

Abstract
BACKGROUND AND PURPOSE
Liver X receptor (LXR) agonists are atheroprotective but often induce hypertriglyceridaemia and liver steatosis. We investigated the effect of a novel high-affinity LXR activator, AZ876, on plasma lipids, inflammation and atherosclerosis, and compared the effects with another LXR agonist, GW3965.
EXPERIMENTAL APPROACH
APOE*3Leiden mice were fed an atherogenic diet alone or supplemented with either AZ876 (5 or 20 µmol·kg−1·day−1) or GW3965 (17 µmol·kg−1·day−1) for 20 weeks. Total cholesterol and triglyceride levels were measured using commercial kits. Plasma cytokines were determined by using bead-based multiplex suspension array kits with the Luminex technology. Atherosclerosis was assessed histochemically and lesion composition was assessed by immunohistochemical methods.
KEY RESULTS
Low-dose AZ876 had no effect on plasma or liver lipids, whereas high-dose AZ876 increased plasma triglycerides (+110%) and reduced cholesterol (−16%) compared with controls. GW3965 increased plasma triglycerides (+70%). Low-dose AZ876 reduced lesion area (−47%); and high-dose AZ876 strongly decreased lesion area (−91%), lesion number (−59%) and severity. In either dose, AZ876 did not affect lesion composition. GW3965 reduced atherosclerosis and collagen content of lesions (−23%; P < 0.01). High-dose AZ876 and GW3965, but not low-dose AZ876, reduced inflammation as reflected by lower cytokine levels and vessel wall activation.
CONCLUSIONS AND IMPLICATIONS
We have identified a novel LXR agonist that when given in a low dose inhibits the progression of atherosclerosis without inducing anti-inflammatory effects, liver steatosis or hypertriglyceridaemia. Therefore, the primary protective action of a low-dose AZ876 is likely to be an increased reverse cholesterol transport.
Keywords: lipids, inflammation, reverse cholesterol transport
Introduction
Atherosclerosis is a dynamic process driven by both inflammation and hyperlipidaemia, and hence treatment should target both aspects of this disease. The current therapies to treat atherosclerosis primarily target hyperlipidaemia. However, different studies have shown that a substantial residual cardiovascular risk remains, even with very aggressive reductions in levels of low-density lipoprotein (LDL)-cholesterol (Baigent et al., 2005). Therefore, new therapies focusing on this unmet need are highly desirable.
The liver X receptors (LXRα or NR1H3 and LXRβ or NR1H2; nomenclature follows Alexander et al., 2009) are members of the nuclear receptor superfamily of transcription factors. It is now appreciated that both LXR isoforms function as intracellular sensors of cholesterol excess. LXRα is predominantly expressed in tissues and cells that play important roles in lipid homeostasis, such as the liver, intestine, adipose tissue and macrophages, while LXRβ is expressed in many cell types (Repa and Mangelsdorf, 2000). The natural ligands for both LXR isoforms include oxidized derivatives of cholesterol (i.e. oxysterols) (Janowski et al., 1996; Lu et al., 2001). Synthetic high-affinity ligands have also been identified, such as T0901317 and GW3965 (Schultz et al., 2000; Collins et al., 2002). The activation of LXR induces numerous genes involved in cholesterol homeostasis, lipogenesis and reverse cholesterol transport (RCT), including those for the ATP binding cassette transporter A1 (ABCA1), ABCG1, ABCG5, ABCG8, scavenger receptor class B type I (SR-B1), cholesteryl ester transfer protein (CETP), apolipoprotein E (apoE), lipoprotein lipase and sterol-response element-binding protein 1-c (SREBP1-c) (Zelcer & Tontonoz, 2006; Rigamonti et al., 2008). RCT is believed to be the main effect of LXR agonists in the prevention or treatment of atherosclerosis. ABCA1, ABCG1, apoE and SR-B1 are the principal proteins involved in RCT to promote cholesterol efflux from macrophage foam cells to high-density lipoprotein (HDL) particles for subsequent delivery of cholesterol to hepatocytes and enterocytes (Brunham et al., 2006; Tall, 2008). In addition to their effects on cholesterol and lipid metabolism, the LXRs repress inflammatory actions of macrophages, by, for instance, interfering with the NF-κB signalling (Rigamonti et al., 2008).
LXR agonists have also been shown to cause hepatic steatosis and hypertriglyceridaemia in animals, primarily resulting from induction of the expression of hepatic SREBP1c (Repa et al., 2000). These effects have hindered the development of LXR agonists for human use until now. However, substantial interest remains and it is believed that more specific or more subtle LXR agonists may reduce atherosclerosis without demonstrating adverse effects (Rader, 2007; Tall, 2008; Kratzer et al., 2009;Quinet et al., 2009).
The aim of the present study was to compare the newly identified LXR agonist AZ876 (Figure 1) (Boström et al., 2006) with GW3965 [a recognized selective LXR agonist with minimal effects on plasma and hepatic triglyceride levels (Joseph et al., 2002; Miao et al., 2004)], with regard to its effects on plasma and liver lipid levels and atherosclerosis development. We used female APOE*3Leiden transgenic mice, which are a well-established mouse model for hyperlipidaemia and atherosclerosis (van Vlijmen et al., 1994). These mice have a lipoprotein profile similar to the profile of patients with familial dys-β-lipoproteinaemia in which the elevated plasma cholesterol and triglyceride levels are mainly confined to the very low-density lipoprotein (VLDL)/LDL-sized lipoprotein fraction. In contrast to other mouse models for dyslipidaemia and/or atherosclerosis (Zadelaar et al., 2007), these mice respond in a similar manner to human patients when treated with human therapies for cardiovascular diseases, such as statins, cholesterol uptake inhibitors, calcium channel blockers, fibrates and angiotensin II receptor antagonists (Delsing et al., 2001; 2003; Kleemann et al., 2003; Verschuren et al., 2005; Kooistra et al., 2006; van der Hoorn et al., 2007).
Figure 1.
Chemical structures of AZ876 and GW3965.
Methods
Binding and transactivation assays
Binding vectors for His-tagged protein production were prepared by inserting the ligand-binding domain cDNA of human LXRα (amino acids 205–447) in pET28 (Novagen, Gibbstown, NJ, USA) and the ligand-binding domain cDNA of LXRβ (amino acids 216–461) in pET24D. Proteins were expressed in Escherichia coli and purified on Ni+ columns. Binding assays using LXRα and LXRβ protein were run by adding reagents to Wallac Isoplate 1450–514. Briefly, each 96 plate well contained assay buffer (20 mM Tris pH 7.5, 80 mM NaCl, 2 mM dithiothreitol, 0.125% Chaps and 10% glycerol), 0.1 mg SPA beads (polylysine-coated yttrium silicate beads, RPNQ0010P, GE Healthcare, Piscataway, NJ, USA), LXRα (0.5 µg) or LXRβ (0.25 µg), 30 nM 3H-ligand (Tularik T0901317, specific activity of 473 Kbq nmol−1) and test compound in a 10-point dose-response dilution. The assay mixture was shaken gently for 2 h on a plate shaker after which the beads were allowed to settle for 1 hour before counting.
Transactivation vectors were prepared by inserting the ligand-binding domain cDNA sequences of human or mouse LXRα and LXRβ in frame with the yeast Gal4 transcription factor DNA binding domain and the nuclear localization signal from the T-antigen of polyoma virus in the eucaryotic expression vector pSG5 (Stratagene, La Jolla, CA, USA). The ligand-binding domain cDNA of human LXRα and LXRβ was the same as mentioned previously. The mouse sequence corresponded to amino acids 203–445 for LXRα and amino acids 201–446 for LXRβ. The vectors were co-transfected with a pGL3 luciferase reporter plasmid containing a minimal SV40 promoter (Promega, Madison, WI, USA) and five copies of the UAS Gal4 recognition site into U2/OS osteosarcoma cells. Ligands were added as 10-point dose-response curves and then luciferase activity was measured after 48 h.
