

Abstract
BACKGROUND AND PURPOSE
The use of non-steroidal anti-inflammatory drugs (NSAIDs) in the treatment of rheumatoid arthritis (RA) is limited by their toxicity. We evaluated the anti-inflammatory efficacy and safety of three novel modified NSAIDs, phospho-aspirin, phospho-ibuprofen and phospho-sulindac.
EXPERIMENTAL APPROACH
We determined the anti-inflammatory effects and gastrointestinal safety of the phospho-NSAIDs in the rat adjuvant arthritis model and studied their mechanism of action in cultured cells, Cytokines were measured with elisa and activation of nuclear factor-κB (NF-κB) by immunohistochemistry.
KEY RESULTS
All three phospho-NSAIDs showed less gastrointestinal toxicity than their parent compounds and demonstrated strong anti-inflammatory effects, essentially reversing joint inflammation and oedema. They have a broad but not uniform effect on the expression of relevant cytokines, in general decreasing IL-6 and IL-1β and increasing IL-10 levels in rat plasma and cultured cells. Phospho-sulindac and phospho-ibuprofen but not phospho-aspirin suppressed PGE2 production in vitro, whereas phospho-aspirin (in contrast to aspirin) showed the same effect in vivo. In joint tissues, phospho-aspirin inhibited NF-κB activation, and suppressed inflammation and bone resorption. Phospho-aspirin also inhibited Jurkat T cell proliferation. In general, phospho-aspirin had greater efficacy but different effects upon inflammatory mediators compared with aspirin. The chemical modification of the parent NSAIDs seems crucial for their safety and efficacy.
CONCLUSIONS AND IMPLICATIONS
Phospho-aspirin, phospho-ibuprofen and phospho-sulindac were safer than their parent NSAIDs, were highly effective in rat adjuvant arthritis and inhibited many key mediators in the pathophysiology of RA. These novel compounds are promising candidate drugs for the treatment of RA and merit further evaluation.
Keywords: arthritis, NSAIDs, anti-inflammation, phospho-aspirin, phospho-ibuprofen, phospho-sulindac, rheumatoid arthritis
Introduction
Rheumatoid arthritis (RA) is a chronic autoimmune disease with a 1% prevalence in the Western world. Chronic inflammation, a hallmark of the disease, causes progressive joint destruction, which leads to significant pain, joint deformities, and functional limitation that can be incapacitating (Alamanos and Drosos, 2005; Eurenius and Stenstrom, 2005). RA is also associated with substantial co-morbidity in the cardiovascular, neurological, and metabolic systems, with the same cytokines that drive synovial pathology being also responsible for generating pathology in extra-articular tissues. Despite substantial progress in recent years, RA remains a formidable clinical problem.
Non-steroidal anti-inflammatory drugs (NSAIDs) are used widely in the treatment of RA to provide symptomatic relief of inflammation and pain; NSAIDs are often used at high doses and on a long-term basis. The main limitation to treatment with NSAIDs is their side effects (Rahme and Bernatsky, 2010; Sostres et al., 2010). Among patients using NSAIDs, up to 4% per year suffer serious gastrointestinal complications. In 1998 the number of deaths in the United States from NSAID-induced GI complications was 16,550, virtually identical to that from AIDS (16 685) (Singh and Triadafilopoulos, 1999). NSAIDs also cause a wide range of tubular, interstitial, glomerular and vascular renal lesions and probably chronic renal failure. Several additional side effects, some serious, have been reported (Roberts and Morrow, 2001).
The rational design of pharmacological agents includes, amongst others, modification of known compounds in order to optimize their pharmacological properties, primarily by increasing their safety profile while maintaining efficacy. Several efforts have been made to modify NSAIDs to reduce their gastrointestinal toxicity while maintaining their anti-inflammatory effect (Cicala et al., 2000; Rigas, 2007; Nemmani et al., 2009). We have recently synthesized a series of modified NSAIDs and assessed their efficacy against cancer, and demonstrated superior efficacy and apparent safety in preclinical models (Mackenzie et al., 2010). Our chemical modification is exemplified by the structures shown in Figure 1, a linker molecule is attached via an ester bond to the carboxylic group of the NSAID, while a phosphate moiety(ies) is also attached to the linker.
Figure 1.
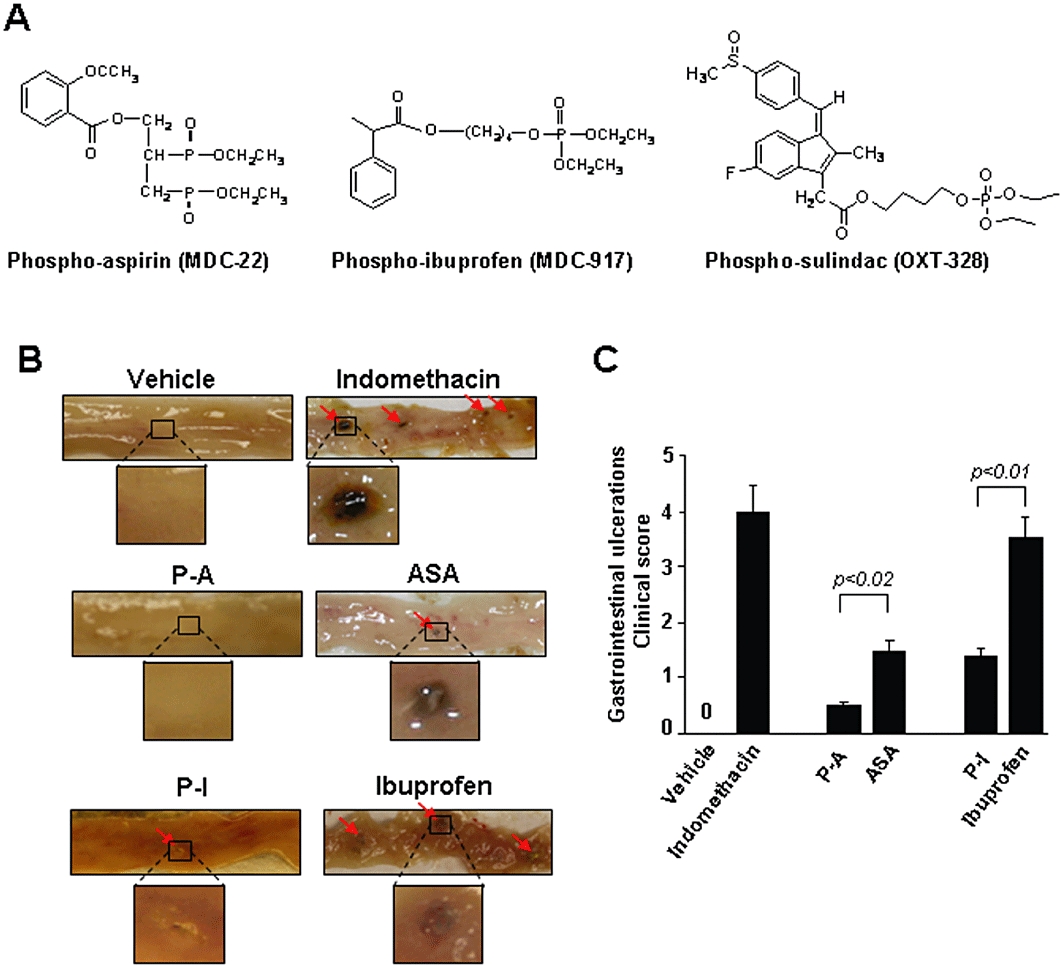
Gastrointestinal toxicity of phospho-aspirin, aspirin, phospho-ibuprofen and ibuprofen. (A) Chemical structures of phospho-aspirin, phospho-ibuprofen and phospho-sulindac. (B and C) The gastrointestinal toxicity of phospho-aspirin (P-A), phospho-ibuprofen (P-I), aspirin (ASA) and ibuprofen was determined in Sprague-Dawley rats. (B) Representative photographs of small intestinal mucosa from the various groups of animals; the small intestine has been cut longitudinally and its mucosal side is viewed. Red arrows: gastrointestinal ulcers. Selected ulcers are magnified for clarity. (C) On day 5, the number and size of small intestinal ulcerations were determined and scored. Values are mean ± standard deviation.
