

Abstract
I here describe a line of research that grew out of studies of spinal cord–damaging decompression sickness, focused on the blood–endothelial interface, that was influenced by the local Shwartzman phenomenon, addressed innate immune and inflammatory mechanisms, and ultimately arrived at mucosal tolerance approaches to prevent stroke. Intranasal instillation of E-selectin is under development as a novel means of targeting immunomodulation to activating blood vessels within the vascular tree supplying the brain. The goal of this form of focused immunomodulation is to prevent recurrent strokes in patients that have previously suffered transient ischemic attacks or strokes.
Keywords: tolerance, stroke, cytokines, immunomodulation, endothelium, E-selectin
I began my research career as a Berry Plan draftee at the Naval Medical Research Institute, National Naval Medical Center in Bethesda Maryland in 1971, after having completed a neurology residency at the University of Michigan. My entrance into stroke research was somewhat unconventional in that I moved from studies of spinal cord damage in decompression sickness to an abiding interest in the mechanisms that induce brain damage in strokes. A major mechanism underlying the predilection for spinal cord damage in central nervous system (CNS) decompression sickness turned out to be the accumulation of bubbles and activated blood products in the epidural vertebral venous system (Batson's Plexus), ultimately leading to venous obstruction, compromised transport of nitrogen out of the lipid-rich spinal cord, and local nucleation of gas bubbles in the cord.1,2 An important factor in the activation of blood and local blood vessels in the venous plexus was the 40–100 Å zone of electrokinetic forces at bubble–blood interfaces (Lee & Hairston) that activated the contact activation system including coagulation, fibrinolysis, complement, kinins, etc.3 Given this background, I viewed stroke from the start as a process in which blood and tissue responses to ischemic stress were likely to be extremely multifactorial and to include inflammatory and immune responses in addition to the direct effects of local energy failure and membrane failure. It seemed highly possible that the blood flowing through an ischemic injury zone could become activated by the damaged or stressed tissue resembling in a general way the activation of blood by bubbles or other foreign surfaces. Further examination of this possibility led to the concept of “blood-damaged tissue interaction,” a forerunner of reperfusion injury that leads to progressive impairment of microvascular perfusion.4,5
An intriguing model of focal blood vessel activation is the localized Shwartzman reaction that was first described by Gregory Shwartzman in 1928.6 There is a preparatory step in which endotoxin acting as a danger-associated molecular pattern (DAMP) is injected intradermally where it acts through toll-like receptors7 to release proinflammatory cytokines and locally activate blood vessels in the skin. This will lead to some local erythema that subsides in the absence of any further stimulation. If, however, there is a provocative step, a critically timed activation of the coagulation system by a small nontoxic dose of endotoxin administered intravenously 18–24 h after the preparatory step, areas of petechial hemorrhage appear in the prepared skin and these enlarge and coalesce into an area of hemorrhagic necrosis that approaches the size of a silver dollar.8 Our group wondered whether established risk factors for stroke could act locally to prepare blood vessels in the brain for a modified Shwartzman reaction in response to a provocative inflammatory stimulus such as intracisternally or intravenously injected endotoxin. Stroke risk factors such as hypertension, diabetes, advanced age, and genetic predisposition to stroke were found sufficient to prepare rat brainstem tissues such that a single intracisternal or intravenous injection of endotoxin provoked the reaction, and affected rats-manifested neurologic deficits accompanied by pathologic lesions (Fig. 1). Brain infarcts developed in only a small proportion of rats without recognized risk factors after endotoxin injection.9 This work suggested that one role of stroke risk factors is to prime vessel activation and endothelial dysfunction as occurs in the Shwartzman reaction, but that a second systemic activation of inflammation and coagulation was necessary to precipitate local thrombosis or hemorrhage and cause a stroke, namely, a “two-hit phenomenon.”
Figure 1.