Macrophage RCT
All animal care and experimental procedures were approved by the Institutional Animal Care and Use Committee of the Netherlands Organization for Applied Scientific Research (TNO) or by the Ethics Review Committee on Animal Experiments (Gothenburg region). The animals received food and water ad libitum. Body weight and food intake were monitored during the study. Macrophage RCT was determined essentially as described by Naik et al. (2006). J774A1 macrophages (obtained from Deutsche Sammlung von Mikroorganismens and Zellkulturen, DSMZ, Braunschweig, Germany) were cultured in RPMI supplemented with 10% fetal bovine serum and radiolabelled with 5 µCi mL−1[3H]cholesterol (specific activity 40–60 Ci/mmol, PerkinElmer, Waltham, MA, USA) together with 100 µg mL−1 acetylated LDL (Intracel, Rockville, MD, USA) and 1% BSA (Sigma, St Louis, MO, USA) for 48 h. The J774A1 cells were then washed with phosphate-buffered saline (PBS) and resuspended in medium. The viability was estimated to be ∼75% using Trypan blue. About 75% of the 3H was found in the cholesteryl ester fraction of the cells before injection into the mice. Individually housed and body weight-matched 7-week-old C57BL/6J males (Harlan, the Netherlands) were treated with vehicle (amorphous nanoparticles with 3.4% dimethylacetamide), AZ876 (20 µmol·kg−1·day−1, ∼9·mg·kg−1·day−1), or GW3965 (34 µmol·kg−1·day−1, ∼20 mg·kg−1·day−1) by oral gavage twice daily for 8 days. On day 6, labelled J774A1 cells (2.5 × 106 viable cells containing 28 × 106 dpm in 0.4 mL medium) were injected intraperitoneally. Faeces were collected between 24 and 48 h after macrophage injection. At termination (48 h after macrophage injection), the animals were anaesthetized with isoflurane (Forene, Abbot Scandinavia AB, Solna, Sweden), plasma was collected and the liver was collected after perfusion with PBS. Faeces (total) and liver (100 mg) were homogenized in PBS and total lipids were extracted from faeces and liver homogenates and plasma according to Folch et al. (1957). Free cholesterol and cholesteryl ester were separated using thin layer chromatography (hexane : diethylether : acetic acid 90:10:1). [3H] in total extracted lipids, free cholesterol and cholesteryl ester were counted in a liquid scintillator and presented as percent dpm injected into the animal. The lipoprotein particles were separated using size-exclusion high-performance liquid chromatography and the [3H] was found in the HDL fraction (data not shown).
Study of treatments for atherosclerosis
Female heterozygous APOE*3Leiden transgenic mice (about 13 weeks of age, bred by TNO), on a C57bl/6 background and characterized by elisa for human apoE (van Vlijmen et al., 1994), were used. During a 3 week run-in period, all animals received a semi-synthetic Western-type diet containing 40.5% sucrose, 15% cacao butter, 0.50% (w/w) cholesterol and 0.33% v/w sunflower oil (necessary to dissolve the compounds). After matching into four groups, based on age, plasma cholesterol and triglyceride levels, the mice received Western-type diet either alone (control group) or supplemented with AZ876 in a low dose (5 µmol·kg−1·day−1; ∼2 mg·kg−1·day−1) or high dose (20 µmol·kg−1·day−1; ∼9 mg·kg−1·day−1) or with GW3965 (17 µmol·kg−1·day−1; ∼10 mg·kd−1·day−1) (Joseph et al., 2002). Initially, the high dose of AZ876 was 10 µmol·kg−1·day−1; however, after 4 weeks of treatment, GW3965 induced hypertriglyceridaemia but AZ876 (10 µmol·kg−1·day−1) did not. Therefore, it was decided to increase the dose of AZ876 to 20 µmol·kg−1·day−1. Both compounds were provided by AstraZeneca (Mölndal, Sweden). After 20 weeks, mice were killed and the hearts, aortic root, livers and small intestine (duodenum) were isolated.
Plasma lipid and lipoprotein analysis and inflammation markers
After a 4 h fasting period from 9 am to 1 pm, blood was collected and EDTA plasma was obtained subsequently (Sarstedt, Nümbrecht, Germany) and lipoproteins were separated by fast protein liquid chromatography (FPLC; Westerterp et al., 2006). Total cholesterol (No-1489437, Roche Diagnostics, Indianapolis, IN, USA) and triglyceride (1488872, Roche Diagnostics) levels were measured. Alanine aminotransferase (ALT) was determined spectrophotometrically using a Reflotron system (Roche Diagnostics). Plasma cytokines were determined by using bead based multiplex suspension array kits with the Luminex technology on a BioPlex (Bio-Rad, Hercules, CA, USA).
7α-Hydroxy-4-cholesten-3-one (C4), lanosterol and β-sitosterol were analysed in serum by HPLC/MS/MS with atmospheric pressure photo ionization (Nilsson et al., 2009). Plasma samples (50 µL) were extracted with isopropanol : heptane 4:1, in which the internal standards were dissolved and injected into the LC/MS/MS system. Samples were ionized in positive mode with multiple reaction monitoring. For C4, the transition at 401 m/z to 177 m/z was monitored. 2H7-7-ketocholesterol was monitored at m/z = 408 to 175. Lanosterol was monitored at transition 409.2 to 191.1, 2H3-lanosterol was monitored at transition 412.2 to 191.1, β-sitosterol was monitored at transition 397.2 to 161 and 2H7-β-sitosterol was monitored at transition 404.2 to 161 at CE = 22 eV. Samples were quantified by external standard calibration using a proxy matrix for calibration samples and deuterated internal standards as volume markers.
Faecal cholesterol and bile acids
Faeces were ground to powder in a mortar and then a 100 mg sample was shaken with 2 mL isopropanol/dioxan (1:1) overnight. Total bile acids and cholesterol in the extracts were measured using commercial reagent systems [Bile Acids-L3K assay (Diagnostic Chemicals Limited, Mansfield, TX, USA) and Chol, Roche/Hitachi 12016630 122 (Roche Diagnostics)]. The assays were performed on a Cobas Mira Analyser (Hoffman-La Roche & Co., Basel, Switzerland).
Histological assessment of atherosclerosis
After the 20 week treatment period, the mice were killed. The hearts with aortic root were dissected, formalin fixed and embedded in paraffin. Serial cross-sections (5 µm thick, spaced 50 µm apart) throughout the entire aortic valve area were used for histological analysis. Sections were stained with haematoxylin-phloxine-saffron. For each mouse, four sections with intervals of 50 µm were used for quantification and qualification of the atherosclerotic lesions. For determination of the severity of atherosclerosis, the lesions were classified into five categories as described previously (Delsing et al. 2001; 2003; Verschuren et al. 2005): (i) early fatty streak; (ii) regular fatty streak; (iii) mild lesion; (iv) moderate lesion; and (v) severe lesion. The percentages of all lesions found in the respective categories were calculated. The total lesion area was calculated per cross-section. In each segment used for lesion assessment, the collagen content of the lesion was quantified morphometrically after Sirius Red staining and the smooth muscle cell (SMC) content after immunostaining with mouse anti-human α-actin (DAKO, Glostrup, Denmark), which cross reacts with mouse α-actin. The number of monocytes adhering to the endothelium was counted and the macrophage area was measured quantitatively after immunostaining with AIA31240 (Accurate Chemical and Scientific Corporation, Westbury, NY, USA). All analyses were performed by the same operator, using a double blind protocol.