In order to assess the safety and efficacy of this new class of compounds, we evaluated three representative compounds, phospho-aspirin (MDC-22), phospho-ibuprofen (MDC-917) and phospho-sulindac (OXT-328), using a rat model of arthritis. Several animal models of RA have been described (Bendele, 2001), including the rat adjuvant arthritis model, type II collagen arthritis, and several transgenic models, such as the human tumor necrosis factor-α (TNF-α) transgenic arthritis model (Feldmann et al., 1996). The rat adjuvant arthritis model, originally described more than 30 years ago (Pearson et al., 1961) is still the most widely used assay to identify chemical agents having a potential therapeutic efficacy in RA (Theisen-Popp and Muller-Peddinghaus, 1994; Whiteley and Dalrymple, 2001). Freund's adjuvant (FA) is an antigen solution, used as an immunopotentiator (Mycobacterium butyricum emulsified in light mineral oil). Following FA injection, a series of cellular changes culminate in severe arthritis, which displays some of the cardinal features of human RA such as swelling of the extremities, cartilage degradation, loss of joint function and lymphocyte infiltration into diseased joints.
We report here that the novel phospho-NSAIDs are highly efficacious in controlling rat adjuvant arthritis, appear safer than their parent NSAIDs, and their anti-inflammatory action includes modulation of cytokine expression and inhibition of the synthesis of PGE2 and of the activation of nuclear factor-κB (NF-κB). Thus, phospho-NSAIDs represent a promising alternative to conventional NSAIDs and deserve further evaluation.
Methods
Cell viability assay
The reduction of the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was determined following the manufacturer's instructions (Roche, Mannheim, Germany) and as previously described (Zhao et al., 2009). Briefly, following incubation of cells for 6 h and 24 h with the test compounds, MTT reagent (10 µL) was added to each well and incubated for an additional 4 h. After that, 100 µL of solubilization/stop solution [10% (w/v) SDS in 0.01 M HCl] was added and the plate was incubated at room temperature overnight to completely solubilize the formazan crystals. The reduction of MTT was monitored at 570 nm, and the amount of reduced MTT was calculated as a percentage of the absorbance determined for the vehicle control treatment.
Cell proliferation assay
The measurement of 5-bromo-2′-deoxyuridine (BrdU) incorporation into newly synthesized cellular DNA was used to determine cell proliferation as previously described (Sun and Rigas, 2008). Briefly, 1 × 106 Jurkat T cells, seeded in 60 mm dishes, were treated with phospho-aspirin or aspirin (1.5 × IC50) for 24 h. After treatment, 10 µM BrdU (BD Biosciences, San Jose, CA, USA) were added to the cell culture media, and cells were incubated for an additional 30 min in the CO2 incubator. Cells were then trypsinized and fixed with 70% ethanol for 30 min on ice. Following one cycle of washing with PBS, DNA was denatured by 2 N HCl/Triton-X100 for 30 min and then neutralized by 0.1 M Na2B4O7, pH 8.5. After incubation with 20 µL anti-BrdU-FITC antibody (BD Biosciences) for 30 min, cells were subjected to flow cytometric analysis.
Animal studies
All animal care and experimental studies were approved by our Institutional Animal Care and Use Committee. The animals (Charles River, Wilmington, MA) were kept under conditions of constant temperature (23 ± 2°C) and humidity (55 ± 15%) with a 12 h light/dark cycle (lights on at 0700 h). Animals had free access to food and water.
Gastrointestinal toxicity
The gastrointestinal toxicity of the novel phospho-NSAIDs (phospho-aspirin, phospho-ibuprofen and phospho-sulindac) and their corresponding parent compounds (aspirin, ibuprofen and sulindac) was determined in rats following a standard protocol (Whiteley and Dalrymple, 1998). Briefly, 6 week-old Sprague Dawley rats (n= 4 per group) were treated for 4 days by gavage with vehicle (corn oil) or indomethacin 4.75 mg·kg−1·day−1 or equimolar amounts of phospho-aspirin (110 mg·kg−1·day−1) and aspirin (35 mg·kg−1·day−1), or equimolar amounts of phospho-sulindac (317 mg·kg−1·day−1) or sulindac (200 mg·kg−1·day−1), or equimolar amounts of phospho-ibuprofen (400 mg·kg−1·day−1) or ibuprofen (200 mg·kg−1·day−1). Indomethacin was used as a reference compound, based on the protocol (Whiteley and Dalrymple, 1998); indomethacin, given once daily for 4 days at a dose of 4.75 mg·kg−1, produces GI toxicity clinical score of approximately 3.75. On day 5, 30 min before killing, animals were injected with 1% Evans blue solution and the number and size of small intestinal ulcerations were recorded after death and the clinical score, ranging between 0 (intact small intestine with no ulcerations or mucosal damage) and 5 (animal dies prior to study's end), was determined (Whiteley and Dalrymple, 1998).
Induction of FA arthritis and treatment with phospho-NSAIDs
We followed the protocol of Whitley et al. (Whiteley and Dalrymple, 2001). Briefly, female LEW/CrlBR Lewis rats (n= 5/group), weighing 110–140 g, were housed with free access to food and water. Arthritis was induced by injecting once 100 µL of 10 mg·mL−1Mycobacterium butyricum (DIFCO Laboratories, Detroit, MI, USA) suspended in incomplete FA into the tail of each rat. Each of the following were given to rats by oral gavage daily for 18 days: aspirin 35 mg·kg−1, phospho-aspirin 110 mg·kg−1 (aspirin and phospho-aspirin doses were equimolar), phospho-ibuprofen 300 mg·kg−1 or phospho-sulindac 150 mg·kg−1 suspended in corn oil or corn oil alone for the control group. No equimolar doses of sulindac and ibuprofen were included because these doses would have been toxic to the rats (Adeyeye et al., 1996; Mackenzie et al., 2010). The administration of the test compounds started on the same day FA was injected. The progression of arthritis was evaluated by scoring the intensity of the oedema in the paws and tail once a week (Whiteley and Dalrymple, 2001). Each paw was assigned a score of 0 to 4 (0 = no swelling or redness in any joint; 4 = very severe swelling and redness in large and small joints; 1–3 = intermediate scores), and the tail of each animal a score of 0 to 3 (0 = no signs of inflammation, even at the site of injection; 3 = >25% of the tail exhibiting oedema and necrosis; 1–2 = intermediate scores). At the completion of the study, animals were killed 1 h after the last administration of the study drug, blood was collected for the determination of levels of cytokines and PGE2 metabolite and their hind paws were transected, weighed and preserved in formalin.
Histology examination and immunohistochemistry
Following 24 h in formalin, hind paws were placed in Surgipath decalcifier (Surgipath, Grayslake, IL) for an additional 24 h. Once the decalcification was complete, the joint was transected along its longitudinal plane to give approximately equal halves and each one was paraffin embedded. A pathologist, unaware of sample identity, evaluated 4 µM-thick joint sections stained with H&E and scored them for inflammation and bone resorption, as previously described (Bendele et al., 1999).
Two additional sections of each sample were placed side-by-side on a microscope slide, with one of them used as negative control; each sample contained synovium, cartilage and bone marrow tissues. Paraffin-embedded sections were deparaffinized, rehydrated, and microwave-heated for 15 min in 0.01 mol·L−1 citrate buffer (pH 6.0) for antigen retrieval, and 3% H2O2 was applied to block endogenous peroxidase activity. After 15 min of blocking with horse serum, the primary antibody or control IgG (dilution 1/50) were applied and incubated overnight at 4°C. Slides were washed thrice with PBS, each for 5 min. The biotinylated secondary antibody and the streptavidin-biotin complex (Vector Laboratories, Burlingame, CA, USA) were applied, each for 30 min at room temperature with an interval washing. After rinsing with PBS, the slides were immersed for 5 min in the substrate 3,3′-diaminobenzidine (DAB; Sigma, St. Louis, MO, USA), then rinsed with distilled water, counterstained with haematoxylin, dehydrated, and coverslipped. We used a phospho-NF-κB p65 (Ser276) antibody, which recognizes activated NF-κB (Cell Signaling Technology, Beverly, MA, USA). Five different fields per slide from synovium, cartilage and bone marrow were counted and scored for NF-κB; positive staining for NF-κB appears brown. Results were expressed as the percentage of positive cells; specificity of binding was verified by isotype control.