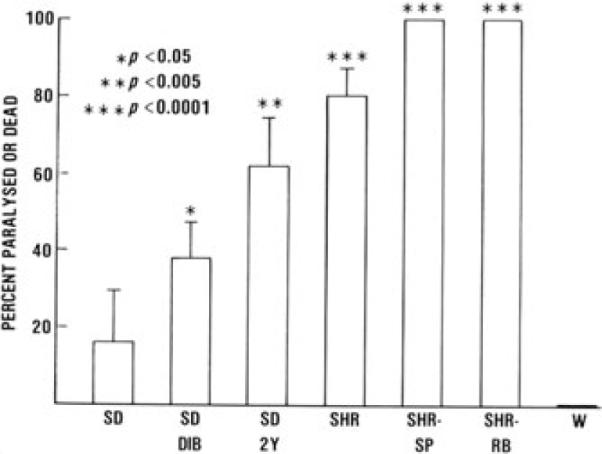
Responses to single 1.8 mg/kg intracisternal doses of endotoxin are compared from groups of rats with and without stroke risk factors. For each group, n is Sprague-Dawley rats (SD) 37,SD diabetic (SDDIB) 26,SD 24-months old (SD2Y) 13, spontaneously hypertensive rats (SHR) 26, stroke-prone spontaneously hypertensive rats (SHR-SP) 4, SHR retired breeder rats (SHR-RB) 4, and Wistar rats (W) 7.
We examined this possibility further with a series of studies that compared the numbers of perivascular macrophages in brain vessels and the endotoxin-induced levels of secreted proinflammatory cytokines from isolated carotid rings among animals with risk factors for stroke and those without such risk factors. The numbers of perivascular ED2-positive macrophages were increased in rats with the stroke risk factor, hypertension (e.g., spontaneously hypertensive rats [SHR] and stroke-prone spontaneously hypertensive rats [SHR-SP]) compared to normotensive controls (e.g., Wistar Kyoto [WKY] rats).10 In addition, in vitro carotid ring production of tumor necrosis factor (TNF)-α in response to a range of endotoxin doses was elevated in SHR versus WKY rats.11 We inferred from these studies that stroke risk factors acted to augment blood vessel activation within the cerebrovascular circulatory network.
Rosenberg and Aird conducted studies of the differences in the molecular mechanisms that provide the primary regulation of hemostasis in vascular beds of different organs and also noted that a variety of different systemic coagulation disorders such as protein C deficiency, heparin-binding site of antithrombin III mutation, factor V Leiden variant, and prothrombin G20210A mutation lead to organ-specific thrombosis rather than a diffuse thrombotic diathesis with disseminated intravascular coagulation. They concluded that hemostatic potential is not regulated by a synchronous, systemic mechanism but is instead controlled asynchronously segment by segment within the vascular tree of an organ.12–14 The local endothelium of a vascular segment was viewed as integrating signals that come from the blood, the blood vessel wall, and the surrounding parenchymal tissue; in response to this ongoing endothelial integration of signals that include cytokines, mechanical forces, circulating lipoproteins, coagulation factors, components of the extracellular matrix, and DAMPs, the properties of the luminal endothelium within a given segment can cycle from an antiinflammatory and anticoagulant phenotype at one extreme to a proinflammatory and procoagulant phenotype at the other extreme. Such cycling occurs innocuously in normal individuals, but if the hemostatic potential in a vascular segment rises above some homeostatic threshold as can occur in the presence of stroke risk factors, local thrombosis or vessel damage with hemorrhage becomes a threat.
Our studies of the patterns of immunoreactive TNF-α (proinflammatory, prothrombotic), heme oxygenase-1 (HO-1, oxidative stress marker), and manganese superoxide dismutase (MnSOD, antioxidant) around the brain vessels of rats with and without risk factors for stroke provide some support for the dynamic segmental control model of hemostasis described above.15 Within brain sections from rats without stroke risk factors, cross-sectioned vessels exhibited scattered coincident vascular and perivascular halos of immunoreactive TNF-α, HO-1, and MnSOD that appeared to periodically begin as thin rings outlining and highlighting a vessel then expanding centrifugally into the surrounding parenchyma before fading. Repeating cycles of the expanding immunoreactivity of these regulatory molecules were clearly vessel centric and were likely to have been responses to cyclic activation and inactivation of discrete vascular segments because the number and intensity of the perivascular rings was significantly increased in rats with stroke risk factors and in risk factor-free rats that received intravenous endotoxin (Fig. 2).
Figure 2.
