

Abstract
Products resulting from oxidation of cell membrane phospholipid 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphorylcholine (OxPAPC) exhibit potent protective effects against lung endothelial cell (EC) barrier dysfunction caused by pathologically relevant mechanical forces and inflammatory agents. These effects were linked to enhancement of peripheral cytoskeleton and cell adhesion interactions mediated by small GTPase Rac and inhibition of Rho-mediated barrier disruptive signaling. However, the mechanism of OxPAPC-induced, Rac-dependent Rho downregulation critical for vascular barrier protection remains unclear. This study tested the hypothesis that Rho negative regulator p190RhoGAP is essential for OxPAPC-induced lung barrier protection against ventilator induced lung injury (VILI), and investigated potential mechanism of p190RhoGAP targeting to adherens junctions (AJ) via p120-catenin. OxPAPC induced peripheral translocation of p190RhoGAP, which was abolished by knockdown of Rac-specific guanine nucleotide exchange factors Tiam1 and Vav2. OxPAPC also induced Rac-dependent tyrosine phosphorylation and association of p190RhoGAP with AJ protein p120-catenin. siRNA-induced knockdown of p190RhoGAP attenuated protective effects of OxPAPC against EC barrier compromise induced by thrombin and pathologically relevant cyclic stretch (18% CS). In vivo, p190RhoGAP knockdown significantly attenuated protective effects of OxPAPC against ventilator-induced lung vascular leak, as detected by increased cell count and protein content in the bronchoalveolar lavage fluid, and tissue neutrophil accumulation in the lung. These results demonstrate for the first time a key role of p190RhoGAP for the vascular endothelial barrier protection in VILI.
Keywords: vascular permeability, lung endothelium, cytoskeleton, p120-catenin, p190RhoGAP, thrombin, cyclic stretch, VILI
INTRODUCTION
Oxidized phospholipids (OxPL) appear in the pulmonary circulation as a result of increased oxidative stress that accompanies acute lung injury, lung inflammation, adult respiratory distress syndrome (ARDS), ventilator-induced lung injury (VILI), systemic inflammatory response syndrome (SIRS) and sepsis [1–3]. Under these conditions, OxPL are mainly represented by lysophospholipids and terminal products of PL oxidation [4], which further contribute to lung vascular barrier dysfunction and inflammation.
However, controlled oxidation of phospholipids such as 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphatidyl choline (OxPAPC) and 1-palmitoyl-2-arachidonoyl-sn-glycero-3- phosphatidyl serine (OxPAPS) in vitro generates a group of bioactive oxidized phospholipid species with potent inhibitory effects on inflammatory signaling and lung injury induced by bacterial lipopolysacharide (LPS) and viral related 2′-deoxyribo(cytidine-phosphate-guanosine) (CpG) DNA [5–7]. In addition, OxPAPC and OxPAPS exhibit direct barrier-protective effects on the pulmonary endothelial cells (EC) and protect against lung vascular hyperpermeability in animal models of VILI [7–13]. These effects of OxPAPC had been linked to the activation of small GTPases Rac and Cdc42, which mediated enhancement of peripheral actin cytoskeleton and increased interactions between cellular adhesive structures essential for EC barrier protective response [8, 11, 12]. It was also reported that barrier-protective effects of OxPAPC against agonist-induced EC barrier dysfunction caused by bioactive molecules and VILI-relevant pathologic cyclic stress are associated with Rac-mediated downregulation of Rho-dependent signaling [10, 14].
Rac and Rho GTPases act as a molecular switch, cycling between the active GTP-bound and the inactive GDP-bound state which is regulated by guanine nucleotide exchange factors (GEFs) facilitating exchange of GDP for GTP, GTPase-activating proteins (GAPs), which increase the intrinsic rate of GTP hydrolysis by Rho GTPases, and by guanine nucleotide dissociation inhibitors (RhoGDI) which associate with inactivated Rho and Rac [15–17]. Rac and Rho play important roles in the regulation of cytoskeletal remodeling, cell-cell adhesive properties, and EC permeability control by mechanical forces and bioactive molecules [8, 18–23]. Rho and Rho-associated kinase may directly catalyze myosin light chain (MLC) phosphorylation or act indirectly via inactivation of MLC phosphatase [24, 25] and cause actomyosin-driven cell contraction and EC barrier dysfunction. In turn, EC barrier enhancement is associated with Rac-mediated formation of a peripheral F-actin rim, enlargement of intercellular adherens junctions, and formation of adherens junction-associated signaling protein complexes [8, 26, 27]. Thus, molecular mechanisms which precisely control the balance between Rho- and Rac siginaling are essential for EC barrier regulation in physiologic and pathologic conditions. Recent studies have suggested a mechanism of Rac-mediated downregulation of Rho pathway in NIH3T3 fibroblasts via Rac-dependent stimulation of negative regulator of Rho signaling, Rho-specific GTPase-activating protein p190RhoGAP [28]. P190RhoGAP becomes activated upon integrin activation, interaction with p120-catenin in fibroblasts [28, 29] or recruitment to the lipid rafts in endothelial cells [30], and may play important role in regulation of cell motility [31, 32] and endothelial barrier regulation [33–35]. However, effects of OxPAPC on p190RhoGAP – p120-catenin interactions, and the role of p190RhoGAP in the mechanisms of Rac-Rho crosstalk and OxPAPC-induced vascular endothelial barrier protection in VILI models have not been yet explored.
This study tested hypothesis that p190RhoGAP may mediate protective effects of OxPAPC against vascular endothelial barrier dysfunction associated with VILI. We characterized effects of OxPAPC on p190RhoGAP tyrosine phosphorylation and association with p120-catenin pulmonary endothelium. Using cell culture models of EC barrier compromise, induced by thrombin and pathological cyclic stretch in vitro, and in vivo two-hit model of VILI we examined involvement of p190RhoGAP in the OxPAPC-mediated inhibition of Rho signaling and preservation of endothelial barrier.
MATERIALS AND METHODS
Reagents and cell culture
Phospho-tyrosine and phospho-MYPT1 antibodies was obtained from Upstate Biotechnology (Lake Placid, NY); di-phospho-MLC, HRP-linked anti-mouse and anti-rabbit IgG were obtained from Cell Signaling (Beverly, MA); antibodies to p190RhoGAP, Rho kinase, and p120 catenin were purchased from BD Transduction Laboratories (San Diego, CA). TRAP6 was obtained from AnaSpec (San Jose, CA). Non oxidized 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphorylcholine (PAPC) was obtained from Avanti Polar Lipids (Alabaster, AL). PAPC was oxidized by exposure to air for 72 hours. The extent of oxidation was measured by positive ion electrospray mass spectrometry (ESI-MS) as previously described [36]. After completion of oxidation, the phospholipids were stored at −70°C dissolved in chloroform and used within 2 weeks after mass spectrometry testing. All oxidized and non-oxidized phospholipid preparations were analyzed by the limulus amebocyte assay (BioWhittaker, Frederick, MD) and shown negative for endotoxin. All reagents for immunofluorescence were purchased from Molecular Probes (Eugene, OR). Human lung pulmonary artery endothelial cells (HPAEC) were obtained from Lonza (Walkersville, MD), cultured according to the manufacturer’s protocol, and used at passages 5–7. Unless specified, biochemical reagents were obtained from Sigma (St. Louis, MO).
Knockdown of endogenous Tiam1, Vav2, Rac and p190RhoGAP
To reduce the content of endogenous Tiam1, Vav2, Rac, or p190RhoGAP, cells were treated with gene-specific siRNA duplexes. Pre-designed Stealth™ siRNAs of standard purity were ordered from Invitrogen (Carlsbad, CA). Transfection of EC with siRNA was performed as described previously [8, 37]. After 48 or 72 hrs cells were harvested and used for experiments. For in vivo experiments, polymer-based administration of non-specific or specific siRNA conjugated with polycation polyethilenimine (PEI-22) shown to promote lung-specific DNA and siRNA delivery [38, 39] was used to deplete p190RhoGAP in the in vivo experiments. Liposome-siRNA polyplexes were formed at ratio 1:10 (1 μg siRNA per 10 μg lipid). The most significant target gene inhibition was achieved at siRNA dose of 4 mg/kg after 72 hrs of transfection, as determined by western blot analysis. Treated mice showed no signs of non-specific siRNA-induced inflammation. Nonspecific, non-targeting siRNA (Dharmacon, Lafayette, CO) was used as a control treatment for both in vitro and in vivo experiments.
Measurement of transendothelial electrical resistance
The cellular barrier properties were analyzed by measurements of transendothelial electrical resistance (TER) across confluent endothelial cell monolayer using the electrical cell-substrate impedance sensing system (Applied Biophysics, Troy, NY).
Immunofluorescence staining
Endothelial cells were grown to confluence, stimulated with agonist of interest, and immunofluorescence staining for F-actin was performed as described elsewhere [20]. Likewise, after 72 hours of transfection with si-p190RhoGAP, static or cyclic stretch-preconditioned EC were stimulated with thrombin with or without OxPAPC pretreatment followed by immunofluorescence staining for F-actin using Texas Red-conjugated phalloidin. Images were processed with Adobe Photoshop 7.0 (Adobe Systems, San Jose, CA) software.