IL-6, IL-10, IL-1β and TNF-α levels
Cytokine levels were assayed in plasma samples using rat IL-1β, IL-6, IL-10 and TNF-αelisa kits (Thermo Fisher Scientific, Rockford, IL, USA) and following the manufacturer's instructions. Cytokines were also measured in RAW 264.7 and NIH 3T3 cell culture media using elisa assays to measure the levels of IL-6 (BD Biosciences) and TNF-α (Abcam, Cambridge, MA, USA), following the manufacturers' protocols. Briefly, 1.5 × 106 cells were pre-incubated with each of the study drugs (1.5 × 24 h–IC50) for 60 min followed by treatment with lipopolysaccharide (LPS; 1 µg·mL−1) for 5 h in under the indicated experimental conditions. After the incubation, the cell culture media were collected and centrifuged at 800× g for 10 min. We determined the various cytokines in the supernatant by elisa following the manufacturers' protocols.
For the time-course, NIH 3T3 cells were pre-treated with phospho-ibuprofen (1 × 24 h–IC50) for 1 h, followed by treatment with LPS (100 ng·mL−1) for 3, 6 or 24 h. Cytokines released into the media were measured after separating the cells by centrifugation at 800× g for 10 min.
Prostaglandin E2 (PGE2)
The levels of PGE2 and 13,14-dihydro-15-keto-PGE2 (PGE2 metabolite) were determined by immunoassay in cell culture media and plasma, respectively, following the manufacturer's instructions (Cayman Chemical, Ann Arbor, MI, USA). Briefly, 1.5 × 106 NIH 3T3 cells were pre-incubated with the various drugs (1.5 × 24 h–IC50) for 30 min followed by treatment with the calcium ionophore A23187 5 µM for 3 h under the indicated experimental conditions.
Data analysis
Results from at least three independent experiments were expressed as mean ± SEM and analysed by one-way analysis of variance followed by Tukey's test for multiple comparisons. To analyse data from the inflammation and histological score systems, we used a non–parametric method (Brunner et al., 2002), where the data were first ranked, and then fitted, using a mixed effects model with an unstructured variance-covariance matrix and P values adjusted for anova-type statistics. P < 0.05 was considered statistically significant.
Materials
The phospho-NSAIDs, phospho-aspirin, phospho-ibuprofen and phospho-sulindac, provided by Medicon Inc, Setauket, NY, USA, were synthesized as reported (Sun and Rigas, 2008). Aspirin, ibuprofen and sulindac and all other chemicals were obtained from Sigma. RAW 264.7 mouse macrophage cells, NIH-3T3 mouse fibroblast cells, and human Jurkat T cells were grown as suggested by American Type Tissue Collection (ATTC, Manassas, VA, USA). These three cell lines represent models of macrophages, fibroblasts and T cells, respectively; each cell type plays an important role in the development of arthritis.
Results
Phospho-NSAIDs are safer than traditional NSAIDs in a rat model of gastrointestinal toxicity
Initially, we evaluated the safety of the novel phospho-NSAIDs using an established protocol that evaluates gastrointestinal side effects of potential anti-inflammatory and other agents (Whiteley and Dalrymple, 1998). Rats were treated for 4 days by oral gavage with vehicle, indomethacin (positive control) or equimolar amounts of each phospho-NSAID or its parent compound as described in the Methods. As expected, treatment with indomethacin (positive control) produced predominantly medium and large ulcerations (>4 mm) in the small intestine (Figure 1). As shown in Figure 1, phospho-aspirin induced 68% less damage than aspirin and phospho-ibuprofen induced 60% less than ibuprofen. Of note, the doses of phospho-aspirin and aspirin were equimolar, as were the doses of phospho-ibuprofen and ibuprofen. We have reported similar results for phospho-sulindac compared with sulindac (Mackenzie et al., 2010). Thus phospho-aspirin and phospho-ibuprofen induced significantly less gastrointestinal toxicity than their parent compounds, aspirin and ibuprofen.
Phospho-NSAIDs inhibit arthritis in rats with FA-induced arthritis
We evaluated the ability of the three phospho-NSAIDs to inhibit the inflammatory response of rat adjuvant arthritis. Following FA injection, rats developed marked arthritis with inflamed hind paws and necrotic tails. We followed the progression of arthritis by evaluating weekly the intensity of the oedema in the paws and tail. Significant oedema was recorded on day 14 (following FA injection). The phospho-NSAIDs prevented the development of oedema in both the tail and paws (Supporting Information Figure S1A). At the end of the experiment on day 18, oedema of the hind paw was significantly reduced by phospho-aspirin and aspirin given at equimolar doses as shown by changes in hind paw weight (Figure 2). Thus, the injection of FA increased paw weight by 49%, phospho-aspirin and aspirin reduced this effect by 87 and 88%, respectively (P < 0.001 for both), essentially normalizing paw weight.
Figure 2.
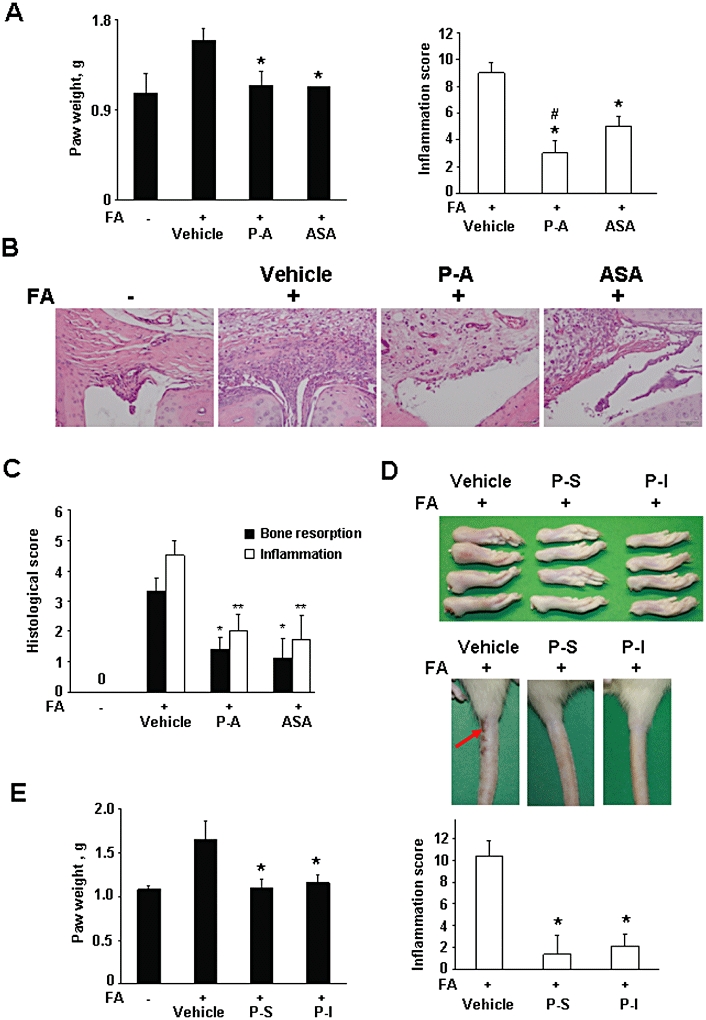
Phospho-non-steroidal anti-inflammatory drugs (NSAIDs) prevent the development of rat adjuvant arthritis. (A–C) Phospho-aspirin prevents the development of rat adjuvant arthritis. Arthritis was induced in rats by injecting Freund's adjuvant (FA) into their tail. Rats were treated daily by oral gavage for 18 days with phospho-aspirin (P-A; 110 mg·kg−1) or aspirin (ASA; 35 mg·kg−1; equimolar doses) suspended in corn oil. (A) The hind paw weight (left) and inflammation (clinical) score (right) of the various study groups. Data are expressed as mean ± standard deviation; n= 5 rats per group. *Significantly different compared with FA/vehicle-treated group; P < 0.03. (B) Histological evaluation of hind paw joints. Formalin-fixed decalcified tissue from affected joints was stained with haematoxylin and eosin and examined under an Olympus BX41 microscope (20×; Olympus, Center Valley, PA, USA) and photographed with an Olympus DP25 digital camera. FA induced oedema and infiltration with inflammatory cells, endothelial cell proliferation and synovial thickening, obliterating the free joint space (compare with normal). phospho-aspirin greatly reduced the inflammatory infiltrate and tissue oedema, restoring the free joint space. aspirin had a similar but less pronounced effect. (C) Bone resorption and inflammation were scored. Values are mean ± SEM. *significantly different compared with FA/vehicle-treated group; *P < 0.03; **P < 0.07. (D, E) Phospho-ibuprofen and phospho-sulindac prevented the development of rat adjuvant arthritis. Arthritis was induced by injecting Freund's adjuvant (FA) into the tail of the rat. Rats were treated with phospho-ibuprofen (P-I; 300 mg·kg−1) or phospho-sulindac (P-S; 150 mg·kg−1), suspended in corn oil, given daily by oral gavage for 18 days. D: Representative photographs of resected hind paws (left panel) and tails (right panel) from the various experimental groups are shown. Red arrow: area of necrosis. E: Hind paw weight (left panel) and (clinical) inflammation score (right panel) of phospho-sulindac and phospho-ibuprofen-treated rats. Values are mean ± standard deviation. *Significantly different compared with FA-treated group; P < 0.01.
