

Abstract
Objective
Studies in humans support a role for the oral pathogen Porphyromonas gingivalis in the development of inflammatory atherosclerosis. The goal of this study was to determine if P. gingivalis infection accelerates inflammation and atherosclerosis in the innominate artery of mice, an artery which has been reported to exhibit many features of human atherosclerotic disease, including plaque rupture.
Methods and Results
Apolipoprotein E-deficient (ApoE−/−) mice were orally infected with P. gingivalis, and Magnetic Resonance Imaging (MRI) was used to monitor the progression of atherosclerosis in live mice. P. gingivalis infected mice exhibited a statistically significant increase in atherosclerotic plaque in the innominate artery as compared to uninfected mice. Polarized light microscopy and immunohistochemistry revealed that the innominate arteries of infected mice had increased lipids, macrophages and T cells as compared to uninfected mice. Increases in plaque, total cholesterol esters and cholesterol monohydrate crystals, macrophages, and T cells were prevented by immunization with heat-killed P. gingivalis prior to pathogen exposure.
Conclusions
These are the first studies to demonstrate progression of inflammatory plaque accumulation in the innominate arteries by in-vivo MRI analysis following pathogen exposure, and to document protection from plaque progression in the innominate artery via immunization.
Keywords: Atherosclerosis, inflammation, P. gingivalis, infection, innominate artery, MRI
Atherosclerosis is a chronic inflammatory disease characterized by sub-endothelial accumulation of inflammatory cells and lipids, which collectively contribute to occlusive disease or to less occlusive plaques at high risk for disruption. The activation of endothelial cells at atherosclerotic lesion-prone sites in the arterial tree results in the up-regulation of cell adhesion molecules and chemokines, which mediate the recruitment of circulating monocytes. Accumulation of monocytes, monocyte-derived phagocytes and T cells contribute to chronic inflammation and atherosclerosis [1]. Epidemiological studies in humans and studies of mouse models of atherosclerosis support a role for infectious agents in inflammatory atherosclerotic plaque accumulation. Infectious agents can induce cellular and molecular changes characteristic of inflammatory processes observed in atherosclerosis, and several studies have implicated the induction of an inflammatory response by infectious agents as a possible mechanism linking infection to the acceleration of atherosclerosis [2–7].
We previously demonstrated that oral infection with Porphyromonas gingivalis, the etiological agent of human periodontal disease, accelerates plaque accumulation in the aortic sinus in an apolipoprotein E (ApoE−/−) mouse model [5, 8]. While these studies in the ApoE−/− mouse model have described atherosclerotic lesions of the aortic sinus by histological analysis, intra-plaque rupture or signs of plaque disruption at the aortic sinus have not been reported [9]. In contrast, recent studies have documented the presence of ruptured plaques in the innominate artery of ApoE−/− mice [10–14]. The innominate artery has been reported to undergo a high degree of lesion progression, and lesions in this artery reported to express features characteristic of clinical disease in humans [10, 11, 15–17]. However, the ability of infectious agents to induce or accelerate inflammatory atherosclerotic plaque has not been evaluated in the innominate artery.
Recently in vivo mouse Magnetic Resonance Imaging (MRI) has been utilized to document atherosclerotic plaque in vascular beds and in small vessels such as carotid, innominate, and subclavian arteries [18]. The objectives of this study were (i) to use serial in vivo MRI to document the progression of atherosclerotic plaque in the innominate artery following P. gingivalis exposure in a longitudinal fashion; (ii) to characterize the inflammatory response in the innominate artery following P. gingivalis exposure, and (iii) to determine if therapeutic intervention could protect mice from P. gingivalis induced inflammatory atherosclerosis in the innominate artery.
Methods
Bacterial Challenge and Immunization
Male six-week-old ApoE−/− mice were divided into 4 groups (Table 1). All mice were cared in accordance with Boston University Institutional Animal Care and Use Committee procedures and received a high fat diet (0.2% of cholesterol, 21.2% of Fat, 13.7% saturated fatty acid, 7.3% total unsaturated fatty acid; Harlan Teklad; TD.88137) throughout the experiment. Mice were challenged with P. gingivalis 381 or PBS in 2% carboxymethyl cellulose [5, 8, 19]. Some groups were immunized subcutaneously 2 times per week for 3 weeks with heat-killed P. gingivalis 381 whole-organism preparations without adjuvant (Supplemental Methods) [8, 19, 20].
Table 1.
Prevention of P. gingivalis infection protects mice from plaque accumulation.
| Group | Immunization | Bacterial Infection | Plaque area (mm2) |
|---|---|---|---|
| i | − | − | 0.48 ± 0.03* |
| ii | + | − | 0.53 ± 0.07 |
| iii | − | + | 0.68 ± 0.10*† |
| iv | + | + | 0.45 ± 0.13† |
Plaque area was measured by MRI at the end of the experimental period (25 wks). Plaque area was manually segmented and calculated as follows: plaque area = outer boundary – inner boundary.
p < 0.05 between group i and iii,
p < 0.01 between group iii and iv, and no significant differences between group i and ii by One-way ANOVA. n = 6 in each group.
