

Abstract
Membrane inlet mass spectrometry (MIMS) has been employed to assay the catalytic activity of oxalate decarboxylase (OxDC), allowing us to demonstrate that nitric oxide (NO) reversibly inhibits the enzyme under dioxygen-depleted conditions. X-band EPR measurements do not provide any direct evidence for the interaction of NO with either of the Mn(II) centers in OxDC raising the possibility that there is a separate dioxygen-binding pocket in the enzyme.
Oxalate decarboxylase catalyzes the conversion of oxalate into carbon dioxide and formate in a reaction that remains incompletely understood.1 It is presently believed that decarboxylation proceeds from an oxalate radical anion intermediate, which is generated in a proton-coupled electron transfer step mediated by the Mn(II) ion.2 In addition, the reported dependence of the enzyme-catalyzed chemistry on the presence of dioxygen3 has led to the proposal that formation of the oxalate radical anion takes place viaa Michaelis complex in which both oxalate and dioxygen are bound to the catalytically active manganese center.4–6 Direct support for this hypothesis has yet to be obtained, however, and there is an absence of unambiguous chemical precedent for the interaction of dioxygen with high-spin Mn(II) inorganic complexes. Efforts to observe the proposed interaction of dioxygen with the metal in OxDC have also been complicated by the fact that the two Mn(II) centers in the enzyme exhibit similar EPR properties.7,8
In an effort to assess the existence of the putative dioxygen binding site in OxDC, we examined the interaction of nitric oxide (NO) with the enzyme using a new continuous assay based on membrane inlet mass spectrometry (MIMS).9 Thus, wild type Bacillus subtilis OxDC bearing a C-terminal His6 tag was expressed and purified using standard procedures.3,5 Addition of the enzyme (1.4 μM) to a solution of 50 mM 13C2-oxalate in 50 mM acetate buffer, pH 4.2, yielded13CO2 (m/z = 45), the amount of which could be monitored by MIMS (data not shown). An assay solution containing 50 mM 13C2-oxalate in 50 mM acetate buffer, pH 4.2, was degassed with He to deplete dioxygen. Air in the head space above the reaction mixture was then removed with He for 2 min prior to the addition of 25 μM MAHMA NONOate10(see supporting information).‡ Under the acidic conditions, this reagent rapidly decomposed to yield 50 μM NO (Figure 1). OxDC (1.4 μM) was then added to the solution at 4 min to initiate the decarboxylation reaction. The presence of NO decreased the initial activity of the enzyme by approximately two orders of magnitude, based on the reduction in observed ion current for 13CO2 (m/z = 45) (Figure 1). Under these conditions, the ion associated with NO (m/z = 30) was also detected. Even in the presence of NO, the enzyme was capable of catalyzing the formation of a small amount of 13CO2 immediately on addition, which likely results from the presence of bound dioxygen on the enzyme added to the reaction mixture. The time-dependent decrease in the ion current for both these species over the next 8 min was due, at least in part, to the loss of CO2 and NO from the reaction solution through the membrane inlet into the mass spectrometer. At 12 min, air was re-introduced, resulting in the restoration of full OxDC catalytic activity, albeit after a time lag of approximately 5 min (Figure 1). Upon introduction of air by bubbling, some CO2 was lost from solution, giving rise to a decrease in the ion current of the m/z 45 peak. Varying the initial amount of MAHMA NONOate under identical experimental conditions established that the extent of OxDC inhibition was dependent on NO concentration, consistent with an NO binding site on the enzyme (Figure 2). A preliminary estimate of the apparent KI for NO is approximately 40 μM, which is of a similar magnitude to the Km for O2 of 28 ± 8 μM (measured from the dependence of decarboxylase activity on dioxygen concentration).11 In order to test the alternate hypothesis that decreased OxDC activity resulted merely from dioxygen depletion due to reaction of the latter gas with NO under the acidic conditions,12 we measured the rate of CO2 formation at similarly low dioxygen levels in the absence of MAHMA NONOate. Under these conditions the intrinsic OxDC decarboxylase activity was higher than that seen when MAHMA NONOate was present, ruling out this possibility (see supporting information). Similarly, control assays using an alternate NO-releasing reagent (diethylammonium (Z)-1-(N,N-diethylamino)diazen-1-ium-1,2-diolate) suggested that the by-product formed after NO release from MAHMA NONOate was not responsible for the observed inhibition (see supporting information).
Fig. 1. Effect of NO on catalysis by C-terminally His6-tagged OxDC.

Time course of changes in ion currents (arbitrary units), determined using MIMS for 13CO2 (red), NO (blue) and O2 (green). Full experimental details are provided as supporting information. (Inset) The same set of data plotted on a linear axis to emphasize the magnitude of the ion currents for 13CO2(red) and NO (blue).