Differential protein fractionation
Sub-cellular protein fractionation was performed as described elsewhere [11]. Confluent HPAEC were stimulated with OxPAPC, then washed with ice cold PBS. The cytosolic fraction was isolated by centrifugation using extraction buffer containing 50 mM Tris-HCl pH 7.4, 100 mM sodium chloride, 0.01% digitonin, protease and phosphatase inhibitors cocktail. Next, pellets were resuspended in extraction buffer containing 50 mM Tris-HCl pH 7.4, 2% Triton X-100, 100 mM sodium chloride, protease and phosphatase inhibitors cocktail and incubated on ice for 30 min. The membrane fraction also containing components of adherens junction complexes and residual components of cortical actin cytoskeleton was separated from insoluble main cytoskeletal fraction by centrifugation 5 min at 16,000 g.
Co-immunoprecipitation and immunoblotting
Co-immunoprecipitation studies were performed using confluent HPAEC as described previously [40, 41]. Protein extracts were subjected to SDS-polyacrylamide gel electrophoresis, transferred to nitrocellulose membrane, and probed with antibodies of interest, as previously described [19, 40, 41].
Cell culture under cyclic stretch
Cyclic stretch (CS) experiments were performed using FX-4000T Flexcell Tension Plus system (Flexcell International, McKeesport, PA) equipped with 25 mm BioFlex Loading station, as previously described [41, 42]. In brief, after siRNA transfection cells were exposed to high magnitude cyclic stretch (18% distension, sinusoidal wave, 25 cycles/min) to recapitulate the mechanical stresses experienced by the alveolar endothelium at high tidal volume mechanical ventilation [42–44]. At two hours, plates were pretreated with vehicle or OxPAPC followed by thrombin treatment with continuous exposure to cyclic stretch. Control BioFlex plates with static EC culture were placed in the same cell culture incubator and processed similarly to CS-preconditioned cells. At the end of experiment, cell lysates were collected for western blot analysis, or CS-exposed endothelial monolayers were fixed with 3.7% formaldehyde and subjected to immunofluorescence staining as previously described [8, 37, 45].
Mechanical ventilation protocol
All experimental protocols involving the use of animals were approved by the University of Chicago Institutional Animal Care & Use Committee for the humane treatment of experimental animals. The animals were housed in pathogen-free conditions in the University of Chicago Animal Care Facilities where they were cared for in accordance with institutional and National Institutes of Health (NIH) guidelines. Adult male C57BL/6J mice, 8–10-week-old, with average weight 20–25 g (Jackson Laboratories, Bar Harbor, ME) were anesthetized with an intraperitoneal injection of ketamine (75 mg/kg) and acepromazine (1.5 mg/kg). Tracheotomy was performed and the trachea was cannulated with a 20-gauge one inch catheter (Penn-Century, Philadelphia, PA), which was tied into place to prevent air leak. The animals were placed on mechanical ventilator (Harvard Apparatus, Boston, MA). Mice were randomized to concurrently receive sterile saline solution or OxPAPC (1.5 mg/kg, intravenous administration) prior to a single dose of TRAP6 (1.5 × 10−5 mol/kg, intratracheal instillation) followed by 4 hours of mechanical ventilation with high tidal volume (30 ml/kg, 75 breaths per minute and 0 PEEP, HTV). Control animals were anesthetized and allowed to breathe spontaneously. After the experiment, BAL was performed using 1 ml of sterile Hanks Balanced Saline Buffer. The BAL protein concentration was determined by BCATM Protein Assay kit (Thermo Scientific, Pittsburg, PA). BAL inflammatory cell counting was performed using a standard hemacytometer technique [10, 46]. For histological assessment of lung injury, the lungs were harvested without lavage collection and fixed in 10% formaldehyde. After fixation, the lungs were embedded in paraffin, cut into 5-μm sections, and stained with hematoxylin and eosin. Sections were evaluated at 40x magnification.
Statistical analysis
Results are expressed as means ± SD of three to six independent experiments. Experimental samples were compared to controls by unpaired Student’s t-test. For multiple-group comparisons, a one-way variance analysis (ANOVA) and post hoc multiple comparisons tests were used. P<0.05 was considered statistically significant.
RESULTS
Oxidized phospholipids induce p190RhoGAP activation
Activation of nucleotide exchange activity of GEFs and GAPs including p190RhoGAP is usually associated with their submembrane translocation. We therefore examined p190RhoGAP intracellular redistribution in OxPAPC-treated endothelium. Immunofluorescence analysis shows peripheral translocation of p190RhoGAP in response to OxPAPC challenge (Figure 1A). Complementary subcellular fractionation experiments with separation of cytosolic and membrane fractions show OxPAPC-induced increase of p190RhoGAP content in the membrane fraction, as compared to non-treated EC (Figure 1B). In contrast, OxPAPC stimulation did not affect intracellular distribution of Rho downstream target Rho kinase (Figure 1B, lower panel), which is not involved in the mediation of OxPAPC effects [8, 9].
Figure 1. Effect of OxPAPC on p190RhoGAP intracellular localization and activation.
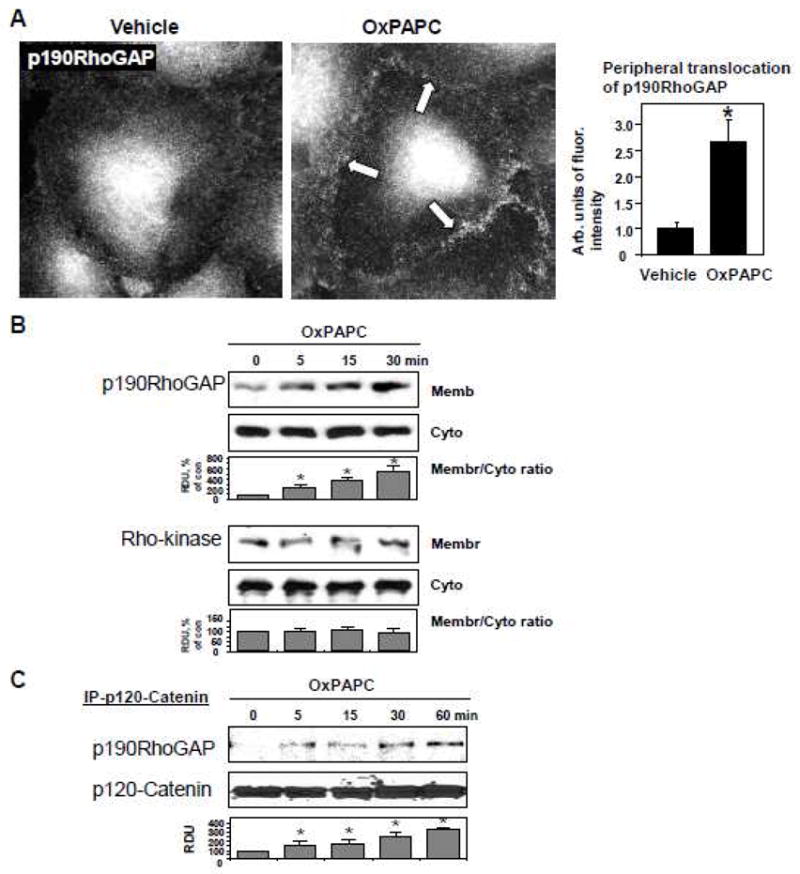
A: Endothelial cells grown on glass coverslips were stimulated with OxPAPC (10 μg/ml) for 15 min followed by immunofluorescence staining for p190RhoGAP. Arrows depict areas of intracellular accumulation of protein. Bar graphs represent quantitative analysis of p190RhoGAP peripheral accumulation in control and treated HPAEC. Data are expressed as mean ± SD of three independent experiments; *p<0.05. B: HPAEC were stimulated with OxPAPC (10 μg/ml) for various periods of time or left untreated. Membrane and cytosolic fractions were isolated as described in Methods section. The content of p190RhoGAP and Rho kinase was determined by western blot analysis of cytosolic and membrane fractions with specific antibodies. C–E: Immunoprecipitation of p120-catenin (C), VE-cadherin (D), or p190RhoGAP (E) under non-denaturing conditions was performed in pulmonary EC stimulated with OxPAPC (10 μg/ml) for various time periods. P190RhoGAP or p120-catenin content in the immunoprecipitates was detected with specific antibodies. Equal protein loadings were confirmed by re-probing of membranes with antibody used for pulldown. F: Immunoprecipitation of p190RhoGAP under denaturing conditions was performed in pulmonary EC stimulated with OxPAPC (10 μg/ml) for various time periods. P190RhoGAP phosphorylation was determined by western blotting with p-tyrosine antibody. Equal protein loading was confirmed by probing of membranes with p190RhoGAP antibody. Result of densitometry shown as mean ± SD, * p<0.05
Because p190RhoGAP may interact with adherens junction protein p120-catenin in submembrane compartments, in the following experiments we tested effect of OxPAPC on p190RhoGAP - p120-catenin interactions in co-immunoprecipitation assays. OxPAPC induced time dependent accumulation of p190RhoGAP in the p120-catenin immunoprecipitates (Figure 1C). In contrast, EC stimulation with edemagenic agonist thrombin, which activates Rho pathway, did not cause p120 catenin - p190RhoGAP association (data not shown). While OxPAPC increased p120-catenin accumulation in VE-cadherin immunoprecipitates, which is consistent with previously described OxPAPC-induced enhancement of adherens junction protein complexes, we did not observe noticeable association of p190RhoGAP with VE-cadherin (Figure 1D). These data indicate that p120-catenin, not VE-cadherin, is a primary binding partner of p190RhoGAP. OxPAPC-induced p120-catenin-p190RhoGAP interaction was further confirmed in reverse co-immunoprecipitation experiments using p190RhoGAP antibody for pulldown (Figure 1E).