RNA preparation and quantitative real-time PCR
Total RNA was extracted using Trizol (Invitrogen, Carlsbad, CA, USA). First-strand cDNA was synthesized from DNAse treated total RNA using the High Capacity cDNA Archive Kit (Applied Biosystems, Foster City, CA, USA). Real-time PCR analysis was performed with an ABI Prism 7900 Sequence Detection System using FAM and TAMRA labelled fluorogenic probes (Applied Biosystems). Expression data were normalized against mouse acidic ribosomal phosphoprotein P0 (m36B4). The relative expression levels were calculated according to the formula 2-ΔCT, where ΔCT is the difference in cycle threshold (CT) values between the target and the m36B4 internal control. ABCA1 sequences; primer 5′-AAGGGTTTCTTTGCTCAGATTGTC- 3′, reverse primer 5′-TGCCAAAGGGTGGCACA-3′ and probe 5′-FAM-CCAGCTGTCTTTGTTTGCATTGCCC-TAMRA-3′. ABCG1 sequences; primer 5′-CCATGAATGCCAGCAGCTACT-3′, reverse primer 5′-CACTGACACGCACACGGACT-3′ and probe 5′-FAM-TGCCGCAATGACGGAGCCC-TAMRA-3′. M36B4 sequences; primer 5′-GAGGAATCAGATGAGGATATGGGA-3′, reverse primer 5′-AAGCAGGCTGACTTGGTTGC-3′ and probe 5′-VIC-TCG GTC TCT TCG ACT AAT CCC GCC AA-TAMRA-3′.
Statistics
Values are expressed as mean ± SD. Comparisons between groups were made by Kruskal–Wallis anova followed by Mann–Whitney U-test. P < 0.05 was considered significant.
Results
Identification and characterization of LXR agonist AZ876
In binding assays, AZ876 was 25-fold and 2.5-fold more potent than GW3965 on human (h)LXRα and hLXRβ respectively. In reporter transactivation assays, AZ876 was 196-fold and fivefold more potent than GW3965 on hLXRα and hLXRβ respectively. AZ876 was also more potent than GW3965 on mouse (m)LXRα (248-fold) and mLXRβ (10.5-fold) (Table 1). Thus, AZ876 is a more potent binder and activator of LXRα and LXRβ than GW3965. In addition, we have data showing that AZ876 is four- to sevenfold more potent than GW3965 on the expression of ABCA1 mRNA in hamster and human blood PMN cells, as determined following incubation with compounds in vitro (data not shown). Therefore, we believe both from human and mouse reporter assays as well as from hamster and human in vitro determination of ABCA1 gene expression data that AZ876 is more potent than GW3965 on RCT genes. Also, AZ876 was highly selective with respect to other nuclear hormone receptors, including retinoid X receptor, farnesoid X receptor, thyroid hormone receptor (TR)α or TRβ, when tested in agonist mode in fluorescence resonance energy transfer assays (data not shown).
Table 1.
Binding affinity (Ki) and transactivation potency (EC50)
| LXRα | LXRβ | |||||
|---|---|---|---|---|---|---|
| Human | Mouse | Human | Mouse | |||
| Ki (µM) | EC50 (µM) | EC50 (µM) | Ki (µM) | EC50 (µM) | EC50 (µM) | |
| AZ876 | 0.007 ± 0.004 | 0.006 ± 0.002 | 0.005 ± 0.002 | 0.011 ± 0.002 | 0.073 ± 0.019 | 0.065 ± 0.020 |
| (n= 3) | (n= 4) | (n= 3) | (n= 3) | (n= 5) | (n= 3) | |
| GW3965 | 0.174 ± 0.069 | 1.175 ± 0.472 | 1.240 ± 0.306 | 0.028 ± 0.014 | 0.374 ± 0.171 | 0.685 ± 0.158 |
| (n= 5) | (n= 15) | (n= 3) | (n= 5) | (n= 22) | (n= 3) | |
Scintillation proximity assay data showing the Ki values of GW3965 and AZ876 to displace [3H]-T0901317 bound to human LXRα or LXRβ protein. Reporter gene data showing EC50 values of GW3965 and AZ876 from U2/OS cells co-transfected with a luciferase reporter gene containing Gal4 DNA-binding sites and Gal4 chimeric LXRα or LXRβ cDNA. Values are means or mean ± SD based on the indicated number of dose-responses (3–4 wells per dose-response).
We also determined the effect of AZ876 and GW3965 on in vivo RCT by following the appearance of radiolabelled lipids in plasma, liver and faeces after injecting radiolabelled J774A1 macrophages into C57Bl/6J mice (Table 2). AZ876 (20 µmol·kg−1) increased [3H]total lipids by 75%, [3H]free cholesterol by 100% (both P < 0.01) and [3H]cholesteryl ester by 126% (P < 0.05) in plasma compared with vehicle controls. Furthermore, AZ876 increased [3H]total lipids recovered from the faeces by 94% (P < 0.05) and [3H]free cholesterol by 195% (P < 0.01) as compared with vehicle control. GW3965 (34 µmol·kg−1) increased macrophage RCT in accordance with the study from Naik et al. (2006) as demonstrated by increased levels of [3H]free cholesterol in plasma and faeces, by 49 and 65% (both P < 0.05), respectively, compared with vehicle controls. Collectively, these data show that AZ876 is a potent activator of the LXRs which is paralleled by a marked effect on in vivo RCT.
Table 2.
In vivo macrophage reverse cholesterol transport
| Control | AZ876 (20 µmol·kg−1·day−1) | GW3965 (34 µmol·kg−1·day−1) | |
|---|---|---|---|
| Plasma lipids (% dpm inj.) | |||
| [3H]Total extracted | 1.275 ± 0.240 | 2.229 ± 0.613** | 1.689 ± 0.539 |
| [3H]Free cholesterol | 0.243 ± 0.069 | 0.487 ± 0.199** | 0.362 ± 0.095* |
| [3H]Cholesteryl ester | 0.686 ± 0.148 | 1.550 ± 0.686* | 0.966 ± 0.354 |
| Liver lipids (% dpm inj.) | |||
| [3H]Total extracted | 2.882 ± 1.883 | 2.177 ± 0.574 | 2.029 ± 0.619 |
| [3H]Free cholesterol | 1.981 ± 0.710 | 1.754 ± 0.451 | 1.748 ± 0.518 |
| [3H]Cholesteryl ester | 0.770 ± 1.161 | 0.330 ± 0.099 | 0.238 ± 0.071 |
| Faecal lipids (% dpm inj.) | |||
| [3H]Total extracted | 0.603 ± 0.141 | 1.17 ± 0.893* | 0.737 ± 0.298 |
| [3H]Free cholesterol | 0.214 ± 0.068 | 0.632 ± 0.570** | 0.353 ± 0.120* |
| [3H]Cholesteryl ester | 0.046 ± 0.013 | 0.044 ± 0.012 | 0.039 ± 0.006 |
Body weight-matched C57BL/6J males were treated with AZ876 (20 µmol·kg−1·day−1, ≈9 mg·kg−1·day−1), GW3965 (34 µmol·kg−1·day−1, ≈20 mg·kg−1·day−1) or vehicle (control) by oral gavage twice daily for 8 days. On the sixth day, mice were injected intraperitoneally with cholesterol-enriched [3H]cholesterol-labeled J774A1 macrophages. [3H]Total extracted lipids, [3H]free cholesterol and [3H]cholesteryl esters were determined in plasma and livers after 48 h and in faeces 24–48 h after injection of labelled J774A1 cells. Values are mean ± SD, n= 7–8,
P < 0.05,
P < 0.01, significantly different from control.