FA-treated rats developed oedema and redness in large and small joints and significant inflammation involving about 25% of the tail (Figure 2D). The clinical signs of inflammation are scored to evaluate the intensity of the oedema in the paws and tail; the resulting inflammation score (or clinical score) ranges between 0 and 4. As shown in Figure 2A, phospho-aspirin and aspirin reduced the mean inflammation score by 67 and 44% respectively (P < 0.03, for both).
We also evaluated the status of bone resorption and the degree of synovial inflammation (Bendele et al., 1999). As shown in Figure 2C, the induction of arthritis by FA was accompanied by a significant amount of bone resorption. Phospho-aspirin and aspirin decreased it to a similar degree (58% for phospho-aspirin, P < 0.04 and 66% for aspirin, P < 0.02). The degree of synovial inflammation, apparent in the histological pictures of Figure 2B, was also decreased by phospho-aspirin (56% for phospho-aspirin, P < 0.03 and 61% for aspirin, P < 0.03).
Phospho-ibuprofen and phospho-sulindac significantly reduced the hind paw weight essentially normalizing it (96% reduction for phospho-sulindac and 86% for phospho-ibuprofen; P < 0.001 for both) (Figure 2E). Their effect on inflammation was equally strong, with 87% reduction for phospho-sulindac and 79% for phospho-ibuprofen; P < 0.001 for both; Figure 2E).
No gastrointestinal toxicity was observed in the FA rats treated with phospho-NSAIDs. After death, the gastrointestinal tract was inspected under a magnifying lens and the mucosa appeared normal, with no evident damage. In addition, the body weight of the phospho-NSAID treated animals was not significantly different from those of the control and FA-treated groups (Supporting Information Figure S1B).
The effect of phospho-NSAIDs on cytokine levels
Proinflammatory cytokines, such as IL-1β, IL-6 and TNF-α, play a central role in the pathophysiology of RA (Feldmann et al., 1996). Thus, we determined the response of several cytokines to our phospho-NSAIDs both in vitro and in vivo.
We investigated if these phospho-NSAIDs could affect the production of TNF-α and IL-6 in RAW 264.7 and NIH 3T3 cells. As the baseline levels of TNF-α and IL-6 were very low or undetectable, we stimulated these cells with LPS (Lichtman et al., 1998). Thus, cells were pre-incubated for 1 h with equipotent concentrations of each of the phospho-NSAIDs or their corresponding parent compound (each at the 1.5 × 24 h–IC50) followed by treatment with LPS for an additional 5 h and the response of these two cytokines was determined at various time intervals after that as indicated. The 24 h–IC50 values for growth inhibition were (i) for RAW 264.7 cells: phospho-aspirin = 406 µM; aspirin = 3.2 mM; phospho-sulindac = 55 µM; phospho-ibuprofen = 173 µM, and (ii) for NIH 3T3 cells: phospho-aspirin = 390 µM; aspirin = 920 µM; phospho-sulindac = 63 µM; and phospho-ibuprofen = 84 µM.
In RAW 264.7 cells, LPS induced a brisk production of both TNF-α and IL-6 (Figure 3A). phospho-aspirin suppressed this increase of TNF-α by 66%, whereas conventional aspirin had a very modest effect (26% reduction). Both phospho-sulindac and phospho-ibuprofen had a dramatic effect, bringing TNF-α levels to zero or nearly zero. Phospho-aspirin, phospho-sulindac and phospho-ibuprofen also brought the levels of IL-6 to near zero, whereas aspirin had again a modest effect (48% reduction). To explore whether this effect was restricted to one cell line, we also examined aspects of the anti-cytokine effect of phospho-ibuprofen in NIH 3T3 cells. As shown in Figure 3B, LPS stimulated IL-6 production and phospho-ibuprofen reduced this effect significantly in a concentration- and time-dependent manner. NIH 3T3 cells responded to the other two compounds in a similar manner in terms of IL-6 production (data not shown). To assess whether the effect of the phospho-NSAIDs on cytokine levels was due to their toxicity, we evaluated cell viability at 6 h, the time when cytokines were measured. At 6 h, the three phospho-NSAIDs and aspirin at 1.5 × IC50 (based on their 24 h–IC50) had a marginal effect on cell viability; viable cells, determined by Trypan blue and MTT, represented 84–100% of the total (Supporting Information Table S1).
Figure 3.

Phospho-non-steroidal anti-inflammatory drugs (NSAIDs) reduce the levels of inflammatory cytokines. (A) RAW 264.7 cells were pre-treated for 1 h with each of the phospho-NSAIDs or aspirin followed by treatment with lipopolysaccharide (LPS; 1 µg·mL−1) for an additional 5 h. The concentration of tumor necrosis factor-α (TNF-α) (left panel) and IL-6 (right panel) in culture media was measured by elisa. Values are mean ± standard deviation of three independent experiments. *Significantly different compared with LPS-stimulated group; P < 0.01. (B) Concentration-dependent (left panel) and time-dependent (right panel) inhibition of IL-6 release to the media by phospho-ibuprofen (P-I) in NIH 3T3 cells stimulated with LPS 100 ng·mL−1. Values are mean ± standard deviation (SD) of three independent experiments. *Significantly different compared with LPS-stimulated group; P < 0.05. (C and D) Cytokines (IL-1β, IL-6 and IL-10) were measured in rat plasma from normal rats or rats with FA-induced arthritis treated with vehicle, phospho-aspirin (P-A; 110 mg·kg−1·day−1), aspirin (ASA; 35 mg·kg−1·day−1), phospho-ibuprofen (P-I; 400 mg·kg−1·day−1) or phospho-sulindac (P-S; 150 mg·kg−1·day−1) for 18 days. Values are mean ± SD (n= 5); *significantly different compared with FA/vehicle-treated group; P < 0.01–0.05.
We also determined the levels of several inflammatory cytokines in the plasma of rats collected on day 18 (Figure 3C and D). FA markedly increased plasma levels of IL-1β and IL-6 (9.4-fold for IL-1β; 27.4% increase for IL-6, as expected (Cicala et al., 2000). phospho-aspirin blocked the increase of IL-1β (93% reduction, P < 0.04) whereas aspirin had a much weaker effect that did not reach statistical significance (46% reduction, P < 0.10). IL-6 levels followed a similar pattern, but the changes were not as pronounced. FA increased IL-6 levels modestly but statistically significantly (27% increase; P < 0.04). phospho-aspirin decreased IL-6 levels by 27% compared with the FA group (P < 0.005), whereas aspirin's effect was less pronounced (15% reduction, P < 0.03). phospho-aspirin and aspirin had no significant effect on IL-10 levels, which FA stimulated by 122% (data not shown).
Of the other two phospho-NSAIDs, phospho-ibuprofen reduced IL-6 by 34% (P < 0.03), stimulated IL-10 by 86% (P < 0.05) (Figure 3D) but had no significant effect on IL-1β (data not shown). phospho-sulindac had no significant effect on IL-1β (data not shown), IL-6 or IL-10. Of note, the plasma concentration of TNF-α was below the level of detection in all groups of rats.
The effect of phospho-NSAIDs on the prostaglandin pathway
PGE2, an important proinflammatory eicosanoid, is involved in the pathogenesis of arthritis (McCoy et al., 2002). We evaluated the effect of phospho-aspirin, aspirin, phospho-sulindac and phospho-ibuprofen on the production of PGE2 by NIH 3T3 cells, both under baseline conditions and following stimulation with the calcium ionophore A23187 (Hyman et al., 1982). In contrast to all other compounds, phospho-aspirin failed to alter significantly the production of PGE2 by these cells, whereas conventional aspirin, as expected, essentially eliminated it (Figure 4A). Phospho-sulindac had a modest effect, decreasing it by 19 and 43%, under baseline and stimulated conditions respectively. Phospho-ibuprofen had a much more pronounced effect, reducing PGE2 production by 85 and 92%, under baseline and stimulated conditions respectively.