Because activation of GAP activity is driven by p190RhoGAP phosphorylation [47–49], we examined phosphorylation status of p190RhoGAP immunoprecipitated from control and OxPAPC-stimulated HPAEC. OxPAPC treatment induced rapid, gradually increasing tyrosine phosphorylation of p190RhoGAP, which remained elevated at least 45 min (Figure 1F).
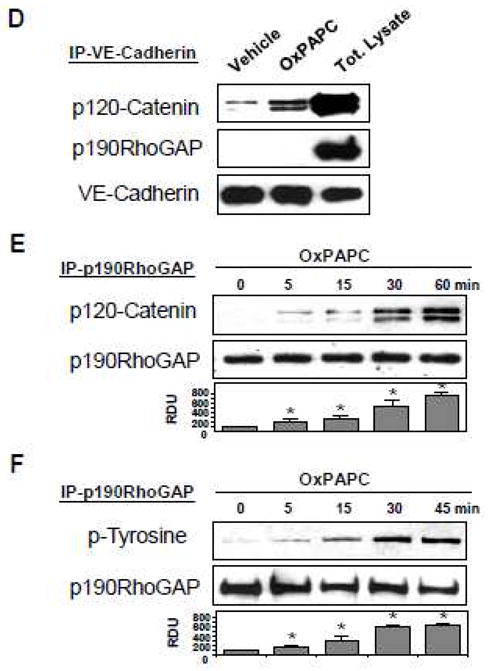
OxPAPC-induced activation of p190RhoGAP is Rac-dependent
Barrier protective effects of OxPAPC are associated with activation of Rac pathway [8, 11]. We tested a role of Rac signaling in p190RhoGAP activation by OxPAPC by depleting endogenous Rac or Rac-specific guanine nucleotide exchange factors (GEFs) Tiam1 and Vav2. Control cells were transfected with non-specific RNA duplexes. After 48 hrs of double transfection with Tiam1- and Vav2-specific siRNAs, EC were stimulated with OxPAPC followed by subcellular fractionation assay. In contrast to non-trasfected cells (Figure 1B) or EC transfected with non-specific RNA, Tiam1 and Vav2 knockdown abolished OxPAPC-induced membrane translocation of p190RhoGAP (Figure 2A, left panel). Specific protein depletion was confirmed by western blot analysis (Figure 2A, right panel). Besides membrane translocation, Rac activity was also required for the OxPAPC-induced formation of p120 catenin - p190RhoGAP complexes, since siRNA-induced Rac depletion inhibited p120 catenin - p190RhoGAP interaction in the OxPAPC-stimulated endothelial cells detected in coimmunoprecipitation assays (Figure 2B). We next tested involvement of Rac signaling in the OxPAPC-induced p190RhoGAP tyrosine phosphorylation. p190RhoGAP was precipitated under denaturing conditions from control and siRac-transfected cells followed by western blot with p190RhoGAP and phospho-tyrosine antibodies. Rac knockdown inhibited OxPAPC-induced p190RhoGAP tyrosine phosphorylation (Figure 2C, upper panel), as compared to non-transfected cells (Figure 1F), or cells transfected with non-specific RNA. Rac protein depletion was verified by western blot (Figure 2C, lower panel).
Figure 2. Involvement of Rac in OxPAPC-induced p190RhoGAP activation.
HPAEC were transfected with non-specific, Tiam1/Vav2-, or Rac-specific siRNAs for 48 hr, followed by OxPAPC stimulation (10 μg/ml, 15 min). A: Cytosolic and membrane fractions were separated according to the protocol described in Methods, and translocation of p190RhoGAP to the membrane fraction was detected with specific antibodies. Inset - western blot analysis of Tiam1 and Vav2 depletion in HPAEC. B: After cell lysis step, protein complexes were immunoprecipitated with p120-catenin antibodies followed by western blot analysis with p190RhoGAP antibody. Equal protein loading was confirmed by reprobing of membranes with p120-catenin antibody. C: p190RhoGAP was immunoprecepitated under denaturing conditions. Phosphorylation status of p190RhoGAP was determined by western blot analysis with p-tyrosine antibody. Equal protein loading was confirmed by probing of membranes with p190RhoGAP antibody. Rac depletion was confirmed by probing of membranes with specific antibody. Result of densitometry shown as mean ± SD, * p<0.05
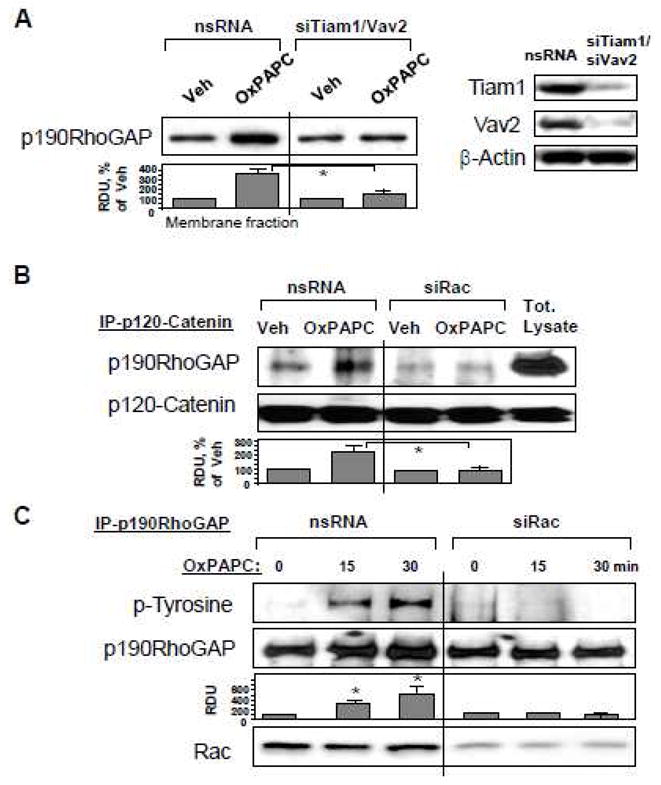
Oxidized phospholipids mediate endothelial barrier protection via p190RhoGAP
Thrombin is an edemagenic agonist released by activated platelets, which triggers Rho- and MLCK-dependent mechanisms of EC permeability [20, 50]. To investigate an involvement of p190RhoGAP in the OxPAPC protective effects against thrombin-induced permeability, we used siRNA-mediated p190RhoGAP knockdown. After 72 hrs of transfection, EC grown on gold microelectrodes for measurements of transendothelial electrical resistance (TER, see Materials and Methods section) were pretreated with OxPAPC or vehicle followed by thrombin challenge, and TER was monitored over the time. Knockdown of p190RhoGAP enhanced thrombin-induced permeability increase and delayed recovery of EC monolayer barrier after thrombin challenge. Importantly, protective effects of OxPAPC against thrombin-induced permeability observed in control cells treated with non-specific RNA were significantly attenuated in EC with depleted p190RhoGAP (Figure 3A). We further examined a role of p190RhoGAP in OxPAPC-induced preservation of EC monolayer integrity in immunofluorescence studies. Control and p190RhoGAP-depleted EC were challenged with thrombin with or without OxPAPC pretreatment. Thrombin induced stress fiber formation (red), and disruption of continuous peripheral VE-cadherin staining (green) in both, control non-specific RNA-treated EC and cells with depleted p190RhoGAP (Figure 3B). OxPAPC preserved monolayer integrity in thrombin-challenged EC treated with nonspecific RNA, but this protective effect of OxPAPC was abolished in EC with p190RhoGAP knockdown (Figure 3B, lower panels). Since Rho pathway of endothelial permeability involves phosphorylation of myosin-binding subunit of myosin-associated phosphatase type 1 (MYPT1) at the Rho kinase specific site Thr-850 [21, 51] leading to increased myosin light chain (MLC) phosphorylation which causes EC actomyosin contraction, we examined effects of OxPAPC and thrombin on MYPT1 and MLC phosphorylation levels in control and p190RhoGAP-depleted cells. Thrombin induced larger increases in MYPT1 and MLC phosphorylation in cells with depleted p190RhoGAP. In turn, OxPAPC inhibited MYPT1 and MLC phosphorylation in control EC transfected with non-specific RNA, but failed to inhibit thrombin-induced MYPT1 and MLC phosphorylation in p190RhoGAP-depleted cells (Figure 3C). Depletion of p190RhoGAP was verified by western blot (Figure 3C, lower panel). phosphorylation of myosin-binding subunit of myosin-associated phosphatase type 1 (MYPT1) at the Rho kinase specific site leading to increased myosin light chain (MLC) phosphorylation
Figure 3. Effect of p190RhoGAP inhibition on OxPAPC-mediated protection against thrombin-induced EC barrier dysfunction.
