

Abstract
BACKGROUND AND PURPOSE
Recently, metformin, a well-known anti-diabetic drug, has been shown to possess anti-inflammatory activities. This study investigated the effect of metformin on the expression of pro-inflammatory cytokines including high mobility group box 1 (HMGB1) in lipopolysaccharide (LPS)-treated animals and cells.
EXPERIMENTAL APPROACH
We investigated whether metformin inhibits the release of HMGB1 in LPS-treated RAW 264.7 cells and increases survival rate in endotoxaemic mice (lethal endotoxaemia was induced by an i.p. injection of LPS). This was achieved by a range of techniques including Western blotting, enzyme-linked immunosorbent assay, specific pharmacological inhibitors, knock out of α1-subunit of AMP-activated protein kinase (AMPK) and recombinant HMGB1.
KEY RESULTS
Both pre- and post-treatment with metformin significantly improved survival of animals during lethal endotoxaemia (survival rate was monitored up to 2 weeks), decreased serum levels of tumour necrosis factor-alpha (TNF-α), interleukin-1β, HMGB1 expression and myeloperoxidase activity in lungs. However, metformin failed to improve survival in endotoxaemic animals that had additionally been treated with recombinant HMGB1. In an in vitro study, metformin dose-dependently inhibited production of pro-inflammatory cytokines and HMGB1 release. Metformin activated AMPK by its phosphorylation. Compound C (pharmacological inhibitor of AMPK) and siAMPKα1 reversed the anti-inflammatory effect of metformin in LPS-treated cells.
CONCLUSIONS AND IMPLICATIONS
Our data indicate that metformin significantly attenuates the pro-inflammatory response induced by LPS both in vivo and in vitro. Metformin improved survival in a mouse model of lethal endotoxaemia by inhibiting HMGB1 release. AMPK activation was implicated as one of the mechanisms contributing to this inhibition of HMGB1 secretion.
Keywords: lipopolysaccharide, sepsis, metformin, AMP-activated protein kinase, high mobility group box 1
Introduction
Sepsis is the third leading cause of death in developed societies and the most common cause of death in many intensive care units. Despite extensive research on the pathophysiology of sepsis and the technical advances, sepsis incidence is constantly rising (1.5–8% per year; Martin et al., 2003). The pathogenesis of sepsis is characterized by overwhelming inflammatory and immune responses that can lead to tissue damage, multiple organ failure and death (Riedemann et al., 2003). Therapies designed to block one single cytokine, such as tumour necrosis factor-alpha (TNF-α) or interleukin-1β (IL-1β), have shown limited efficacy probably due to the early and transient kinetics of these inflammatory cytokines. In the past few years, different lines of evidence indicate that high mobility group box 1 (HMGB1) is a necessary and sufficient late mediator of severe sepsis and therefore, targeting it provides a wide window for clinical intervention (Lotze and Tracey, 2005). HMGB1, a non-histone DNA-binding protein, was recently found to be secreted by activated monocytes/macrophages and epithelial cells, acting as a late pro-inflammatory factor in many disorders including sepsis (see HMGB1 reviews; Furugen et al., 2008; Fujii et al., 2009; Zhang et al., 2009; Abdulahad et al., 2010; Andersson and Harris, 2010; Sims et al., 2010; Stros, 2010). Once released, HMGB1 can bind to cell-surface receptors, such as the receptor for advanced glycation end products, Toll-like receptor (TLR) 2 and TLR 4, and mediate various cellular responses, chemotactic cell movement and release of pro-inflammatory cytokines (Andersson et al., 2000; Park et al., 2004; Rouhiainen et al., 2007). High levels of systemic HMGB1 are present in humans and animals with sepsis and endotoxaemia (Wang et al., 1999; Karlsson et al., 2008). Administration of recombinant HMGB1 to mice causes gut barrier dysfunction and lethal multiple organ failure (Wang et al., 1999; Sappington et al., 2002). In contrast, passive immunization with neutralizing antibodies against HMGB1 improves survival and prevents organ failure in septic mice (Yang et al., 2004).
Metformin (1,1-dimethylbiguanide hydrochloride) is one of the most widely prescribed drugs for the treatment of type 2 diabetes (Hardie, 2007). The main molecular target of metformin is AMP-activated protein kinase (AMPK) activation. AMPK is a highly conserved heterotrimeric kinase that functions as a metabolic switch, thereby coordinating the cellular enzymes involved in carbohydrate and fat metabolism to enable ATP conservation and synthesis. AMPK is activated by conditions that increase the adenosine monophosphate (AMP) : adenosine triphosphate (ATP) ratio, such as exercise and metabolic stress. The effects of stress, exercise, hypoxia and ischaemia on AMPK activation have been extensively examined. When the adenosine monophosphate (AMP) : adenosine triphosphate (ATP) ratio increases, AMPK is activated by AMPK kinase, and a conformational change is induced by it combining with AMP, thereby decreasing the AMP : ATP ratio by switching off ATP-consuming pathways and switching on ATP-generating pathways (Hardie et al., 1998). Recently, it has been found that AMPK plays an important role in inflammation, and metformin can serve as a potential drug to treat inflammation-related disorders (Bergheim et al., 2006; Hattori et al., 2006; Isoda et al., 2006; Sag et al., 2008; Nath et al., 2009). Metformin prevented endotoxin-induced liver injury in a model of post-surgical sepsis in rats (Bergheim et al., 2006), but did not affect the survival rate in an Escherichia coli-induced model of sepsis in mice (Gras et al., 2006). Therefore, it is not clear whether metformin can affect HMGB1 release, a late-phase cytokine, in lethal endotoxaemia and thereby impact on survival outcome. Thus, in this study, we investigated the effect of metformin on HMGB1 release in both lipopolysaccharides (LPS)-treated cells and an animal model of endotoxaemia; the importance of AMPK activation in the metformin-mediated anti-inflammatory effect was also assessed.