In vivo Mouse Magnetic Resonance Angiography (MRA) and MRI
In vivo imaging of the innominate artery was performed using a vertical-bore Bruker 11.7-T Avance spectometer (Bruker; Billerica, MA). Details are described below and in Supplemental Methods. Data acquisition and reconstruction were performed with ParaVision software provided by the vendor. Mice were placed headfirst in a supine position in a vertical 30 mm probe (Micro 2.5). The animals were maintained at room temperature (23°C) for the imaging experiments. Mice were anesthetized with 0.5–2% inhaled isoflurane and immobilized using a holder with a bite bar and wrapped with parafilm to reduce motion. Respiration was monitored with a respiration pillow placed on the abdomen using a small animal monitoring and gating system (SA Instruments, Wahkesha, WI).
The un-gated 3D gradient echo MRA was acquired as scout images. A fast low-angle shot (FLASH) sequence was used. Respiration-gated T1-weighted (T1W) black-blood (T1BB) Magnetic Resonance (MR) images were acquired with a 2D axial gradient echo flow compensation (GEFC) sequence. Continuous axial images of the innominate artery were acquired 0.3mm below the branch. A 8 mm saturation band was placed 0.5 mm inferior to the imaging plane to suppress the blood signal. The total scan time was ~ 20 min.
Image analysis
Visualization of the vasculature was achieved by 3D maximum intensity projections (MIP) of angiographic images reconstructed using Paravisiontm. Black-blood images were used to calculate the plaque area. By manually segmenting the lumen and the outer wall boundaries with Image J (NIH), the outer wall and luminal cross-sectional areas were measured on both the MR images and the histological sections. Plaque area was calculated from the area of the outer wall boundary (total area) minus the lumen area. The intra reader reliability was excellent with interclass correlation coefficient values of 0.87. The maximum plaque thickness was measured by drawing a line across the thickest region of the vessel wall on the cross-sectional images. The right subclavian artery was used as an internal anatomical landmark for registration of MRI images acquired at different imaging sessions and for histology.
Histology
Cryosections obtained from the innominate artery (obtained from 50% of mice from each group) were stained with hematoxylin and eosin, and plaque area was quantified from on-screen images using IPLabs (Scanalytics, Inc, Rockville, MD). Polarized light photomicrographs were taken at 25°C and polarized light microscopy using unstained sections was used to detect lipids (cholesterol monohydrate crystals and cholesterol esters) based on their birefringecfnce [21, 22] and Supplemental Methods. The total area of cholesterol monohydrate crystals and cholesterol esters was calculated using Image J. Sections obtained from the innominate artery and the spleen were examined by immunohistochemistry using anti-mouse F4/80 (Serotec, Raleigh, NC), CD3 (abcam, Cambridge, MA), iNOS (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), Arginase-I (Arg-I) (BD Transduction Laboratories, Sparks, MD), actin (Sigma-Aldrich, St. Louis, MO) antibodies, or isotype-matched antibodies. Quantitative immunohistochemistry was performed as previously described (Supplemental Reference 1). Verhoeff-van Gieson staining for elastin was performed using the Accustain Elastic Stain kit (Sigma-Aldrich, St. Louis, MO) according to the manufacturer’s recommendations. Picrosirius red staining was used for the histological assessment of the total collagen content (Electron Microscopy Sciences, Hatfield, PA). Collagen Type I and III were visualized in circularly polarized light, quantified using IPLabs (Scanalytics Inc., Fairfax, VA), and the collagen Type I / III ratio calculated.
Statistical analyses
Analyses were performed using SPSS 11.0 (Systat software, Chicago, IL). One-way ANOVA with Tukey-Kramer multiple-comparisons test was performed to assess the differences in plaque, lipid, macrophage, and T cell accumulation. Two independent observers blinded to the histological findings (N.H. and C.H.) analyzed the black-blood T1W images to calculate the plaque area. The inter-observer variability was assessed by using the inter-class correlation coefficient (ICC). The data are presented as the mean ± SEM. Probability values of p <0.05 were considered significant.
Results
P. gingivalis infection accelerates atherosclerosis in the innominate artery and this is prevented by immunization prior to challenge
To examine atherosclerotic plaque accumulation in the subclavian and innominate arteries following P. gingivalis infection we first utilized MRA. This analysis of blood flow revealed that P. gingivalis exposure resulted in the progression of occlusive plaque in the innominate arteries but not in the subclavian arteries as measured at 27 wks following infection (data not shown). Based on these results we subsequently designed experiments using MRI to measure atherosclerotic plaque accumulation in the innominate artery following P. gingivalis challenge (Supplemental Figure 1). Furthermore, to determine if prevention of P. gingivalis infection via therapeutic intervention provided protection from plaque progression in the innominate artery, we included one group of mice immunized with a heat-killed preparation of P. gingivalis prior to infection (Table 1 and Supplemental Figure 1) [8]. We have previously determined that immunization prior to P. gingivalis infection prevents P. gingivalis induced oral inflammatory bone loss and atherosclerosis in the aortic arch region [5]. MRI analysis revealed that mice infected with P. gingivalis exhibited an increase in plaque area in the innominate artery at 25 wks after the start of P. gingivalis challenge (Figure 1 and Table 1). Representative MR images revealed progression of plaque in the innominate artery at 11wks and 25 wks after the start of P. gingivalis challenge (Figure 1C and 1D). MRI analysis revealed a statistically significant increase in plaque area in the innominate arteries of infected mice as compared to uninfected mice as determined at 25 wks after infection (Table 1). Importantly, immunization with a heat-killed preparation of P. gingivalis prior to infection with live bacteria was found to prevent the increase in plaque area observed in the innominate arteries of P. gingivalis infected mice (Table 1).