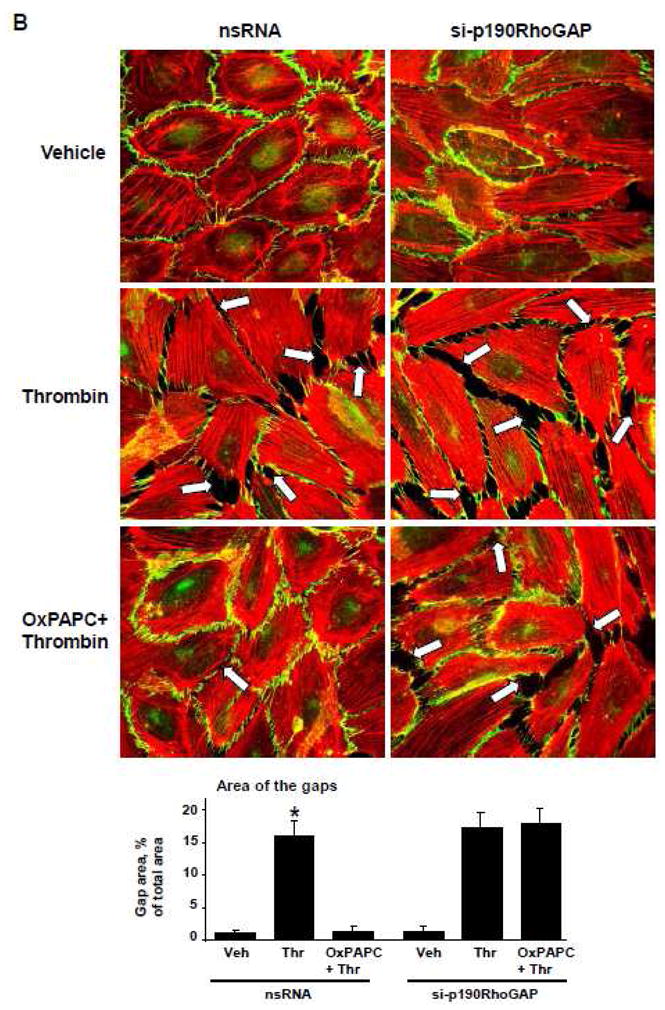
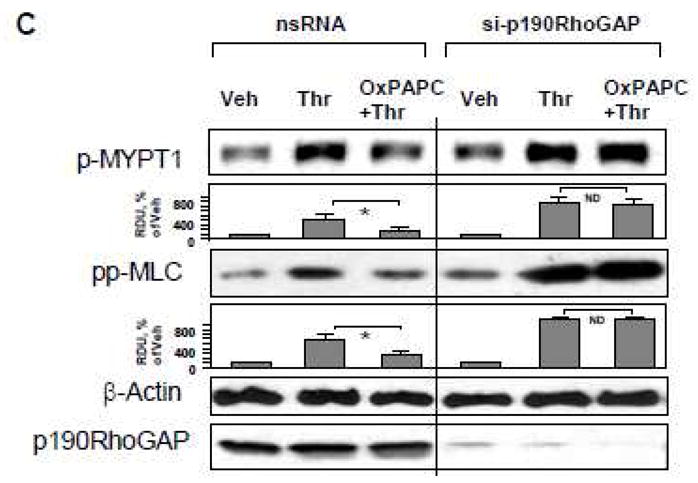
A – C: HPAEC were transfected with p190RhoGAP -specific siRNA or non-specific RNA duplexes for 72 hr. A - EC were treated with OxPAPC (10 μg/ml, 15 min) or left untreated prior to thrombin (0.1 U/ml) challenge, and TER was monitored over the time. B and C - EC were subjected to thrombin challenge (0.1 U/ml, 15 min) with or without OxPAPC pretreatment (10 μg/ml, 15 min). Analysis of actin cytoskeletal remodeling was performed by immunofluorescence staining with Texas Red phalloidin. Paracellular gaps are marked by arrows. Bar graphs represent quantitative analysis of gap formation in control and treated HPAEC. Data are expressed as mean ± SD of three independent experiments; *p<0.05. (B). Phosphorylation status of MYPT1 or MLC was determined by western blot with specific antibodies. Equal protein loading was confirmed by re-probing of membranes with antibody to housekeeping protein β-tubulin. P190RhoGAP depletion was confirmed by probing of membranes with specific antibody (C). Result of densitometry shown as mean ± SD, * p<0.05
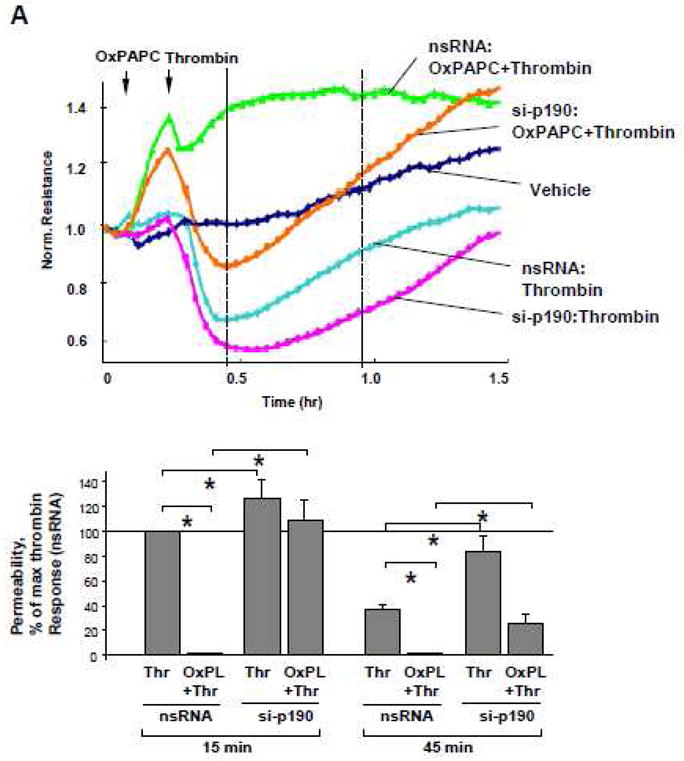
p190RhoGAP is required for protective effects of OxPAPC in the in vitro model of VILI
We used previously established cell culture two-hit model of VILI [37, 42] and exposed pulmonary EC to pathologically-relevant levels of cyclic stretch (18% CS) with or without thrombin stimulation to evaluate a role of p190RhoGAP in the OxPAPC-mediated protection against EC barrier dysfunction. Effects of pathological CS were judged by enhanced paracellular gap formation and activation of actomyosin contraction indicated by increased MLC phosphorylation. After 72 hours of HPAEC transfection with p190RhoGAP-specific or non-specific siRNAs, cells were subjected to 18% CS for 2 hrs followed by addition of OxPAPC or vehicle prior to thrombin challenge with continuous CS exposure.
In agreement with published data [37, 52], preconditioning of lung EC transfected with non-specific RNA at 18% CS caused cytoskeletal reorientation without affecting EC monolayer integrity. Thrombin challenge caused pronounced monolayer disruption and increased actin stress fiber formation in CS-preconditioned EC, which was dramatically reduced by pretreatment with OxPAPC (Figure 4A, left panels). In contrast, protective effects of OxPAPC against EC monolayer disruption induced by 18% CS and thrombin were abolished by p190RhoGAP knockdown (Figure 4A, right panels).
Figure 4. Effects of p190RhoGAP knockdown on protective effects by OxPAPC in the in vitro model of VILI.