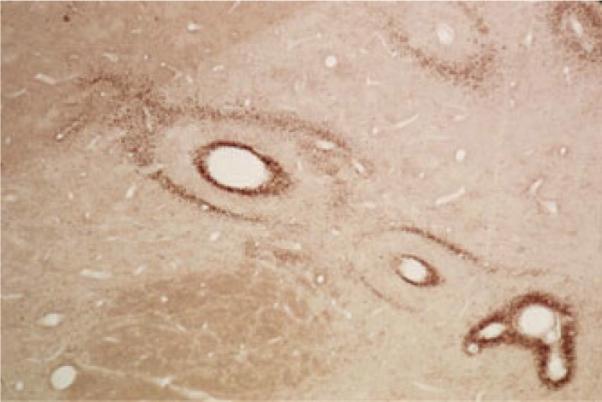
Patterns of manganese superoxide dismutase (MnSOD) expression around brain vessels of spontaneously hypertensive stroke-prone rats. These patterns were coincident with the expression patterns of immunoreactive TNF-α and hemoxygenase-1 in serial sections. The concentric rings of immunoractive MnSOD appear to expand radially from scattered vessel epicenters out into the brain parenchyma in a non-synchronous manner. The concentric circles suggest that these responses occur in waves of activation and deactivation of individual vascular segments.
Mediators of the anticoagulant/antiinflammatory phenotype of the luminal endothelium in a vascular segment can include prostacyclin, nitric oxide, heparin-like glycosaminoglycans, thrombomodulin, protein C, protein S, tissue-type plasminogen activator, tissue factor pathway inhibitor, hemoxygenase-1, MnSOD, and adenosine diphosphatase/5′-nucleotidase.16 In response to proinflammatory mediators like TNF-α, interleukin (IL)-1, and lipopolysaccharide (LPS), luminal endothelium can make a “Jekyll to Hyde” change in phenotype and express tissue factor, IL-1, TNF-α, coagulation factor binding/activation, adhesion receptors, plasminogen activator inhibitor-1, platelet activating factor, endothelin, chemokines, and von Willebrand factor.17
One could easily dichotomize the molecular processes that regulate the phenotype of the luminal endothelium into a subset that is potentially harmful, the procoagulant and proinflammatory mediators, and a subset that is potentially salutary, the anticoagulant and anti-inflammatory mediators. It turns out not to be that simple.18 For instance, TNF-α would seem to be an archetypal “bad guy” that induces both inflammation and cytotoxicity. This cytokine, however, can be harmful19–22 or beneficial23,24 or act as a preconditioning stimulus to induce stress tolerance25–28 depending on the circumstances. Indeed, an examination of TNF-α function within the conventional “ischemic cascade” can provide a semi-quantitative insight into the complexity and multifactorality of the regulatory network in cells. A widely accepted series of general pathobiological mechanisms that are set into motion by brain ischemia would include metabolic failure, membrane failure, excitotoxicity, apoptosis, oxidative/nitrative stress, pathological gene expression, neurovascular unit dysfunction, and inflammatory/immune responses.29 Cytokines constitute only one subset of the inflammatory/immune mediators and there are over 100 of these signaling molecules. Drilling down further, one of these cytokines is TNF-α, which reacts with two different receptors. A study mapping the protein interaction network that links TNF-α receptor activation with activation of only one of its transcription factor targets, nuclear factor κB (NFκB), showed that there are 221 protein–protein interactions that modulate NFκB activation.30 Once activated, NFκB can influence the expression of more than 150 genes.31 By going through the mental exercise of scaling this level of complexity back through other recognized stroke pathobiological mechanisms, one can gain a greater appreciation of the extraordinary complexity and multifactorality of the dynamic network that controls the cellular responses related to ischemia.