Figure 1. Magnetic Resonance Angiography (MRA) and Imaging (MRI) analysis of plaque progression following P. gingivalis infection.
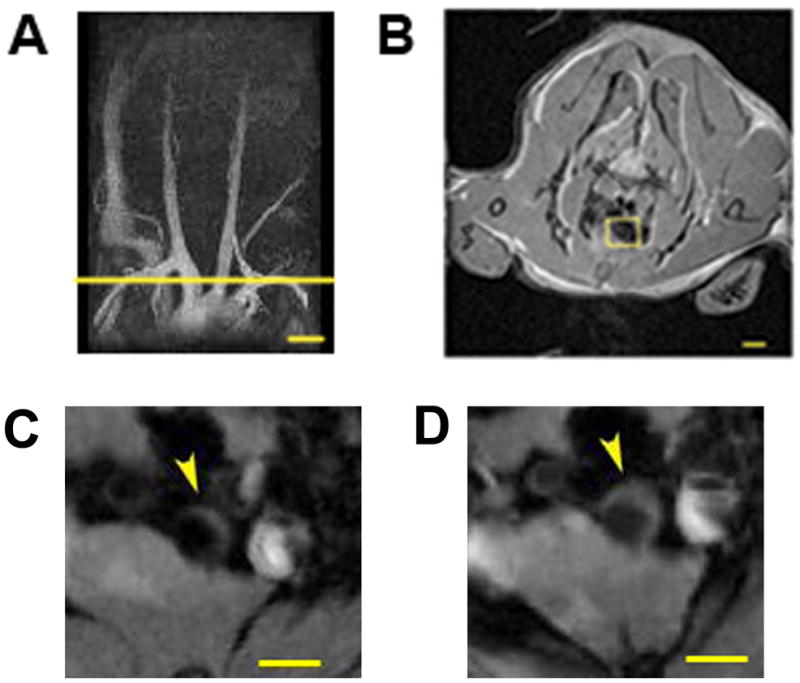
(A) Representative Magnetic Resonance (MR) angiogram of aortic arch and major vessels of a P. gingivalis infected mouse at 34 weeks of age. (B) Axial MR image from the yellow line in Figure 1A of the innominate artery of a mouse, 0.3mm below its bifurcation. MRI of the innominate artery of one mouse at 11wks (C) and 25 wks (D) after the start of bacterial challenge are shown. Bar represents 500 μm.
Histological analysis confirms that P. gingivalis infection accelerates plaque accumulation in the innominate artery
To confirm and expand the results obtained by MRI, mice were sacrificed following the final MRI analysis for histological analysis of the extracted innominate arteries. Cryosections were obtained from the region corresponding to the greatest plaque size (approximately 0.3 mm below the bifurcation) and representative sections stained with Hematoxylin & Eosin. As shown in Figure 2, the innominate arteries obtained from non-immunized mice infected with P. gingivalis had increased plaque areas as compared to that observed in the innominate arteries of non-immunized uninfected mice (p <0.01). However, in immunized mice we did not observe differences in the plaque area between infected mice and uninfected mice (Figure 2E). As observed by MRI, immunization with a heat-killed preparation of P. gingivalis prior to challenge with live bacteria was found to prevent the increase in plaque area in the innominate arteries of P. gingivalis infected mice (p < 0.01). Likewise the plaque area in the innominate arteries of uninfected mice was similar to that observed in immunized mice, indicating that immunization alone did not alter atherosclerotic plaque area (Figure 2E).
Figure 2. Histological analysis of plaque in the innominate artery following P. gingivalis infection.

Representative sections obtained from the region corresponding to greatest plaque size in the innominate artery (0.3 mm below the bifurcation) of (A) group i: uninfected / non-immunized, (B) group ii: uninfected / immunized, (C) group iii: P. gingivalis infected / non-immunized, and (D) group iv: P. gingivalis infected / immunized ApoE−/− mouse. Bar represents 200 μm. (E) Plaque area was quantified from on-screen images using IPLabs (Scanalytics, Inc). In non-immunized ApoE−/− mice P. gingivalis infection increased plaque area compared to uninfected ApoE−/− mice. In infected ApoE−/− mice the plaque area was significantly decreased in the immunized ApoE−/− mice compared to non-immunized ApoE−/− mice. ** p < 0.01, One-way ANOVA. NS indicates no significant differences.