Methods
Materials
Anti-HMGB1 was purchased from Abcam (Cambridge, MA, USA), anti-inducible nitric oxide synthase from Transduction Laboratories (Lexington, KY, USA), anti-cyclooxygenase-2 and anti-β-actin from Santa Cruz Biotechnology (Santa Cruz, CA, USA), anti-phosphor-AMPKα (Thr172) and anti-AMPKα from Cell Signaling Technology (Beverly, MA, USA). Compound C was obtained from Calbiochem (San Diego, CA, USA). Enhanced chemiluminescence (ECL) Western blotting detection reagent was from Amersham (Buckinghamshire, UK). All other chemicals, including LPS (E. coli 0111:B4) and metformin were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Cell culture and stimulation
RAW 264.7 cells were obtained from American Type Culture Collection (ATCC, Rockville, MD, USA). The cells were grown in RPMI-1640 medium supplemented with 25 mM N-(2-hydroxyethyl) piperazine-N-2-ethanesulphonic acid, 100 U·mL−1 penicillin, 100 µg·mL−1 streptomycin and 10% heat-inactivated foetal calf serum. RAW 264.7 cells were plated at a density of 1 × 104 cells per 100 mm dish. The cells were stimulated with LPS from E. coli 0111: B4 (1 µg·mL−1), in the presence or absence of different concentrations of metformin (1, 5, 10 mM). All control samples were treated with distilled water. To detect inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2) or nitric oxide (NO) and prostaglandin E2 (PGE2), cells were incubated for 8 or 16 h, respectively, after stimulation as previously described (Kim et al., 2007; Tsoyi et al., 2008). To detect HMGB1, cells were incubated for 24 h after stimulation as previously described (Tsoyi et al., 2009).
Assay for NOx production
NO was measured by its stable oxidative metabolite, nitrite (NOx) with Griess reagent as described previously (Kang et al., 1999). At the end of the incubation, 100 µL of the culture medium was mixed with an equal volume of Griess reagent (0.1% naphthylethylenediamine dihydrochloride and 1% sulphanilamide in 5% phosphoric acid). Light absorbance was measured at 550 nm, and the nitrite concentration was determined using a curve calibrated with sodium nitrite standards.
Cytokines
The levels of IL-1β, TNF-α, interleukin 6 (IL-6) and PGE2 were measured by commercially available enzyme-linked immunosorbent assay (elisa) kits from R&D systems (Minneapolis, MN, USA) according to the manufacturer's instructions.
siRNA technique
siRNAs against mouse AMPKα1 and scramble siRNA were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Transient transfections were performed using Lipofectin (Gibco-BRL, Rockville, MD, USA). 5 × 105 cells were plated on 60 mm plates the day before transfection and grown to about 70% confluence. Cells were transfected with scramble siRNA (100 nM) and siAMPKα1 (100 nM). Transfections were allowed to proceed for 4 h. After this, the culture media was changed and the incubation proceeded for a further 16 h. The transfected cells were washed with 4 mL of 1 × phosphate-buffered saline (pH 7.4) and then stimulated with 1 µg·mL−1 LPS for an additional 8 h. Cells were harvested and subjected to immunoblotting.
HMGB1 analysis
Culture medium samples were briefly centrifuged to remove cellular debris. The same volumes of samples were then concentrated 40-fold with Amicon Ultra-4-10000 NMWL (Millipore, Billerica, MA, USA). Centrifugation conditions were fixed angle (35 degrees) and 7500×g for 20 min at 4°C. Then concentrated samples were mixed with 2× loading dye and boiled at 95°C for 5 min. Proteins were separated on 12% sodium dodecyl sulphate (SDS)-polyacrylamide gels and transferred to immunoblot membranes. Membranes were blocked with 5% bovine serum albumin (BSA) overnight at 4°C, then washed with Tris-buffered saline/Tween 20 (TBS-T) buffer for 1 h. at room temperature (RT). Next membranes were incubated with anti-HMGB1 antibody (Abcam, 1:1000) at 4°C for 16 h, and then washed with TBS-T for 1 h at RT and incubated with goat anti-rabbit IgG-HRP secondary antibody (1:5000 dilution in TBS-T containing 1% BSA). The signals were detected by ECL (Amersham, Piscataway, NJ, USA).
Recombinant human HMGB1
For the recombinant human HMGB1 protein (rHMGB1), the full-length coding sequence of human HMGB1 (GenBank accession no.X12597) was inserted into T&A Cloning Vector (RBC, Chung Ho, Taipei, Taiwan) and a Nhe I/Xho I fragment subcloned into pET-28a vector (Novagen, Madison, WI, USA). Positive clones were selected and confirmed by DNA sequencing. The plasmids were transformed into protease deficient E. coli strain BL21 (DE3) pLysE (Novagen) and induced with 0.5 mM isopropyl-D-thiogalactopyranoside for 3 h. The rHMGB1 was purified with Ni-NTA agarose column (Qiagen, Santa Clara, CA, USA) and ion-exchange chromatography (GE Healthcare Bio-Sciences AB, Piscataway, NJ, USA). Endotoxin was removed by detergent phase separation with Triton X-114 (Sigma).
Western blot
The cytoplasmic/nuclear fractionation was performed using nuclear/cytosol fractionation kit (Cat # K266-25, BioVision, Mountain View, CA, USA) according to manufacturer's manual. Whole cell lysate were performed using buffer containing 0.5% SDS, 1% Nonidet P-40, 1% sodium deoxycholate, 150 mM NaCl, 50 mM Tris-Cl (pH 7.5) and protease inhibitors. Concentrated supernatants (to detect HMGB1) and whole cell lysates (to detect iNOS, COX-2, p-AMPK, AMPK, β-actin) as well as nuclear, cytosol lysates were subjected to electrophoresis in different percentage polyacrylamide gels, depending on the size of protein of interest. The gels were transferred to polyvinylidene difluoride (PVDF) membranes by semidry electrophoretic transfer at 15 V for 60 to 75 min. The membranes was stained with Ponceau S solution (2 µg·mL−1) for 5 min to determine efficiency of transfer or/and protein loading levels per track. Then the PVDF membranes were blocked overnight at 4°C in 5% BSA. The cells were incubated with primary antibodies diluted 1:500 in TBS-T containing 5% BSA for overnight in 4°C and then incubated with secondary antibody at RT for 1 h. The signals were detected by ECL.
