnifications, ×400
Fenofibrate inhibited up-regulation of SPP-1 and MCP-1 in the diabetic kidneys
SPP-1 and MCP-1 are potent proinflammatory cytokines that play an important role in the progression of diabetic nephropathy. Using quantitative PCR, we found that renal cortical SPP-1 and MCP-1 gene expression was up-regulated by 12- and 2.5-fold in ZD rats compared with ZL controls (Figure 2). This increase in renal SPP-1 and MCP-1 transcripts was markedly suppressed by fenofibrate treatment in the diabetic animals (Figure 2). We also performed immunohistochemistry analysis and confirmed that SPP-1 protein paralleled mRNA expression. SPP-1 protein was dramatically increased in the tubular epithelium throughout the cortex of the diabetic kidneys compared with ZL controls (Figure 3). Treatment with fenofibrate markedly reduced SPP-1 staining in the diabetic kidneys (Figure 3).
Fenofibrate suppressed interstitial macrophage infiltration
Figure 4 presents the representative immunohistochemical images of macrophage cell infiltration in vehicle- or fenofibrate-treated diabetic kidneys. Compared with the ZL rats, the number of ED-1-positive cells was significantly increased in the interstitial space of ZD kidneys, whereas macrophage infiltration only slightly increased in the glomeruli. Diabetes-related increase in interstitial macrophage accumulation was greatly attenuated by fenofibrate treatment (Figure 4).
Figure 4.
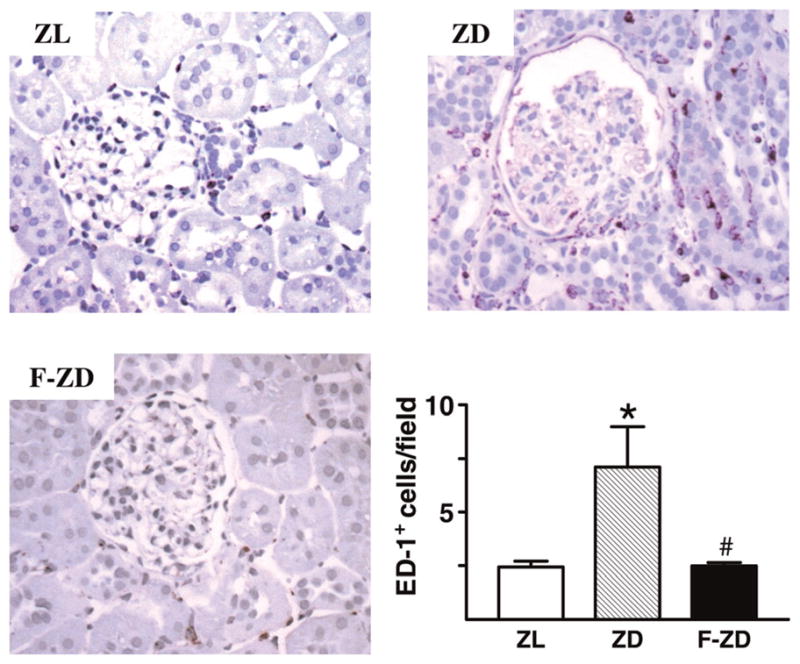
Immunolocalization of macrophage-specific antigen (CD68) in the glomeruli and interstitia of Zucker lean (ZL), Zucker diabetic fatty (ZD) and fenofibrate-treated Zucker diabetic rats (F-ZD) kidneys. Macrophage infiltration (dark brown) was dramatically increased in the interstitial spaces of ZD rats, which was reduced by fenofibrate treatment (F-ZD). Bar graph presents averaged data for CD68-positive cells in the interstitial space. Values are mean ± standard error of mean. n = 5–6 animals/group. *P < 0.05 versus ZL animals; #P < 0.05 versus vehicle-treated ZD group. Original magnifications, ×400
Fenofibrate suppressed renal NF-κB activity
Renal cortical NF-κB DNA-binding activity was evaluated by EMSA analysis. As shown in Figure 5, NF-κB was activated in the kidney cortex of ZD rats compared with ZL controls. Although fenofibrate did not affect NF-κB-binding activity in the lean rats, renal activation of NF-κB in the diabetic animals was significantly attenuated by fenofibrate treatment (Figure 5).
Figure 5.