Figure 4.
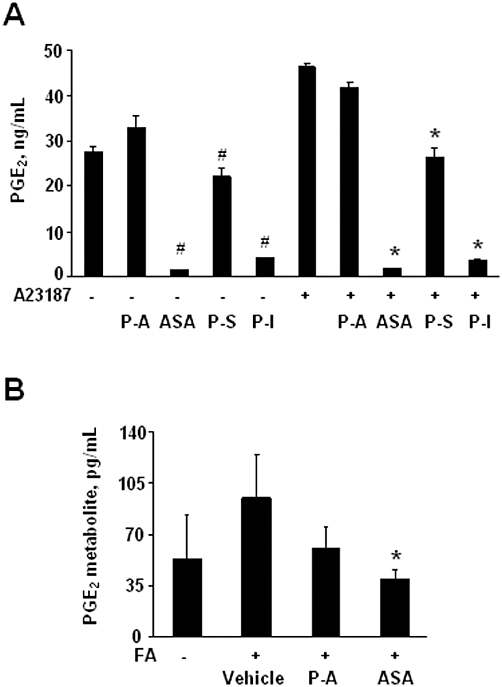
Effect of phospho-non-steroidal anti-inflammatory drugs (NSAIDs) on PGE2. (A) PGE2 levels in NIH 3T3 cells pre-treated with 1.5 × IC50 phospho-aspirin (P-A), aspirin (ASA), phospho-ibuprofen (P-I) and phospho-sulindac (P-S) for 30 min followed by treatment with A23187 5 µM for 3 h. Values are mean ± standard deviation of three independent experiments; #significantly different compared with control non-stimulated group, P < 0.05; *significantly different compared with A23187-stimulated group; P < 0.01. (B) PGE2 levels was measured by elisa in rat plasma from normal or FA/vehicle-treated rats, or rats treated with phospho-aspirin (110 mg·kg−1·day−1) or aspirin (35 mg·kg−1·day−1) for 18 days. Values are mean ± SEM (n= 5); *significantly different compared with FA/vehicle-treated group; P < 0.05).
In vivo PGE2 is rapidly converted to its 13,14-dihydro-15-keto metabolite (Hamberg and Samuelsson, 1971). Therefore, measuring this metabolite (instead of PGE2 itself) provides a more reliable estimate of actual PGE2 production by the whole animal (Hamberg and Samuelsson, 1971). FA treatment increased the levels of the PGE2 metabolite by 77% (P < 0.06), compared with normal rats (Figure 4B). Phospho-aspirin decreased the plasma levels of PGE2 metabolite by 36%, compared with the FA/vehicle-treated group but this difference did not reach statistical significance (P= 0.09). In contrast, aspirin restored the levels of the PGE2 metabolite to baseline values, similar to those in normal controls (P < 0.04).
Phospho-aspirin suppressed the activation of NF-κB in rat joints
NF-κB is activated in RA, being involved in its inflammatory stages (Brown et al., 2008). For this reason, we evaluated its activation by immunohistochemistry in three anatomical regions of the joint: synovium, cartilage and bone marrow. While the synovium of the normal, non-treated group showed a single layer of epithelial cells, the FA/vehicle-treated group had an increased number of layers of epithelial cells and was accompanied by increased NF-κB activation. As shown in Figure 5, following administration of FA, the greatest NF-κB activation occurred in the synovium (263% increase), followed by that in cartilage (80% increase), while it was very limited in the bone marrow. Phospho-aspirin suppressed significantly the activation of NF-κB by FA in both the synovium and cartilage (41 and 48%, respectively, P < 0.05 for both). In contrast, aspirin had weaker effects on both the synovium (29% decrease, P < 0.05) and cartilage (24%, statistically not significant). Neither compound significantly affected the NF-κB status in bone marrow cells.
Figure 5.
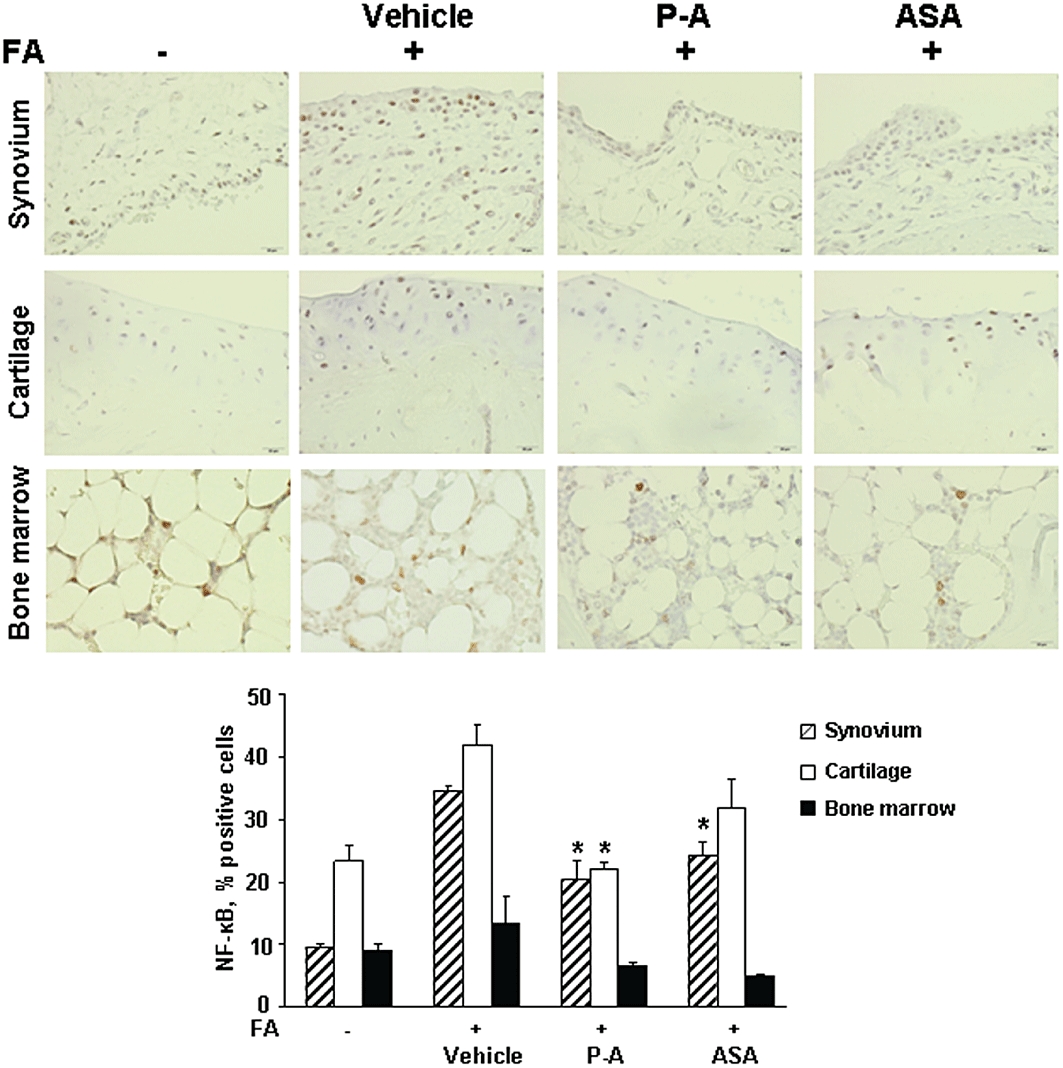
Effect of phospho-aspirin and aspirin on activated nuclear factor-κB (NF-κB). Immunohistochemical staining for activated NF-κB (p65 subunit) in synovium, cartilage and bone marrow from hind paw joint tissue samples from normal controls, and rats with Freund's adjuvant (FA)-induced arthritis treated with vehicle, phospho-aspirin (P-A; 110 mg·kg−1·day−1) or aspirin (ASA; 35 mg·kg−1·day−1). Tissue sections were examined using an Olympus BX41 microscope (40×) and photographed with an Olympus DP25 digital camera. NF-κB positive cells stain brown; specificity of binding was verified by using an isotypic control antibody. Upper panel: The synovium in the control consists of a monolayer of cells, which became multilayered in response to FA; phospho-aspirin reduced it significantly as did aspirin, but to a lesser degree. In cartilage, FA increased the number of NF-κB positive cells compared with control; phospho-aspirin reduced markedly NF-κB activation and aspirin had a similar but less pronounced effect. The bone marrow samples show essentially similar levels of NF-κB positivity. Lower panel: Five different fields per slide were scored as in the Methods and results for NF-κB staining in synovium, cartilage and bone marrow are shown as mean ± SEM; *significantly different compared with FA/vehicle-treated group; P < 0.05).