In the mid-1990s, Kyra Becker, an outstanding postdoctoral fellow with a strong interest in immunology, joined our group. She decided to address the stroke cytoprotection problem by augmenting endogenous immune regulatory ensembles that function broadly within the dynamic network rather than by identifying and targeting a single inflammatory or immune mechanism that would have a relatively circumscribed network function. Specifically, she proposed to apply to studies of brain ischemia the mucosal tolerance model32 that was known to modulate immune responses and had shown efficacy in experimental autoimmune encephalitis models. In the mucosal tolerance model, serial transmucosal administration of “low dose” antigen in the microgram range will either encounter gut-associated lymphoid tissue if delivered orally or will encounter nasal-associated lymphoid tissue if instilled intranasally. CD4+ lymphocytes can be primed by antigen presenting cells (APCs) to differentiate into regulatory T cells that will traffic through the body and secrete IL-10 (Tr1 cells) or TGFβ (Th3 cells) when the antigen to which they have been primed is again presented to them by APCs. If the priming antigen can be presented either locally or within draining lymph nodes (from which the regulatory T cells can return to the locus of inflammation), these regulatory T cells can release immunomodulatory cytokines and locally suppress inflammation and immune responses regardless of whether or not the presented antigen is actually causing the inflammation. This process is termed “bystander suppression.” A brain antigen, myelin basic protein (MBP) was fed by gavage to rats, and the animals were subsequently protected from transient middle cerebral artery occlusion-induced ischemic brain damage compared to rats fed a nonspecific, nonmammalian protein, ovalbumin. In addition, MBP sensitization in another animal group led to increased ischemic brain damage. Further studies that included demonstration of antigen-specific suppression of delayed-type hypersensitivity validated the presence of an antigen-specific state of immune tolerance.33 This work showed that endogenous immunomodulation mechanisms could be harnessed to change immune network dynamics in acute stroke in order to suppress brain cell damage.
A limiting factor in the application of this approach to acute stroke treatment is that the induction of mucosal tolerance by serial exposure of antigens to a mucosal surface such as the gut or the nasal mucosa takes several days to develop. We became aware in the late 1990s that Protein Design Laboratories had produced a recombinant human E-selectin polypeptide that included much of the extramembranous portion that leukocyte receptor. E-selectin expression is virtually confined to luminal endothelium and it is not constitutively expressed. It is only expressed in vessel segments that are becoming activated. We were struck (a) because local release of immune and inflammatory mediators contributes to local vessel activation, appropriately targeted local immunomodulation could counter that activation and (b) because T-lymphocytes are key players in the regulation of the immune system, induction of mucosal tolerance to E-selectin could potentially serve to target regulatory T cells to activating blood vessel segments, provide local immunomodulation, and reduce the risk of stroke in individuals with stroke risk factors.
Our initial study was in spontaneously hypertensive, stroke-prone rats (SHRSP/Izm) that had been kindly supplied by Y. Yamori from the Disease Model Cooperative Research Association to Maria Spatz in consideration of her many years as a mentor for Japanese visiting fellows at the NIH. After initial dose–response studies we instilled in the nonbooster group, 5 mg of recombinant human E-selectin (rhES) intranasally on an every other day schedule for five doses and then followed the animals until they developed strokes, other complications of severe hypertension such as heart failure or renal failure, or until completion of the 56-week study. We also gave repeated five-dose schedules of E-selectin intranasally at 21-day intervals to a booster group of animals and followed them as described above. Control animals received single or booster schedules of intranasal instillation of either phosphate buffered saline (PBS) or a nonmammalian protein, ovalbumin, and also had corresponding follow-ups. In comparison to the other groups, the animals that received booster instillations of rhES showed a massively reduced incidence of ischemic stroke and the absence of parenchymal hemorrhages34 (Fig. 3).
Figure 3.

Comparisons are shown of the average number of brain infarcts per animal (A), the average area of brain infarcts per animal (B), the average number of brain intraparenchymal hemorrhages per animal (C), and the average area of brain intraparenchymal hemorrhages per animal (D) in each of 4 experimental groups. The experimental groups received either a single schedule of five every other day 5 μg doses of intranasally instilled ovalbumen or E-selectin, or they received booster repetition of these dosage schedules every three weeks to maintain any mucosal tolerance that had been induced.
On the basis of this study, we began to further develop intranasal instillation of E-selectin for the secondary prevention of stroke. We reasoned that if a stroke-preventive agent that is already on board had some cytoprotective capacity, it would confer added value because the earlier a therapeutic can be administered after a stroke, the higher the likelihood of a beneficial response. We therefore performed permanent middle cerebral artery occlusion (MCAO) studies in SHR-SP rats and found that E-selectin tolerization strongly reduced infarct volumes compared to PBS controls.35 In addition, we replicated this finding by adoptive transfer of spleen cells from E-selectin tolerized donor animals into naive SHR-SP showing that E-selectin tolerization is a cell-mediated phenomenon.