Fenofibrate suppressed DNA-binding activity of nuclear factor (NF)-κB in the kidney cortex of the diabetic rats. Activated NF-κB in Zucker lean (ZL), Zucker diabetic fatty (ZD), fenofibrate-treated Zucker lean (F-ZL) and fenofibrate-treated Zucker diabetic rat (Z-FD) groups was detected by electrophoretic mobility shift assay. Nuclear extracts (lanes 1–4) were incubated with a double-stranded NF-κB probe labeled with biotin. Competition experiment was performed by adding a 200-fold molar excess of unlabeled NF-κB probe as competitor (lane 5). Bar graph depicts relative optical density for NF-κB DNA-binding bands. Values are mean ± standard error of mean (n = 4 animals/group). *P < 0.05 versus ZL controls; #P < 0.05 versus vehicle-treated ZD group
Fenofibrate reduced TGF-β1 expression and Smad3 phosphorylation
Western blots were performed to evaluate TGF-β1, total Smad3 and phospho-Smad3 protein levels in the kidney cortex isolated from vehicle- or fenofibrate-treated Zucker rats. As expected, TGF-β1 protein was significantly up-regulated in the ZD kidneys (Figure 6). This increase in TGF-β1 was accompanied by a dramatic induction of Smad3 phosphorylation in the diabetic kidneys (Figure 6). Chronic treatment with fenofibrate significantly reduced TGF-β1 and phospho-Smad3 proteins in the diabetic kidneys, whereas their levels were not altered in the F-ZL group (Figure 6). Real-time PCR analysis demonstrated that TGF-β1 mRNA expression was also markedly inhibited by fenofibrate in the diabetic kidneys (Figure 6).
Figure 6.

(a) Representative Western blots of transforming growth factor (TGF)-β1, Smad3 and phospho-Smad3 (P-Smad3) protein in the kidney cortex of Zucker lean (ZL), fenofibrate-treated Zucker lean (F-ZL), Zucker diabetic fatty (ZD) and fenofibrate-treated Zucker diabetic animals (F-ZD). (b) Left bar graph depicts relative optical density for renal cortical levels of TGF-β1 protein. Right bar graph depicts the real-time polymerase chain reaction results of TGF-β1 mRNA. Values are mean ± standard error of mean. n = 5–6 animals/group. *P < 0.05 versus ZL controls; #P < 0.05 versus vehicle-treated ZD group
Discussion
The present study demonstrated that fenofibrate attenuated tubulointerstitial fibrosis by reducing collagen and SMA deposition in the diabetic kidneys. In addition, fenofibrate ameliorated tubular injury as evidenced by the observation that diabetes-associated renal expression of KIM-1 was inhibited after treatment. We also found that fenofibrate decreased SPP-1 and MCP-1 expression in the kidneys of diabetic animals. Accompanying the decrease in renal expression of proinflammatory factors, interstitial macrophage infiltration was significantly reduced in the F-ZD animals. Furthermore, renal NF-κB and TGF-β1/Smad3, which were activated in vehicle-treated diabetic rats, were significantly suppressed by fenofibrate treatment. These results suggest that the renoprotective effects of fenofibrate are likely to be achieved, at least partly, by suppressing NF-κB and TGF-β1/Smad3 signaling pathways in diabetes mellitus.
Diabetic nephropathy is characterized by a series of ultra-structural changes, including basement membrane thickening, glomerular and tubular hypertrophy, mesangial expansion, glomerulosclerosis and tubulointerstitial fibrosis. Glomerular proteinuria and glomerulopathy resulting in microalbuminuria have been recognized as early consequences of both type 1 and type 2 diabetes. Leaked protein in the tubular lumen stimulates proinflammatory and profibrotic responses that directly contribute to chronic tubulointerstitial damage. On the other hand, tubular damage may occur early in the course of the disease not only secondary to glomerular damage. Some previous studies have shown that renal dysfunction is better correlated with tubulointerstitial changes than with glomerular abnormalities.27–30 Yaqoob et al.31 reported that the diabetic patients with microalbuminuria had higher glomerular proteinuria, tubular proteinuria and enzymuria than those with normoalbuminuria and healthy non-diabetic controls. They also revealed that the diabetic patients with normoalbuminuria had significantly higher tubular proteinuria and tubular enzymuria than non-diabetic controls,31 suggesting that tubular damage may even precede microalbuminuria in diabetic nephropathy. We12 and others13 have demonstrated that the PPARα ligand fenofibrate reduces glomerular hypertrophy, mesangial matrix production and urinary protein excretion in the diabetic animals. Here, we further report that fenofibrate attenuates tubular injury by decreasing KIM-1 renal production.
KIM-1 is a type I cell membrane glycoprotein that contains, in its extracellular portion, a novel six-cysteine immunoglobulin-like domain, two N-glycosylation sites and a T/SP-rich domain characteristic of mucin-like O-glycosylated proteins.32,33 KIM-1 level reflects the severity of kidney damage and has been recognized as a sensitive bio-marker of both acute and chronic kidney tubular damage. Increased KIM-1 has also been established in experimental models such as protein-overload nephropathy,34 adriamycin-induced nephropathy35,36 and angiotensin II-induced renal damage in homozygous Ren2 rats.37 Using immunostaining, we found that KIM-1 protein was undetectable, although low KIM-1 gene was present, in the normal kidneys of ZL rats. However, KIM-1 mRNA was dramatically induced and protein was robustly generated and localized on the apical membrane of the proximal tubules in the ZD animals. Interestingly, chronic treatment with fenofibrate markedly blunted renal expression of KIM-1 in the diabetic rats, suggesting that fenofibrate could protect against tubular damage in diabetes mellitus.