P. gingivalis infection leads to lipid accumulation in the innominate artery
We next characterized the accumulation of lipids in the innominate artery of infected mice using polarized light microscopy of unstained histology sections (Figure 3). In the innominate arteries of non-immunized mice infected with P. gingivalis we observed increased lipids as compared to non-immunized uninfected mice (p < 0.05) (Figure 3E). Immunization with a heat-killed preparation of P. gingivalis prior to challenge prevented the increase in lipid accumulation observed in the innominate arteries of P. gingivalis infected mice (p < 0.05). Thus the innominate arteries of P. gingivalis infected mice exhibited both larger atherosclerotic plaque areas and higher levels of lipids. The area of lipids observed in the innominate arteries of non-immunized uninfected mice was similar to that observed in immunized uninfected mice, indicating that immunization alone did not alter lipid accumulation (Figure 3E).
Figure 3. Lipid accumulation in the innominate arteries following P. gingivalis infection.

Representative images of birefringence in plaque corresponding to lipids (total cholesterol esters and cholesterol monohydrate crystals) are shown. (A) group i: uninfected / non-immunized, (B) group ii: uninfected / immunized, (C) group iii: P. gingivalis infected / non-immunized, and (D) group iv: P. gingivalis infected / immunized ApoE−/− mouse. Bar represents 200 μm. (E) Lipid area. In non-immunized ApoE−/− mice P. gingivalis infection increased lipid area compared to uninfected ApoE−/− mice. In P. gingivalis infected ApoE−/− mice, the lipid area was significantly decreased in the immunized group compared to non-immunized group. *p < 0.01, One-way ANOVA. NS indicates no significant difference.
P. gingivalis infection leads to macrophage accumulation in the innominate artery
We next characterized the composition of the inflammatory lesions in the innominate arteries of P. gingivalis infected mice by immunohistochemistry for detection of macrophages. The innominate arteries obtained from non-immunized mice infected with P. gingivalis exhibited increased staining for the macrophage marker F4/80 as compared to non-immunized uninfected mice (Figure 4). Immunization with a heat-killed preparation of P. gingivalis prior to challenge with live bacteria was found to prevent the increase in macrophage specific staining observed in the innominate arteries of P. gingivalis infected mice (Figure 4). We also observed that the innominate arteries obtained from P. gingivalis infected mice exhibited enhanced staining for the M1 macrophage marker iNOS as compared to uninfected mice (Supplemental Figure 2). Immunization with a heat-killed preparation of P. gingivalis prior to challenge with live bacteria was found to prevent the increase in iNOS specific staining observed in the innominate arteries of P. gingivalis infected mice (Supplemental Figure 2). We also observed that immunized uninfected mice exhibited increased Arginase-I staining in both the innominate artery and spleen samples as compared to non-immunized uninfected mice. These results suggest that immunization itself may alter macrophage subtypes, and future studies will examine polarized macrophage populations in P. gingivalis induced plaque formation and protection by immunization.
Figure 4. Macrophage accumulation in the innominate arteries following P. gingivalis infection.

Representative images of plaque sections. The macrophage marker F4/80 (A–I) was detected immunohistochemically in the innominate artery. (A and F) group i: uninfected / non-immunized, (B and G) group ii: uninfected / immunized, (C and H) group iii: P. gingivalis infected / non-immunized, and (D and I) group iv: P. gingivalis infected / immunized ApoE−/− mouse. (E) Isotype control. F, G, H and I are the magnified image in the boxes in A, B, C and D respectively. Bar represents 200 μm (A–E) and 50 μm (F–I). (J) F4/80 area in the innominate artery. In non-immunized ApoE−/− mice P. gingivalis infection increased macrophage expression compared to uninfected ApoE−/− mice. In P. gingivalis infected ApoE−/− mice, macrophage accumulation was significantly decreased in immunized ApoE−/− mice compared to non-immunized ApoE−/− mice. *p < 0.05, One-way ANOVA. NS indicates no significant differences.
P. gingivalis infection enhanced T cell accumulation in the innominate artery
As shown in Figure 5, the innominate arteries obtained from non-immunized P. gingivalis infected mice exhibited increased staining for the T cell marker CD3 as compared to the innominate arteries obtained from non-immunized uninfected mice, although this was not statistically significant. We also observed an increase in T cell specific staining in the innominate arteries of uninfected mice that were immunized as compared to uninfected immunized mice (Figure 5). However, T cell specific staining observed in the innominate arteries of non-immunized uninfected mice was similar to those observed in immunized uninfected mice, indicating that immunization alone did not alter T cell accumulation (Figure 5E).
Figure 5. T cell accumulation in the innominate arteries following P. gingivalis infection.

Representative images of plaque sections. The T cell marker CD3 was detected immunohistochemically in the innominate artery. (A and F) group i: uninfected / non-immunized, (B and G) group ii: uninfected / immunized, (C and H) group iii: P. gingivalis infected / non-immunized, (D and I) group iv: P. gingivalis infected / immunized ApoE−/− mouse, and (E) isotype control. Bar represents 200 μm (A–E) and 50 μm (F–I). F, G, H, and I are the magnified images in the boxes in A, B, C, and D, respectively. Arrows point the positive staining of CD3 area in the innominate artery. (J) CD3 area. In non-immunized ApoE−/− mice P. gingivalis infection increased CD3 expression as compared to uninfected ApoE−/− mice. In P. gingivalis infected ApoE−/− mice, CD3 expression was decreased in immunized ApoE−/− mice compared to non-immunized ApoE−/− mice. NS indicates no significant differences.