We have also shown the efficacy of mucosal tolerization to E-selectin in reducing white matter damage and functional/behavioral deficits in models of vascular cognitive impairment,36 in augmenting adult neurogenesis and improving functional recovery after stroke,37 in suppressing subarachnoid hemorrhage-delayed vasospasm,38 and (in unpublished work) in improving neurological outcome in an experimental autoimmune encephalomyelitis model of multiple sclerosis, and in reducing of aortic plaque burden in a model of atherosclerosis in apolipoprotein E (ApoE)-null mice. These studies have involved different sets of investigators and have been conducted in several different labs.
In our current work on this project, we are carrying out the necessary bridging studies to show equivalence between homologous rat or murine E-selectin antigen in rodents and heterologous human E-selectin in rodents. We are also conducting safety and dose studies that examine both intended biological/immunomodulatory responses and potential toxicity/immunotoxicity consequences of extended periods of E-selectin nasal instillation. An additional goal is to validate techniques that can be applied in our future clinical trials to provide accessible biomarkers of functional activity and immunotoxicity. This work will lead into nonhuman primate studies that pose that the final hurdle before filing an IND and conducting a Phase I clinical trial.
We hope to immunomodulate the intricately interconnected ensembles of molecular mechanisms in the dynamic network that function in a stroke-prone individual and can lead to a stroke. We have chosen an approach that has relatively plurifunctional effects in the network and is consonant with a prescient quote from Oscar Ratnoff (who identified Factor XII Hageman factor) that influenced me greatly early in my career. In a comprehensive review that integrated hemostasis, fibrinolysis, immunity, and inflammation,39 he concluded his article with the insight that investigators interested in any given system (e.g., complement or kinins) tend to break the system out and study it in isolation, as if it operated in a discrete and disarticulated fashion. In the body, however, these systems are intricately interconnected and, in Ratnoff's vivid phrase, “form a seamless web.” This is both a beautiful and a useful concept for those interested in improving the prevention and treatment of stroke.
Footnotes
Conflicts of interest J.H. has a patent on the use of E-selectin for immunomodulating vessel activation and preventing stroke.
References
- 1.Hallenbeck JM. Cinephotomicrography of dog spinal vessels during cord-damaging decompression sickness. Neurology. 1976;26:190–199. doi: 10.1212/wnl.26.2.190. [DOI] [PubMed] [Google Scholar]
- 2.Hallenbeck JM, Bove AA, Elliott DH. Mechanisms underlying spinal cord damage in decompression sickness. Neurology. 1975;25:308–316. doi: 10.1212/wnl.25.4.308. [DOI] [PubMed] [Google Scholar]
- 3.Bennett PB, Elliott DH. The Physiology and Medicine of Diving and Compressed Air Work. 2nd ed. Baillière Tindall; London: 1975. p. 566. [Google Scholar]
- 4.Hallenbeck JM. Prevention of postischemic impairment of microvascular perfusion. Neurology. 1977;27:3–10. doi: 10.1212/wnl.27.1.3. [DOI] [PubMed] [Google Scholar]
- 5.Hallenbeck JM, Furlow TW., Jr. Prostaglandin I2 and indomethacin prevent impairment of post-ischemic brain reperfusion in the dog. Stroke. 1979;10:629–637. doi: 10.1161/01.str.10.6.629. [DOI] [PubMed] [Google Scholar]
- 6.Shwartzman G. Studies on Bacillus typhosus toxic substances-I. Phenomenon of local skin reactivity to B. typhosus culture filtrate. J. Exp. Med. 1928;48:247–268. doi: 10.1084/jem.48.2.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Motegi A, et al. An in vitro Shwartzman reaction-like response is augmented age-dependently in human peripheral blood mononuclear cells. J. Leukoc. Biol. 2006;79:463–472. doi: 10.1189/jlb.0705396. [DOI] [PubMed] [Google Scholar]
