

Abstract
BACKGROUND AND PURPOSE
Adipocyte fatty acid-binding protein (A-FABP) is up-regulated in regenerated endothelial cells and modulates inflammatory responses in macrophages. Endothelial dysfunction accompanying regeneration is accelerated by hyperlipidaemia. Here, we investigate the contribution of A-FABP to the pathogenesis of endothelial dysfunction in the aorta of apolipoprotein E-deficient (ApoE−/−) mice and in cultured human endothelial cells.
EXPERIMENTAL APPROACH
A-FABP was measured in aortae of ApoE−/−mice and human endothelial cells by RT-PCR, immunostaining and immunoblotting. Total and phosphorylated forms of endothelial nitric oxide synthase (eNOS) were measured by immunoblotting. Changes in isometric tension were measured in rings of mice aortae
KEY RESULTS
A-FABP was expressed in aortic endothelium of ApoE−/− mice aged 12 weeks and older, but not at 8 weeks or in C57 wild-type mice. Reduced endothelium-dependent relaxations to acetylcholine, UK14304 (selective α2-adrenoceptor agonist) and A23187 (calcium ionophore) and decreased protein presence of phosphorylated and total eNOS were observed in aortae of 18 week-old ApoE−/− mice compared with age-matched controls. A 6 week treatment with the A-FABP inhibitor, BMS309403, started in 12 week-old mice, improved endothelial function, phosphorylated and total eNOS and reduced plasma triglyceride levels but did not affect endothelium-independent relaxations. The beneficial effect of BMS309403 on UK14304-induced relaxations was attenuated by Pertussis toxin. In cultured human microvascular endothelial cells, lipid-induced A-FABP expression was associated with reduced phosphorylated eNOS and NO production and was reversed by BMS309403.
CONCLUSIONS AND IMPLICATIONS
Elevated expression of A-FABP in endothelial cells contributes to their dysfunction both in vivo and in vitro.
Keywords: A-FABP, endothelium-dependent relaxation, eNOS, endothelial dysfunction, nitric oxide, atherosclerosis
Introduction
Endothelial dysfunction occurs early in vascular disease (Vanhoutte, 2004; 2009;). It is characterized by a reduced bioavailability of nitric oxide (NO) in the intimal layer of the arterial wall and an increased production of endothelium-derived vasoconstrictor prostanoids (Vanhoutte, 2004; 2009; Vanhoutte and Tang, 2008; Tang and Vanhoutte, 2009). The constitutive form of NO synthase (eNOS) is the enzyme responsible for the conversion of L-arginine into NO in normal endothelial cells. Both Gi (Pertussis toxin-sensitive) and Gq proteins couple endothelial cell membrane receptors to eNOS. The former mediate the responses to α2-adrenoceptor agonists and serotonin (5-HT) and the latter those to bradykinin and ADP (Flavahan et al., 1989; Shimokawa et al., 1991; Vanhoutte, 2004; 2009;). Accelerated turnover of the endothelium is observed in aging and involves apoptotic death and desquamation, followed by regeneration of endothelial cells (Vanhoutte, 2004; 2009; Hoenig et al., 2008). Endothelial regeneration also contributes to the natural repair process in response to vascular injury, which results in an altered endothelial phenotype exhibiting selective dysfunction of the Gi-proteins coupled to eNOS and premature senescence (Shimokawa et al., 1991; Taddei et al., 2001; Kennedy et al., 2003). When endothelial regeneration and hypercholesterolemia are combined (a condition known to accelerate atherosclerosis), endothelium-dependent relaxations are impaired further, suggesting exacerbation of the pathological changes in regenerated endothelium (Shimokawa et al., 1991). The conjunction of the reduced production of NO, the increased generation of reactive oxygen species (ROS), and the increased generation/presence of oxidized forms of low-density lipoprotein (LDL) in the regenerated endothelium sets the stage for the inflammatory response that leads to atherosclerosis (Vanhoutte and Shimokawa, 1989; Taddei et al., 2001; Kennedy et al., 2003; Vanhoutte, 2004; 2009; Lee et al., 2007; Libby, 2007; Yildiz, 2007; Pierce et al., 2009; Rodríguez-Mañas et al., 2009).