Fig. 2. Dependence of OxDC inhibition on initial MAHMA NONOate concentration.

Progress curves for the generation of 12CO2 under anaerobic conditions, as measured by ion current in the MIMS procedure (arbitrary units), in the presence of MAHMA NONOate at an initial concentration of 0 μM (black), 13 μM (green), 25 μM (blue) and 100 μM (red). The C-terminally His6-tagged OxDC was used at a concentration of 1.4 μM. Other reaction conditions were those used to generate the data shown in Figure 1. (Inset) An expanded plot of the data generated during the first minute of reaction.
Given that NO-dependent OxDC inhibition could be reversed by the re-introduction of dioxygen, we postulated that NO inhibited decarboxylase activity by binding to the Mn(II) center, possibly at a coordination site occupied by dioxygen during catalytic turnover,4 or by nitrosylating the side chain of Cys-383 in the protein,13 which is located at the C-terminus distant from the putative catalytic site.5,14 The latter proposal was ruled out, however, by the fact that the C383S OxDC mutant exhibited identical catalytic behavior to the wild type enzyme, including reversible inhibition by NO under the conditions of our earlier experiments (see supporting information). We next undertook a series of EPR measurements on frozen solutions of OxDC containing NO with the expectation that NO might bind to one, or both, of the high-spin Mn(II) centers present in OxDC.7 As a result, we anticipated that the intensity observed for the EPR signal(s) associated with the Mn(II) centers would decrease because the interaction of NO with the enzyme would yield a Mn(III) species possessing an integer spin, which would be “silent” in our EPR measurements. Briefly, wild type OxDC (0.12 mM) was dissolved in 110 mM KOAc buffer, pH 4.1, which had been carefully degassed by sparging with argon(120 μL total volume). After freezing in liquid isopentane pre-cooled in liquid N2, CW-EPR spectra were taken at 4.2 K (Figure 3). The initial degassed OxDC sample showed the usual X-band spectrum for the bound Mn(II) ions,5 i.e. six relatively sharp lines exhibiting the average hyperfine coupling constant of 92 G typical of hexa-coordinated manganese.15 In addition, the spectrum shows a shoulder at 3050 G and a broad maximum centered at 2340 G. Using multi-frequency EPR measurements, we have interpreted this spectrum as arising from two different Mn(II) sites with fine structure parameters of |D| = 1200 MHz and 2150 MHz.7 Recent work has suggested that the X-band EPR spectrum of the enzyme is likely sensitive only to the Mn(II) site possessing the smallest fine structure constant D (see simulations in supporting information). This sample was then mixed with 0.5 mM MAHMA NONOate (full details are provided in supporting information) and we examined whether there was a change in the intensity of all, or parts of, the Mn(II) signal when OxDC was exposed to NO. Perhaps unexpectedly, the Mn(II) signal in the EPR spectrum of OxDC under these conditions was of approximately the same intensity as that observed in the absence of NO (Figure 3). Although there was some background signal from MAHMA NONOate (presumably arising from free NO), subtraction of the relevant spectra showed a reduction in Mn(II) signal intensity of only 10–20% in conditions under which NO completely inhibited decarboxylase activity (Figure 3). Such a decrease is expected to be associated solely with OxDC sample dilution resulting from addition of the MAHMA NONOate solution. A sample of OxDC that contained both MAHMA NONOate and oxalate also gave similar results when analyzed using our standard EPR procedures (Figure 3). These observations seem to indicate that NO does not exert its inhibitory effect on decarboxylase activity by displacing dioxygen from the metal center or by forming a heptacoordinate Mn(II) species, at least for the site that is visible in the X-band EPR spectrum. High-frequency EPR analysis of OxDC by other workers has also suggested that the Mn(II) site exhibiting the smaller fine structure value Disactually located in the N-terminal domain of the enzyme,8 this assignment being based on spectroscopic evidence for the presence of a pentacoordinate Mn(II) center in OxDC at high solution pH.5 If this interpretation is correct then our findings would seem to exclude NO binding at the solvent accessible catalytically active N-terminal site.14 These observations are also consistent with the remarkably small number of well characterized mononuclear {Mn-NO}6 complexes that have been reported.16,17 We also note that only circumstantial evidence for the formation of Mn(III) during catalytic turnover (a metal species that might bind NO) has been reported.18 On the other hand, it remains possible that any Mn(II)/NO interaction is masked by the complexity of the X-band EPR spectrum for OxDC.7,8
Fig. 3. Overlaid CW-EPR spectra of the Mn(II) centers in OxDC in the presence and absence of NO released from MAHMA NONOate.