- 8.Thomas L. The Youngest Science : Notes of a Medicine-Watcher. In: Alfred P, editor. Sloan Foundation Series. Viking Press; New York: 1983. p. 270. [Google Scholar]
- 9.Hallenbeck JM, et al. Stroke risk factors prepare rat brainstem tissues for modified local Shwartzman reaction. Stroke. 1988;19:863–869. doi: 10.1161/01.str.19.7.863. [DOI] [PubMed] [Google Scholar]
- 10.Liu Y, et al. Quantitation of perivascular monocytes and macrophages around cerebral blood vessels of hypertensive and aged rats. J. Cereb. Blood Flow Metab. 1994;14:348–352. doi: 10.1038/jcbfm.1994.43. [DOI] [PubMed] [Google Scholar]
- 11.Liu Y, et al. Evidence for activation of endothelium and monocytes in hypertensive rats. Am. J. Physiol. 1996;270(6 Pt 2):H2125–H2131. doi: 10.1152/ajpheart.1996.270.6.H2125. [DOI] [PubMed] [Google Scholar]
- 12.Aird WC. Spatial and temporal dynamics of the endothelium. J. Thromb. Haemost. 2005;3:1392–1406. doi: 10.1111/j.1538-7836.2005.01328.x. [DOI] [PubMed] [Google Scholar]
- 13.Edelberg JM, Christie PD, Rosenberg RD. Regulation of vascular bed-specific prothrombotic potential. Circ. Res. 2001;89:117–124. doi: 10.1161/hh1401.093954. [DOI] [PubMed] [Google Scholar]
- 14.Rosenberg RD, Aird WC. Vascular-bed-specific hemostasis and hypercoagulable states. N. Engl. J. Med. 1999;340:1555–1564. doi: 10.1056/NEJM199905203402007. [DOI] [PubMed] [Google Scholar]
- 15.Ruetzler CA, et al. Brain vessels normally undergo cyclic activation and inactivation: evidence from tumor necrosis factor-alpha, heme oxygenase-1, and manganese superoxide dismutase immunostaining of vessels and perivascular brain cells. J. Cereb. Blood Flow Metab. 2001;21:244–252. doi: 10.1097/00004647-200103000-00008. [DOI] [PubMed] [Google Scholar]
- 16.Hallenbeck JM. Inflammatory reactions at the blood–endothelial interface in acute stroke. Adv. Neurol. 1996;71:281–297. discussion 297–300. [PubMed] [Google Scholar]
- 17.Goldberg RB. Cytokine and cytokine-like inflammation markers, endothelial dysfunction, and imbalanced coagulation in development of diabetes and its complications. J. Clin. Endocrinol. Metab. 2009;94:3171–3182. doi: 10.1210/jc.2008-2534. [DOI] [PubMed] [Google Scholar]
- 18.Hallenbeck JM. The many faces of tumor necrosis factor in stroke. Nat. Med. 2002;8:1363–1368. doi: 10.1038/nm1202-1363. [DOI] [PubMed] [Google Scholar]
- 19.Barone FC, et al. Tumor necrosis factor-alpha. A mediator of focal ischemic brain injury. Stroke. 1997;28:1233–1244. doi: 10.1161/01.str.28.6.1233. [DOI] [PubMed] [Google Scholar]
- 20.Dawson DA, Martin D, Hallenbeck JM. Inhibition of tumor necrosis factor-alpha reduces focal cerebral ischemic injury in the spontaneously hypertensive rat. Neurosci. Lett. 1996;218:41–44. doi: 10.1016/0304-3940(96)13116-5. [DOI] [PubMed] [Google Scholar]
- 21.Nawashiro H, Martin D, Hallenbeck JM. Inhibition of tumor necrosis factor and amelioration of brain infarction in mice. J. Cereb. Blood Flow Metab. 1997;17:229–232. doi: 10.1097/00004647-199702000-00013. [DOI] [PubMed] [Google Scholar]
- 22.Shohami E, et al. Inhibition of tumor-necrosis-factor-alpha (Tnf-Alpha) activity in rat -brain is associated with cerebroprotection after closed-head injury. J. Cereb. Blood Flow Metab. 1996;16:378–384. doi: 10.1097/00004647-199605000-00004. [DOI] [PubMed] [Google Scholar]
- 23.Bruce AJ, et al. Altered neuronal and microglial responses to excitotoxic and ischemic brain injury in mice lacking TNF receptors. Nat. Med. 1996;2:788–794. doi: 10.1038/nm0796-788. [DOI] [PubMed] [Google Scholar]