The gene encoding for adipocyte fatty acid-binding protein (A-FABP) is not expressed in native endothelial cells, but appears in cell cultures derived from porcine regenerated coronary endothelial cells (Lee et al., 2007). A-FABP activates both the NFκB pathway and c-Jun NH2-terminal kinase (JNK), which in turn up-regulates the transcriptional activity of inflammatory genes such as the chemokine CCL2 (MCP-1), interleukins and tumor necrosis factor (TNFα) in macrophages (Makowski et al. 2001; 2005; Furuhashi et al., 2007; Hui et al., 2010). A-FABP contributes to the formation of foam cells and atherosclerotic plaques (Makowski and Hotamisligil, 2005; Furuhashi et al., 2008; Krusinová and Pelikánová, 2008). In humans, high circulating levels of A-FABP are associated with an increased risk of developing atherosclerosis (Yeung et al., 2007). By contrast, in apolipoprotein E−/− (ApoE−/−) mice, the targeted disruption of the A-FABP gene or pharmacological inhibition of A-FABP activity reduces the expression of several chemoattractant and inflammatory cytokines in macrophages, and inhibits the transformation of macrophages into foam cells, thereby leading to a significant attenuation of atherosclerosis (Boord et al., 2002; Furuhashi et al., 2007).
In the ApoE−/− mice, hyperlipidemic conditions exacerbate endothelial dysfunction, as reflected by an impairment of acetylcholine-induced, endothelium-dependent relaxations (Williams et al., 2000; Crauwels et al., 2003; Kawashima, 2004; Hans et al., 2009). The present study was designed to test the hypothesis that A-FABP plays a role in the endothelial dysfunction of the ApoE−/− mouse.
Methods
ApoE−/− mice
All animal care and experimental procedures were approved by the institutional animal ethics committee (Committee on the Use of Live Animals in Teaching & Research) of The University of Hong Kong. C57BL/6J mice (wild-type strain; ApoE+/+ mice) and ApoE−/− mice were studied. Mice homozygous for the Apoetm1Unc mutation were provided by the Jackson Laboratory (Bar Harbor, Maine, ME, USA). The breeding line was maintained by directly pairing male and female homozygous mutated Apoetm1Unc mice. The mice were maintained under pathogen-free conditions in filter-topped cages in an air-conditioned room at constant temperature (23 ± 1°C), fed a standard laboratory diet (LabDiet, 5053, PMI; St. Louis, MO, USA) and given water ad libitum. To study endothelial function, ApoE−/− mice 8 to 18 weeks old, and age-matched wild-type mice were compared. To determine the effects of pharmacological inhibition of the actions of A-FABP, either the A-FABP inhibitor BMS309403 (15 mg·kg−1·day−1) (Furuhashi et al., 2007) or vehicle (4% Tween 80) were administered chronically by daily oral gavage for 6 weeks in ApoE−/− mice (starting at weeks 12 of age). Mice were anaesthetized with a bolus injection of pentobarbitone sodium (230 mg·kg−1) and their aorta removed and dissected for ex vivo studies.
Blood samples from mice with or without BMS309403 treatment were collected at the time of death by direct puncture of the heart. They were centrifuged at 1500× g for 15 min at 15°C and the plasma was collected. The triglyceride concentration was determined with 20 µL plasma using a commercially available measurement kit (WAKO, Osaka, Japan). Plasma levels of LDL and high density lipoprotein (HDL) cholesterol were determined using another commercially available HDL and LDL/VLDL Cholesterol Quantification Kit (BioVision, Mountain View, CA, USA). Glucose was measured with a commercial biochemical assay kit from Stanbio Laboratory (Boerne, TX, USA) and insulin was measured with a commercial elisa kit from Mercodia (Uppsala, Sweden).
Isometric tension measurements
Mice were anaesthetized with a bolus injection of pentobarbitone sodium (230 mg·kg−1) and their aorta removed and dissected free of adherent connective tissues. Rings (2 mm in length) with endothelium were suspended in a Halpern-Mulvany myograph (Model 610 M, Danish Myo Technology A/S, Denmark) using 40 µm stainless steel wires for recording of isometric contractile force (PowerLab 4SP, ADInstrruments, Colorado Springs, CO USA) (Tang et al., 2005). The preparations were immersed in Krebs' Ringer bicarbonate solution (37°C, aerated with a 95% O2/5% CO2 gas mixture, pH7.4) of the following composition (in mM): NaCl 118, KCl 4.7, CaCl2 2.5, MgSO4 1.2, KH2PO4 1.2, NaHCO3 25, and glucose 11.1 (control solution). The rings were allowed to equilibrate in control solution for one hour at a resting tension of 500 mN. After stabilization, they were then stretched step by step and exposed to 60 mM KCl (15 min) until a maximal, reference contraction was obtained. To demonstrate the presence of endothelium, relaxations to 10 µM acetylcholine were obtained during a contraction to 0.5 µM U46619 (thromboxane TP receptor agonist); if the relaxation to acetylcholine was less than 80%, the preparations were excluded from further experimentation.