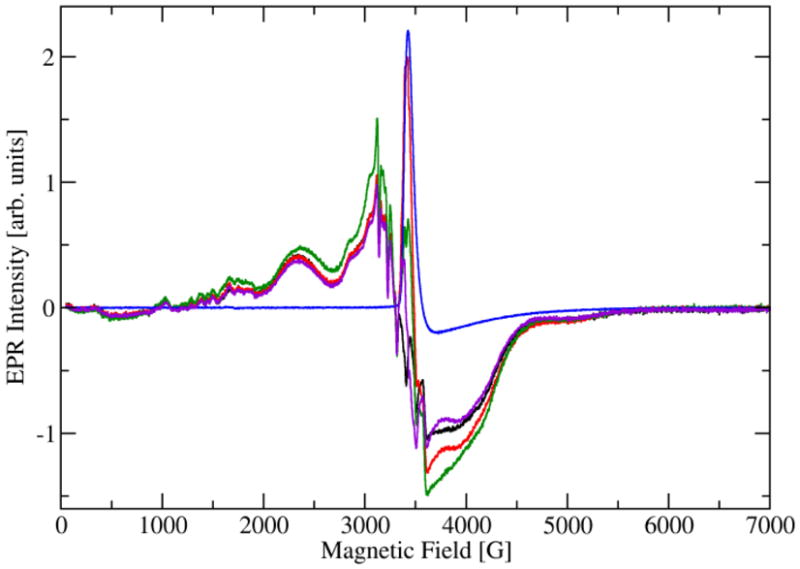
(A) Degassed (Ar) sample of WT OxDC in 110 mM acetate buffer, pH 4.1 (black). (B) Same sample as in (A) to which MAHMA NONOate was added (red). (C) Degassed (Ar) sample of WT OxDC in 110 mM acetate buffer, pH 4.1, containing MAHMA NONOate and oxalate (green). (D) Metal-free 110 mM acetate buffer, pH 4.1, containing released NO (blue), and the difference spectrum (purple) generated by subtraction of (D) from (A). All spectra were recorded at 4.2 K (full experimental details are provided as supporting information).
If NO is exerting its inhibitory effect without binding to either of the two Mn centers, other explanations need to be considered for the molecular interaction of NO with OxDC leading to inhibition. It seems unlikely that covalent modification of tyrosine residues (such as Tyr-200 in the putative catalytic site of OxDC) as observed for Mn-dependent superoxide dismutase,19 is responsible for OxDC inhibition because such changes would be expected to be irreversible. However, the Y200F OxDC mutant exhibited identical catalytic behavior to the wild type enzyme, including reversible inhibition by NO under our standard conditions (Moral & Tu, unpublished data). A third possibility is that NO reacts with intermediates formed during catalytic turnover, such as superoxide or the putative oxalate radical anion.2 Such reactions would, however, yield nitrite, nitrate and/or carboxylate anion products, which cannot be detected by MIMS, and CO2 that would appear as a reaction product. Moreover, any interruption of the catalytic cycle would be expected to form Mn(III) in the active site thereby reducing Mn(II) signal intensity in the EPR spectrum when both MAHMA NONOate and oxalate are present, which is not observed(sample (C) in Figure 3). Therefore, our spectroscopic findings argue against reaction of NO with catalytic intermediates in the absence of other data, although this possibility cannot be ruled out unequivocally on the basis of these X-band studies. Given that the effects of NO are reversed by the re-introduction of dioxygen into the assay mixture, our working hypothesis is that there is a specific dioxygen binding site in the protein that can be occupied by NO at low dioxygen concentrations. Locating this putative binding site is the subject of ongoing experiments.
Supplementary Material
Acknowledgments
Funding for this work was provided by the National Institutes of Health (DK061666 to N.G.J.R. and GM025154 to D.N.S.) and the National Science Foundation (CHE0809729 to A.A.). A plasmid containing the gene encoding C-terminally, His-tagged wild type Bacillus subtilis OxDC was very generously provided by Dr. Stephen Bornemann (John Innes Centre, Norwich, UK).
Footnotes
Electronic Supplementary Information (ESI) available: Full experimental details for the MIMS and EPR experiments together with procedures for enzyme expression and purification. Data are also included for control experiments and X-band EPR spectral simulations. See DOI: 10.1039/b000000x/
MAHMA NONOate: (Z)-1-(N-methyl-N-[6-(N-methylammonio hexyl)amino]diazen-1-ium-1,2-diolate.