- 24.Cheng B, Christakos S, Mattson MP. Tumor necrosis factors protect neurons against metabolic-excitotoxic insults and promote maintenance of calcium homeostasis. Neuron. 1994;12:139–153. doi: 10.1016/0896-6273(94)90159-7. [DOI] [PubMed] [Google Scholar]
- 25.Eddy LJ, Goeddel DV, Wong GHW. Tumor necrosis factor-α pretreatment is protective in a rat model of myocardial ischemia-reperfusion injury. Biochem. Biophys. Res. Comm. 1992;184:1056–1059. doi: 10.1016/0006-291x(92)90698-k. [DOI] [PubMed] [Google Scholar]
- 26.Liu J, et al. Hypoxic preconditioning protects cultured neurons against hypoxic stress via TNF-alpha and ceramide. Am. J. Physiol. Cell Physiol. 2000;278:C144–C153. doi: 10.1152/ajpcell.2000.278.1.C144. [DOI] [PubMed] [Google Scholar]
- 27.Nawashiro H, et al. TNF-alpha pretreatment induces protective effects against focal cerebral ischemia in mice. J. Cereb. Blood Flow Metab. 1997;17:483–490. doi: 10.1097/00004647-199705000-00001. [DOI] [PubMed] [Google Scholar]
- 28.Tasaki K, et al. Lipopolysaccharide pre-treatment induces resistance against subsequent focal cerebral ischemic damage in spontaneously hypertensive rats. Brain Res. 1997;748:267–270. doi: 10.1016/s0006-8993(96)01383-2. [DOI] [PubMed] [Google Scholar]
- 29.Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22:391–397. doi: 10.1016/s0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- 30.Bouwmeester T, et al. A physical and functional map of the human TNF-alpha/NF-kappa B signal transduction pathway. Nat. Cell Biol. 2004;6:97–105. doi: 10.1038/ncb1086. [DOI] [PubMed] [Google Scholar]
- 31.Pahl HL. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene. 1999;18:6853–6866. doi: 10.1038/sj.onc.1203239. [DOI] [PubMed] [Google Scholar]
- 32.Faria AM, Weiner HL. Oral tolerance: mechanisms and therapeutic applications. Adv. Immunol. 1999;73:153–264. doi: 10.1016/s0065-2776(08)60787-7. [DOI] [PubMed] [Google Scholar]
- 33.Becker KJ, et al. Immunologic tolerance to myelin basic protein decreases stroke size after transient focal cerebral ischemia. Proc. Natl. Acad. Sci. USA. 1997;94:10873–10878. doi: 10.1073/pnas.94.20.10873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Takeda H, et al. Induction of mucosal tolerance to E-selectin prevents ischemic and hemorrhagic stroke in spontaneously hypertensive genetically stroke-prone rats. Stroke. 2002;33:2156–2163. doi: 10.1161/01.str.0000029821.82531.8b. [DOI] [PubMed] [Google Scholar]
- 35.Chen Y, et al. Mucosal tolerance to E-selectin provides cell-mediated protection against ischemic brain injury. Proc. Natl. Acad. Sci. USA. 2003;100:15107–15112. doi: 10.1073/pnas.2436538100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wakita H, et al. Mucosal tolerization to E-selectin protects against memory dysfunction and white matter damage in a vascular cognitive impairment model. J. Cereb. Blood Flow Metab. 2008;28:341–353. doi: 10.1038/sj.jcbfm.9600528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ishibashi S, et al. Mucosal tolerance to E-selectin promotes the survival of newly generated neuroblasts via regulatory T-cell induction after stroke in spontaneously hypertensive rats. J. Cereb. Blood Flow Metab. 2009;29:606–620. doi: 10.1038/jcbfm.2008.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nakayama T, et al. Intranasal administration of E-selectin to induce immunological tolerization can suppress subarachnoid hemorrhage-induced vasospasm implicating immune and inflammatory mechanisms in its genesis. Brain Res. 2007;1132:177–184. doi: 10.1016/j.brainres.2006.09.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ratnoff OD. Some relationships among hemostasis, fibrinolytic phenomena, immunity, and the inflammatory response. Adv. Immunol. 1969;10:145–227. doi: 10.1016/s0065-2776(08)60417-4. [DOI] [PubMed] [Google Scholar]