To study endothelium-dependent relaxations, cumulative concentration–relaxation curves to acetylcholine (0.1 nM to 0.1 mM), the calcium ionophore A23187 (0.1 nM to 1 mM) and the α2-adrenoceptor agonist UK14304 (0.1 nM to 10 mM) were obtained [in the absence or presence of NG-nitro-L-arginine methyl ester (L-NAME) (10 mM)] during sustained contractions to U46619 (10 to 50 nM; to achieve a level of contraction approximating 90% of the reference response to 60 mM KCl). To study endothelium-independent relaxations, cumulative concentration–relaxation curves to sodium nitroprusside (1 nM to 0.1 mM) were obtained also during sustained contractions to U46619. Some preparations were incubated with Pertussis toxin (PTX; 400 ng·mL−1) or its solvent (0.1 mM sodium phosphate; pH 7.0) for 90 min before obtaining a concentration–response curve to UK14304 (Shimokawa et al., 1989; 1991;).
RNA preparation and real-time PCR
The endothelial layer of the aorta of wild-type and ApoE−/− mice was collected by gently scraping as described (Lee et al., 2007). Total RNA was extracted directly with RNA lysis thiocyanate (RLT) buffer (containing 25–50% guanidinium thiocyanate) using a RNeasy Mini kit (Qiagen, Valencia, CA, USA). Samples were stored immediately at −70°C until use. Total RNA was added to a reverse transcription mixture [(40 µL; first strand buffer, 10 mM DTT, 0.5 mM DNTPs, 10 ng·µL−1 Oligo(dT) (Gibco-BRL, Grand Island, NY, USA), 1 unit·µL−1 Rnasin, 1 unit·µL−1 Moloney murine leukemia virus reverse transcriptase (M-MLV RT; Gibco-BRL)] for 10 min at room temperature followed by 37°C for 60 min. The product was denatured by exposing the samples to 94°C for 7 min to produce the first-strand cDNAs. Two microliters of the total reverse transcription product were added to a reaction mixture (20 µL) containing 10 µL 2X SYBER Green master mix (Applied Biosystems, Warrington, UK) and the primers (sense and anti-sense; 1 µM) for the polymerase chain reaction (PCR). Real-time PCR technique was used to determine the mRNA expression. The PCR products were amplified using a primer pair specific for mouse A-FABP: 5′-CCGCAGACGACAGGA-3′ and 5′-CTCATGCCCTTTCATAAACT-3′ (177 bp) (Furuhashi et al., 2007), smooth muscle alpha actin: 5′-AGCAGAACAGAGGAATGCAGTGGAAGAGAC-3′ and 5′-CCTCCCACTCGCCTCCCAAACAAGGAGC-3′ (146 bp) (Wamhoff et al., 2008) and GAPDH, 5′-AATGACCCCTTCATTGACCTCC-3′ and 5′-GCTTCCCATTCTCAGCCTTGAC-3′ (100 bp). The purity of the endothelial mRNA was demonstrated by a minimal expression level of smooth muscle α-actin (<1:10 when compared with aortic smooth muscle samples).
Real-time PCR analysis was performed using a 7900HT Fast Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). The cycling conditions to amplify PCR products of A-FABP, smooth muscle α-actin and GAPDH were 50°C for 2 min; 95°C for 10 min; 95°C for 15 s, 60°C for 1 min; and 95°C for 15 s. Each sample was analyzed in duplicate. Results were normalized to the copy numbers of GAPDH gene products. The PCR products were visualized on 1.2% (w/v) agarose gels using ethidium bromide to ensure that they formed a single band of the expected size.