Effect of AZ876 and GW3965 on plasma lipid levels
The effect of both compounds on plasma lipid levels was investigated in hyperlipidaemic APOE*3Leiden mice. The mice were fed a Western-type diet, to induce hyperlipidaemia, giving on average plasma cholesterol levels of 18.2 ± 3.9 mmol L−1 and triglyceride levels of 2.0 ± 0.4 mmol L−1 in the control group (Figure 2A,B). AZ876 in low dose (5 µmol·kg−1·day−1) tended to decrease cholesterol levels by 12% [not significant (NS)], whereas it did not affect triglyceride levels. High-dose AZ876 (20 µmol·kg−1·day−1) significantly decreased total cholesterol levels by 16% (P < 0.05) and increased triglyceride levels by 110% (P < 0.001). GW3965 tended to decrease cholesterol levels by 12% (NS) and increased plasma triglycerides by 70% (P < 0.001). The reduction in plasma cholesterol was confined to the VLDL fractions as measured after plasma separation by FPLC. Large HDL-1 particles in the lipoprotein profile were observed in the groups of mice treated with 20 µmol·kg−1·day−1 AZ876 or with GW3965, but not in the mice treated with the lower dose of AZ876 (5 µmol·kg−1·day−1) (Figure 2C). This effect is likely to be related to the increased abca1 mRNA expression in the intestine in the two groups (+410% in the high AZ876 group and +330% in the GW3965 group, both P < 0.01) as intestinal ABCA1 directly contributes to the HDL-raising effect of LXR agonists (Brunham et al., 2006). Supporting this idea, we found that the aortic mRNA expression of both abca1 (+101% and +120%, both P < 0.05) and abcg1 (+164%, P < 0.001 and +154%, P < 0.05) were increased in these groups (Figure 3).
Figure 2.
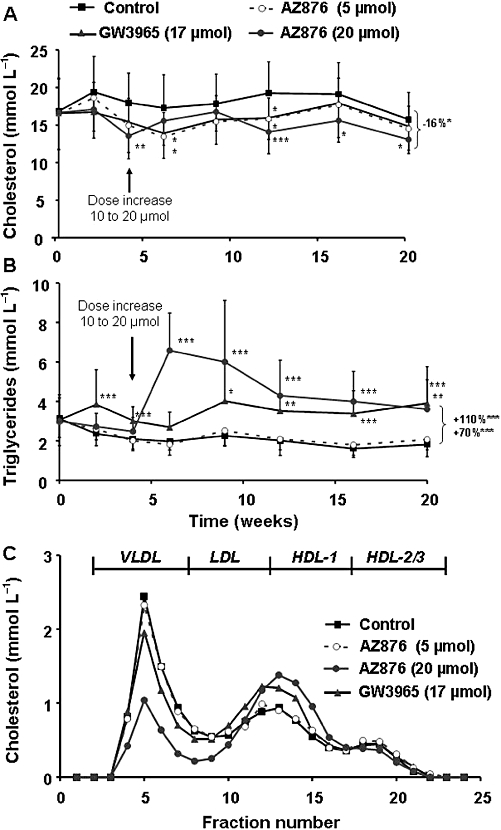
APOE*3Leiden mice were treated for 20 weeks with a Western-type diet alone (control) or supplemented with AZ876 (5 or 20 µmol·kg−1·day−1), or with GW3965 (17 µmol·kg−1·day−1). The plasma cholesterol levels (A) and triglyceride levels (B) over 20 weeks are shown. Lipoproteins were separated by fast protein liquid chromatography and cholesterol profiles were measured in pooled plasma per group at the end of the study (C). Dose of AZ876 was increased from 10 to 20 µmol 4 weeks after treatment as indicated (arrow). Values are mean ± SD. n= 15, *P < 0.05, **P < 0.01, ***P < 0.001, significantly different from control.
Figure 3.
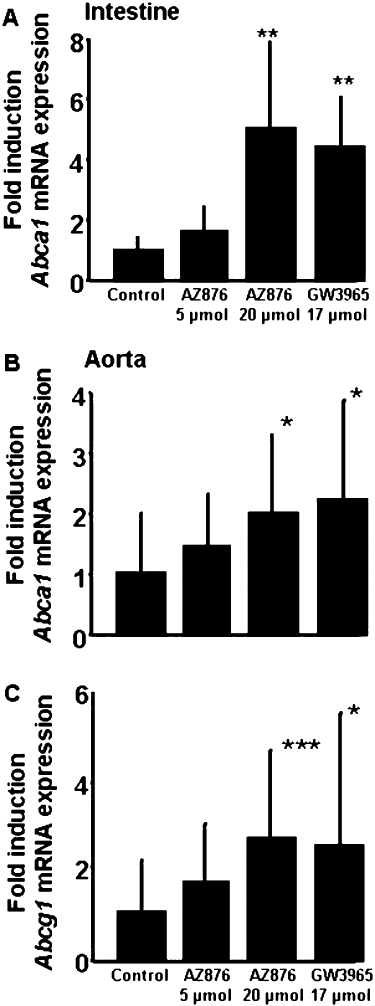
APOE*3Leiden mice were treated for 20 weeks with a Western-type diet alone (control) or supplemented with AZ876 (5 or 20 µmol·kg−1·day−1), or with GW3965 (17 µmol·kg−1·day−1). Intestinal mRNA expression of abca1 (A) and aortic mRNA expression of abca1 (B) and abcg1 (C) are shown. Values are mean ± SD. n= 15, *P < 0.05, **P < 0.01, ***P < 0.001, significantly different from control.
Effect of AZ876 and GW3965 on synthesis, intestinal uptake and faecal output of cholesterol
We determined whether an altered intestinal cholesterol uptake, changed cholesterol synthesis or faecal output of cholesterol or bile acids could have contributed to the changes in plasma cholesterol levels. Serum levels of lanosterol, reflecting the cholesterol synthesis (Bjorkhem et al., 1987), were increased in the high-dose AZ876 (+310%, P < 0.01) and GW3965 (+141%, P < 0.001), but not in the low AZ876 group (+29%, NS).
Food intake and body weight showed no difference between the groups, reflecting a similar cholesterol intake. In addition, β-sitosterol was measured as a marker for intestinal cholesterol uptake. β-Sitosterol levels were not affected by any of the treatments, indicating no change in intestinal cholesterol uptake. We then investigated the faecal excretion of cholesterol and bile acids. Faecal cholesterol output was 0.12 ± 0.02 mmol/100 g bw/day in the control group and unchanged by either LXR agonist. The faecal bile acid output was also unchanged (11.1 ± 2.4 mmol/100 g bw/ day in the control group), which was in line with the unaffected serum levels of the marker of bile acid biosynthesis, 7α−hydroxy-4-cholesten-3-one (C4). In summary, the LXR agonists induce cholesterol synthesis, but it was not possible to detect changes in cholesterol absorption or losses as demonstrated by no change in markers of cholesterol absorption or faecal loss of cholesterol or bile acids.
Effect on liver triglyceride content
LXR agonists are notorious for their induction of hepatosteatosis and we therefore measured liver weight and triglyceride content. The Western-type diet induced a slight steatosis in the control group (Table 3). Treatment with 5 µmol·kg−1·day−1 AZ876 or GW3965 did not affect liver weight and triglyceride content, whereas the high dose of AZ876 (20 µmol·kg−1) increased both liver weight (+29%, P < 0.05) and triglyceride content (+53%, P < 0.01). Concomitantly, plasma ALT levels, as a marker for liver damage, were on average 114% increased (P < 0.01) after treatment with the higher dose of AZ876, as measured in pooled samples per group. No ALT increase was observed in the 5 µmol·kg−1 AZ876- and GW3965-treated animals (Table 3).