Notes and references
- 1.Emiliani E, Bekes P. Arch Biochem Biophys. 1964;105:488. doi: 10.1016/0003-9861(64)90040-2. [DOI] [PubMed] [Google Scholar]; Tanner A, Bornemann S. J Bacteriol. 2000;182:5271. doi: 10.1128/jb.182.18.5271-5273.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]; Mehta A, Datta A. J Biol Chem. 1991;266:23548. [PubMed] [Google Scholar]
- 2.Reinhardt LA, Svedruzic D, Chang CH, Cleland WW, Richards NGJ. J Am Chem Soc. 2003;125:1244. doi: 10.1021/ja0286977. [DOI] [PubMed] [Google Scholar]
- 3.Tanner A, Bowater L, Farihurst SA, Bornemann S. J Biol Chem. 2001;276:43627. doi: 10.1074/jbc.M107202200. [DOI] [PubMed] [Google Scholar]
- 4.Chang CH, Richards NGJ. J Chem Theor Comput. 2005;1:994. doi: 10.1021/ct050063d. [DOI] [PubMed] [Google Scholar]
- 5.Just VJ, Stevenson CEM, Bowater L, Tanner A, Lawson DM, Bornemann S. J Biol Chem. 2004;279:19867. doi: 10.1074/jbc.M313820200. [DOI] [PubMed] [Google Scholar]
- 6.Moomaw EW, Angerhofer A, Moussatche P, Ozarowski A, Garcia-Rubio I, Richards NGJ. Biochemistry. 2009;48:6116. doi: 10.1021/bi801856k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Angerhofer A, Moomaw EW, Garcia-Rubio I, Ozarowski A, Krzystek J, Weber RT, Richards NGJ. J Phys Chem B. 2007;111:5043. doi: 10.1021/jp0715326. [DOI] [PubMed] [Google Scholar]
- 8.Tabares LC, Gatjens J, Hureau C, Burrell MR, Bowater L, Pecoraro VL, Bornemann S, Un S. J Phys Chem B. 2009;113:9016. doi: 10.1021/jp9021807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tu CK, Swensen ER, Silverman DN. Free Rad Biol Med. 2007;43:1453. doi: 10.1016/j.freeradbiomed.2007.07.026. [DOI] [PubMed] [Google Scholar]; Mikulski R, Tu CK, Swensen ER, Silverman DN. Free Rad Biol Med. 2010;48:325. doi: 10.1016/j.freeradbiomed.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davies KM, Wink DA, Saavedra JE, Keefer LK. J Am Chem Soc. 2001;123:5473. doi: 10.1021/ja002899q. [DOI] [PubMed] [Google Scholar]; Hrabie JA, Klose JR, Wink DA, Keefer LK. J Org Chem. 1993;58:1472. [Google Scholar]
- 11.Burrell MR, Just VJ, Bowater L, Fairhurst SA, Requena L, Lawson DM, Bornemann S. Biochemistry. 2007;46:12327–12336. doi: 10.1021/bi700947s. [DOI] [PubMed] [Google Scholar]
- 12.Koppenol WH. Free Rad Biol Med. 1998;25:385. doi: 10.1016/s0891-5849(98)00093-8. [DOI] [PubMed] [Google Scholar]
- 13.Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Nat Rev Mol Cell Biol. 2005;6:150. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- 14.Anand R, Dorrestein PC, Kinsland C, Begley TP, Ealick SE. Biochemistry. 2002;41:7659–7669. doi: 10.1021/bi0200965. [DOI] [PubMed] [Google Scholar]
- 15.Reed GH, Markham GD. In: Biological Magnetic Resonance. Berliner LJ, Reuben J, editors. Vol. 6. Plenum Press; New York: 1984. p. 73. [Google Scholar]
- 16.Hoffman-Luca CG, Eroy-Reveles AA, Alvarenga J, Mascharak PK. In org Chem. 2009;48:9104–9111. doi: 10.1021/ic900604j. [DOI] [PMC free article] [PubMed] [Google Scholar]; Eroy-Reveles AA, Leung Y, Beavers CM, Olmstead MM, Mascharak PK. J Am Chem Soc. 2008;130:4447. doi: 10.1021/ja710265j. [DOI] [PubMed] [Google Scholar]; Ford PC, Fernandez BO, Lim MD. Chem Rev. 2005;105:2439. doi: 10.1021/cr0307289. [DOI] [PubMed] [Google Scholar]
- 17.The notation used to describe these complexes is that of Feltham and Enemark. See: Enemark J, Feltham RD. Coord Chem Rev. 1974;13:339.
- 18.Chang CH, Svedruzic D, Ozarowski A, Walker L, Yeagle G, Britt RD, Angerhofer A, Richards NGJ. J Biol Chem. 2004;279:52840. doi: 10.1074/jbc.M402345200. [DOI] [PubMed] [Google Scholar]
- 19.Quint P, Reutzel R, Milkulski R, McKenna R, Silverman DN. Free Rad Biol Med. 2006;40:453. doi: 10.1016/j.freeradbiomed.2005.08.045. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.