Immunostaining
Fixed sections of aortic samples were re-hydrated in serial concentrations of ethanol (100 to 50%). Endogenous peroxidase activity was blocked by incubation with 3% hydrogen peroxide in methanol for 20 min with constant shaking. Following incubation of the slides with IgG blocking reagent, the sections were incubated with a primary antibody against A-FABP (1:50) or von Willebrand factor (1:100) overnight at 4°C. For immunohistochemistry, after washing in phosphate buffer solution containing 0.01% Triton X-100, the sections were incubated for 30 min in biotinylated antibody against IgG (Vector). After washing, they were exposed to a solution of Vectastain Elite ABC [horseradish peroxidase (HRP)] until color developed. Double immunoflorescent staining of A-FABP and von Willebrand factor was achieved by incubating the slides with Alexa Fluor® 488 (Ex495/Em519, green) and Alexa Fluor® 594 (Ex590/Em617, red) for anti-goat and anti-rabbit IgG secondary antibodies (1:1500), respectively, after incubation with the primary antibodies and extensive washing according to the manufacturer's instruction. Immunoflorescence of the sections was visualized using an inverted fluorescent microscope (Leica Microsystems GmbH, Wetzlar, Germany).
Western blotting
Total protein from aortae of ApoE−/− and C57 wild-type mice at 18 weeks of age or cultures of human microvascular endothelial cells (HMECs) were collected and then lysed in ice-cold lysis buffer (20 mM Tris-HCl, 1% Triton X-100, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM sodium orthovanadate) containing a cocktail of protease inhibitors (1 mM phenylmethylsulfonyl fluoride, 100 ng·mL−1 trypsin inhibitor, 20 µg·mL−1 leupeptin and 1 µM pepstatin). Protein lysates were sonicated further for 15 s on ice to facilitate homogenization. The protein concentration was determined using the Bradford assay. Total protein (40–50 µg) was separated on a polyacrylamide gel (7.5% or 15%) and blotted on polyvinylidene difluoride (PVDF) membranes (180 mA, 1.5 h). The blot was incubated for one hour in TBS containing 5% fat-free milk (Lee et al., 2007). Membranes were incubated with antibodies against A-FABP, phosphorylated eNOS (Ser1177) and total eNOS at 4°C overnight. This was followed by incubation of the HRP-labelled secondary antibody (Amersham, Freiburg, Germany) prior to image detection by enhanced chemiluminescence using a commercially available kit (Amersham) (Lee et al., 2007).
Human endothelial cells
HMECs were obtained from Lonza (Basel, Switzerland) and grown at 37°C in humidified 5% CO2 95% air. The medium was changed every two days. Cells [at passage 6 to 8] were treated with various concentrations of palmitate (0.1 to 0.3 mM) dissolved in bovine serum albumin as control for 24 h. Some cells were treated with 0.3 mM palmitate with or without 50 µM BMS309403 for 24 h followed by stimulation with 100 nM insulin for 20 min. Samples of protein (40 µg) from total lysates were used for the detection of A-FABP, phospho-eNOS (Ser1177) and total eNOS using Western blotting (Lee et al., 2007). In other cell cultures, the cGMP level was measured using a commercial elisa kit as described (Wang et al., 2009).
Data analysis
Concentration–relaxation curves are shown [calculated using GraphPad Prism 5 (La Jolla, CA, USA)] to compare the responses with various agonists. The maximal relaxation and the concentrations causing 50% of the maximal response (EC50) are also reported. The intensity of Western blot images was calculated with a computerized program (Multi-Analyst version 1.1, Bio-Rad Laboratories, Inc., Hercules, CA, USA). Data are expressed as means ± SEM. Statistical analysis was performed using Student's unpaired t-test for single comparison, and/or two-way anova followed by Bonferroni post hoc test for multiple comparisons. P values equal to or less than 0.05 were considered to indicate statistically significant differences.