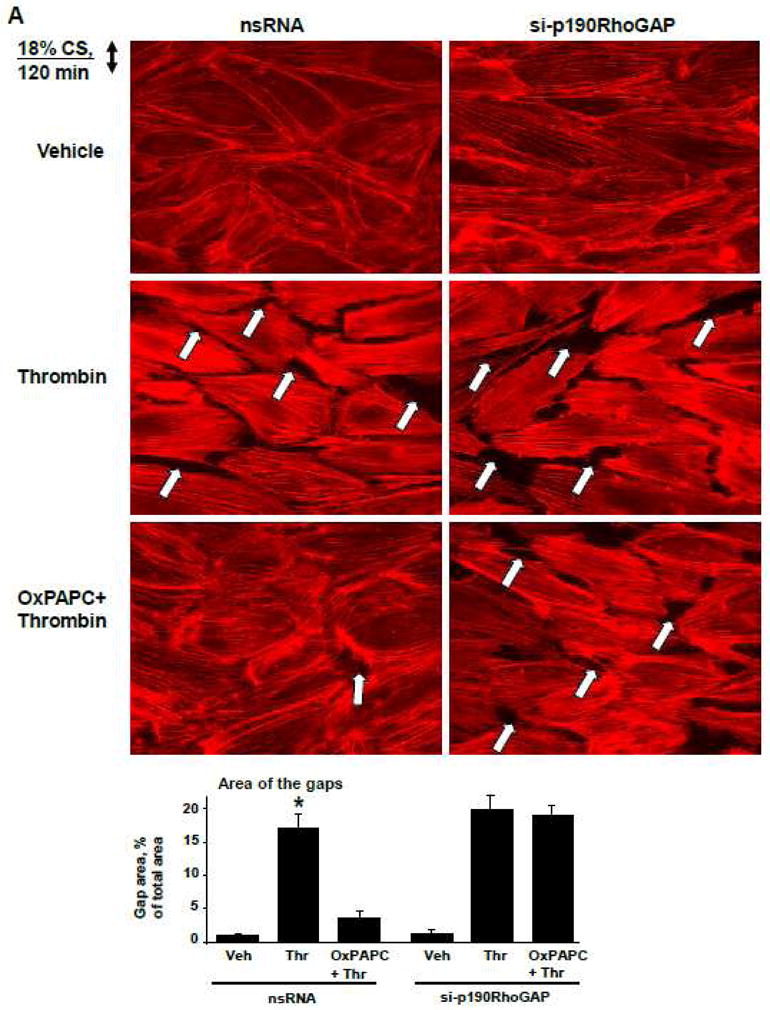
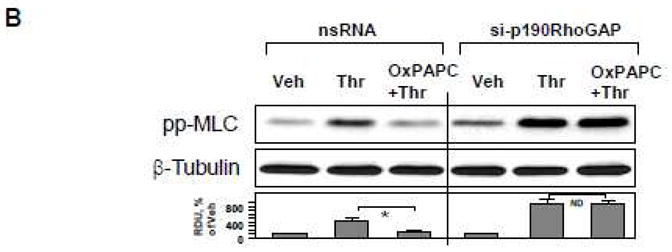
HPAEC grown on Flexcell plates were transfected with p190RhoGAP-specific siRNA or non-specific RNA duplexes. After 72 hr cell monolayers were preconditioned at pathologic (18%) levels of CS for 2 hr and stimulated with thrombin (0.1 U/ml, 15 min) with or without OxPAPC pretreatment (10 μg/ml, 15 min). A: F-actin was visualized by immunofluorescence staining with Texas-Red phalloidin. Paracellular gaps are marked by arrows. Bar graphs represent quantitative analysis of gap formation in control and treated HPAEC. Data are expressed as mean ± SD of three independent experiments; *p<0.05. B: Phosphorylation of MLC was detected by western blot with phospho-specific antibodies. Equal protein loading was confirmed by re-preobing of membranes with β-tubulin antibody. Result of densitometry shown as mean ± SD, * p<0.05
Western blot analysis of MLC phosphorylation showed that thrombin increased MLC phosphorylation in both, control and p190RhoGAP-depleted CS-preconditioned cells, but OxPAPC failed to inhibit thrombin-induced MLC phosphorylation in p190RhoGAP-depleted cells, as compared to controls transfected with nonspecific RNA (Figure 4B).
P190RhoGAP knockdown attenuates protective effects by oxidized phospholipids in the in vivo two-hit model of VILI
To investigate whether p190RhoGAP is involved in OxPAPC-activated signaling in vivo, we examined effects of intravenous OxPAPC administration on p190RhoGAP phosphorylation reflecting its activation in lungs. Mice were treated with OxPAPC for 30 min or 4 hrs followed by lung harvesting, immunoprecipitation of p190RhoGAP under denaturing conditions and western blot analysis with phosphor-tyrosine antibody. Activation of p190RhoGAP was assessed by analysis of its tyrosine phosphorylation in control and OxPAPC-treated lung samples. OxPAPC induced phosphorylation of p190RhoGAP at both time points, which became robust at 4 hrs (Figure 5A).
Figure 5. Involvement of p190RhoGAP in protective effects by OxPAPC against ventilator-induced lung injury.
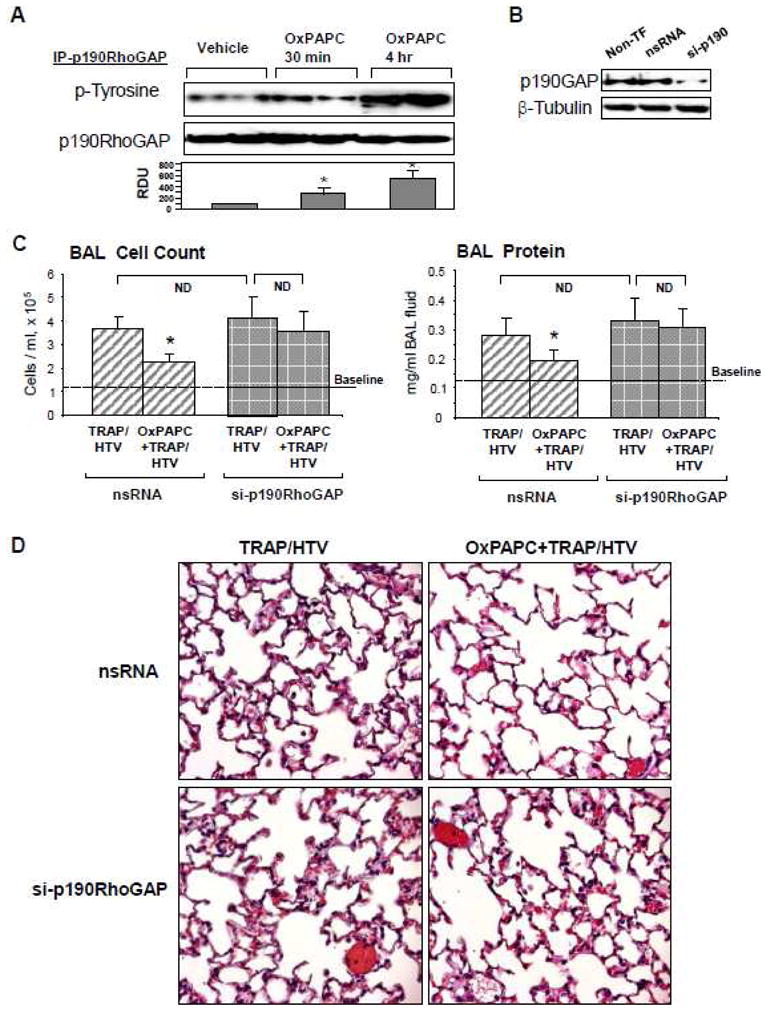
A: Mice were treated with OxPAPC (1.5 mg/kg, i/v) for indicated time points. p190RhoGAP was immunoprecepitated deom lung tissue samples under denaturing conditions. Phosphorylation status of p190RhoGAP was determined by western blot analysis with p-tyrosine antibody. Equal protein loading was confirmed by probing of membranes with p190RhoGAP antibody. Result of densitometry shown as mean ± SD, * p<0.05 B – D: Mice were tranfected with non-specific or p190RhoGAP-specific siRNAs for 72 hrs followed by TRAP6 injection (1.5 × 10−5 mol/kg, i/t) and mechanical ventilation at high tidal volume (HTV, 30 ml/kg, 4 hr) with or without OxPAPC treatment (1.5 mg/kg, i/v). P190RhoGAP depletion in lung samples was assessed by western blot (B). Cell count and measurements of protein concentration were performed in BAL fluid. Data are presented as mean ± SD (n=6–8 mice per group); *p<0.05 (C). Whole lungs were fixed in 10% formalin, and used for histological evaluation by hematoxylin and eosin staining. Images are representative of 4–6 lung specimens for each condition with ×40 magnification (D).
In the next experiments we attempted to reproduce two-hit model of lung injury and exposed mice to ventilation at high tidal volume (HTV, 30 ml/kg) mechanical ventilation and TRAP6, the thrombin-derived non-thrombogenic peptide that serves as a PAR1 receptor ligand [53]. To examine whether p190RhoGAP is involved in OxPAPC-mediated protective effects in vivo, endogenous p190RhoGAP was depleted using siRNA approach outlined in Materials and Methods. Mice were transfected with non-specific or p190RhoGAP-specific siRNA for 72 hr followed by TRAP6 administration and HTV with or without OxPAPC pretreatment. After four hours of ventilation, BAL was performed as described in Methods. p190RhoGAP protein depletion was confirmed by western blot analysis of lung tissue (Figure 5B). Knockdown of p190RhoGAP had no statistically significant effects on TRAP6/HTV-induced elevation of BAL cell counts and protein content (Figure 5B, inset), but attenuated protective effects of OxPAPC against TRAP6/HTV-induced increases in BAL cell count (Figure 5C, left panels) and protein concentration (Figure 5C, right panels), observed in control animals transfected with non-specific siRNA. Histological analysis of lung sections confirmed that p190RhoGAP knockdown suppressed the ability of oxidized phospholipids to inhibit TRAP6/HTV-mediated alveolar fluid accumulation and leukocyte infiltration (Figure 5D). Collectively, these data strongly suggest the importance of p190RhoGAP in the protective effects of OxPAPC in vivo.
DISCUSSION
Oxidized phospholipids exhibit potent barrier-protective effects on endothelial monolayers and in the animal models of endotoxin- and ventilator-induced lung injury [7, 14]. Barrier-protective effects of OxPAPC under these conditions depend on activation of Rac signaling and Rac-mediated suppression of Rho pathway of endothelial permeability [8–10, 12, 14]. However, the nature of Rac-dependent Rho downregulation induced by OxPAPC remains unclear. This study investigated a role of p190RhoGAP as a switch between Rac and Rho signaling activated by OxPAPC which is a critical mechanism regulating lung vascular endothelial barrier properties in the models of acute lung injury.