Table 3.
Effects of AZ876 and GW3965 on liver weight, triglycerides and plasma ALT levels
| Control | AZ876 (5 µmol·kg−1·day−1) | AZ876 (20 µmol·kg−1·day−1) | GW3965 (17 µmol·kg−1·day−1) | |
|---|---|---|---|---|
| Liver weight (g) | 1.43 ± 0.22 | 1.34 ± 0.22 | 1.76*± 0.35 | 1.37 ± 0.16 |
| Triglycerides (g/100 g tissue) | 9.7 ± 1.5 | 10.7 ± 1.6 | 14.8**± 3.5 | 11.3 ± 2.3 |
| ALT (U/L) | 105 ± 19 | 94 ± 14 | 225**± 43 | 87 ± 7 |
Plasma alanine aminotransferase (ALT) levels as measured in plasma pooled per group. Values are mean in time (n= 5 per group) ± SD,
P < 0.05,
P < 0.01, significantly different from control.
Effect of GW3965 and AZ876 on atherosclerosis
In order to determine the effect of both LXR agonists on atherosclerosis development, we measured the number of lesions and the total lesion area. We also phenotyped the severity of these lesions in the aortic root after a 20 week treatment period. About half (54 ± 24%) of the lesions in the control group (Figure 4) were severe types IV–V lesions. This figure also shows that 5 µmol·kg−1·day−1 AZ876 did not affect lesion number, whereas it decreased lesion area by 47% (P < 0.05) and tended to decrease the amount of severe lesions (36 ± 24%, P= 0.056). AZ876 (20 µmol·kg−1) markedly reduced atherosclerosis development with regard to lesion number (−59%), lesion area (−91%) and the abundance of severe lesions (17 ± 23%, all P < 0.001). GW3965 was more potent in inhibiting atherosclerosis than the low dose of AZ876, resulting in reductions in the number of lesions (by 38%, P < 0.05) and lesion area (by 74%, P < 0.001) and less severe lesions (28 ± 21%, P < 0.01) as compared with control. We observed no effect of 5 µmol·kg−1 AZ876 on the amount of undiseased segments (14 ± 21% in the control). However, 20 µmol·kg−1·day−1 AZ876 and GW3965 clearly increased the amount of undiseased segments (to 58 ± 23%, P < 0.001 and 44 ± 28%, P < 0.01, respectively). Both high-dose AZ876 and GW3965 were significantly (P < 0.001 and P < 0.05, respectively) more potent in preventing lesion development than the low-dose AZ876 treatment.
Figure 4.
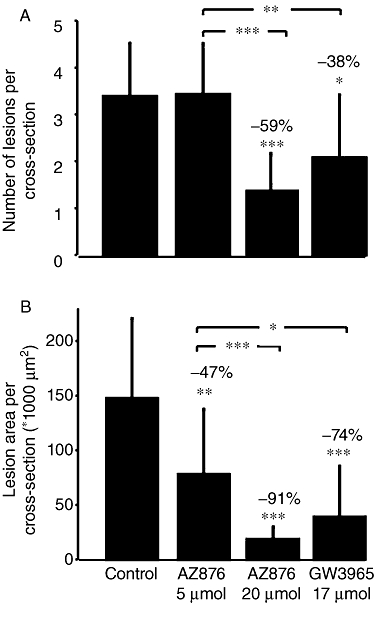
APOE*3Leiden mice were treated for 20 weeks with a Western-type diet alone (control) or supplemented with AZ876 (5 or 20 µmol·kg−1·day−1) or GW3965 (17 µmol·kg−1·day−1). The amount of lesions in the aortic root was counted (A) and the lesion area was measured (B). Values are mean ± SD. n= 15, *P < 0.05, **P < 0.01, ***P < 0.001, significantly different from control.
We then assessed the lesion composition by measuring the collagen and the SMC content of the lesions, both considered to stabilize the lesions, and the amount of macrophages, known to be a destabilizing component in the lesions (Delsing et al., 2001; Libby, 2002). As shown in Figure 5, treatment with AZ876 in either dose did not affect the collagen or SMC content, relative to values in the control lesions. GW3965, however, reduced the collagen content by 23% (P < 0.01) as compared with control, resulting in significantly less collagen content than in the 20 µmol·kg−1·day−1 AZ876 group, without affecting the SMC content. Treatment with either dose of AZ876 or GW3965 did not significantly affect the macrophage content (Figure 5). In summary, GW3965 induced a less stable lesion phenotype as compared with the control, whereas AZ876 in either dose did not affect the lesion composition.
Figure 5.
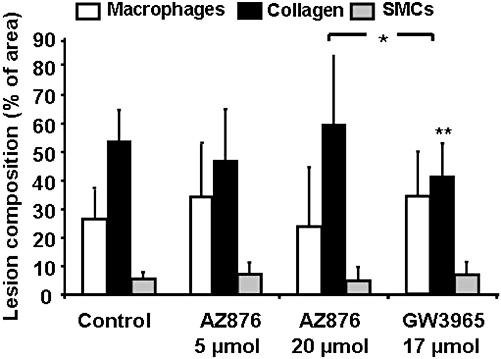
The effect of 5 or 20 µmol·kg−1·day−1 AZ876 or GW3965 (17 µmol·kg−1·day−1) on lesion composition was assessed by measuring the macrophage, collagen and smooth muscle cell (SMC) area, all calculated as percentage of the total lesion area. Values are mean ± SD. n= 15, *P < 0.05, **P < 0.01, ***P < 0.001, significantly different from control.
As a functional parameter for vessel wall inflammation, we also investigated the numbers of monocytes adhering to the activated endothelium of the aortic root, which is considered the first step in lesion development. A representative image of adhering monocytes is presented in Figure 6A. The summary data (Figure 6B) show that the number of adherent macrophages was reduced by 72% upon treatment with 20 µmol·kg−1·day−1 AZ876 and by 53% in the GW3965-treated mice. These anti-inflammatory effects of both LXR agonists were confirmed by plasma analysis of inflammatory cytokines TNF-α, IL-1β and IL-6 (Table 4), which were decreased upon treatment with 20 µmol·kg−1 AZ876 and with GW3965. Low-dose AZ876 did not affect monocyte adherence or plasma cytokine levels (Figure 6B and Table 4).
Figure 6.
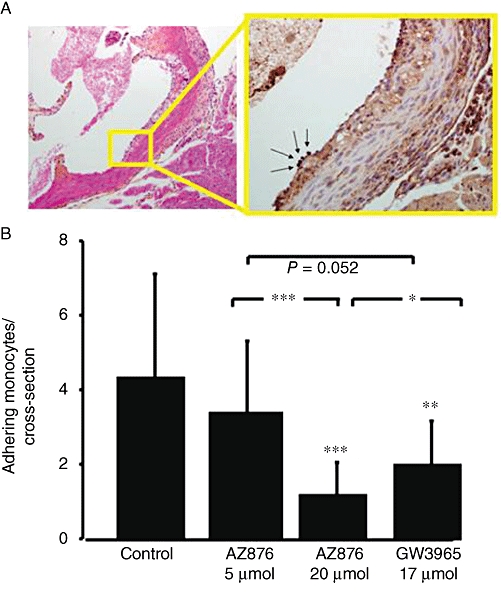
To investigate the effect of 5 or 20 µmol·kg−1·day−1 AZ876 or GW3965 (17 µmol·kg−1·day−1) on vessel wall inflammation, monocytes were stained in the aortic root (A). The number of monocytes adhering to the endothelium was counted in each animal (B). Values are mean ± SD. n= 15, *P < 0.05, **P < 0.01, ***P < 0.001, significantly different from control.