Effect of phospho-aspirin on Jurkat T cell proliferation
In RA, there is an increase in inflammatory cell growth and proliferation, including T and B lymphocytes, macrophages and mast cells (Monaco et al., 2004). Thus, we investigated the capacity of phospho-aspirin and aspirin to inhibit Jurkat T cell growth. After 48 h of incubation, phospho-aspirin decreased cell growth in a concentration-dependent manner, with an IC50= 144 µM. aspirin, on the other hand, only significantly decreased Jurkat T cell growth at concentrations higher than 500 µM, with an IC50= 1056 µM (Figure 6). In this respect, phospho-aspirin was 7.3-fold more potent than aspirin. The respective IC50 values for phospho-sulindac and phospho-ibuprofen were 38 and 22 µM. Finally, we assessed the capacity of phospho-aspirin and aspirin to prevent cell proliferation in Jurkat T cells. At 24 h, treatment with phospho-aspirin 1.5 × IC50 reduced their rate of proliferation by 20% whereas aspirin had no effect (Figure 6B).
Figure 6.
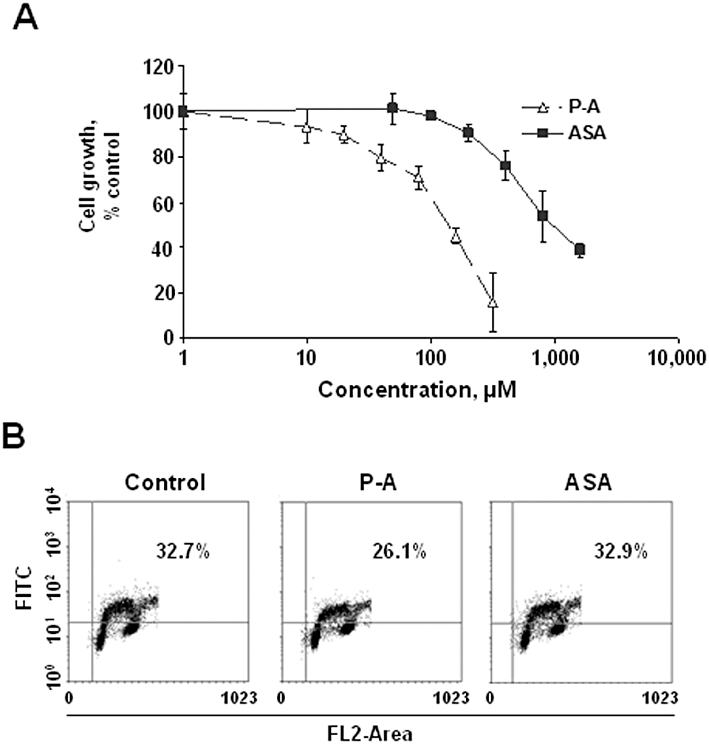
Effect of phospho-aspirin and aspirin on Jurkat T cell growth and proliferation. (A) Jurkat T cells were incubated for 48 h with various concentrations of phospho-aspirin (P-A) or aspirin (ASA) and their growth was determined by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. (B) Jurkat T cells were grown overnight and treated with phospho-aspirin or aspirin 1.5 × IC50 for 24 h. Cell proliferation was assayed based on 5-bromo-2′-deoxyuridine (BrdU) incorporation into DNA during the S-phase of the cell cycle. The percentage of BrdU positive cells is shown.
Discussion
Our findings have demonstrated two potentially important properties of these phospho-NSAIDs, representing a wider group of similarly modified compounds: their profound anti-inflammatory effects on FA-induced arthritis and their apparent gastrointestinal safety. The evidence that we have obtained suggests a complex multi-targeted effect that includes cytokines, eicosanoids and NF-κB, three pivotal players in the pathogenesis of RA, all of which are also key regulators of inflammation in general.
First of all, our study addressed the issue of gastrointestinal safety of these three compounds. Their safety is of paramount importance, for, as already mentioned, gastrointestinal toxicity is the most limiting toxicity of NSAIDs (Rahme and Bernatsky, 2010; Sostres et al., 2010), especially when they are used long-term, as is the case with RA, and length of treatment is, in fact, associated with increased incidence of complications (Silverstein et al., 2000). In agreement with our previous report on phospho-sulindac (Mackenzie et al., 2010), phospho-aspirin and phospho-ibuprofen have significantly reduced gastrointestinal toxicity compared with their parent compounds, as assessed by our acute toxicity study and deduced by the absence of any morbidity during the 18 days of daily treatment of our rats. These findings, encouraging as they are, should not, however, be considered as definitive; more detailed studies and of longer duration will be required. Nevertheless, the notion that the carboxylic group in the molecule of the conventional NSAIDs may account to a large extent for their gastrointestinal toxicity is significantly advanced (Piazza et al., 2009). All our compounds have their –COOH groups esterified with the linker and thus our findings are congruent with this theory.
All three compounds showed remarkable efficacy against arthritis in the rat adjuvant arthritis model. All essentially normalized hind paw oedema, preventing the associated impairment in mobility, and reduced the inflammation score, with phospho-sulindac and phospho-ibuprofen bringing it down to near zero. Bone resorption and synovial inflammation, evaluated in the case of phospho-aspirin, were also reduced significantly indicating a broad drug activity effect. These are highly relevant effects, as RA is characterized by extensive inflammation and proliferation of the synovium of diarthrodial joints leading to articular destruction.
It is now clear that a large number of cytokines play a fundamental role in the processes that cause inflammation, articular destruction, and the co-morbidities associated with RA (Dayer, 2003). In fact, the success of TNF-α blockade as a treatment for RA has made other cytokines realistic targets for therapeutic intervention. Our compounds have strong in vitro and in vivo effects on cytokines currently considered pivotal to the pathogenesis of RA.
Our in vitro studies using two cell lines demonstrate extensive suppression or elimination of the levels of TNF-α and IL-6 by all three compounds. Similarly, in the rat model, phospho-aspirin had a very strong effect on IL-1β, suppressing its levels to those of animals without arthritis. Several members of the IL-1 superfamily, including IL-1β, have been implicated in the pathogenesis of RA (Dayer, 2003). phospho-aspirin and phospho-ibuprofen (but not phospho-sulindac) also normalized the levels of IL-6, with phospho-ibuprofen actually suppressing them below those of non-arthritic animals. IL-6 mediates pleiotropic functions, including effects on the maturation and activation of B and T cells, macrophages, osteoclasts, and chondrocytes and probably drives the hepatic acute phase response in RA (Kishimoto, 2005). Of interest, the levels of the anti-inflammatory IL-10 were significantly increased by phospho-ibuprofen, somewhat by phospho-sulindac, but not by phospho-aspirin. IL-10 is a potent anti-inflammatory cytokine that regulates endogenous proinflammatory cytokine production in RA synovial tissue, including that of TNF-α and IL-1β (see Kunz and Ibrahim, 2009). Of note, in our study, TNF-α plasma concentration was below the level of detection in all groups of rats. Although several reports indicate that TNF-α levels are elevated in RA rats (Conway et al., 2001; Hayashida et al., 2004), others are in agreement with our findings. For example, a detailed time-course study of adjuvant arthritis in rats showed that plasma TNF-α levels increased 6 h after the adjuvant injection, peaked at 12 h, returned to near basal control concentrations on day 2 and slightly increased until day 20 post injection (Philippe et al., 1997).
PGs were among the first lipid mediators implicated in the inflammation of RA (Akaogi et al., 2006; Hikiji et al., 2008). PGE2, present in large amounts at sites of inflammation, is by far the major prostanoid synthesized in the joint, playing an important role in the pathogenesis of arthritis (McCoy et al., 2002). Increased levels of PGE2 are frequently measured in serum and synovial fluid from RA patients. The bone-resorptive activity of PGE2 is mediated by receptor activator of NF-κB ligand, whereas stimulation of bone resorption by IL-1- and IL-6 involves PGE2 production. Traditional NSAIDs elicit their anti-RA effects by blocking the production of PGE2, and several groups are focusing their attention in regulating this pathway, primarily by modulating COX and PGE synthase (Fahmi, 2004).