Previous studies also support that KIM-1 may be involved in the pathogenesis of interstitial fibrosis. For example, Kuehn et al.38 have reported that the interstitium surrounding KIM-1-expressing tubules shows high proliferative activity and staining for SMA, characteristic of myofibroblasts. In addition, renal and urinary KIM-1 correlated with proteinuria and interstitial damage in adriamycin nephrosis.39 The increase in renal KIM-1 was significantly attenuated after antiproteinuric treatment with renin–angiotensin system blockade in patients with non-diabetic proteinuric kidney disease40 and animals injected with adriamycin.39 This is also supported by our finding that KIM-1 was mainly located in dilated and flattened proximal tubules. Moreover, the present study provides a strong association between reduction of KIM-1 and improvement of tubulointerstitial fibrosis after PPARα activation, although we cannot conclude from our study if KIM-1 is only a consequence of tubular injury or if it also plays a role in kidney damage and repair. It will be interesting to explore further the role of KIM-1 in progressive diabetic nephropathy by specifically silencing or overexpressing KIM-1 gene.
It is well known that macrophage infiltration is implicated in the pathogenesis and progression of diabetic nephropathy.41–45 A correlation between peritubular myofibroblastic cells and interstitial macrophage infiltration has been demonstrated in both diabetic patients43,44 and db/db animals.41 In agreement with these previous findings, we found that the number of CD68-positive cells was significantly increased in the interstitia of ZD rats, although there is no significant change in macrophage infiltration in the diabetic glomeruli. In addition, a significant decrease in the number of interstitial CD68-positive cells was observed in fenofibrate-treated diabetic kidneys. Overall, our results support that suppression of macrophage infiltration may contribute to the renoprotective effect of fenofibrate in progressive diabetic nephropathy.
SPP-1 and MCP-1/CC-CCL2 are important promoters of inflammation, renal injury and fibrosis in diabetic nephropathy. In response to high glucose, human and mouse mesangial cells have been shown to produce MCP-1 through a pathway that involves activation of protein kinase C, increased levels of oxidative stress and the activation/nuclear translocation of the transcription factor NF-κB.46–49 A recent study indicated that high glucose also stimulated MCP-1 secretion from the cultured rat proximal tubular cells.50 They found that the cortical tubules became the predominant source of MCP-1 in overt diabetic nephropathy. Macrophage accumulation around these tubules was associated with tubular injury and myofibroblast accumulation, which lead to progressive decline of kidney function.41 In the present study, we observed that diabetes-induced SPP-1 and MCP-1 were significantly reduced by fenofibrate, which was accompanied by a decrease in macrophage infiltration and collagen deposition in the kidney of F-ZD animals. Previous in vitro study indicated that PPARα overexpression inhibited adriamycin-induced activity of NF-κB in the renal tubular cells, which was associated with an interaction between PPARα and NF-κB p65 subunit as revealed in immunoprecipitation assays.51 As expected, we found that NF-κB DNA-binding activity was activated in the kidney cortex of diabetic rats. Interestingly, chronic treatment with fenofibrate suppressed renal NF-κB activity, suggesting that fenofibrate may exert its anti-inflammatory action by directly or indirectly regulating NF-κB signaling pathway. This result is also consistent with a previous report that PPARα and PPARδ are able to mitigate cardiomyocyte hypertrophy in vitro by inhibiting NF-κB activation.52
In addition to the NF-κB pathway, the TGF-β/Smad pathway also plays a critical role in renal pathophysiology by promoting ECM deposition and fibrosis. TGF-β1 promotes fibroblast proliferation and increases the synthesis of a number of ECM proteins, including collagens via binding to serine/threonine kinase receptors on the plasma membrane and subsequently activating Smad molecules and additional signaling proteins that coordinately regulate gene expression or cytoplasmic processes.21 PPARα ligands have been shown to inhibit TGF-β1 activation in cultured mesangial cells treated with H2O222 or high glucose.11 Our in vivo study indicated that renal production of TGF-β1 was greatly inhibited by fenofibrate in the diabetic animals. Moreover, an increase in renal Smad3 phosphorylation was almost abolished by fenofibrate in these animals. Recent studies suggest that Smad3 is a key molecule mediating TGF-β activity leading to renal fibrosis, inflammation and apoptosis.53,54 Mice deficient in Smad3 demonstrated attenuated collagen deposition and macrophage infiltration, and reduced numbers of myofibroblasts and tubular apoptotic cells in the kidneys after unilateral ureteral obstruction.54 Therefore, a reduction of phospho-Smad3 by fenofibrate may directly lead to an attenuation of inflammatory and fibrotic responses in the diabetic kidneys. Another possibility would be that the suppression of TGF-β/Smad3 may reduce NF-κB activity, resulting in an amelioration of renal inflammation and fibrosis. This notion is supported by the previous findings that overexpression of Smad3 enhances transaction of κB sites in Mv1Lu cells and co-expression of Smad3 together with NF-κB subunits, especially p52 and p65, further increases those responses.55 Together, our data suggest that PPARα agonist attenuates diabetic renal damage, at least partly, by inhibiting TGF-β1/Smad3 and NF-κB signaling pathways with subsequent suppression of inflammation and fibrosis (Figure 7).