Table 4.
Effects of AZ876 and GW3965 on plasma cytokines
| Control | AZ876 (5 µmol·kg−1·day−1) | AZ876 (20 µmol·kg−1·day−1) | GW3965 (17 µmol·kg−1·day−1) | |
|---|---|---|---|---|
| TNF-α (pg mL−1) | 1110 ± 1294 | 955 ± 865 | 521*± 454 | 362**± 322 |
| IL-1β (pg mL−1) | 191 ± 141 | 213 ± 166 | 114*± 52 | 94**± 44 |
| IL-6 (pg mL−1) | 124 ± 130 | 102 ± 68 | 65 ± 47 | 43**± 18 |
Plasma levels of cytokines as measured by luminex assay. Values are mean ± SD,
P < 0.05,
P < 0.01, significantly different from control.
Discussion
In this study, we evaluated the effect of the novel LXR agonist AZ876 on plasma lipid levels and atherosclerosis in hyperlipidaemic APOE*3Leiden transgenic mice. AZ876 and GW3965 both markedly inhibited atherosclerosis development, whereas AZ876 induced a more stable lesion phenotype. Additionally, AZ876 showed no adverse effects when given in low dose, pointing to dose-dependency of the effects induced by AZ876.
The results of the present study confirm the findings of previous studies in APOE*3Leiden (Grefhorst et al., 2002;Verschuren et al., 2009), LDLr–/– (Terasaka et al., 2003) and apoE–/– (Dai et al., 2007) mice with another LXR agonist, T0901317, with regard to elevation of plasma triglyceride and HDL levels and reduction of atherosclerosis. In wild-type mice, GW3965 (20–100 mg·kg−1·day−1) has been reported to be a tissue- and gene-selective modulator of LXR activity with less lipogenic effects than T0901317 after a 3 day treatment (Miao et al., 2004). Also in LDLr–/– mice, GW3965 (10 mg·kg−1·day−1) did not affect plasma or liver lipid levels when administered for 8 weeks (Quinet et al., 2009). In contrast to these latter reports, we showed that in a more sensitive model of hyperlipidaemia, the APOE*3Leiden mouse, even a relatively low dose of GW3965 given for 20 weeks (17 µmol·kg−1·day−1, ∼10 mg·kg−1·day−1) increased plasma triglyceride levels. The low dose of 5 µmol·kg−1·day−1 AZ876 did not affect plasma triglycerides; however, the high dose of 20 µmol·kg−1·day−1 AZ876 did induce hypertriglyceridaemia indicative of a dose-dependent effect. Next to the undesired induction of hypertriglyceridaemia, GW3965 and 20 µmol·kg−1·day−1 AZ876 increased HDL levels, thereby resembling findings described for T0901317 and fenofibrate-treated APOE*3Leiden mice (Kooistra et al., 2006;Verschuren et al., 2009), as reflected by the appearance of a large cholesteryl ester-rich HDL-1 particle in the lipoprotein profile, containing mainly the apoE, and to a minor extent apoB and no apoAI (Gruen et al., 2005). The appearance of this large HDL-1 particle upon LXR agonist treatment is specific for species without CETP and may have atheroprotective effects by supporting apoAI-independent cholesterol efflux (Jiang et al., 1992). In species containing CETP, like hamsters and cynomolgus monkeys (Groot et al., 2005; Quinet et al., 2009) and APOE*3Leiden.CETP transgenic mice (PCN Rensen and HMG Princen, unpubl. data), this HDL is absent via the action of CETP. In our study, the increased HDL levels were in accordance with an increase in intestinal abca1 mRNA expression in the GW3965 and 20 µmol·kg−1·day−1 AZ876 group, as intestinal ABCA1 directly contributes to the HDL raising effect of LXR agonists (Brunham et al., 2006; Tang et al., 2008).
Additionally, we observed increased lanosterol levels, suggesting an increased cholesterol synthesis. The most likely explanation for this finding is an increased cholesterol loss. However, we could not detect any signs of increased cholesterol loss by measuring biomarkers for cholesterol uptake (sitosterol) or bile acid synthesis (C4) or changed cholesterol or bile acid content in faeces. Another explanation could be the down-regulation of the cholesterologenic enzyme lanosterol 14α-demethylase (CYP51A1) by LXR activation, which in turn leads to an accumulation of lanosterol. However, as opposing effects of distinct ligands (natural oxysterols vs. synthetic ligands) have been described for the regulation of CYP51A1 expression, this remains unclear (Wang et al., 2008).
Whereas the changes in lipoprotein profiles (reduction in VLDL and rise in HDL) per se are anti-atherogenic, we also observed reductions in circulating levels of the cytokines TNFα, IL-1β and IL-6, reflecting reduced inflammation brought about by an equimolar dose of AZ876 and GW3965. Furthermore, we showed that both agonists are potent inducers of RCT in vivo. This was studied in male C57BL/6 mice on standard chow diet in order to compare results with already published GW3965 data (Naik et al., 2006). The percent of radioactivity from injected 3H-labelled cholesterol found in plasma, liver and faeces in the present study is similar to the latter study. Importantly, the [3H]–radioactivity detected in blood was found in the HDL fraction. Thus, the radioactivity detected in blood most likely did not derive from circulating macrophages but involved an active transport of labelled cholesterol in HDL. It can only be speculated that most of the labelled cholesteryl ester still resides in macrophages within the peritoneal cavity after 48 h. In addition to this in vivo RCT experiment, we have in-house data to show that AZ876 reduces atherosclerosis in male apoE-deficient mice (data not shown). Moreover, LXR activation has been found by other groups to reduce atherosclerosis in both male and female apoE-deficient mice (Kratzer et al., 2009). Thus, although not shown in the present study, there are data to support anti-atherogenic effects of LXR agonists including AZ876 in both male and female mice.
We then investigated the effects of the compounds on atherosclerosis development. Both GW3965 and AZ876 at equimolar doses were very potent in reducing all parameters of atherosclerosis (i.e. lesion number, area and severity). The low dose of AZ876 (5 µmol·kg−1·day−1) reduced the lesion area and tended to reduce abundance of severe lesions without affecting the number of lesions. Additionally, and in line with the reduction in plasma cytokine levels, both GW3965 and high-dose AZ876 decreased the amount of monocytes adhering to the vessel wall, which is considered as a functional parameter for vessel wall inflammation. Looking into further detail into lesion composition, we observed a discrepancy between the effect of 17 µmol·kg−1·day−1 GW3965 and of the similar dose of 20 µmol·kg−1·day−1 AZ876. Treatment with GW3965 appeared to induce a less stable lesion phenotype than the equimolar dose of AZ876, as reflected by lower collagen content.
In line with our data in APOE*3Leiden mice, Joseph et al. (2002) reported that GW3965 prevented atherosclerosis development in LDLr–/– and apoE–/– mice, which was attributed mainly to a direct effect on cholesterol efflux from macrophages in the vessel wall. The latter study, unfortunately, did not show data on the lesion composition. Extending these findings, the same group demonstrated that GW3965 also has anti-inflammatory effects in the vessel wall, which also may have contributed to the observed atheroprotective effect. Our study in APOE*3Leiden mice confirms the anti-inflammatory effect of GW3965 as reflected by reduced levels of plasma cytokines and a decreased adherence of monocytes.