Animal model of endotoxaemia
Endotoxaemia was induced in BALB/c mice (male 7–8 weeks, 20–25 g) by injection of bacterial endotoxin (LPS 15 mg·kg−1, i.p.). To evaluate the effect of metformin on survival rate in endotoxaemic mice, mice were both pretreated and post-treated with metformin. For the pretreatment protocol, mice were pretreated with either saline (i.p., n= 20), metformin (50 mg·kg−1, i.p., n= 20) or metformin (100 mg·kg−1, i.p., n= 20) 2 h prior to the injection of LPS (15 mg·kg−1, i.p.). At 12, 24, 48, 72 and 96 h after the onset of endotoxaemia, animals were administered with either saline or metformin, 50 mg·kg−1 or 100 mg·kg−1. Survival was monitored daily for up to 2 weeks. For the post-treatment protocol, mice were injected with LPS, 15 mg·kg−1 i.p., and then 12 h later, saline (i.p., n= 20), metformin (50 mg·kg−1, i.p., n= 20) or metformin (100 mg·kg−1, i.p., n= 20) were administered. These treatments were repeated at 24, 48, 72 and 96 h after induction of endotoxaemia. Animals were monitored up to 2 weeks to evaluate survival rate after LPS challenge following the different treatment schedules with metformin. To further evaluate the link between HMGB1 and effect of metformin on survival rate, mice were pretreated with saline (i.p., n= 10) or metformin (100 mg·kg−1, i.p., n= 10). Then, mice were subjected to lethal endotoxaemia (LPS, 15 mg·kg−1, i.p.). At 12, 24, 48, 72 and 96 h after the onset of endotoxaemia, animals were administered either saline or metformin (100 mg·kg−1) + rHMGB1 (100 µg per mouse). Survival was monitored daily for up to 2 weeks. In addition, to evaluate the cytokine and HMGB1 levels in blood, 15 mice were randomly divided into three groups: (i) saline (n= 5); (ii) LPS (n= 5); and (iii) LPS + metformin (n= 5). Metformin (100 mg·kg−1, i.p.) was administered 2 h before LPS (15 mg·kg−1, i.p.) injection. Twenty-four hours after LPS treatment, mice were anaesthetized with ketamine (30 mg·kg−1) and xylazine (6 mg·kg−1), and blood samples were collected by cardiac puncture and kept at RT for 2 h before being centrifuged at 2000×g for 20 min. Serum was collected and analysed for TNF-α, IL-1β by elisa or for HMGB1 analysis as described above. Mice were maintained in accordance with the Guide for the Care and Use of Laboratory Animals (NIH publication 85-23, revised 1996) and were treated ethically. The protocol was approved in advance by the Animal Research Committee of the Gyeongsang National University.
Myeloperoxidase (MPO) activity
Fifteen mice were randomly divided into three groups: (i) saline (n= 5); (ii) LPS (n= 5); and (iii) LPS + metformin. Metformin (100 mg·kg−1, i.p.) was administrated 2 h before LPS (15 mg·kg−1, i.p.) injection. Twenty-four hours after LPS treatment, mice were anaesthetized with ketamine (30 mg·kg−1) and xylazine (6 mg·kg−1) and lungs were extracted. Lung samples were homogenized and MPO activity was measured by specific elisa (HK210 MPO elisa kit, HyCult biotechnology, Uden, the Netherlands).
Statistical evaluation
Data are expressed as the mean ± SD of results obtained from n number of replicate treatments. Differences between data sets were assessed by one-way analysis of variance followed by Newman–Keuls tests. The Kaplan–Meier method was used to compare the differences in mortality rates between groups. P < 0.05 was accepted as statistically significant.
Results
Metformin attenuates pro-inflammatory response in LPS-stimulated macrophages
We showed that metformin dose-dependently inhibited cytokine release such as IL-1β, TNF-α and IL-6 induced by LPS(Figure 1A–C). iNOS and COX-2 are potent, inducible pro-inflammatory gene products (Vane et al., 1994). Metformin significantly attenuated iNOS and COX-2 protein levels and also levels of their respective metabolites NO and PGE2 (Figure 1D,E).
Figure 1.

Effect of metformin on the release of various cytokines, iNOS (NO) and COX-2 (PGE2) expression in LPS-activated macrophages. (A to E) Cells were pretreated with metformin (Met) 1, 5 and 10 mM for 1 h, then stimulated with LPS (1 µg·mL−1) for other 16 h. After incubation, culture medium samples were collected and subjected to elisas for IL-1β (A), TNF-α (B), IL-6 (C), PGE2 (E) and nitric oxide production by NO assay as described in Methods. Cells were harvested and iNOS and COX-2 protein levels were determined by Western blot. Data are presented as mean ± SD of three independent experiments. One-way analysis of variance was used to compare multiple group means followed by Newman–Keuls test (significance compared with control, **P < 0.01; significance compared with LPS, †P < 0.05 or ††P < 0.01). COX-2, cyclooxygenase-2; ELISA, enzyme-linked immunosorbent assay; IL-1β, interleukin-1β; IL-6, interleukin-6; iNOS, inducible nitric oxide synthase; PGE2, prostaglandin E2; TNF-α, tumour necrosis factor-alpha.
Metformin inhibits LPS-stimulated HMGB1 extracellular release and its nuclear/cytosolic translocation
Extracellular HMGB1 plays a critical role in the development of sepsis (Lotze and Tracey, 2005). Thus, we determined whether metformin can affect HMGB1 extracellular levels induced by LPS. Figure 2A and B clearly show that metformin inhibited HMGB1 cytosolic translocation from the nucleus and subsequently attenuated extracellular levels of HMGB1 induced by LPS in macrophages. These data indicate that metformin possesses potent anti-inflammatory effects in an in vitro model of endotoxaemia.
Figure 2.

Effect of metformin on HMGB1 release in LPS-activated macrophages. (A and B) Cells were treated with metformin (1, 5 and 10 mM) for 1 h, then stimulated with LPS (1 µg·mL−1) for another 24 h. After incubation, culture medium was collected and subjected to HMGB1 anylasis (A). Cells were subjected to nuclear/cytosol fractionation and immunoblotted against HMGB1 as described in Methods. Blot bands are representative of three independent experiments. Data are presented as mean ± SD of three independent experiments. One-way analysis of variance was used to compare multiple group means followed by Newman–Keuls test (significance compared with control, **P < 0.01; significance compared with LPS, †P < 0.05 or ††P < 0.01). HMGB1, high mobility group box 1; LPS, lipopolysaccharides.