Both phospho-sulindac and phospho-ibuprofen inhibited the production of PGE2 by cultured cells as did conventional aspirin. Phospho-aspirin was, however, unable to suppress PGE2 production by both cultured cells and rats with arthritis, raising the possibility of a PG-independent mode of action. Interestingly, aspirin inhibited FA-induced PGE2 metabolite levels, serving as an important methodological control. The combined action of PGs and cytokines cause the osteopenia of RA and, in the absence of anti-resorptive therapy, osteopenia affects all patients with RA. As phospho-aspirin, however, does not affect PGE2 production, it is likely that its anti-resorptive effect may be through its effect on cytokines.
NF-κB, the master switch of inflammation (Karin, 2009), was the third important signaling cascade to be suppressed by the phospho-NSAIDs. NF-κB, constitutively activated in RA, is strongly implicated in the inflammatory stages of RA, due to its involvement in the induction of inflammatory cytokines and other mediators of inflammation that drive the pathology (see Boyce et al., 2010). This has been demonstrated by the ectopic expression of IκBα (the main inhibitor of classical NF-κB activation) in human macrophages and primary RA synoviocytes, in which it inhibited the production of IL-1β, IL-6 and TNF-α (Brown et al., 2008). Our findings in the involved joints confirmed the activation of NF-κB in the synovium and cartilage and established the ability phospho-aspirin to suppress it drastically in both. Interestingly, the bone marrow showed no activation of NF-κB in response to FA and the effect of phospho-aspirin was not significant. Given the role of NF-κB in inflammation and its suppression by phospho-aspirin, it appears plausible that phospho-aspirin's anti-inflammatory effect may be mediated, at least in part, by the NF-κB pathway.
Finally, the antiproliferative effect of phospho-aspirin in the Jurkat T cells far exceeds that of conventional aspirin. Although these results cannot be extrapolated to animals or humans, they raise the possibility that such an effect may take place in vivo and help explain the greater in vivo anti-inflammatory efficacy of phospho-aspirin compared with aspirin.
The modulation of these signalling cascades may be part of the mechanism of anti-inflammatory action of the three phospho-NSAIDs. All these effects are biologically plausible, but their individual contribution, if any, to the final result is unclear. Whether one of them is the only or dominant mechanism of action or all three are required to obtain the therapeutic effect cannot be inferred from our data. As already alluded to, all three can cross-talk to each other, making, as often is the case, the underlying mode of action even more complicated.
There are three interesting aspects concerning the action of these compounds, indicating that the chemical modification of their parent NSAIDs is functionally significant and has a sustained effect. First, phospho-ibuprofen and phospho-sulindac tend to have similar effects, whereas those of phospho-aspirin differs substantially from these. Although our studies were not designed to evaluate these compounds in terms of structure-activity effects, it is nonetheless tempting to consider that some of these differences may be due to the differences in their modification; phospho-ibuprofen and phospho-sulindac have a butano-diethylphosphate moiety whereas phospho-aspirin has a propano-bisdi-ethylphosphate moiety attached to it. This indicates that their mechanism of action may be dependent, to some extent, on their chemical modification. Second, there is an often striking difference between phospho-aspirin and conventional aspirin, for example, different effects on NF-κB or PGE2. Third, the difference in gastrointestinal toxicity between the parent and the modified NSAIDs is clear-cut and further underscores the significance of their chemical modification.
In summary, we have shown that three phospho-NSAIDs have greatly reduced gastrointestinal toxicity compared with traditional NSAIDs and exhibit strong potential therapeutic efficacy in RA. The latter appears to be mediated through a multi-targeted effect involving several cytokines, PGE2, NF-κB and lymphocyte proliferation, leading to reduced articular inflammation and oedema, and joint bone resorption. Our findings suggest that phospho-NSAIDs may represent a new class of anti-RA drugs that merit further investigation.
Acknowledgments
Grant support: NIH grants, R01-CA139453, NIH RCA153662A and R01CA13945402.
Glossary
Abbreviations
- BrdU
5-bromo-2′-deoxyuridine
- FA
Freund's adjuvant
- LPS
lipopolysaccharide
- NF-κB
nuclear factor-κB
- NSAID
non-steroidal anti-inflammatory drug
- RA
rheumatoid arthritis
Conflicts of interest
The authors have no conflicts of interest except for BR, who has an equity position in Medicon, Inc.
Supplementary material
Additional Supporting Information may be found in the online version of this article:
Supporting Information: Teaching Materials; Figs 1–6 as PowerPoint slide.
Figure S1 Phospho-NSAIDs prevent the development of rat adjuvant arthritis. Arthritis was induced in rats by injecting into their tail Freund's adjuvant (FA). Rats were treated daily by oral gavage for 18 days with P-A 110 mg·kg−1 or ASA 35 mg·kg−1 (equimolar) or P-I 300 mg·kg−1 or P-S 150 mg·kg−1, each suspended in corn oil. (A) Table showing the time-dependent progression of the Inflammation (clinical) score among the various study groups. The arthritis in the FA Group is evident starting on day 14 (end of second week). (B) Body weight progression for the various study groups. The differences between any of the groups at all time points are not statistically significant. Error bars have been omitted for clarity.
Table S1 Effect of phospho-NSAIDs and aspirin on cell viability.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Adeyeye CM, Bricker JD, Vilivalam VD, Smith WI. Acute gastrointestinal toxic effects of suspensions of unencapsulated and encapsulated ibuprofen in rats. Pharm Res. 1996;13:784–793. doi: 10.1023/a:1016068104462. [DOI] [PubMed] [Google Scholar]
- Akaogi J, Nozaki T, Satoh M, Yamada H. Role of PGE2 and EP receptors in the pathogenesis of rheumatoid arthritis and as a novel therapeutic strategy. Endocr Metab Immune Disord Drug Targets. 2006;6:383–394. doi: 10.2174/187153006779025711. [DOI] [PubMed] [Google Scholar]
- Alamanos Y, Drosos AA. Epidemiology of adult rheumatoid arthritis. Autoimmun Rev. 2005;4:130–136. doi: 10.1016/j.autrev.2004.09.002. [DOI] [PubMed] [Google Scholar]
- Bendele A. Animal models of rheumatoid arthritis. J Musculoskelet Neuronal Interact. 2001;1:377–385. [PubMed] [Google Scholar]
- Bendele A, McComb J, Gould T, McAbee T, Sennello G, Chlipala E, et al. Animal models of arthritis: relevance to human disease. Toxicol Pathol. 1999;27:134–142. doi: 10.1177/019262339902700125. [DOI] [PubMed] [Google Scholar]
- Boyce BF, Yao Z, Xing L. Functions of nuclear factor kappaB in bone. Ann N Y Acad Sci. 2010;1192:367–375. doi: 10.1111/j.1749-6632.2009.05315.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown KD, Claudio E, Siebenlist U. The roles of the classical and alternative nuclear factor-kappaB pathways: potential implications for autoimmunity and rheumatoid arthritis. Arthritis Res Ther. 2008;10:212. doi: 10.1186/ar2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunner E, Domhof S, Langer F. Nonparametric Analysis of Longitudinal Data in Factorial Designs. New York: Wiley; 2002. [Google Scholar]