The data presented here demonstrate that OxPAPC engages p190RhoGAP to protect against ventilator-induced lung vascular leakage and attenuate endothelial hyper-permeability in pulmonary EC cultures induced by edemagenic agonists and VILI- relevant cyclic stretch. Our data show that OxPAPC signals to Tiam1 and Vav2 to stimulate Rac1, which in turn induces p190RhoGAP tyrosine phosphorylation, p190RhoGAP association with p120-catenin, and p190RhoGAP-dependent inhibition of RhoA signaling, leading to EC barrier preservation in pulmonary EC exposed to inflammatory agonists and pathologic cyclic stretch.
OxPAPC stimulated time-dependent accumulation of p190RhoGAP at the cell periphery, which correlates with the time course of OxPAPC-induced EC barrier enhancement and p190RhoGAP - p120-catenin association. This interaction was regulated by Rac GTPase. Rac specific GEF Tiam1 has been previously implicated in the OxPAPC-induced Rac activation and barrier enhancement [11]. The current results show involvement of Tiam1 and Vav2 in the Rac-dependent p190RhoGAP submembrane accumulation. Tyrosine phosphorylation of p190RhoGAP appears to be important for its interaction with binding partners in fibroblasts [28, 32]. Our data show Rac-dependent tyrosine phosphorylation of p190RhoGAP, which also coincided with p120-catenin – p190RhoGAP complex formation. A number of protein tyrosine kinases such as Src [54], Fyn [55] and FAK [33] may directly phosphorylate and activate p190RhoGAP. This is a plausible mechanism of p190RhoGAP phosphorylation, because OxPAPC activates Src and FAK, and Src mediates OxPAPC barrier enhancement in pulmonary EC [9].
Our data show that OxPAPC-induced tyrosine phosphorylation of p190RhoGAP is Rac-dependent. This is another recently described possible mechanism, which involves Rac-dependent activation of NADPH oxidase complex, stimulation of reactive oxygen species (ROS) production, and ROS-induced inhibition of the low-molecular-weight protein tyrosine phosphatase (LMW-PTP) leading to increased tyrosine phosphorylation and activation of p190RhoGAP [56]. Tyrosine phosphorylation and targeting of p190RhoGAP to specific subcellular domains may be also important mechanisms regulating its GAP activity towards Rho GTPase [28, 32]. p190RhoGAP may also localize to focal adhesions, where it dissociates from p120RasGAP and binds tyrosine-phosphorylated paxillin leading to local suppression of Rho activity during cell adhesion [31]. In addition, p190RhoGAP may localize to lipid rafts, where it mediates Rho inactivation during cell spreading [30]. Previous study [28] and our data strongly suggest that p120-catenin directs localization of p190RhoGAP to adherens junctions. Depletion of p190RhoGAP binding partner p120-catenin in NIH3T3 fibroblasts caused constitutive activation of Rho [28]. Depletion of p190RhoGAP in our studies impaired OxPAPC-enhanced recovery of monolayer integrity in pulmonary EC exposed to thrombin and pathologically relevant levels of cyclic stretch. Taken together, these results strongly suggest involvement of p120-catenin – p190RhoGAP complex in protecting endothelial barrier under pathologic conditions. Our results and other studies also suggest that localized p190RhoGAP activation at different subcellular compartments (focal adhesion, adherens junctions, lipid rafts) may provide additional mechanisms contributing to peripheral actin cytoskeletal and cell adhesion remodeling critical for barrier-protective effects of OxPAPC. Elucidation of these mechanisms awaits further investigation.
Importantly, OxPAPC-induced tyrosine phosphorylation of p190RhoGAP observed in pulmonary EC cultures (Figures 1,2) was also detected in the lung, which further supports p190RhoGAP involvement in OxPAPC-induced effects in vivo. Although OxPAPC alone does not affect baseline of lung vascular permeability [7, 14], OxPAPC-induced p190RhoGAP phosphorylation observed in the lungs may reflect mobilization of vascular barrier-protective mechanisms, which then become evident in the lungs exposed to pathologic mechanical ventilation and TRAP (Figure 5).
The present study demonstrates for the first time that p190RhoGAP knockdown in vivo abolishes barrier protective effects of OxPAPC in the two-hit model of ventilator induced lung injury. We have previously described protective effect of OxPAPC in the model of ventilator-induced lung barrier dysfunction via downregulation of Rho signaling [14]. Protective effects of OxPAPC against agonist-induced EC barrier disruption were associated with inhibition of Rho activation and accelerated recovery of Rac signaling in pulmonary EC monolayers [9, 10, 14]. However, more precise mechanisms regulating Rac-Rho balance remained unclear.
p190RhoGAP has been implicated in the protective effects of angiopoietin-1 (Ang-1) against endotoxin-mediated vascular leakage. Ang-1 prevented endotoxin-induced cytoskeletal rearrangements in the EC by activating a PI3-kinase and Rac1, which were also linked to phosphorylation of p190 RhoGAP and inhibition of RhoA activity [35]. Our results also show that siRNA-induced knockdown of p190RhoGAP abolished inhibitory effects of OxPAPC on MLC phosphorylation in pulmonary EC induced by thrombin and high magnitude cyclic stretch suggesting a role for p190RhoGAP in attenuating the Rho pathway of EC barrier dysfunction associated with VILI. These data suggest a mechanism of Rac-Rho crosstalk via OxPAPC-triggered, Rac-p120-catenin mediated activation and recruitment of p190RhoGAP to the cell-cell junction area, where it may locally downregulate Rho signaling and attenuate barrier-disruptive effects.
Although molecular inhibition of p190RhoGAP did not significantly alter baseline endothelial monolayer integrity, basal EC permeability, and in vivo lung structural integrity, p190RhoGAP expression and activation became essential for reduction of Rho signaling activated by barrier disruptive agonists and mechanical forces. Similar results showing lack of p190RhoGAP knockdown effects in animals without additional treatments were observed in the other study by Mammoto et al. [35]. Lack of significant effects of p190RhoGAP knockdown on the basal Rho activation and EC barrier properties may reflect low levels of basal p190RhoGAP activity in non-stimulated EC monolayers. In contrast, in thrombin-stimulated EC, p190RhoGAP depletion potentiated MLC and MYPT1 phosphorylation, promoted thrombin-induced permeability increase and delayed recovery of EC monolayer barrier. These findings are consistent with report by Holinstat et al. [33], who demonstrated the p190RhoGAP role in EC monolayer recovery after thrombin challenge and showed stimulation of p190RhoGAP activity by FAK-mediated tyrosine phosphorylation leading to suppression of Rho signaling during recovery after thrombin.
Our results suggest that barrier-disruptive Rho signaling triggered by VILI-relevant mechanical forces and inflammatory mediators can be blocked by OxPAPC-induced activation of p190 RhoGAP. Therefore, although p190 RhoGAP may be dispensable for control of endothelial barrier integrity at baseline, it is crucial for prevention of cell-cell contact disruption and cytoskeletal rearrangements in pulmonary EC induced by Rho activators.
In addition to Rac, OxPAPC also activates Cdc42 GTPase, which together with Rac participates in OxPAPC-induced barrier protective EC responses. Downregulation of Cdc42 alone or in combination with Rac disturbed peripheral cytoskeletal enhancement and attenuated protective effects of OxPAPC [8, 12]. Report by Ramchandran et al. shows that similar to Rac, barrier protecitve effects of Cdc42 involve negative crosstalk with Rho [57]. However, involvement of Cdc42 in the mechanism of p190RhoGAP activation remains to be evaluated. Activation of Cdc42 may also contribute to AJ disassembly and barrier disruption. In vivo studies show that coexpression of dominant-negative Cdc42 (N17Cdc42) prevented the increase in K(f,c) induced by expression of VE-cadherin mutant lacking extracellular domain [58]. Collectively, these data illustrate a complex role of Cdc42 in control of lung vascular permeability.
In summary, based on published data and results of this study we propose a scheme of OxPAPC-induced protective effects in the models of VILI, in which OxPAPC triggers Rac activation by Rac-specific GEFs, Tiam1 and Vav2 leading to Rac-dependent activation of tyrosine phosphorylation of p190RhoGAP. Phosphorylated p190RhoGAP associates with p120-catenin, localizes to cell-cell junctions and locally inhibits stretch- and agonist-induced barrier-disruptive Rho signaling in the pulmonary endothelium. Such inhibition improves lung vascular endothelial barrier properties and prevents lung vascular leak induced by high tidal volume mechanical ventilation and inflammatory agonists. We speculate that this paradigm which also illustrates the mechanism of Rac-Rho crosstalk triggered by OxPAPC, may be generally applicable to other barrier-protective compounds (i.e. hepatocyte growth factor and sphingosine 1-phosphate) known to activate Rac GTPase. Thus, stimulation of p190RhoGAP function using molecular engineering approaches or target-oriented drug design may be a promising direction for new therapeutic strategies aimed at the treatment of ALI and its devastating complication, ARDS.