Anti-inflammatory effect of metformin is mediated through AMPK activation
The primary molecular target of metformin is AMPK (Hardie, 2007). We investigated whether the anti-inflammatory effect of metformin on the response to LPS is mediated through activation of AMPK. Firstly, we demonstrated that metformin induced phosphorylation of AMPK in a dose- and time-dependent manner (Figure 3). Next, we used a pharmacological inhibitor of AMPK (compound C) to determine whether the anti-inflammatory effect of metformin is mediated through AMPK. Figure 4A and B show that compound C significantly reversed the inhibition of iNOS, and HMGB1 induced release by metformin. AMPK is a heterotrimeric enzyme consisting of a catalytic subunit α (α1 and α2) and two regulatory subunits β (β1 and β2) and γ (γ1, γ2, and γ3). AMPK is activated by both AMP and phosphorylation of Thr172 of the α subunit by upstream kinase and differential regulatory mechanisms. Previously, it has been observed that macrophages contain the α1 but not the α2 subunit (Jhun et al., 2004). Thus, we targeted to knock-out α1 subunit in macrophages by specific siRNA. Figure 5 clearly indicates that metformin is unable to attenuate LPS-induced inflammatory response when AMPK is inhibited. We did not observe any effect of scramble siRNA or compound C administration on iNOS expression (Supporting Information Figure S1A).
Figure 3.

Metformin activates AMPK in macrophages. (A) Cells were treated with metformin (1, 2.5, 5, 10 mM) for 8 h. After incubation, phosphor- and total AMPK were assessed by Western blot. (B) Cells were incubated with metformin (5 mM) for 1, 2, 4, 8, and 12 h. After incubation, cells were lysed and subjected to Western blot for phosphor-AMPK and AMPK detection. Data are presented as mean ± SD of three independent experiments. One-way analysis of variance was used to compare multiple group means followed by Newman–Keuls test (significance compared with control, *P < 0.05; **P < 0.01). AMPK, AMP-activated protein kinase.
Figure 4.

Pharmacological inhibition of AMPK reversed metformin-mediated anti-inflammatory effect in LPS-activated macrophages. Cells were treated with metformin (10 mM) in the presence or absence of compound C (comp C; 12 µM) for 1 h. Then cells were stimulated with LPS (1 µg·mL−1) for the next 24 h. After incubation, cells were lysed and subjected to Western blot (A); culture medium samples were extracted for HMGB1 detection by HMGB1 analysis (B) as described in Methods. Data are presented as mean ± SD of three independent experiments. One-way analysis of variance was used to compare multiple group means followed by Newman–Keuls test. Significance compared with control, **P < 0.01 and *P < 0.05; significance compared with LPS, ††P < 0.01; significance compare to Met (10 mM), §§P < 0.01. AMPK, AMP-activated protein kinase; HMGB1, high mobility group box 1; iNOS, inducible nitric oxide synthase; LPS, lipopolysaccharides.
Figure 5.

Knock down of AMPK-α1 attenuates anti-inflammatory effect of metformin in LPS-activated macrophages. (A) Cells were transfected with scramble siRNA (ssiRNA, 100 nM) or with siAMPK-α1 (100 nM) and incubated for 16 h. After incubation, cells were harvested and AMPK or β-actin was detected by Western blot. (B and C) Cells were transfected with ssiRNA or siAMPK-α1. After transfection, cells were stimulated with LPS (1 µg·mL−1) in the presence or absence of metformin (10 mM). Cells were lysed, 24 h later, for iNOS detection. Culture medium was subjected to HMGB1 analysis as described in Methods. Data are presented as ±SD of three independent experiments. One-way analysis of variance was used to compare multiple group means followed by Newman–Keuls test. Significance compared with control, **P < 0.01 and *P < 0.05; significance compared with LPS, ††P < 0.01; significance compare to Met (10 mM), §§P < 0.01. AMPK, AMP-activated protein kinase; HMGB1, high mobility group box 1; iNOS, inducible nitric oxide synthase; LPS, lipopolysaccharides.
Metformin administration improves survival and attenuates the inflammatory response in endotoxaemic animals
Next, we tried to elucidate whether the anti-inflammatory effect of metformin can be beneficial in an animal model of endotoxaemia. Firstly, we observed survival in untreated and metformin-treated animals. Figure 6A shows that metformin, 100 mg·kg−1, significantly improved survival rate compared with untreated mice (75% vs. 17%). As the production of inflammatory cytokines is rapid, for example, TNF-α and IL-1β reach maximum levels within 4 h after LPS infusion (Chorny et al., 2008), we next determined whether delayed administration of metformin could still protect mice from death in conditions of established endotoxaemia. Indeed, metformin administered 12 h after LPS treatment still significantly protected animals from death (Figure 6B). Furthermore, improved survival was tightly associated with a decreased inflammatory response to LPS: decreased circulating cytokine (TNF-α and IL-1β) and HMGB1 levels in metformin-treated mice compared with untreated mice (Figure 7A–C). Previously, it has been reported that AMPK activation can attenuate neutrophil activity and lung injury in response to LPS in mice (Zhao et al., 2008). Consistent with this, we observed that metformin decreased MPO activity (a marker of neutrophil activation) during endotoxaemia in lung tissue (Figure 7D).
Figure 6.

Metformin improves survival in lipopolysaccharides (LPS)-induced mouse model of sepsis. (A) BALB/c mice were pretreated with either saline (i.p., n= 20), metformin (50 mg·kg−1, i.p., n= 20) or metformin (100 mg·kg−1, i.p., n= 20) 2 h prior to LPS challenge (15 mg·kg−1, i.p.). At 12, 24, 48, 72 and 96 h after the onset of endotoxaemia, animals were administered (i.p.) either saline or metformin 50 mg·kg−1 or 100 mg·kg−1. (B) BALB/c mice were subjected to lethal endotoxaemia (LPS, 15 mg·kg−1, i.p.) and 12 h later treated with either saline (i.p., n= 20), metformin (50 mg·kg−1, i.p., n= 20) or metformin (100 mg·kg−1, i.p., n= 20). Treatments were repeated at 24 h, 48 h, 72 h, and 96 h after induction of endotoxaemia. Survival was monitored daily, for up to two weeks. The Kaplan–Meier program was utilized to compare the differences in mortality rates between groups. Significance compared with saline, *P < 0.05.
Figure 7.

Metformin attenuates inflammation and neutrophil infiltration into lungs induced by LPS. BALB/c mice were treated with either saline (i.p., n= 5); saline (i.p.) +LPS (15 mg·kg−1, i.p.) (n= 5) or metformin (100 mg·kg−1) +LPS (15 mg·kg−1, i.p.) (n= 5); 24 h after LPS treatment mice were anaesthetized, blood and lungs were extracted. Serum levels of IL-1β and TNF-α were detected by elisa, HMGB1 by HMGB1 analysis. Lungs were homogenized and MPO activity was measured by elisa. Data are presented as mean ± SD of three independent experiments. One-way analysis of variance was used to compare multiple group means followed by Newman–Keuls test (significance compared with control, *P < 0.05 or **P < 0.01; significance compared with LPS, †P < 0.05 or ††P < 0.01). IL-1β, interleukin-1 β; LPS, lipopolysaccharide; MPO, myeloperoxidase; TNF-α, tumour necrosis factor α.