- Cicala C, Ianaro A, Fiorucci S, Calignano A, Bucci M, Gerli R, et al. NO-naproxen modulates inflammation, nociception and downregulates T cell response in rat Freund's adjuvant arthritis. Br J Pharmacol. 2000;130:1399–1405. doi: 10.1038/sj.bjp.0703449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway JG, Andrews RC, Beaudet B, Bickett DM, Boncek V, Brodie TA, et al. Inhibition of tumor necrosis factor-alpha (TNF-alpha) production and arthritis in the rat by GW3333, a dual inhibitor of TNF-alpha-converting enzyme and matrix metalloproteinases. J Pharmacol Exp Ther. 2001;298:900–908. [PubMed] [Google Scholar]
- Dayer JM. The pivotal role of interleukin-1 in the clinical manifestations of rheumatoid arthritis. Rheumatology. 2003;42(Suppl 2):ii3–i10. doi: 10.1093/rheumatology/keg326. [DOI] [PubMed] [Google Scholar]
- Eurenius E, Stenstrom CH. Physical activity, physical fitness, and general health perception among individuals with rheumatoid arthritis. Arthritis Rheum. 2005;53:48–55. doi: 10.1002/art.20924. [DOI] [PubMed] [Google Scholar]
- Fahmi H. mPGES-1 as a novel target for arthritis. Curr Opin Rheumatol. 2004;16:623–627. doi: 10.1097/01.bor.0000129664.81052.8e. [DOI] [PubMed] [Google Scholar]
- Feldmann M, Brennan FM, Maini RN. Role of cytokines in rheumatoid arthritis. Annu Rev Immunol. 1996;14:397–440. doi: 10.1146/annurev.immunol.14.1.397. [DOI] [PubMed] [Google Scholar]
- Hamberg M, Samuelsson B. On the metabolism of prostaglandins E 1 and E 2 in man. J Biol Chem. 1971;246:6713–6721. [PubMed] [Google Scholar]
- Hayashida K, Kaneko T, Takeuchi T, Shimizu H, Ando K, Harada E. Oral administration of lactoferrin inhibits inflammation and nociception in rat adjuvant-induced arthritis. J Vet Med Sci. 2004;66:149–154. doi: 10.1292/jvms.66.149. [DOI] [PubMed] [Google Scholar]
- Hikiji H, Takato T, Shimizu T, Ishii S. The roles of prostanoids, leukotrienes, and platelet-activating factor in bone metabolism and disease. Prog Lipid Res. 2008;47:107–126. doi: 10.1016/j.plipres.2007.12.003. [DOI] [PubMed] [Google Scholar]
- Hyman BT, Stoll LL, Spector AA. Prostaglandin production by 3T3-L1 cells in culture. Biochim Biophys Acta. 1982;713:375–385. doi: 10.1016/0005-2760(82)90256-9. [DOI] [PubMed] [Google Scholar]
- Karin M. NF-kappaB as a critical link between inflammation and cancer. Cold Spring Harb Perspect Biol. 2009;1:a000141. doi: 10.1101/cshperspect.a000141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishimoto T. Interleukin-6: from basic science to medicine – 40 years in immunology. Annu Rev Immunol. 2005;23:1–21. doi: 10.1146/annurev.immunol.23.021704.115806. [DOI] [PubMed] [Google Scholar]
- Kunz M, Ibrahim SM. Cytokines and cytokine profiles in human autoimmune diseases and animal models of autoimmunity. Mediators Inflamm. 2009;2009:979258. doi: 10.1155/2009/979258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtman SN, Wang J, Lemasters JJ. LPS receptor CD14 participates in release of TNF-alpha in RAW 264.7 and peritoneal cells but not in kupffer cells. Am J Physiol. 1998;275:G39–G46. doi: 10.1152/ajpgi.1998.275.1.G39. [DOI] [PubMed] [Google Scholar]
- McCoy JM, Wicks JR, Audoly LP. The role of prostaglandin E2 receptors in the pathogenesis of rheumatoid arthritis. J Clin Invest. 2002;110:651–658. doi: 10.1172/JCI15528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie GG, Sun Y, Huang L, Xie G, Ouyang N, Gupta RC, et al. Phospho-sulindac (OXT-328), a novel sulindac derivative, is safe and effective in colon cancer prevention in mice. Gastroenterology. 2010;139:1320–1332. doi: 10.1053/j.gastro.2010.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monaco C, Andreakos E, Kiriakidis S, Feldmann M, Paleolog E. T-cell-mediated signalling in immune, inflammatory and angiogenic processes: the cascade of events leading to inflammatory diseases. Curr Drug Targets Inflamm Allergy. 2004;3:35–42. doi: 10.2174/1568010043483881. [DOI] [PubMed] [Google Scholar]
- Nemmani KV, Mali SV, Borhade N, Pathan AR, Karwa M, Pamidiboina V, et al. NO-NSAIDs: gastric-sparing nitric oxide-releasable prodrugs of non-steroidal anti-inflammatory drugs. Bioorg Med Chem Lett. 2009;19:5297–5301. doi: 10.1016/j.bmcl.2009.07.142. [DOI] [PubMed] [Google Scholar]
- Pearson CM, Waksman BH, Sharp JT. Studies of arthritis and other lesions induced in rats by injection of mycobacterial adjuvant. V. Changes affecting the skin and mucous membranes. Comparison of the experimental process with human disease. J Exp Med. 1961;113:485–510. doi: 10.1084/jem.113.3.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippe L, Gegout-Pottie P, Guingamp C, Bordji K, Terlain B, Netter P, et al. Relations between functional, inflammatory, and degenerative parameters during adjuvant arthritis in rats. Am J Physiol. 1997;273:R1550–R1556. doi: 10.1152/ajpregu.1997.273.4.R1550. [DOI] [PubMed] [Google Scholar]
- Piazza GA, Keeton AB, Tinsley HN, Gary BD, Whitt JD, Mathew B, et al. A novel sulindac derivative that does not inhibit cyclooxygenases but potently inhibits colon tumor cell growth and induces apoptosis with antitumor activity. Cancer Prev Res. 2009;2:572–580. doi: 10.1158/1940-6207.CAPR-09-0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahme E, Bernatsky S. NSAIDs and risk of lower gastrointestinal bleeding. Lancet. 2010;376:146–148. doi: 10.1016/S0140-6736(10)60839-2. [DOI] [PubMed] [Google Scholar]
- Rigas B. The use of nitric oxide-donating nonsteroidal anti-inflammatory drugs in the chemoprevention of colorectal neoplasia. Curr Opin Gastroenterol. 2007;23:55–59. doi: 10.1097/MOG.0b013e32801145b0. [DOI] [PubMed] [Google Scholar]
- Roberts J, II, Morrow J. Analgesic-antipyretic and antiinflammatory agents and drugs employed in the treatment gout. In: Hardman JG, Limbird LE, editors. Goodman and Gilman's The Pharmacological Basis of Therapeutics. 10th edn. New York: McGraw-Hill; 2001. pp. 687–731. [Google Scholar]
- Silverstein FE, Faich G, Goldstein JL, Simon LS, Pincus T, Whelton A, et al. Gastrointestinal toxicity with celecoxib vs nonsteroidal anti-inflammatory drugs for osteoarthritis and rheumatoid arthritis: the CLASS study: a randomized controlled trial. Celecoxib Long-term Arthritis Safety Study. JAMA. 2000;284:1247–1255. doi: 10.1001/jama.284.10.1247. [DOI] [PubMed] [Google Scholar]
- Singh G, Triadafilopoulos G. Epidemiology of NSAID induced gastrointestinal complications. J Rheumatol. 1999;26(Suppl 56):18–24. [PubMed] [Google Scholar]
- Sostres C, Gargallo CJ, Arroyo MT, Lanas A. Adverse effects of non-steroidal anti-inflammatory drugs (NSAIDs, aspirin and coxibs) on upper gastrointestinal tract. Best Pract Res Clin Gastroenterol. 2010;24:121–132. doi: 10.1016/j.bpg.2009.11.005. [DOI] [PubMed] [Google Scholar]
- Sun Y, Rigas B. The thioredoxin system mediates redox-induced cell death in human colon cancer cells: implications for the mechanism of action of anticancer agents. Cancer Res. 2008;68:8269–8277. doi: 10.1158/0008-5472.CAN-08-2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theisen-Popp P, Muller-Peddinghaus R. Antirheumatic drug profiles evaluated in the adjuvant arthritis of rats by multiparameter analysis. Agents Actions. 1994;42:50–55. doi: 10.1007/BF02014300. [DOI] [PubMed] [Google Scholar]
- Whiteley PE, Dalrymple SA. Models of inflammation: measuring gastrointestinal ulcerations in the rat. In: Enna SJ, editor. Current Protocols in Pharmacology. Vol. 3. New York: John Wiley & Sons, Inc; 1998. pp. 10.2.1–10.2.4. [DOI] [PubMed] [Google Scholar]
- Whiteley PE, Dalrymple SA. Models of inflammation: adjuvant-induced arthritis in the rat. In: Enna SJ, editor. Current Protocols in Pharmacology. Vol. 2. New York: John Wiley & Sons, Inc; 2001. pp. 5.5.1–5.5.5. [DOI] [PubMed] [Google Scholar]
- Zhao W, Mackenzie GG, Murray OT, Zhang Z, Rigas B. Phosphoaspirin (MDC-43), a novel benzyl ester of aspirin, inhibits the growth of human cancer cell lines more potently than aspirin: a redox-dependent effect. Carcinogenesis. 2009;30:512–519. doi: 10.1093/carcin/bgp015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.