Acknowledgments
Supported by National Heart, Lung, and Blood Institutes grants HL87823, HL76259, and HL58064 for KGB; HL89257 and the American Heart Association Midwest Affiliate Grant-in-Aid for AAB
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chabot F, Mitchell JA, Gutteridge JM, Evans TW. Reactive oxygen species in acute lung injury. Eur Respir J. 1998;11:745–757. [PubMed] [Google Scholar]
- 2.Wood LG, Gibson PG, Garg ML. Biomarkers of lipid peroxidation, airway inflammation and asthma. Eur Respir J. 2003;21:177–186. doi: 10.1183/09031936.03.00017003a. [DOI] [PubMed] [Google Scholar]
- 3.Lang JD, McArdle PJ, O’Reilly PJ, Matalon S. Oxidant-antioxidant balance in acute lung injury. Chest. 2002;122:314S–320S. doi: 10.1378/chest.122.6_suppl.314s. [DOI] [PubMed] [Google Scholar]
- 4.Bochkov VN, Oskolkova OV, Birukov KG, Levonen AL, Binder CJ, Stockl J. Generation and biological activities of oxidized phospholipids. Antioxid Redox Signal. 2010;12:1009–1059. doi: 10.1089/ars.2009.2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bochkov VN, Kadl A, Huber J, Gruber F, Binder BR, Leitinger N. Protective role of phospholipid oxidation products in endotoxin-induced tissue damage. Nature. 2002;419:77–81. doi: 10.1038/nature01023. [DOI] [PubMed] [Google Scholar]
- 6.Ma Z, Li J, Yang L, Mu Y, Xie W, Pitt B, Li S. Inhibition of LPS- and CpG DNA-induced TNF-alpha response by oxidized phospholipids. Am J Physiol Lung Cell Mol Physiol. 2004;286:L808–816. doi: 10.1152/ajplung.00220.2003. [DOI] [PubMed] [Google Scholar]
- 7.Nonas SA, Miller I, Kawkitinarong K, Chatchavalvanich S, Gorshkova I, Bochkov VN, Leitinger N, Natarajan V, Garcia JG, Birukov KG. Oxidized phospholipids reduce vascular leak and inflammation in rat model of acute lung injury. Am J Respir Crit Care Med. 2006;173:1130–1138. doi: 10.1164/rccm.200511-1737OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Birukov KG, Bochkov VN, Birukova AA, Kawkitinarong K, Rios A, Leitner A, Verin AD, Bokoch GM, Leitinger N, Garcia JG. Epoxycyclopentenone-containing oxidized phospholipids restore endothelial barrier function via Cdc42 and Rac. Circ Res. 2004;95:892–901. doi: 10.1161/01.RES.0000147310.18962.06. [DOI] [PubMed] [Google Scholar]
- 9.Birukova AA, Chatchavalvanich S, Oskolkova O, Bochkov VN, Birukov KG. Signaling pathways involved in OxPAPC-induced pulmonary endothelial barrier protection. Microvasc Res. 2007;73:173–181. doi: 10.1016/j.mvr.2006.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Birukova AA, Fu P, Chatchavalvanich S, Burdette D, Oskolkova O, Bochkov VN, Birukov KG. Polar head groups are important for barrier protective effects of oxidized phospholipids on pulmonary endothelium. Am J Physiol Lung Cell Mol Physiol. 2007;292:L924–935. doi: 10.1152/ajplung.00395.2006. [DOI] [PubMed] [Google Scholar]
- 11.Birukova AA, Malyukova I, Mikaelyan A, Fu P, Birukov KG. Tiam1 and betaPIX mediate Rac-dependent endothelial barrier protective response to oxidized phospholipids. J Cell Physiol. 2007;211:608–617. doi: 10.1002/jcp.20966. [DOI] [PubMed] [Google Scholar]
- 12.Birukova AA, Malyukova I, Poroyko V, Birukov KG. Paxillin - {beta}-catenin interactions are involved in Rac/Cdc42-mediated endothelial barrier-protective response to oxidized phospholipids. Am J Physiol Lung Cell Mol Physiol. 2007;293:L199–211. doi: 10.1152/ajplung.00020.2007. [DOI] [PubMed] [Google Scholar]
- 13.Nonas SA, Miller IL, Birukova AA, Chatchavalvanich S, Garcia JG, Birukov KG. Protective effects of oxidized phospholipids on ventilator-induced lung injury. Proc Am Thorac Soc. 2006;3:A245. [Google Scholar]
- 14.Nonas S, Birukova AA, Fu P, Xing J, Chatchavalvanich S, Bochkov VN, Leitinger N, Garcia JG, Birukov KG. Oxidized phospholipids reduce ventilator-induced vascular leak and inflammation in vivo. Crit Care. 2008;12:R27. doi: 10.1186/cc6805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boguski MS, McCormick F. Proteins regulating Ras and its relatives. Nature. 1993;366:643–654. doi: 10.1038/366643a0. [DOI] [PubMed] [Google Scholar]
- 16.Zheng Y. Dbl family guanine nucleotide exchange factors. Trends Biochem Sci. 2001;26:724–732. doi: 10.1016/s0968-0004(01)01973-9. [DOI] [PubMed] [Google Scholar]
- 17.Bishop AL, Hall A. Rho GTPases and their effector proteins. Biochem J 348 Pt. 2000;2:241–255. [PMC free article] [PubMed] [Google Scholar]
- 18.Birukova AA, Adyshev D, Gorshkov B, Birukov KG, Verin AD. ALK5 and Smad4 are involved in TGF-beta1-induced pulmonary endothelial permeability. FEBS Lett. 2005;579:4031–4037. doi: 10.1016/j.febslet.2005.06.018. [DOI] [PubMed] [Google Scholar]
- 19.Birukova AA, Adyshev D, Gorshkov B, Bokoch GM, Birukov KG, Verin AA. GEF-H1 is involved in agonist-induced human pulmonary endothelial barrier dysfunction. Am J Physiol Lung Cell Mol Physiol. 2006;290:L540–548. doi: 10.1152/ajplung.00259.2005. [DOI] [PubMed] [Google Scholar]
- 20.Birukova AA, Smurova K, Birukov KG, Kaibuchi K, Garcia JGN, Verin AD. Role of Rho GTPases in thrombin-induced lung vascular endothelial cells barrier dysfunction. Microvasc Res. 2004;67:64–77. doi: 10.1016/j.mvr.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 21.Essler M, Amano M, Kruse HJ, Kaibuchi K, Weber PC, Aepfelbacher M. Thrombin inactivates myosin light chain phosphatase via Rho and its target Rho kinase in human endothelial cells. J Biol Chem. 1998;273:21867–21874. doi: 10.1074/jbc.273.34.21867. [DOI] [PubMed] [Google Scholar]
- 22.Majumdar M, Seasholtz TM, Goldstein D, de Lanerolle P, Brown JH. Requirement for Rho-mediated myosin light chain phosphorylation in thrombin-stimulated cell rounding and its dissociation from mitogenesis. J Biol Chem. 1998;273:10099–10106. doi: 10.1074/jbc.273.17.10099. [DOI] [PubMed] [Google Scholar]
- 23.Sanders LC, Matsumura F, Bokoch GM, de Lanerolle P. Inhibition of myosin light chain kinase by p21-activated kinase [see comments] Science. 1999;283:2083–2085. doi: 10.1126/science.283.5410.2083. [DOI] [PubMed] [Google Scholar]
- 24.Vouret-Craviari V, Boquet P, Pouyssegur J, Van Obberghen-Schilling E. Regulation of the actin cytoskeleton by thrombin in human endothelial cells: role of Rho proteins in endothelial barrier function. Mol Biol Cell. 1998;9:2639–2653. doi: 10.1091/mbc.9.9.2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van Nieuw Amerongen GP, van Delft S, Vermeer MA, Collard JG, van Hinsbergh VW. Activation of RhoA by thrombin in endothelial hyperpermeability: role of Rho kinase and protein tyrosine kinases. Circ Res. 2000;87:335–340. doi: 10.1161/01.res.87.4.335. [DOI] [PubMed] [Google Scholar]
- 26.Garcia JG, Liu F, Verin AD, Birukova A, Dechert MA, Gerthoffer WT, Bamberg JR, English D. Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. J Clin Invest. 2001;108:689–701. doi: 10.1172/JCI12450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu F, Schaphorst KL, Verin AD, Jacobs K, Birukova A, Day RM, Bogatcheva N, Bottaro DP, Garcia JG. Hepatocyte growth factor enhances endothelial cell barrier function and cortical cytoskeletal rearrangement: potential role of glycogen synthase kinase-3beta. FASEB J. 2002;16:950–962. doi: 10.1096/fj.01-0870com. [DOI] [PubMed] [Google Scholar]
- 28.Wildenberg GA, Dohn MR, Carnahan RH, Davis MA, Lobdell NA, Settleman J, Reynolds AB. p120-catenin and p190RhoGAP regulate cell-cell adhesion by coordinating antagonism between Rac and Rho. Cell. 2006;127:1027–1039. doi: 10.1016/j.cell.2006.09.046. [DOI] [PubMed] [Google Scholar]
- 29.Bradley WD, Hernandez SE, Settleman J, Koleske AJ. Integrin signaling through Arg activates p190RhoGAP by promoting its binding to p120RasGAP and recruitment to the membrane. Mol Biol Cell. 2006;17:4827–4836. doi: 10.1091/mbc.E06-02-0132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mammoto A, Huang S, Ingber DE. Filamin links cell shape and cytoskeletal structure to Rho regulation by controlling accumulation of p190RhoGAP in lipid rafts. J Cell Sci. 2007;120:456–467. doi: 10.1242/jcs.03353. [DOI] [PubMed] [Google Scholar]