P. gingivalis infection induces inflammatory cell recruitment in spleens
Consistent with results obtained in the innominate arteries of P. gingivalis infected mice, the accumulation of macrophages and T cells was also observed in spleen samples (Supplemental Figure 3). We also observed that P. gingivalis infection resulted in an increased expression of F4/80 and iNOS in spleen samples, and that immunization prior to infection with P. gingivalis resulted in diminished F4/80 and iNOS as compared to that observed in non-immunized P. gingivalis infected mice. Likewise, Arg-I positive cells were increased in spleen samples of uninfected immunized mice as compared to uninfected non-immunized mice (Supplemental Figure 3).
P. gingivalis infection also resulted in increased levels of T cells in the spleen samples as compared to that observed in uninfected mice (p < 0.001; Supplemental Figure 3). Immunization with a heat-killed preparation of P. gingivalis prior to challenge with live bacteria prevented the increase in T cell specific staining observed in the spleens of P. gingivalis infected mice (p < 0.001; Supplemental Figure 3V). The increase in inflammatory cells in the spleens of infected mice also correlated with increased spleen / body weight ratios (Supplemental Figure 4). P. gingivalis infection did not alter the serum levels of IL-6, TNF-α, IL-1α, IL-1β, GM-CSF or IFN-γ (data not shown). However, uninfected immunized mice exhibited elevated levels of serum IL-6 as compared to uninfected non-immunized mice (data not shown).
Characterization of collagen deposition and elastic laminae in the innominate arteries of P. gingivalis infected mice
The area of total collagen was slightly increased in P. gingivalis infected mice as compared to uninfected mice, although this was not statistically significant (Supplemental Figure 5I). We did however observe a statistically significant increase in the ratio of collagen type I/III in P. gingivalis infected immunized mice as compared to non-immunized mice (Supplemental Figure 5J). These results suggest that immunization is associated with alterations in collagen deposition in the innominate artery which may be associated with plaque stability [23]. We also observed some degree of degradation of elastic laminae in all groups of mice examined (Figure 6). Mice infected with P. gingivalis exhibited larger discontinuities as compared to uninfected mice. Differences in elastic discontinuities correlated with smooth muscle cell penetration into the intima in P. gingivalis infected mice. These observed changes in P. gingivalis infected mice were not observed in mice that were first immunized prior to P. gingivalis infection (Figure 6E–L).
Figure 6. Characterization of elastic laminae in the innominate arteries of P. gingivalis infected mice.

Representative Verhoeff-van Gieson staining for elastin shows elastic laminae (A–H) and α-actin staining for smooth muscle cells (arrows in I–L) in the innominate artery are shown. Discontinuities in the elastic lamina were observed (arrows in E–H). α-actin was detected at the sites of elastin degradation (E–L). (A, E, and I) group i: uninfected / non-immunized, (B, F, and J) group ii: uninfected / immunized, (C, G, and K) group iii: P. gingivalis infected / non-immunized, and (D, H, and L) group iv: P. gingivalis infected / immunized ApoE−/− mouse. Bar represents 200 μm (A–D) and 50 μm (E–L). E and I, F and J, G and K, H and L are the higher magnification of the framed area in A, B, C, and D, respectively.
Discussion
A hallmark of infection with P. gingivalis is the induction of a chronic inflammatory response [24, 25]. P. gingivalis induces a local inflammatory response that results in oral bone destruction, which is manifested as periodontal disease, an inflammatory disease that affects approximately 100 million people in the US [25]. In addition to chronic inflammation at the initial site of infection, mounting evidence has accumulated supporting a role for P. gingivalis-mediated periodontal disease as a risk factor for systemic diseases including, diabetes, pre-term birth, stroke, acute cerebrovascular ischemia, and atherosclerotic cardiovascular disease [24–30]. Case control studies have concluded that there is correlation between cardiovascular disease and periodontal disease after adjusting for confounding factors including cholesterol levels, smoking, hypertension, social class, and body mass index [31, 32]. Results from the Oral Infections and Vascular Disease Epidemiology Study revealed an association between periodontal disease pathogens and sub-clinical atherosclerosis [33]. P. gingivalis has also been detected in human atherosclerotic plaque [2, 3].
Plaque rupture is the basis for the coronary thrombosis in acute ischemia [34]. In humans plaques with extensive macrophage accumulation and highly active inflammation have a greater likelihood of disruption at their luminal surface and formation of a life-threatening thrombus [34]. In ApoE−/− mice the innominate artery exhibits vessel narrowing characterized by atrophic media and perivascular inflammation and plaque disruption [10]. It has also been reported that spontaneous plaque rupture may occur in the innominate artery in ApoE−/− mice [10]. However, the unknown timing of disruption precludes MR imaging of characteristics of the plaque just before disruption, as it has been done with a rabbit model of controlled plaque disruption [35].