In conclusion, we have shown that, in contrast to the higher dose of AZ876, low-dose AZ876 did not adversely affect plasma and liver lipid levels, the lipoprotein profile and did not reduce inflammation, but still inhibited lesion development. This indicates that the presence of the large HDL-1 particle and anti-inflammatory effects, as observed after high-dose AZ876 treatment, is not the sole explanation of its anti-atherosclerotic effect. AZ876 (5 µmol·kg−1·day−1)did not affect the amount of adhering monocytes and the number of lesions, and the amount of undiseased segments were unaffected. Thus, the compound did not inhibit the onset of lesion development; instead it inhibited progression of the lesion area. As we showed that AZ876 is a very potent inducer of RCT in vivo, we suggest that this reduced lesion area is primarily due to an increased cholesterol efflux from the lesions by RCT. This idea is supported by the increased expression of ABCA1 and ABCG1 mRNA in the aorta in animals treated with the high dose of AZ876. Although the low dose only tended to increase the expression of these genes, it is possible that, over longer periods of time, such small changes might result in a physiologically relevant effect on RCT to influence progression of atherosclerosis.
Our data demonstrate the feasibility of identifying LXR agonists that inhibit the progression of atherosclerosis and, depending on the dose, without having an effect on inflammation and without inducing the detrimental lipogenic effects in liver and plasma. Identification of LXR modulators with a greater separation between beneficial and adverse effects remains an important challenge for the development of such agents for the future treatment of atherosclerosis.
Acknowledgments
Ria van den Hoogen and Erik Offerman at TNO BioSciences and Maj-Lis Hermansson, Lena William-Olsson, Ulla Karlsson, Gunilla Andersson, Britt-Marie Andersson, Markus Sennelöv and Lisa Jinton at AstraZeneca are thanked for their excellent technical assistance. Peter Åkerblad and Krister Bamberg at AstraZeneca are thanked for method development. The study was supported in part by a grant from AstraZeneca, Mölndal, Sweden.
Glossary
Abbreviations
- ABC
ATP binding cassette transporter
- ALT
alanine aminotransferase
- apoE
apolipoprotein E
- C4
7α−hydroxy-4-cholesten-3-one
- CE
cholesteryl ester
- CETP
cholesteryl ester transfer protein
- CT
cycle threshold
- LXR
liver X receptor
- RCT
reverse cholesterol transport
- SMC
smooth muscle cell
- SR-BI
scavenger receptor class B type I
- SREBP1-c
sterol-response element-binding protein 1-c
Conflicts of interest
DL, UL, RN, JO and ELL are employees of AstraZeneca R&D, Sweden.
Supplementary material
Supporting Information: Teaching Materials; Figs 1–6 as PowerPoint slide.
References
- Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 4th edn. Br J Pharmacol. 2009;158(Suppl. 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baigent C, Keech A, Kearney PM, Blackwell L, Buck G, Pollicino C, et al. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet. 2005;366:1267–1278. doi: 10.1016/S0140-6736(05)67394-1. [DOI] [PubMed] [Google Scholar]
- Bjorkhem I, Miettinen T, Reihner E, Ewerth S, Angelin B, Einarsson K. Correlation between serum levels of some cholesterol precursors and activity of HMG-CoA reductase in human liver. J Lipid Res. 1987;28:1137–1143. [PubMed] [Google Scholar]
- Boström J, Brickmann K, Broo A, Holm P, Li L, Sandberg P, et al. Derivatives of isothiazol-3(2H)-one 1,1-dioxides as liver X receptor modulators. 2006 WO2006073363. [Google Scholar]
- Brunham LR, Kruit JK, Iqbal J, Fievet C, Timmins JM, Pape TD, et al. Intestinal ABCA1 directly contributes to HDL biogenesis in vivo. J Clin Invest. 2006;116:1052–1062. doi: 10.1172/JCI27352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins JL, Fivush AM, Watson MA, Galardi CM, Lewis MC, Moore LB, et al. Identification of a nonsteroidal liver X receptor agonist through parallel array synthesis of tertiary amines. J Med Chem. 2002;45:1963–1966. doi: 10.1021/jm0255116. [DOI] [PubMed] [Google Scholar]
- Dai X, Ou X, Hao X, Cao D, Tang Y, Hu Y, et al. Effect of T0901317 on hepatic proinflammatory gene expression in apoE-/- mice fed a high-fat/high-cholesterol diet. Inflammation. 2007;30:105–117. doi: 10.1007/s10753-007-9026-2. [DOI] [PubMed] [Google Scholar]
- Delsing DJ, Offerman EH, van Duyvenvoorde W, van der Boom H, de Wit EC, Gijbels MJ, et al. Acyl-CoA:cholesterol acyltransferase inhibitor avasimibe reduces atherosclerosis in addition to its cholesterol-lowering effect in ApoE*3-Leiden mice. Circulatio. 2001;103:1778–1786. doi: 10.1161/01.cir.103.13.1778. [DOI] [PubMed] [Google Scholar]
- Delsing DJ, Jukema JW, Van De Wiel MA, Emeis JJ, van der Laarse A, Havekes LM, et al. Differential effects of amlodipine and atorvastatin treatment and their combination on atherosclerosis in apoE*3-Leiden transgenic mice. J Cardiovasc Pharmacol. 2003;42:63–70. doi: 10.1097/00005344-200307000-00010. [DOI] [PubMed] [Google Scholar]
- Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem. 1957;226:497–509. [PubMed] [Google Scholar]
- Grefhorst A, Elzinga BM, Voshol PJ, Plosch T, Kok T, Bloks VW, et al. Stimulation of lipogenesis by pharmacological activation of the liver X receptor leads to production of large, triglyceride-rich very low density lipoprotein particles. J Biol Chem. 2002;277:34182–34190. doi: 10.1074/jbc.M204887200. [DOI] [PubMed] [Google Scholar]
- Groot PH, Pearce NJ, Yates JW, Stocker C, Sauermelch C, Doe CP, et al. Synthetic LXR agonists increase LDL in CETP species. J Lipid Res. 2005;46:2182–2191. doi: 10.1194/jlr.M500116-JLR200. [DOI] [PubMed] [Google Scholar]
- Gruen ML, Plummer MR, Zhang W, Posey KA, Linton MF, Fazio S, et al. Persistence of high density lipoprotein particles in obese mice lacking apolipoprotein A-I. J Lipid Res. 2005;46:2007–2014. doi: 10.1194/jlr.M500181-JLR200. [DOI] [PubMed] [Google Scholar]
- van der Hoorn JW, Kleemann R, Havekes LM, Kooistra T, Princen HM, Jukema JW. Olmesartan and pravastatin additively reduce development of atherosclerosis in APOE*3Leiden transgenic mice. J Hypertens. 2007;25:2454–2462. doi: 10.1097/HJH.0b013e3282ef79f7. [DOI] [PubMed] [Google Scholar]
- Janowski BA, Willy PJ, Devi TR, Falck JR, Mangelsdorf DJ. An oxysterol signalling pathway mediated by the nuclear receptor LXR alpha. Nature. 1996;383:728–731. doi: 10.1038/383728a0. [DOI] [PubMed] [Google Scholar]