Figure 7.

Schematic depiction of an attenuation of renal inflammation and injury by fenofibrate via the nuclear factor-κB and transforming growth factor-β/Smad3 pathways in Zucker diabetic fatty rats
Previous studies have suggested that both systemic and local actions play a role in the renoprotective effects of PPARα ligands in diabetic kidney diseases.8,13,56–58 At present, we cannot distinguish between an activation of PPARα by fenofibrate in the kidney from its direct systemic and/or local effects. It is interesting to note that 10-week treatment with fenofibrate did not reduce blood glucose levels in ZD rats. Our result suggests that the renoprotective action of fenofibrate is independent of the blood glucose-lowering effect in these diabetic animals. This result is also consistent with the previous reports showing that fenofibrate (at 30–300 mg/kg/day) did not improve hyperglycemia in db/db mice57 or streptozotocin-induced diabetic rats.8
In summary, fenofibrate treatment inhibited renal KIM-1 expression and reduced tubulointerstitial fibrosis in the ZD animals. An increase in renal expression of SPP-1 and MCP-1 and macrophage infiltration was greatly attenuated by fenofibrate in this animal model of type 2 diabetes. Moreover, the anti-inflammatory and antifibrotic effects of PPARα activator were accompanied by a suppression of NF-κB and TGF-β1/Smad3 in the diabetic kidneys. Therefore, our data further support that the administration of exogenous fibrates may slow the progression of diabetic kidney disease by inhibiting the inflammatory and fibrotic processes.
Acknowledgments
The authors would like to thank Andrew Shaw and Dr Yan Xiao for their technical assistance in immunostaining preparation. This work was supported by the American Heart Association Scientist Development Grant, Satellite Healthcare Norman S Coplon Extramural Research Grant, NIH/NCRR Grant 5U54RR022814 and NIH/NCRR/RCMI Grant 5G12RR03034-23.
Footnotes
Author contributions: Dr Li conducted all experiments described in the accepted manuscript, except for the real-time PCR analysis. She also participated in data analysis and manuscript preparation. Dr Emmett and Dr Mann participated in the design of the experiments, interpretation of the data, and preparation and revision of the accepted manuscript. Dr Zhao (the corresponding author) was responsible for the design of the experiments, performance of real-time PCR, analysis and interpretation of the data, and preparation and revision of the accepted manuscript.
References
- 1.Backes JM, Gibson CA, Ruisinger JF, Moriarty PM. Fibrates: what have we learned in the past 40 years? Pharmacotherapy. 2007;27:412–24. doi: 10.1592/phco.27.3.412. [DOI] [PubMed] [Google Scholar]
- 2.Capell WH, DeSouza CA, Poirier P, Bell ML, Stauffer BL, Weil KM, Hernandez TL, Eckel RH. Short-term triglyceride lowering with fenofibrate improves vasodilator function in subjects with hypertriglyceridemia. Arterioscler Thromb Vasc Biol. 2003;23:307–13. doi: 10.1161/01.atv.0000046230.02211.b4. [DOI] [PubMed] [Google Scholar]
- 3.Despres JP, Lemieux I, Salomon H, Delaval D. Effects of micronized fenofibrate versus atorvastatin in the treatment of dyslipidaemic patients with low plasma HDL-cholesterol levels: a 12-week randomized trial. J Intern Med. 2002;251:490–9. doi: 10.1046/j.1365-2796.2002.00988.x. [DOI] [PubMed] [Google Scholar]
- 4.Keating GM, Croom KF. Fenofibrate: a review of its use in primary dyslipidaemia, the metabolic syndrome and type 2 diabetes mellitus. Drugs. 2007;67:121–53. doi: 10.2165/00003495-200767010-00013. [DOI] [PubMed] [Google Scholar]