In this study, we demonstrate that P. gingivalis infection accelerates atherosclerotic plaque accumulation in the innominate artery. In vivo MRI imaging revealed that each of the mice exposed to P. gingivalis exhibited a greater degree of progressive encroachment of atherosclerotic plaque into the lumen of the innominate arteries as compared to uninfected mice, with increases in areas of plaques found in these arteries following pathogen challenge over a 14 week period. Polarized light microscopy and immunohistochemistry revealed that the innominate arteries and spleens of infected mice had higher levels of total cholesterol esters and cholesterol monohydrate crystals, macrophages, and T cells as compared to uninfected mice. Furthermore, increases in mean plaque area, total cholesterol esters and cholesterol monohydrate crystals, macrophage, and T cell accumulation were prevented by immunization with a heat-killed preparation of P. gingivalis prior to challenge with live bacteria. Collectively these results demonstrate that MRI is an effective tool to measure atherosclerotic plaque accumulation in the innominate arteries in response to P. gingivalis exposure.
Importantly in the present study, we confirmed that histological analysis in the innominate artery correlated with plaque area measurements determined by in vivo MRI. The use of an inferior pre-saturation band together with respiratory-gating sufficiently reduced phase-ghosting artifacts and decreased intraluminal signal. This approach allowed for the delineation of the vessel wall and visualization of atherosclerotic plaque in the innominate artery.
Histological and immunohistochemical analysis of the innominate artery revealed that P. gingivalis exposure correlated with a higher inflammatory infiltrate with high numbers of macrophages and T cells, and increases in total cholesterol esters and cholesterol monohydrate crystals accumulation. Although the presence of T cells in atherosclerotic lesions is well documented, the presence of T cells or macrophages in the innominate arteries following P. gingivalis exposure has not previously been demonstrated. We also confirmed that P. gingivalis infection resulted in enhanced staining for the M1 macrophage marker iNOS and that this was prevented by immunization. M1 macrophages typically participate as inducer and effector cells in polarized Th1 responses and mediate resistance against intracellular parasites [36]. The ability of P. gingivalis to induce iNOS staining in plaque samples is consistent with the ability of this pathogen to be internalized in various host cells including macrophages [37] and to previous observations of a Th1 induced response in the aortic arch [38]. It will be important in future studies to determine if P. gingivalis infection also modifies levels of Ly-6Chi circulating leukocytes and macrophage populations, as increased levels of Ly-6Chi cells have been proposed as a proinflammatory marker associated with atherosclerosis [39]. Finally, P. gingivalis infection was also demonstrated to increase collagen and smooth muscle cell accumulation in the innominate arteries. These results suggest that P. gingivalis infection can modify smooth muscle cell proliferation in the innominate artery [40].
In conclusion, using in vivo MRI analysis together with ex vivo immunohistochemistry, our studies demonstrate that P. gingivalis exposure results in an increase of atherosclerotic plaque accumulation in the innominate artery that is associated with the accumulation of lipids and macrophages. Furthermore, increases in mean plaque area, lipids, and macrophage accumulation were prevented by immunization with a heat-killed preparation of P. gingivalis prior to challenge with live bacteria. An important question is whether P. gingivalis accelerates atherosclerotic plaque formation in the innominate artery leading to increased numbers of vulnerable plaques, and possibly enhanced plaque rupture. Future studies will explore this possibility as well as the testing of new therapeutic strategies to prevent P. gingivalis-induced atherosclerotic disease.
Supplementary Material
Acknowledgments
We would like to acknowledge Zifang Guo for technical assistance.
Sources of Funding
This work was supported by National Institutes of Health grant HL-RO1-80387 (C. A. G.) and PO1-AI078894 (C. A. G. and J. A. H.).
Footnotes
Disclosure
None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Chie Hayashi, Email: hayashi@bu.edu.
Jason Viereck, Email: ViereckJ@neneuro.com.
Ning Hua, Email: huaning@bu.edu.
Alkystis Phinikaridou, Email: alkystisp@gmail.com.
Andres G. Madrigal, Email: madrigal@bu.edu.
Frank C. Gibson, III, Email: fgibson@bu.edu.
James A. Hamilton, Email: jhamilt@bu.edu.
Caroline A. Genco, Email: cgenco@bu.edu.