- Jiang XC, Agellon LB, Walsh A, Breslow JL, Tall A. Dietary cholesterol increases transcription of the human cholesteryl ester transfer protein gene in transgenic mice. Dependence on natural flanking sequences. J Clin Invest. 1992;90:1290–1295. doi: 10.1172/JCI115993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph SB, McKilligin E, Pei L, Watson MA, Collins AR, Laffitte BA, et al. Synthetic LXR ligand inhibits the development of atherosclerosis in mice. Proc Natl Acad Sci USA. 2002;99:7604–7609. doi: 10.1073/pnas.112059299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleemann R, Princen HM, Emeis JJ, Jukema JW, Fontijn RD, Horrevoets AJ, et al. Rosuvastatin reduces atherosclerosis development beyond and independent of its plasma cholesterol-lowering effect in APOE*3-Leiden transgenic mice: evidence for antiinflammatory effects of rosuvastatin. Circulation. 2003;108:1368–1374. doi: 10.1161/01.CIR.0000086460.55494.AF. [DOI] [PubMed] [Google Scholar]
- Kooistra T, Verschuren L, de Vries-Van der Weij J, Koenig W, Toet K, Princen HM, et al. Fenofibrate reduces atherogenesis in ApoE*3Leiden mice: evidence for multiple antiatherogenic effects besides lowering plasma cholesterol. Arterioscler Thromb Vasc Biol. 2006;26:2322–2330. doi: 10.1161/01.ATV.0000238348.05028.14. [DOI] [PubMed] [Google Scholar]
- Kratzer A, Buchebner M, Pfeifer T, Becker TM, Uray G, Miyazaki M, et al. Synthetic LXR agonist attenuates plaque formation in apoE-/- mice without inducing liver steatosis and hypertriglyceridemia. J Lipid Res. 2009;50:312–326. doi: 10.1194/jlr.M800376-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–874. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- Lu TT, Repa JJ, Mangelsdorf DJ. Orphan nuclear receptors as eLiXiRs and FiXeRs of sterol metabolism. J Biol Chem. 2001;276:37735–37738. doi: 10.1074/jbc.R100035200. [DOI] [PubMed] [Google Scholar]
- Miao B, Zondlo S, Gibbs S, Cromley D, Hosagrahara VP, Kirchgessner TG, et al. Raising HDL cholesterol without inducing hepatic steatosis and hypertriglyceridemia by a selective LXR modulator. J Lipid Res. 2004;45:1410–1417. doi: 10.1194/jlr.M300450-JLR200. [DOI] [PubMed] [Google Scholar]
- Naik SU, Wang X, Da Silva JS, Jaye M, Macphee CH, Reilly MP, et al. Pharmacological activation of liver X receptors promotes reverse cholesterol transport in vivo. Circulation. 2006;113:90–97. doi: 10.1161/CIRCULATIONAHA.105.560177. [DOI] [PubMed] [Google Scholar]
- Nilsson R, Iöfgren L, Hansson GI. 2009. Automated extraction and analysis of plasma biomarkers for cholesterol absorbtion, cholesterol synthesis and synthesis of bile acids by HPLC-APPI tandem mass spectrometry.
- Quinet EM, Basso MD, Halpern AR, Yates DW, Steffan RJ, Clerin V, et al. LXR ligand lowers LDL cholesterol in primates, is lipid neutral in hamster, and reduces atherosclerosis in mouse. J Lipid Res. 2009;50:2358–2370. doi: 10.1194/jlr.M900037-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rader DJ. Liver X receptor and farnesoid X receptor as therapeutic targets. Am J Cardiol. 2007;100:n15–n19. doi: 10.1016/j.amjcard.2007.08.008. [DOI] [PubMed] [Google Scholar]
- Repa JJ, Mangelsdorf DJ. The role of orphan nuclear receptors in the regulation of cholesterol homeostasis. Annu Rev Cell Dev Biol. 2000;16:459–481. doi: 10.1146/annurev.cellbio.16.1.459. [DOI] [PubMed] [Google Scholar]
- Repa JJ, Liang G, Ou J, Bashmakov Y, Lobaccaro JM, Shimomura I, et al. Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRalpha and LXRbeta. Genes Dev. 2000;14:2819–2830. doi: 10.1101/gad.844900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigamonti E, Chinetti-Gbaguidi G, Staels B. Regulation of macrophage functions by PPAR-alpha, PPAR-gamma, and LXRs in mice and men. Arterioscler Thromb Vasc Biol. 2008;28:1050–1059. doi: 10.1161/ATVBAHA.107.158998. [DOI] [PubMed] [Google Scholar]
- Schultz JR, Tu H, Luk A, Repa JJ, Medina JC, Li L, et al. Role of LXRs in control of lipogenesis. Genes Dev. 2000;14:2831–2838. doi: 10.1101/gad.850400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tall AR. Cholesterol efflux pathways and other potential mechanisms involved in the athero-protective effect of high density lipoproteins. J Intern Med. 2008;263:256–273. doi: 10.1111/j.1365-2796.2007.01898.x. [DOI] [PubMed] [Google Scholar]
- Tang W, Ma Y, Jia L, Ioannou YA, Davies JP, Yu L. Niemann-Pick C1-like 1 is required for an LXR agonist to raise plasma HDL cholesterol in mice. Arterioscler Thromb Vasc Biol. 2008;28:448–454. doi: 10.1161/ATVBAHA.107.160465. [DOI] [PubMed] [Google Scholar]
- Terasaka N, Hiroshima A, Koieyama T, Ubukata N, Morikawa Y, Nakai D, et al. T-0901317, a synthetic liver X receptor ligand, inhibits development of atherosclerosis in LDL receptor-deficient mice. FEBS Lett. 2003;536:6–11. doi: 10.1016/s0014-5793(02)03578-0. [DOI] [PubMed] [Google Scholar]
- Verschuren L, Kleemann R, Offerman EH, Szalai AJ, Emeis SJ, Princen HM, et al. Effect of low dose atorvastatin versus diet-induced cholesterol lowering on atherosclerotic lesion progression and inflammation in apolipoprotein E*3-Leiden transgenic mice. Arterioscler Thromb Vasc Biol. 2005;25:161–167. doi: 10.1161/01.ATV.0000148866.29829.19. [DOI] [PubMed] [Google Scholar]
- Verschuren L, de Vries-Van der Weij J, Zadelaar S, Kleemann R, Kooistra T. LXR agonist suppresses atherosclerotic lesion growth and promotes lesion regression in apoE*3Leiden mice: time course and mechanisms. J Lipid Res. 2009;50:301–311. doi: 10.1194/jlr.M800374-JLR200. [DOI] [PubMed] [Google Scholar]
- van Vlijmen BJ, van den Maagdenberg AM, Gijbels MJ, van der Boom H, HogenEsch H, Frants RR, et al. Diet-induced hyperlipoproteinemia and atherosclerosis in apolipoprotein E3-Leiden transgenic mice. J Clin Invest. 1994;93:1403–1410. doi: 10.1172/JCI117117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Rogers PM, Su C, Varga G, Stayrook KR, Burris TP. Regulation of cholesterologenesis by the oxysterol receptor, LXRalpha. J Biol Chem. 2008;283:26332–26339. doi: 10.1074/jbc.M804808200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerterp M, van der Hoogt CC, de Haan W, Offerman EH, Dallinga-Thie GM, Jukema JW, et al. Cholesteryl ester transfer protein decreases high-density lipoprotein and severely aggravates atherosclerosis in APOE*3-Leiden mice. Arterioscler Thromb Vasc Biol. 2006;26:2552–2559. doi: 10.1161/01.ATV.0000243925.65265.3c. [DOI] [PubMed] [Google Scholar]
- Zadelaar S, Kleemann R, Verschuren L, de Vries-Van der Weij J, van der Hoorn JW, Princen HM, et al. Mouse models for atherosclerosis and pharmaceutical modifiers. Arterioscler Thromb Vasc Biol. 2007;27:1706–1721. doi: 10.1161/ATVBAHA.107.142570. [DOI] [PubMed] [Google Scholar]
- Zelcer N, Tontonoz P. Liver X receptors as integrators of metabolic and inflammatory signalling. J Clin Invest. 2006;116:607–614. doi: 10.1172/JCI27883. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.