- 5.Liang B, McMaster JC, Kroeger EA, Hatch GM, Mymin D, Dembinski T, Arthur G, Shen G, Man RY, Choy PC. The effect of fenofibrate treatment on endothelium-dependent relaxation induced by oxidative modified low density lipoprotein from hyperlipidemic patients. Mol Cell Biochem. 2000;207:123–9. doi: 10.1023/a:1007019019911. [DOI] [PubMed] [Google Scholar]
- 6.Ansquer JC, Foucher C, Rattier S, Taskinen MR, Steiner G. Fenofibrate reduces progression to microalbuminuria over 3 years in a placebo-controlled study in type 2 diabetes: results from the Diabetes Atherosclerosis Intervention Study (DAIS) Am J Kidney Dis. 2005;45:485–93. doi: 10.1053/j.ajkd.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 7.Chen LL, Zhang JY, Wang BP. Renoprotective effects of fenofibrate in diabetic rats are achieved by suppressing kidney plasminogen activator inhibitor-1. Vasc Pharmacol. 2006;44:309–15. doi: 10.1016/j.vph.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 8.Chen YJ, Quilley J. Fenofibrate treatment of diabetic rats reduces nitrosative stress, renal cyclooxygenase-2 expression, and enhanced renal prostaglandin release. J Pharmacol Exp Ther. 2008;324:658–63. doi: 10.1124/jpet.107.129197. [DOI] [PubMed] [Google Scholar]
- 9.Ginsberg HN, Bonds DE, Lovato LC, Crouse JR, Elam MB, Linz PE, O’Connor PJ, Leiter LA, Weiss D, Lipkin E, Fleg JL. Evolution of the lipid trial protocol of the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial. Am J Cardiol. 2007;99:56i–67i. doi: 10.1016/j.amjcard.2007.03.024. [DOI] [PubMed] [Google Scholar]
- 10.Molitch ME. Management of dyslipidemias in patients with diabetes and chronic kidney disease. Clin J Am Soc Nephrol. 2006;1:1090–9. doi: 10.2215/CJN.00780306. [DOI] [PubMed] [Google Scholar]
- 11.Park CW, Kim HW, Ko SH, Chung HW, Lim SW, Yang CW, Chang YS, Sugawara A, Guan Y, Breyer MD. Accelerated diabetic nephropathy in mice lacking the peroxisome proliferator-activated receptor alpha. Diabetes. 2006;55:885–93. doi: 10.2337/diabetes.55.04.06.db05-1329. [DOI] [PubMed] [Google Scholar]
- 12.Zhao X, Li LY. PPAR-alpha agonist fenofibrate induces renal CYP enzymes and reduces blood pressure and glomerular hypertrophy in Zucker diabetic fatty rats. Am J Nephrol. 2008;28:598–606. doi: 10.1159/000116885. [DOI] [PubMed] [Google Scholar]
- 13.Park CW, Zhang Y, Zhang X, Wu J, Chen L, Cha DR, Su D, Hwang MT, Fan X, Davis L, Striker G, Zheng F, Breyer M, Guan Y. PPARalpha agonist fenofibrate improves diabetic nephropathy in db/db mice. Kidney Int. 2006;69:1511–7. doi: 10.1038/sj.ki.5000209. [DOI] [PubMed] [Google Scholar]
- 14.Hirata K, Shikata K, Matsuda M, Akiyama K, Sugimoto H, Kushiro M, Makino H. Increased expression of selectins in kidneys of patients with diabetic nephropathy. Diabetologia. 1998;41:185–92. doi: 10.1007/s001250050888. [DOI] [PubMed] [Google Scholar]
- 15.Maeda S. Do inflammatory cytokine genes confer susceptibility to diabetic nephropathy? Kidney Int. 2008;74:413–5. doi: 10.1038/ki.2008.291. [DOI] [PubMed] [Google Scholar]
- 16.Sassy-Prigent C, Heudes D, Mandet C, Belair MF, Michel O, Perdereau B, Bariety J, Bruneval P. Early glomerular macrophage recruitment in streptozotocin-induced diabetic rats. Diabetes. 2000;49:466–75. doi: 10.2337/diabetes.49.3.466. [DOI] [PubMed] [Google Scholar]
- 17.Utimura R, Fujihara CK, Mattar AL, Malheiros DM, Noronha IL, Zatz R. Mycophenolate mofetil prevents the development of glomerular injury in experimental diabetes. Kidney Int. 2003;63:209–16. doi: 10.1046/j.1523-1755.2003.00736.x. [DOI] [PubMed] [Google Scholar]
- 18.Lee FT, Cao Z, Long DM, Panagiotopoulos S, Jerums G, Cooper ME, Forbes JM. Interactions between angiotensin II and NF-kappaB-dependent pathways in modulating macrophage infiltration in experimental diabetic nephropathy. J Am Soc Nephrol. 2004;15:2139–51. doi: 10.1097/01.ASN.0000135055.61833.A8. [DOI] [PubMed] [Google Scholar]