References
- 1.Swirski FK, Pittet MJ, Kircher MF, et al. Monocyte accumulation in mouse atherogenesis is progressive and proportional to extent of disease. Proc Natl Acad Sci U S A. 2006;103:10340–10345. doi: 10.1073/pnas.0604260103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Haraszthy VI, Zambon JJ, Trevisan M, et al. Identification of periodontal pathogens in atheromatous plaques. J Periodontol. 2000;71:1554–1560. doi: 10.1902/jop.2000.71.10.1554. [DOI] [PubMed] [Google Scholar]
- 3.Padilla C, Lobos O, Hubert E, et al. Periodontal pathogens in atheromatous plaques isolated from patients with chronic periodontitis. J Periodontal Res. 2006;41:350–353. doi: 10.1111/j.1600-0765.2006.00882.x. [DOI] [PubMed] [Google Scholar]
- 4.Brodala N, Merricks EP, Bellinger DA, et al. Porphyromonas gingivalis bacteremia induces coronary and aortic atherosclerosis in normocholesterolemic and hypercholesterolemic pigs. Arterioscler Thromb Vasc Biol. 2005;25:1446–1451. doi: 10.1161/01.ATV.0000167525.69400.9c. [DOI] [PubMed] [Google Scholar]
- 5.Gibson FC, 3rd, Hong C, Chou HH, et al. Innate immune recognition of invasive bacteria accelerates atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2004;109:2801–2806. doi: 10.1161/01.CIR.0000129769.17895.F0. [DOI] [PubMed] [Google Scholar]
- 6.Lalla E, Lamster IB, Hofmann MA, et al. Oral infection with a periodontal pathogen accelerates early atherosclerosis in apolipoprotein E-null mice. Arterioscler Thromb Vasc Biol. 2003;23:1405–1411. doi: 10.1161/01.ATV.0000082462.26258.FE. [DOI] [PubMed] [Google Scholar]
- 7.Li L, Messas E, Batista EL, Jr, et al. Porphyromonas gingivalis infection accelerates the progression of atherosclerosis in a heterozygous apolipoprotein E-deficient murine model. Circulation. 2002;105:861–867. doi: 10.1161/hc0702.104178. [DOI] [PubMed] [Google Scholar]
- 8.Miyamoto T, Yumoto H, Takahashi Y, et al. Pathogen-accelerated atherosclerosis occurs early after exposure and can be prevented via immunization. Infect Immun. 2006;74:1376–1380. doi: 10.1128/IAI.74.2.1376-1380.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jackson CL. Defining and defending murine models of plaque rupture. Arterioscler Thromb Vasc Biol. 2007;27:973–977. doi: 10.1161/01.ATV.0000261545.53586.f0. [DOI] [PubMed] [Google Scholar]
- 10.Rosenfeld ME, Polinsky P, Virmani R, et al. Advanced atherosclerotic lesions in the innominate artery of the ApoE knockout mouse. Arterioscler Thromb Vasc Biol. 2000;20:2587–2592. doi: 10.1161/01.atv.20.12.2587. [DOI] [PubMed] [Google Scholar]
- 11.Rosenfeld ME, Carson KG, Johnson JL, et al. Animal models of spontaneous plaque rupture: the holy grail of experimental atherosclerosis research. Curr Atheroscler Rep. 2002;4:238–242. doi: 10.1007/s11883-002-0025-3. [DOI] [PubMed] [Google Scholar]
- 12.Calara F, Silvestre M, Casanada F, et al. Spontaneous plaque rupture and secondary thrombosis in apolipoprotein E-deficient and LDL receptor-deficient mice. J Pathol. 2001;195:257–263. doi: 10.1002/path.915. [DOI] [PubMed] [Google Scholar]
- 13.Falk E, Schwartz SM, Galis ZS, et al. Putative murine models of plaque rupture. Arterioscler Thromb Vasc Biol. 2007;27:969–972. doi: 10.1161/01.ATV.0000261572.33474.e0. [DOI] [PubMed] [Google Scholar]
- 14.Schwartz SM, Galis ZS, Rosenfeld ME, et al. Plaque rupture in humans and mice. Arterioscler Thromb Vasc Biol. 2007;27:705–713. doi: 10.1161/01.ATV.0000261709.34878.20. [DOI] [PubMed] [Google Scholar]
- 15.Getz GS. Mouse model of unstable atherosclerotic plaque? Arterioscler Thromb Vasc Biol. 2000;20:2503–2505. doi: 10.1161/01.atv.20.12.2503. [DOI] [PubMed] [Google Scholar]
- 16.Reardon CA, Getz GS. Mouse models of atherosclerosis. Curr Opin Lipidol. 2001;12:167–173. doi: 10.1097/00041433-200104000-00010. [DOI] [PubMed] [Google Scholar]
- 17.Reardon CA, Blachowicz L, Lukens J, et al. Genetic background selectively influences innominate artery atherosclerosis: immune system deficiency as a probe. Arterioscler Thromb Vasc Biol. 2003;23:1449–1454. doi: 10.1161/01.ATV.0000079793.58054.2E. [DOI] [PubMed] [Google Scholar]
- 18.Tu P, Bhasin S, Hruz PA, et al. Genetic disruption of myostatin reduces the development of proatherogenic dyslipidemia and atherogenic lesions in Ldlr null mice. Diabetes. 2009 doi: 10.2337/db09-0349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gibson FC, 3rd, Genco CA. Prevention of Porphyromonas gingivalis-induced oral bone loss following immunization with gingipain R1. Infect Immun. 2001;69:7959–7963. doi: 10.1128/IAI.69.12.7959-7963.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gibson FC, 3rd, Gonzalez DA, Wong J, et al. Porphyromonas gingivalis-Specific Immunoglobulin G Prevents P. gingivalis-Elicited Oral Bone Loss in a Murine Model. Infect Immun. 2004;72:2408–2411. doi: 10.1128/IAI.72.4.2408-2411.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Waugh DA, Small DM. Identification and detection of in situ cellular and regional differences of lipid composition and class in lipid-rich tissue using hot stage polarizing light microscopy. Lab Invest. 1984;51:702–714. [PubMed] [Google Scholar]