Metformin improves survival in lethal endotoxaemia through HMGB1
Although our results showed that metformin has a potent therapeutic effect in endotoxaemia, it is unclear whether this beneficial effect of metformin is mediated through inhibition of HMGB1 release alone or together with early cytokines (TNF-α, IL-1β). To resolve this question, we treated endotoxaemic animals with metformin alone or in combination with rHMGB1. As shown in Figure 8, metformin improved survival in LPS-treated mice but failed to do so in combination with rHMGB1. Thus, inhibition of HMGB1 release by metformin plays a critical role in metformin-mediated improved survival of endotoxaemic animals.
Figure 8.
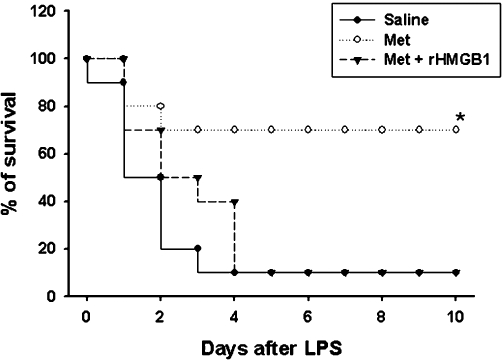
rHMGB1 reversed the therapeutic effect of metformin on lethal endotoxaemia. BALB/c mice were pretreated with saline (i.p., n= 10), metformin (100 mg·kg−1, i.p., n= 10) 2 h prior to LPS injection (15 mg·kg−1, i.p.). At 12, 24, 48, 72 and 96 h after the onset of endotoxaemia, animals were administered (i.p.) saline or metformin (100 mg·kg−1) + rHMGB1 (100 µg). Survival was monitored daily, for up to 2 weeks. The Kaplan–Meier program was utilized to compare the differences in mortality rates between groups. Significance compared with saline, *P < 0.05. HMGB1, high mobility group box 1; rHMGB1, recombinant human high mobility group box 1.
Discussion
In the present study, we demonstrated that metformin significantly decreased inflammatory gene expression (iNOS, COX-2) and pro-inflammatory cytokines such as TNF-α, IL-1β and HMGB1 in LPS-treated RAW 264.7 cells. In addition, administration of metformin increased the survival rate of endotoxaemic mice by a mechanism that involves inhibition of HMGB1 release. Metformin was discovered in the 1920s in a search for guanidine-containing compounds with anti-diabetic activities and was introduced clinically in Europe in the 1950s and later in the USA in 1995 (Bailey and Turner, 1996). Metformin is often the first drug used for newly diagnosed type 2 diabetic patients and in 2006, it accounted for 37% of the non-insulin diabetic prescriptions in the USA (Strack, 2008). Indeed, metformin is used clinically as an anti-diabetic drug. More recently, from results obtained in in vivo and in vitro models, it has been suggested that metformin can be used in other pathophysiological conditions such as oncological, auto-immune and cardiovascular disorders (Bergheim et al., 2006; Hattori et al., 2006; Isoda et al., 2006; Nath et al., 2009; Tan et al., 2009; Gonzalez-Angulo and Meric-Bernstam, 2010).
Several reports have suggested that AMPK can serve as an anti-inflammatory molecule. For example, activation of AMPK by aminoimidazole carboxamide ribonucleotide or metformin attenuates infiltration of monocytes in the central nervous system in an experimental model of multiple sclerosis by inhibition of pro-inflammatory cytokines (Nath et al., 2005; 2009; Paintlia et al., 2006). Moreover, genetic regulation of AMPK activity clearly demonstrated that AMPK has the ability to regulate macrophage functional polarization. It has been suggested that AMPK is able to switch macrophages from a pro- to an anti-inflammatory functional state by differential regulation of transcription factors, inhibition of NF-κB (pro-inflammatory) and activation of Akt and cAMP response element binding (CREB) (anti-inflammatory), which subsequently decrease pro-inflammatory genes but increase anti-inflammatory ones, for example, IL-10 (Sag et al., 2008). Recently, it has been reported that metformin activates AMPK in macrophages and this results in the inhibition of biosynthesis of phospholipids as well as neutral lipids and down-regulates the expression of LPS-induced pro-inflammatory cytokines (iNOS and COX-2) and their mediators (NO and PGE2) (Nath et al., 2009). In addition, metformin attenuates the increase in iNOS and COX-2 expression induced by IFN-γ and IL-17 in RAW267.4 cells (Nath et al., 2009), supporting our observations that metformin attenuates detrimental inflammation in an LPS-induced model of sepsis through activation of AMPK.
HMGB1 is a critical regulator of sepsis severity. HMGB1 can be actively released from activated monocytes and macrophages, and it has been reported that the increases in HMGB1 levels in blood correlate with increased death incidence in animal models of sepsis (Wang et al., 1999; Yang et al., 2004; Lotze and Tracey, 2005; Tsoyi et al., 2009). Indeed, HMGB1 is lethal to mice that develop signs of endotoxaemia, which include lethargy, piloerection, diarrhoea, shivering and microthrombi formation in the lungs and liver (Wang et al., 1999). Mice have been shown to have increased serum levels of HMGB1 after endotoxin exposure, and HMGB1 potentiates the inflammatory process, such as sepsis or acute lung injury (Wang et al., 1999). The level of serum HMGB1 is highly correlated with the severity of sepsis in rats (Hou et al., 2009). Therapy with anti-HMGB1 antibodies before or after endotoxin exposure confers that significant protection against lethality and administration of HMGB1 itself was lethal, suggesting that HMGB1 is an endogenous mediator of endotoxin lethality (Wang et al., 1999; Andersson et al., 2000). The importance of HMGB1 levels and severity of sepsis is not confined to animal studies, clinical studies have also demonstrated that HMGB1 is a late mediator of sepsis in amplifying the inflammatory response that follows acute tissue damage, and it has been reported that serum/plasma HMGB1 concentrations are elevated in patients with sepsis (Ueno et al., 2004). Patients with sepsis who succumbed to the infection had higher serum HMGB1 levels than those that survived (Wang et al., 1999; Karlsson et al., 2008), indicating that HMGB1 can be a possible target molecule for treatment of sepsis.