- 19.Yang F, Chung AC, Ru Huang X, Lan HY. Angiotensin II induces connective tissue growth factor and collagen I expression via transforming growth factor-{beta}-dependent and -independent Smad pathways. The role of Smad3. Hypertension. 2009;54:877–84. doi: 10.1161/HYPERTENSIONAHA.109.136531. [DOI] [PubMed] [Google Scholar]
- 20.Murphy M, Docherty NG, Griffin B, Howlin J, McArdle E, McMahon R, Schmid H, Kretzler M, Droguett A, Mezzano S, Brady HR, Furlong F, Godson C, Martin F. IHG-1 amplifies TGF-beta1 signaling and is increased in renal fibrosis. J Am Soc Nephrol. 2008;19:1672–80. doi: 10.1681/ASN.2007101080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moustakas A, Heldin CH. Dynamic control of TGF-beta signaling and its links to the cytoskeleton. FEBS Lett. 2008;582:2051–65. doi: 10.1016/j.febslet.2008.03.027. [DOI] [PubMed] [Google Scholar]
- 22.Wilmer WA, Dixon CL, Hebert C, Lu L, Rovin BH. PPAR-alpha ligands inhibit H2O2-mediated activation of transforming growth factor-beta1 in human mesangial cells. Antioxid Redox Signal. 2002;4:877–84. doi: 10.1089/152308602762197416. [DOI] [PubMed] [Google Scholar]
- 23.Sardo MA, Campo S, Mandraffino G, Saitta C, Bonaiuto A, Castaldo M, Cinquegrani M, Pizzimenti G, Saitta A. Tissue factor and monocyte chemoattractant protein-1 expression in hypertensive individuals with normal or increased carotid intima–media wall thickness. Clin Chem. 2008;54:814–23. doi: 10.1373/clinchem.2007.095547. [DOI] [PubMed] [Google Scholar]
- 24.Zhao X, Yamamoto T, Newman JW, Kim IH, Watanabe T, Hammock BD, Stewart J, Pollock JS, Pollock DM, Imig JD. Soluble epoxide hydrolase inhibition protects the kidney from hypertension-induced damage. J Am Soc Nephrol. 2004;15:1244–53. [PubMed] [Google Scholar]
- 25.Iwano M, Fischer A, Okada H, Plieth D, Xue C, Danoff TM, Neilson EG. Conditional abatement of tissue fibrosis using nucleoside analogs to selectively corrupt DNA replication in transgenic fibroblasts. Mol Ther. 2001;3:149–59. doi: 10.1006/mthe.2000.0251. [DOI] [PubMed] [Google Scholar]
- 26.Taggart CC, Cryan SA, Weldon S, Gibbons A, Greene CM, Kelly E, Low TB, O’Neill SJ, McElvaney NG. Secretory leucoprotease inhibitor binds to NF-kappaB binding sites in monocytes and inhibits p65 binding. J Exp Med. 2005;202:1659–68. doi: 10.1084/jem.20050768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eddy AA. Proteinuria and interstitial injury. Nephrol Dial Transplant. 2004;19:277–81. doi: 10.1093/ndt/gfg533. [DOI] [PubMed] [Google Scholar]
- 28.Howie AJ, Lote CJ. Interactions between renal tubules and interstitium. J Clin Pathol. 1996;49:783–6. doi: 10.1136/jcp.49.10.783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marcussen N, Jacobsen NO. The progression of cisplatin-induced tubulointerstitial nephropathy in rats. Apmis. 1992;100:256–68. doi: 10.1111/j.1699-0463.1992.tb00869.x. [DOI] [PubMed] [Google Scholar]
- 30.Taft JL, Nolan CJ, Yeung SP, Hewitson TD, Martin FI. Clinical and histological correlations of decline in renal function in diabetic patients with proteinuria. Diabetes. 1994;43:1046–51. doi: 10.2337/diab.43.8.1046. [DOI] [PubMed] [Google Scholar]
- 31.Yaqoob M, McClelland P, Patrick AW, Stevenson A, Mason H, Bell GM. Tubulopathy with macroalbuminuria due to diabetic nephropathy and primary glomerulonephritis. Kidney Int Suppl. 1994;47:S101–4. [PubMed] [Google Scholar]
- 32.Bailly V, Zhang Z, Meier W, Cate R, Sanicola M, Bonventre JV. Shedding of kidney injury molecule-1, a putative adhesion protein involved in renal regeneration. J Biol Chem. 2002;277:39739–48. doi: 10.1074/jbc.M200562200. [DOI] [PubMed] [Google Scholar]
- 33.Bonventre JV. Kidney injury molecule-1 (KIM-1): a urinary biomarker and much more. Nephrol Dial Transplant. 2009;24:3265–8. doi: 10.1093/ndt/gfp010. [DOI] [PubMed] [Google Scholar]