- 22.Phinikaridou A, Hallock KJ, Qiao Y, et al. A robust rabbit model of human atherosclerosis and atherothrombosis. J Lipid Res. 2009 doi: 10.1194/jlr.M800460-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dong B, Zhang C, Feng JB, et al. Overexpression of ACE2 enhances plaque stability in a rabbit model of atherosclerosis. Arterioscler Thromb Vasc Biol. 2008;28:1270–1276. doi: 10.1161/ATVBAHA.108.164715. [DOI] [PubMed] [Google Scholar]
- 24.Hayashi C, Gudino CV, Gibson FC, 3rd, et al. Pathogen-Induced Chronic Inflammation at Sites Distant from Oral Infection: Bacterial Persistence and Modulation of Cell Specific Innate Immune Inflammatory Pathways. Mol Oral Microbiol. 2010 doi: 10.1111/j.2041-1014.2010.00582.x. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gibson FC, 3rd, Ukai T, Genco CA. Engagement of specific innate immune signaling pathways during Porphyromonas gingivalis induced chronic inflammation and atherosclerosis. Front Biosci. 2008;13:2041–2059. doi: 10.2741/2822. [DOI] [PubMed] [Google Scholar]
- 26.Amar S, Gokce N, Morgan S, et al. Periodontal disease is associated with brachial artery endothelial dysfunction and systemic inflammation. Arterioscler Thromb Vasc Biol. 2003;23:1245–1249. doi: 10.1161/01.ATV.0000078603.90302.4A. [DOI] [PubMed] [Google Scholar]
- 27.Dasanayake AP, Russell S, Boyd D, et al. Preterm low birth weight and periodontal disease among African Americans. Dent Clin North Am. 2003;47:115–125. x–xi. doi: 10.1016/s0011-8532(02)00056-3. [DOI] [PubMed] [Google Scholar]
- 28.Morrison HI, Ellison LF, Taylor GW. Periodontal disease and risk of fatal coronary heart and cerebrovascular diseases. J Cardiovasc Risk. 1999;6:7–11. doi: 10.1177/204748739900600102. [DOI] [PubMed] [Google Scholar]
- 29.Pussinen PJ, Alfthan G, Jousilahti P, et al. Systemic exposure to Porphyromonas gingivalis predicts incident stroke. Atherosclerosis. 2006 doi: 10.1016/j.atherosclerosis.2006.06.027. [DOI] [PubMed] [Google Scholar]
- 30.Tonetti MS, D'Aiuto F, Nibali L, et al. Treatment of periodontitis and endothelial function. N Engl J Med. 2007;356:911–920. doi: 10.1056/NEJMoa063186. [DOI] [PubMed] [Google Scholar]
- 31.Seinost G, Wimmer G, Skerget M, et al. Periodontal treatment improves endothelial dysfunction in patients with severe periodontitis. Am Heart J. 2005;149:1050–1054. doi: 10.1016/j.ahj.2004.09.059. [DOI] [PubMed] [Google Scholar]
- 32.Wu T, Trevisan M, Genco RJ, et al. Periodontal disease and risk of cerebrovascular disease: the first national health and nutrition examination survey and its follow-up study. Arch Intern Med. 2000;160:2749–2755. doi: 10.1001/archinte.160.18.2749. [DOI] [PubMed] [Google Scholar]
- 33.Desvarieux M, Demmer RT, Rundek T, et al. Periodontal microbiota and carotid intima-media thickness: the Oral Infections and Vascular Disease Epidemiology Study (INVEST) Circulation. 2005;111:576–582. doi: 10.1161/01.CIR.0000154582.37101.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shah PK. Mechanisms of plaque vulnerability and rupture. J Am Coll Cardiol. 2003;41:15S–22S. doi: 10.1016/s0735-1097(02)02834-6. [DOI] [PubMed] [Google Scholar]
- 35.Phinikaridou A, Ruberg FL, Hallock KJ, et al. In vivo detection of vulnerable atherosclerotic plaque by MRI in a rabbit model. Circ Cardiovasc Imaging. 3:323–332. doi: 10.1161/CIRCIMAGING.109.918524. [DOI] [PubMed] [Google Scholar]
- 36.Mantovani A, Sica A, Locati M. Macrophage polarization comes of age. Immunity. 2005;23:344–346. doi: 10.1016/j.immuni.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 37.Dorn BR, Dunn WA, Jr, Progulske-Fox A. Porphyromonas gingivalis traffics to autophagosomes in human coronary artery endothelial cells. Infect Immun. 2001;69:5698–5708. doi: 10.1128/IAI.69.9.5698-5708.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hayashi C, Madrigal AG, Liu X, et al. Pathogen-mediated inflammatory atherosclerosis is mediated in part via Toll-like receptor 2-induced inflammatory responses. J Innate Immun. 2:334–343. doi: 10.1159/000314686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Libby P, Nahrendorf M, Pittet MJ, et al. Diversity of denizens of the atherosclerotic plaque: not all monocytes are created equal. Circulation. 2008;117:3168–3170. doi: 10.1161/CIRCULATIONAHA.108.783068. [DOI] [PubMed] [Google Scholar]
- 40.Andreeva ER, Pugach IM, Orekhov AN. Collagen-synthesizing cells in initial and advanced atherosclerotic lesions of human aorta. Atherosclerosis. 1997;130:133–142. doi: 10.1016/s0021-9150(96)06056-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.