The most striking finding of the present study is that metformin inhibited endotoxin-induced HMGB1 release, both in vitro and in vivo. Moreover, we demonstrated that additional treatment with rHMGB1 did not prevent metformin-treated endotoxaemic animals from dying, suggesting that metformin's protective mechanism involves inhibition of HMGB1 release (Figure 8). How does metformin decrease HMGB1 release in LPS-activated RAW 264.7 cells and in the serum of endotoxaemic mice? At the present, the exact mechanism by which metformin inhibits the release of HMGB1 is not known. The inhibitory effect of metformin on HMGB1 release can come from different ways, e.g. inhibition of TNF-α and IL-1β or iNOS/NO (Chen et al., 2004; Jiang and Pisetsky, 2006). Moreover, metformin has been shown to inhibit NF-κB activation in LPS-treated macrophages (Supporting Information Figure S1B), which implicates an effect on NF-κB as a possible mechanism for its inhibitory effect on HMGB1 release (Bonaldi et al., 2003). Nevertheless, in this study, we showed that the activation of AMPK is responsible for metformin's anti-inflammatory action; treatment with either the pharmacological inhibitor of AMPK, compound C or the genetically knock-out α1 subunit of AMPK, specific AMPKα1 siRNA, significantly reversed the reduction of HMGB1 release induced by LPS in vitro.
Interestingly, Gras et al. (2006) reported that the administration of metformin at different doses neither increased nor decreased survival rate in an animal model of E. coli-induced sepsis; even administration of a high dose (500 mg·kg−1) of metformin had no effect on the mortality rate. However, our results showed that 100 mg·kg−1 metformin increased the survival rate in LPS-treated endotoxaemic mice. Furthermore, delayed administration of 100 mg·kg−1 metformin, in conditions of established endotoxaemia, could still protect the mice from death. We have no adequate explanation as to why our results differ from those of Gras et al. (2006), but the discrepancy could be due to the different models of sepsis investigated; we used LPS to induce endotoxaemia, whereas they used E. coli.
Although the mechanism by which activation of AMPK (metformin) decreases HMGB1 release and translocation from the nucleus remains to be explored, we clearly showed that metformin protected animals in established endotoxaemia from death by reducing HMGB1 release (Figure 6B). Inhibition of JAK2 with AG490 has been shown to inhibit extracellular release of HMGB1 from LPS-treated macrophages (Kim et al., 2007; Peña et al., 2010; Tsoyi et al., 2010) and prevent the increase in serum HMGB1 levels that occur during polymicrobial sepsis (Peña et al., 2010); therefore, it would be interesting to investigate whether activation of AMPK by metformin is linked to JAK/STAT signals in macrophages activated with LPS or in polymicrobial sepsis.
In summary, our results demonstrate, for the first time, that metformin has a therapeutic effect in an LPS-induced animal model of endotoxaemia and this effect is mediated by reducing HMGB1 release. In particular, we demonstrated that metformin treatment significantly attenuated the inflammatory response induced by LPS both in vitro and in vivo via AMPK activation. Because metformin significantly inhibited HMGB1 release in endotoxaemic mice, this effect could be the critical step for the protection of endotoxaemic animals from death. Our results indicate new targets for clinical research for the development of an effective therapy for sepsis.
Acknowledgments
This work was supported by a grant from NRF (03-2010-0298) and Korea Research Foundation (R13-2005-012-01003-0).
Glossary
Abbreviations
- AMPK
AMP-activated protein kinase
- ECL
enhanced chemoluminescence
- HMGB1
high mobility group box 1
- IL-1β
interleukin-1β
- iNOS
inducible nitric oxide synthase
- LPS
lipopolysaccharide
- MPO
myeloperoxidase
- TNF-α
tumour necrosis factor α
Conflict of interest
No conflict of interest to report.
Supplementary material
Additional Supporting Information may be found in the online version of this article:
Supporting Information: Teaching Materials; Figs 1–8 as PowerPoint slide.
Figure S1 (A) Cells were transfected with ssiRNA or treated with compC as described in Figure 5. After transfection cells were treated with lipopolysaccharides (LPS) (1 µg·mL−1) for 8 h. Then cells were harvested and subjected to immunoblotting for iNOS and β-actin detection. Blot bands are representative of three independent experiments. (B) Cells were transfected with 1 µg of NF-κB-luciferase plus 0.5 µg of pRL-TK-luciferase. Cells were allowed to recover overnight and were then treated with 1 µg·mL−1 of LPS with/without metformin (5 and 10 mM). Cells were harvested 6 h after treatment. Luciferase activities are presented as fold activation relative to that of the untreated cells. Data are presented as ±SD of three independent experiments. One-way analysis of variance was used to compare multiple group means followed by Newman–Keuls test (significance compared with control, **P < 0.01; significance compared with LPS,††P < 0.01).