- 34.van Timmeren MM, Bakker SJ, Vaidya VS, Bailly V, Schuurs TA, Damman J, Stegeman CA, Bonventre JV, van Goor H. Tubular kidney injury molecule-1 in protein-overload nephropathy. Am J Physiol Renal Physiol. 2006;291:F456–64. doi: 10.1152/ajprenal.00403.2005. [DOI] [PubMed] [Google Scholar]
- 35.Kramer AB, van der Meulen EF, Hamming I, van Goor H, Navis G. Effect of combining ACE inhibition with aldosterone blockade on proteinuria and renal damage in experimental nephrosis. Kidney Int. 2007;71:417–24. doi: 10.1038/sj.ki.5002075. [DOI] [PubMed] [Google Scholar]
- 36.Kramer AB, van Timmeren MM, Schuurs TA, Vaidya VS, Bonventre JV, van Goor H, Navis G. Reduction of proteinuria in adriamycin-induced nephropathy is associated with reduction of renal kidney injury molecule (Kim-1) over time. Am J Physiol Renal Physiol. 2009;296:F1136–45. doi: 10.1152/ajprenal.00541.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.de Borst MH, van Timmeren MM, Vaidya VS, de Boer RA, van Dalen MB, Kramer AB, Schuurs TA, Bonventre JV, Navis G, van Goor H. Induction of kidney injury molecule-1 in homozygous Ren2 rats is attenuated by blockade of the rennin–angiotensin system or p38 MAP kinase. Am J Physiol Renal Physiol. 2007;292:F313–20. doi: 10.1152/ajprenal.00180.2006. [DOI] [PubMed] [Google Scholar]
- 38.Kuehn EW, Park KM, Somlo S, Bonventre JV. Kidney injury molecule-1 expression in murine polycystic kidney disease. Am J Physiol Renal Physiol. 2002;283:F1326–36. doi: 10.1152/ajprenal.00166.2002. [DOI] [PubMed] [Google Scholar]
- 39.Kramer AB, van Timmeren MM, Schuurs TA, Vaidya VS, Bonventre JV, van Goor H, Navis G. Reduction of proteinuria in adriamycin-induced nephropathy is associated with reduction of renal Kidney injury molecule-1 (Kim-1) over time. Am J Physiol Renal Physiol. 2009;296:F1136–45. doi: 10.1152/ajprenal.00541.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Waanders F, Vaidya VS, van Goor H, Leuvenink H, Damman K, Hamming I, Bonventre JV, Vogt L, Navis G. Effect of renin–angiotensin–aldosterone system inhibition, dietary sodium restriction, and/or diuretics on urinary kidney injury molecule 1 excretion in nondiabetic proteinuric kidney disease: a post hoc analysis of a randomized controlled trial. Am J Kidney Dis. 2009;53:16–25. doi: 10.1053/j.ajkd.2008.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chow FY, Nikolic-Paterson DJ, Atkins RC, Tesch GH. Macrophages in streptozotocin-induced diabetic nephropathy: potential role in renal fibrosis. Nephrol Dial Transplant. 2004;19:2987–96. doi: 10.1093/ndt/gfh441. [DOI] [PubMed] [Google Scholar]
- 42.Lavaud S, Michel O, Sassy-Prigent C, Heudes D, Bazin R, Bariety J, Chevalier J. Early influx of glomerular macrophages precedes glomerulosclerosis in the obese Zucker rat model. J Am Soc Nephrol. 1996;7:2604–15. doi: 10.1681/ASN.V7122604. [DOI] [PubMed] [Google Scholar]
- 43.Lewis A, Steadman R, Manley P, Craig K, de la Motte C, Hascall V, Phillips AO. Diabetic nephropathy, inflammation, hyaluronan and interstitial fibrosis. Histol Histopathol. 2008;23:731–9. doi: 10.14670/HH-23.731. [DOI] [PubMed] [Google Scholar]
- 44.Yonemoto S, Machiguchi T, Nomura K, Minakata T, Nanno M, Yoshida H. Correlations of tissue macrophages and cytoskeletal protein expression with renal fibrosis in patients with diabetes mellitus. Clin Exp Nephrol. 2006;10:186–92. doi: 10.1007/s10157-006-0426-7. [DOI] [PubMed] [Google Scholar]
- 45.Young BA, Burdmann EA, Johnson RJ, Alpers CE, Giachelli CM, Eng E, Andoh T, Bennett WM, Couser WG. Cellular proliferation and macrophage influx precede interstitial fibrosis in cyclosporine nephrotoxicity. Kidney Int. 1995;48:439–48. doi: 10.1038/ki.1995.312. [DOI] [PubMed] [Google Scholar]