- 31.Tsubouchi A, Sakakura J, Yagi R, Mazaki Y, Schaefer E, Yano H, Sabe H. Localized suppression of RhoA activity by Tyr31/118-phosphorylated paxillin in cell adhesion and migration. J Cell Biol. 2002;159:673–683. doi: 10.1083/jcb.200202117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tomar A, Lim ST, Lim Y, Schlaepfer DD. A FAK-p120RasGAP-p190RhoGAP complex regulates polarity in migrating cells. J Cell Sci. 2009;122:1852–1862. doi: 10.1242/jcs.046870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Holinstat M, Knezevic N, Broman M, Samarel AM, Malik AB, Mehta D. Suppression of RhoA activity by focal adhesion kinase-induced activation of p190RhoGAP: role in regulation of endothelial permeability. J Biol Chem. 2006;281:2296–2305. doi: 10.1074/jbc.M511248200. [DOI] [PubMed] [Google Scholar]
- 34.Harrington EO, Shannon CJ, Morin N, Rowlett H, Murphy C, Lu Q. PKCdelta regulates endothelial basal barrier function through modulation of RhoA GTPase activity. Exp Cell Res. 2005;308:407–421. doi: 10.1016/j.yexcr.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 35.Mammoto T, Parikh SM, Mammoto A, Gallagher D, Chan B, Mostoslavsky G, Ingber DE, Sukhatme VP. Angiopoietin-1 requires p190 RhoGAP to protect against vascular leakage in vivo. J Biol Chem. 2007;282:23910–23918. doi: 10.1074/jbc.M702169200. [DOI] [PubMed] [Google Scholar]
- 36.Watson AD, Leitinger N, Navab M, Faull KF, Horkko S, Witztum JL, Palinski W, Schwenke D, Salomon RG, Sha W, Subbanagounder G, Fogelman AM, Berliner JA. Structural identification by mass spectrometry of oxidized phospholipids in minimally oxidized low density lipoprotein that induce monocyte/endothelial interactions and evidence for their presence in vivo. J Biol Chem. 1997;272:13597–13607. doi: 10.1074/jbc.272.21.13597. [DOI] [PubMed] [Google Scholar]
- 37.Birukova AA, Chatchavalvanich S, Rios A, Kawkitinarong K, Garcia JG, Birukov KG. Differential regulation of pulmonary endothelial monolayer integrity by varying degrees of cyclic stretch. Am J Pathol. 2006;168:1749–1761. doi: 10.2353/ajpath.2006.050431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thomas M, Ge Q, Lu JJ, Chen J, Klibanov AM. Cross-linked small polyethylenimines: while still nontoxic, deliver DNA efficiently to mammalian cells in vitro and in vivo. Pharm Res. 2005;22:373–380. doi: 10.1007/s11095-004-1874-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Singleton PA, Chatchavalvanich S, Fu P, Xing J, Birukova AA, Fortune JA, Klibanov AM, Garcia JG, Birukov KG. Akt-mediated transactivation of the S1P1 receptor in caveolin-enriched microdomains regulates endothelial barrier enhancement by oxidized phospholipids. Circ Res. 2009;104:978–986. doi: 10.1161/CIRCRESAHA.108.193367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shikata Y, Birukov KG, Birukova AA, Verin AD, Garcia JG. Involvement of site-specific FAK phosphorylation in sphingosine-1 phosphate- and thrombin-induced focal adhesion remodeling: role of Src and GIT. FASEB J. 2003;17:2240–2249. doi: 10.1096/fj.03-0198com. [DOI] [PubMed] [Google Scholar]
- 41.Shikata Y, Rios A, Kawkitinarong K, DePaola N, Garcia JG, Birukov KG. Differential effects of shear stress and cyclic stretch on focal adhesion remodeling, site-specific FAK phosphorylation, and small GTPases in human lung endothelial cells. Exp Cell Res. 2005;304:40–49. doi: 10.1016/j.yexcr.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 42.Birukov KG, Jacobson JR, Flores AA, Ye SQ, Birukova AA, Verin AD, Garcia JG. Magnitude-dependent regulation of pulmonary endothelial cell barrier function by cyclic stretch. Am J Physiol Lung Cell Mol Physiol. 2003;285:L785–797. doi: 10.1152/ajplung.00336.2002. [DOI] [PubMed] [Google Scholar]
- 43.Tschumperlin DJ, Oswari J, Margulies AS. Deformation-induced injury of alveolar epithelial cells. Effect of frequency, duration, and amplitude. Am J Respir Crit Care Med. 2000;162:357–362. doi: 10.1164/ajrccm.162.2.9807003. [DOI] [PubMed] [Google Scholar]
- 44.Tschumperlin DJ, Margulies SS. Alveolar epithelial surface area-volume relationship in isolated rat lungs. J Appl Physiol. 1999;86:2026–2033. doi: 10.1152/jappl.1999.86.6.2026. [DOI] [PubMed] [Google Scholar]
- 45.Birukova AA, Cokic I, Moldobaeva N, Birukov KG. Paxillin is Involved in the Differential Regulation of Endothelial Barrier by HGF and VEGF. Am J Respir Cell Mol Biol. 2008 doi: 10.1165/rcmb.2008-0099OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fu P, Birukova AA, Xing J, Sammani S, Murley JS, Garcia JG, Grdina DJ, Birukov KG. Amifostine reduces lung vascular permeability via suppression of inflammatory signalling. Eur Respir J. 2009;33:612–624. doi: 10.1183/09031936.00014808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brouns MR, Matheson SF, Settleman J. p190 RhoGAP is the principal Src substrate in brain and regulates axon outgrowth, guidance and fasciculation. Nat Cell Biol. 2001;3:361–367. doi: 10.1038/35070042. [DOI] [PubMed] [Google Scholar]
- 48.Peacock JG, Miller AL, Bradley WD, Rodriguez OC, Webb DJ, Koleske AJ. The Abl-related gene tyrosine kinase acts through p190RhoGAP to inhibit actomyosin contractility and regulate focal adhesion dynamics upon adhesion to fibronectin. Mol Biol Cell. 2007;18:3860–3872. doi: 10.1091/mbc.E07-01-0075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Grande-Garcia A, Echarri A, de Rooij J, Alderson NB, Waterman-Storer CM, Valdivielso JM, del Pozo MA. Caveolin-1 regulates cell polarization and directional migration through Src kinase and Rho GTPases. J Cell Biol. 2007;177:683–694. doi: 10.1083/jcb.200701006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Garcia JG, Davis HW, Patterson CE. Regulation of endothelial cell gap formation and barrier dysfunction: role of myosin light chain phosphorylation. J Cell Physiol. 1995;163:510–522. doi: 10.1002/jcp.1041630311. [DOI] [PubMed] [Google Scholar]
- 51.Velasco G, Armstrong C, Morrice N, Frame S, Cohen P. Phosphorylation of the regulatory subunit of smooth muscle protein phosphatase 1M at Thr850 induces its dissociation from myosin. FEBS Lett. 2002;527:101–104. doi: 10.1016/s0014-5793(02)03175-7. [DOI] [PubMed] [Google Scholar]
- 52.Birukova AA, Rios A, Birukov KG. Long-term cyclic stretch controls pulmonary endothelial permeability at translational and post-translational levels. Exp Cell Res. 2008;314:3466–3477. doi: 10.1016/j.yexcr.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Storck J, Zimmermann ER. Regulation of the thrombin receptor response in human endothelial cells. Thromb Res. 1996;81:121–131. doi: 10.1016/0049-3848(95)00220-0. [DOI] [PubMed] [Google Scholar]
- 54.Herbrand U, Ahmadian MR. p190-RhoGAP as an integral component of the Tiam1/Rac1-induced downregulation of Rho. Biol Chem. 2006;387:311–317. doi: 10.1515/BC.2006.041. [DOI] [PubMed] [Google Scholar]
- 55.Groger M, Pasteiner W, Ignatyev G, Matt U, Knapp S, Atrasheuskaya A, Bukin E, Friedl P, Zinkl D, Hofer-Warbinek R, Zacharowski K, Petzelbauer P, Reingruber S. Peptide Bbeta(15-42) preserves endothelial barrier function in shock. PLoS One. 2009;4:e5391. doi: 10.1371/journal.pone.0005391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nimnual AS, Taylor LJ, Bar-Sagi D. Redox-dependent downregulation of Rho by Rac. Nat Cell Biol. 2003;5:236–241. doi: 10.1038/ncb938. [DOI] [PubMed] [Google Scholar]
- 57.Ramchandran R, Mehta D, Vogel SM, Mirza MK, Kouklis P, Malik AB. Critical role of Cdc42 in mediating endothelial barrier protection in vivo. Am J Physiol Lung Cell Mol Physiol. 2008;295:L363–369. doi: 10.1152/ajplung.90241.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Broman MT, Kouklis P, Gao X, Ramchandran R, Neamu RF, Minshall RD, Malik AB. Cdc42 regulates adherens junction stability and endothelial permeability by inducing alpha-catenin interaction with the vascular endothelial cadherin complex. Circ Res. 2006;98:73–80. doi: 10.1161/01.RES.0000198387.44395.e9. [DOI] [PubMed] [Google Scholar]