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Abdulahad DA, Westra J, Limburg PC, Kallenberg CG, Bijl M. HMGB1 in Systemic Lupus Erythematosus: its role in cutaneous lesions development. Autoimmun Rev. 2010;9:661–665. doi: 10.1016/j.autrev.2010.05.015. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Andersson U, Harris HE. The role of HMGB1 in the pathogenesis of rheumatic disease. Biochim Biophys Acta. 2010;1799:141–148. doi: 10.1016/j.bbagrm.2009.11.003. [DOI] [PubMed] [Google Scholar]
- Andersson U, Wang H, Palmblad K, Aveberger AC, Bloom O, Erlandsson-Harris H, et al. High mobility group 1 protein (HMG-1) stimulates proinflammatory cytokine synthesis in human monocytes. J Exp Med. 2000;192:565–570. doi: 10.1084/jem.192.4.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey CJ, Turner RC. Metformin. N Engl J Med. 1996;334:574–579. doi: 10.1056/NEJM199602293340906. [DOI] [PubMed] [Google Scholar]
- Bergheim I, Luyendyk JP, Steele C, Russell GK, Guo L, Roth RA, et al. Metformin prevents endotoxin-induced liver injury after partial hepatectomy. J Pharmacol Exp Ther. 2006;316:1053–1061. doi: 10.1124/jpet.105.092122. [DOI] [PubMed] [Google Scholar]
- Bonaldi T, Talamo F, Scaffidi P, Ferrera D, Porto A, Bachi A, et al. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J. 2003;22:5551–5560. doi: 10.1093/emboj/cdg516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Li J, Ochani M, Rendon-Mitchell B, Qiang X, Susarla S, et al. Bacterial endotoxin stimulates macrophages to release HMGB1 partly through CD14- and TNF-dependent mechanisms. J Leukoc Biol. 2004;76:994–1001. doi: 10.1189/jlb.0404242. [DOI] [PubMed] [Google Scholar]
- Chorny A, Anderson P, Gonzalez-Rey E, Delgado M. Ghrelin protects against experimental sepsis by inhibiting high-mobility group box 1 release and by killing bacteria. J Immunol. 2008;180:8369–8377. doi: 10.4049/jimmunol.180.12.8369. [DOI] [PubMed] [Google Scholar]
- Fujii K, Luo Y, Sasahira T, Denda A, Ohmori H, Kuniyasu H. Co-treatment with deoxycholic acid and azoxymethane accelerates secretion of HMGB1 in IEC6 intestinal epithelial cells. Cell Prolif. 2009;42:701–709. doi: 10.1111/j.1365-2184.2009.00624.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furugen M, Higa F, Hibiya K, Teruya H, Akamine H, Haranaga S, et al. Legionella pneumophila infection induces programmed cell death, caspase activation, and release of high mobility group box 1 protein in A549 alveolar epithelial cells. Respir Res. 2008;9:39. doi: 10.1186/1465-9921-9-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Angulo AM, Meric-Bernstam F. Metformin: a therapeutic opportunity in breast cancer. Clin Cancer Res. 2010;16:1695–1700. doi: 10.1158/1078-0432.CCR-09-1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gras V, Bouffandeau B, Montravers PH, Lalau JD. Effect of metformin on survival rate in experimental sepsis. Diabetes Metab. 2006;32:147–150. doi: 10.1016/s1262-3636(07)70261-6. [DOI] [PubMed] [Google Scholar]
- Hardie DG. AMP-activated protein kinase as a drug target. Annu Rev Pharmacol Toxicol. 2007;47:185–210. doi: 10.1146/annurev.pharmtox.47.120505.105304. [DOI] [PubMed] [Google Scholar]
- Hardie DG, Carling D, Carlson M. The AMP-activated/SNF1 protein kinase subfamily: metabolic sensors of the eukaryotic cell? Annu Rev Biochem. 1998;67:821–855. doi: 10.1146/annurev.biochem.67.1.821. [DOI] [PubMed] [Google Scholar]
- Hattori Y, Suzuki K, Hattori S, Kasai K. Metformin inhibits cytokine-induced nuclear factor kappaB activation via AMP-activated protein kinase activation in vascular endothelial cells. Hypertension. 2006;47:1183–1188. doi: 10.1161/01.HYP.0000221429.94591.72. [DOI] [PubMed] [Google Scholar]
- Hou LC, Qin MZ, Zheng LN, Lu Y, Wang Q, Peng DR, et al. Severity of sepsis is correlated with the elevation of serum high-mobility group box 1 in rats. Chin Med J. 2009;122:449–454. [PubMed] [Google Scholar]
- Isoda K, Young JL, Zirlik A, MacFarlane LA, Tsuboi N, Gerdes N, et al. Metformin inhibits proinflammatory responses and nuclear factor-kappaB in human vascular wall cells. Arterioscler Thromb Vasc Biol. 2006;26:611–617. doi: 10.1161/01.ATV.0000201938.78044.75. [DOI] [PubMed] [Google Scholar]
- Jhun BS, Jin Q, Oh YT, Kim SS, Kong Y, Cho YH, et al. 5-Aminoimidazole-4-carboxamide riboside suppresses lipopolysaccharide-induced TNF-alpha production through inhibition of phosphatidylinositol 3-kinase/Akt activation in RAW 264.7 murine macrophages. Biochem Biophys Res Commun. 2004;318:372–380. doi: 10.1016/j.bbrc.2004.04.035. [DOI] [PubMed] [Google Scholar]
- Jiang W, Pisetsky DS. The role of IFN-alpha and nitric oxide in the release of HMGB1 by RAW264.7 cells stimulated with polyinosinic-polycytidylic acid or lipopolysaccharide. J Immunol. 2006;177:3337–3343. doi: 10.4049/jimmunol.177.5.3337. [DOI] [PubMed] [Google Scholar]
- Kang YJ, Koo EB, Lee YS, Yun-Choi HS, Chang KC. Prevention of the expression of inducible nitric oxide synthase by a novel positive inotropic agent, YS 49, in rat vascular smooth muscle and RAW 264.7 macrophages. Br J Pharmacol. 1999;128:357–364. doi: 10.1038/sj.bjp.0702787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson S, Pettila V, Tenhunen J, Laru-Sompa R, Hynninen M, Ruokonen E. HMGB1 and a predictor of organ dysfunction and outcome in patients with severe sepsis. Intensive Care Med. 2008;34:1046–1053. doi: 10.1007/s00134-008-1032-9. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Tsoyi K, Heo JM, Kang YJ, Park MK, Lee YS, et al. Regulation of lipopolysaccharide-induced inducible nitric-oxide synthase expression through the nuclear factor-kappaB pathway and interferon-beta/tyrosine kinase 2/Janus tyrosine kinase 2-signal transducer and activator of transcription-1 signaling cascades by 2-naphthylethyl-6,7-dihydroxy-1,2,3,4-tetrahydroisoquinoline (THI 53), a new synthetic isoquinoline alkaloid. J Pharmacol Exp Ther. 2007;320:782–789. doi: 10.1124/jpet.106.112052. [DOI] [PubMed] [Google Scholar]