- 46.Ha H, Yu MR, Choi YJ, Kitamura M, Lee HB. Role of high glucose-induced nuclear factor-kappaB activation in monocyte chemoattractant protein-1 expression by mesangial cells. J Am Soc Nephrol. 2002;13:894–902. doi: 10.1681/ASN.V134894. [DOI] [PubMed] [Google Scholar]
- 47.Ihm CG, Park JK, Hong SP, Lee TW, Cho BS, Kim MJ, Cha DR, Ha H. A high glucose concentration stimulates the expression of monocyte chemotactic peptide 1 in human mesangial cells. Nephron. 1998;79:33–7. doi: 10.1159/000044988. [DOI] [PubMed] [Google Scholar]
- 48.Tesch GH. MCP-1/CCL2: a new diagnostic marker and therapeutic target for progressive renal injury in diabetic nephropathy. Am J Physiol Renal Physiol. 2008;294:F697–701. doi: 10.1152/ajprenal.00016.2008. [DOI] [PubMed] [Google Scholar]
- 49.Zhang Z, Yuan W, Sun L, Szeto FL, Wong KE, Li X, Kong J, Li YC. 1,25-Dihydroxyvitamin D3 targeting of NF-kappaB suppresses high glucose-induced MCP-1 expression in mesangial cells. Kidney Int. 2007;72:193–201. doi: 10.1038/sj.ki.5002296. [DOI] [PubMed] [Google Scholar]
- 50.Chow FY, Nikolic-Paterson DJ, Ozols E, Atkins RC, Rollin BJ, Tesch GH. Monocyte chemoattractant protein-1 promotes the development of diabetic renal injury in streptozotocin-treated mice. Kidney Int. 2006;69:73–80. doi: 10.1038/sj.ki.5000014. [DOI] [PubMed] [Google Scholar]
- 51.Lin H, Hou CC, Cheng CF, Chiu TH, Hsu YH, Sue YM, Chen TH, Hou HH, Chao YC, Cheng TH, Chen CH. Peroxisomal proliferator-activated receptor-alpha protects renal tubular cells from doxorubicin-induced apoptosis. Mol Pharmacol. 2007;72:1238–45. doi: 10.1124/mol.107.037523. [DOI] [PubMed] [Google Scholar]
- 52.Smeets PJ, Teunissen BE, Planavila A, de Vogel-van den Bosch H, Willemsen PH, van der Vusse GJ, van Bilsen M. Inflammatory pathways are activated during cardiomyocyte hypertrophy and attenuated by peroxisome proliferator-activated receptors PPARalpha and PPARdelta. J Biol Chem. 2008;283:29109–18. doi: 10.1074/jbc.M802143200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.ten Dijke P, Hill CS. New insights into TGF-beta-Smad signalling. Trends Biochem Sci. 2004;29:265–73. doi: 10.1016/j.tibs.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 54.Inazaki K, Kanamaru Y, Kojima Y, Sueyoshi N, Okumura K, Kaneko K, Yamashiro Y, Ogawa H, Nakao A. Smad3 deficiency attenuates renal fibrosis, inflammation, and apoptosis after unilateral ureteral obstruction. Kidney Int. 2004;66:597–604. doi: 10.1111/j.1523-1755.2004.00779.x. [DOI] [PubMed] [Google Scholar]
- 55.Lopez-Rovira T, Chalaux E, Rosa JL, Bartrons R, Ventura F. Interaction and functional cooperation of NF-kappa B with Smads. Transcriptional regulation of the junB promoter. J Biol Chem. 2000;275:28937–46. doi: 10.1074/jbc.M909923199. [DOI] [PubMed] [Google Scholar]
- 56.Jeong S, Yoon M. Fenofibrate inhibits adipocyte hypertrophy and insulin resistance by activating adipose PPARalpha in high fat diet-induced obese mice. Exp Mol Med. 2009;41:397–405. doi: 10.3858/emm.2009.41.6.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen X, Matthews J, Zhou L, Pelton P, Liang Y, Xu J, Yang M, Cryan E, Rybczynski P, Demarest K. Improvement of dyslipidemia, insulin sensitivity, and energy balance by a peroxisome proliferator-activated receptor alpha agonist. Metabolism. 2008;57:1516–25. doi: 10.1016/j.metabol.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 58.Idzior-Walus B, Sieradzki J, Rostworowski W, Zdzienicka A, Kawalec E, Wojcik J, Zarnecki A, Blane G. Effects of comicronised fenofibrate on lipid and insulin sensitivity in patients with polymetabolic syndrome X. Eur J Clin Invest. 2000;30:871–8. doi: 10.1046/j.1365-2362.2000.00734.x. [DOI] [PubMed] [Google Scholar]