- Lotze MT, Tracey KJ. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol. 2005;5:331–342. doi: 10.1038/nri1594. [DOI] [PubMed] [Google Scholar]
- Martin GS, Mannino DM, Eaton S, Moss M. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med. 2003;348:1546–1554. doi: 10.1056/NEJMoa022139. [DOI] [PubMed] [Google Scholar]
- Nath N, Giri S, Prasad R, Salem ML, Singh AK, Singh I. 5-aminoimidazole-4-carboxamide ribonucleoside: a novel immunomodulator with therapeutic efficacy in experimental autoimmune encephalomyelitis. J Immunol. 2005;175:566–574. doi: 10.4049/jimmunol.175.1.566. [DOI] [PubMed] [Google Scholar]
- Nath N, Khan M, Paintlia MK, Singh I, Hoda MN, Giri S. Metformin attenuated the autoimmune disease of the central nervous system in animal models of multiple sclerosis. J Immunol. 2009;182:8005–8014. doi: 10.4049/jimmunol.0803563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paintlia AS, Paintlia MK, Singh I, Singh AK. Immunomodulatory effect of combination therapy with lovastatin and 5-aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside alleviates neurodegeneration in experimental autoimmune encephalomyelitis. Am J Pathol. 2006;169:1012–1025. doi: 10.2353/ajpath.2006.051309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JS, Svetkauskaite D, He H, Kim J, Strassheim D, Ishizaka A, et al. Involvement of TLR2 and TLR4 in cellular activation by high mobility group box 1 protein (HMGB1) J Biol Chem. 2004;279:7370–7376. doi: 10.1074/jbc.M306793200. [DOI] [PubMed] [Google Scholar]
- Peña G, Cai B, Deitch EA, Ulloa L. JAK2 inhibition prevents innate immune responses and rescues animals from sepsis. J Mol Med. 2010;88:851–859. doi: 10.1007/s00109-010-0628-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedemann NC, Guo RF, Ward PA. The enigma of sepsis. J Clin Invest. 2003;112:460–467. doi: 10.1172/JCI19523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouhiainen A, Tumova S, Valmu L, Kalkkinen N, Rauvala H. Pivotal advance: analysis of proinflammatory activity of highly purified eukaryotic recombinant HMGB1 (amphoterin) J Leukoc Biol. 2007;81:49–58. doi: 10.1189/jlb.0306200. [DOI] [PubMed] [Google Scholar]
- Sag D, Carling D, Stout RD, Suttles J. Adenosine 5'-monophosphate-activated protein kinase promotes macrophage polarization to an anti-inflammatory functional phenotype. J Immunol. 2008;181:8633–8641. doi: 10.4049/jimmunol.181.12.8633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sappington PL, Yang R, Yang H, Tracey KJ, Delude RL, Fink MP. HMGB1 B box increases the permeability of Caco-2 enterocytic monolayers and impairs intestinal barrier function in mice. Gastroenterology. 2002;123:790–802. doi: 10.1053/gast.2002.35391. [DOI] [PubMed] [Google Scholar]
- Sims GP, Rowe DC, Rietdijk ST, Herbst R, Coyle AJ. HMGB1 and RAGE in inflammation and cancer. Annu Rev Immunol. 2010;28:367–388. doi: 10.1146/annurev.immunol.021908.132603. [DOI] [PubMed] [Google Scholar]
- Strack T. Metformin: a review. Drugs Today (Barc) 2008;44:303–314. doi: 10.1358/dot.2008.44.4.1138124. [DOI] [PubMed] [Google Scholar]
- Stros M. HMGB proteins: interactions with DNA and chromatin. Biochim Biophys Acta. 2010;1799:101–113. doi: 10.1016/j.bbagrm.2009.09.008. [DOI] [PubMed] [Google Scholar]
- Tan BK, Adya R, Chen J, Farhatullah S, Heutling D, Mitchell D, et al. Metformin decreases angiogenesis via NF-kappaB and Erk1/2/Erk5 pathways by increasing the antiangiogenic thrombospondin-1. Cardiovasc Res. 2009;83:566–574. doi: 10.1093/cvr/cvp131. [DOI] [PubMed] [Google Scholar]
- Tsoyi K, Kim HJ, Shin JS, Kim DH, Cho HJ, Lee SS, et al. HO-1 and JAK-2/STAT-1 signals are involved in preferential inhibition of iNOS over COX-2 gene expression by newly synthesized tetrahydroisoquinoline alkaloid, CKD712, in cells activated with lipopolysacchride. Cell Signal. 2008;20:1839–1847. doi: 10.1016/j.cellsig.2008.06.012. [DOI] [PubMed] [Google Scholar]
- Tsoyi K, Lee TY, Lee YS, Kim HJ, Seo HG, Lee JH, et al. Heme-oxygenase-1 induction and carbon monoxide-releasing molecule inhibit lipopolysaccharide (LPS)-induced high-mobility group box 1 release in vitro and improve survival of mice in LPS- and cecal ligation and puncture-induced sepsis model in vivo. Mol Pharmacol. 2009;76:173–182. doi: 10.1124/mol.109.055137. [DOI] [PubMed] [Google Scholar]
- Tsoyi K, Nizamutdinova IT, Jin Jang H, Mun L, Kim HJ, Seo HG, et al. Carbon monoxide from CORM-2 reduces HMGB1 release through regulation of IFN-beta/JAK-2/STAT-1/INOS/NO signaling but not COX-2 in TLR-activated macrophages. Shock. 2010;34:608–614. doi: 10.1097/SHK.0b013e3181e46f15. [DOI] [PubMed] [Google Scholar]
- Ueno H, Matsuda T, Hashimoto S, Amaya F, Kitamura Y, Tanaka M, et al. Contributions of high mobility group box protein in experimental and clinical acute lung injury. Am J Respir Crit Care Med. 2004;170:1310–1316. doi: 10.1164/rccm.200402-188OC. [DOI] [PubMed] [Google Scholar]
- Vane JR, Mitchell JA, Appleton I, Tomlinson A, Bishop-Bailey D, Croxtall J, et al. Inducible isoforms of cyclooxygenase and nitric-oxide synthase in inflammation. Proc Natl Acad Sci USA. 1994;91:2046–2050. doi: 10.1073/pnas.91.6.2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285:248–251. doi: 10.1126/science.285.5425.248. [DOI] [PubMed] [Google Scholar]
- Yang H, Ochani M, Li J, Qiang X, Tanovic M, Harris HE, et al. Reversing established sepsis with antagonists of endogenous high-mobility group box 1. Proc Natl Acad Sci USA. 2004;101:296–301. doi: 10.1073/pnas.2434651100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Zhong J, Yang P, Gong F, Wang CY. HMGB1, an innate alarmin, in the pathogenesis of type 1 diabetes. Int J Clin Exp Pathol. 2009;3:24–38. [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Zmijewski JW, Lorne E, Liu G, Park YJ, Tsuruta Y, et al. Activation of AMPK attenuates neutrophil proinflammatory activity and decreases the severity of acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2008;295:L497–L504. doi: 10.1152/ajplung.90210.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.