Materials
The selective A-FABP inhibitor BMS309403 {2-[2′-(5-ethyl-3,4-diphenyl-1H-pyrazol-1-yl)biphenyl-3-yloxy] acetic acid} was manufactured as described (Hui et al., 2010). Acetylcholine, A23187, insulin, L-NAME, palmitate, sodium nitroprusside and UK14304 were purchased from Sigma Chemical Company (St. Louis, MO, USA) and PTX was purchased from List Biological (Campbell, CA, USA). U46619 was purchased from Biomol (Plymouth Meeting, PA, USA). Antibody against mouse A-FABP (which shows less than 5% cross-reactivity with recombinant mFABP5, rFABP2 and rFABP1 according to the product insert) was purchased from R & D Systems (AF1443; Minneapolis, MN, USA) and that against von Willebrand factor from Sigma Chemical Company. Alexa Fluor® 488 and Alexa Fluor® 594 anti-goat and anti-rabbit IgG secondary antibodies were purchased from Invitrogen (Carlsbad, California, USA). The antibodies against phosphorylated eNOS (Ser1177) (p-eNOS; 1:1000) and total eNOS (eNOS; 1:2500) were from Transduction Laboratories (Lexington, KY, USA). Drug concentrations are expressed as final molar concentrations in the bath solution.
Results
Plasma levels of lipids, glucose and insulin
At 18 weeks of age, the body weight of wild-type, vehicle-treated and BMS309403-treated ApoE−/− mice was 28.1 ± 0.6, 25.7 ± 1.2 and 26.7 ± 0.7 g, respectively. The plasma levels of triglycerides and LDL-C were significantly elevated in 18 week-old ApoE−/− mice with or without BMS309403 treatment (15 mg·kg−1·day−1) while the HDL-C was reduced significantly compared with the wild-type mice of the same age (Table 1). The glucose and insulin levels were comparable in the wild-type and ApoE−/− mice. The only significant effect of 6 weeks of treatment with BMS309403 was to reduce the triglyceride level.
Table 1.
Plasma lipid, glucose and insulin levels in wild-type, ApoE−/− mice without (vehicle) or with BMS309403 (BMS) treatment at 18 weeks of age
| Plasma level (mmol·L−1) | C57 wk18 | ApoE−/−+Vehicle | ApoE−/−+BMS |
|---|---|---|---|
| Triglyceride | 0.66 ± 0.18 | 2.77 ± 0.27** | 1.47 ± 0.25**† |
| LDL-C | 0.36 ± 0.08 | 2.91 ± 0.27** | 2.51 ± 0.04*ns |
| HDL-C | 0.60 ± 0.06 | 0.35 ± 0.05* | 0.33 ± 0.08*ns |
| Glucose | 195.30 ± 15.7ns | 218.07 ± 9.0ns | 220.62 ± 9.40ns |
| Insulin (µg·L−1) | 1.76 ± 0.26ns | 1.23 ± 0.17ns | 1.33 ± 0.17ns |
Data are shown as means ± SEM
P < 0.05;
P < 0.01; compared with the wild-type control.
P < 0.05; compared with the ApoE−/− mice without BMS309403 treatment.
ns, not significant. n= 5.
Presence of A-FABP in the endothelium
The mRNA expression of A-FABP was minimal in the aorta of 18 weeks old wild-type and 8 weeks old ApoE−/− mice, but it increased progressively in the aortic endothelial cell layer of ApoE−/− mice from 12 to 18 weeks of age (Figure 1).
Figure 1.
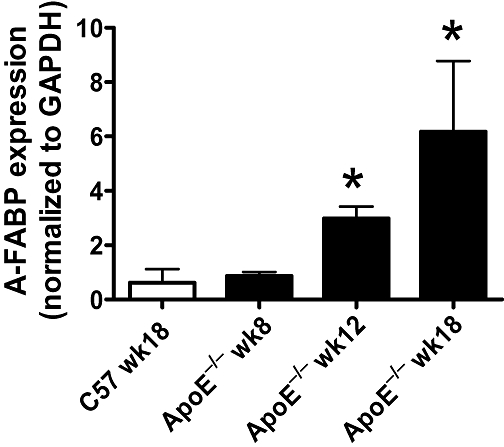
Quantitative real-time PCR for mRNA expression of adipocyte fatty acid-binding protein (A-FABP) in aortae of wild-type and apolipoprotein E-deficient (ApoE−/−) mice. Data are shown as means ± SEM *P < 0.05 compared with the wild-type control (C57 wk18). n= 3–4.
The immunohistochemical staining revealed the presence of A-FABP protein in the endothelium of ApoE−/− mice at the age of 12 weeks but not in that of younger animals (Figure 2). The immunofluorescent staining analysis showed co-localization of A-FABP and von Willebrand factor in the endothelial cells of 12 weeks old ApoE−/− mice but not in the wild-type controls (Figure 3).
Figure 2.
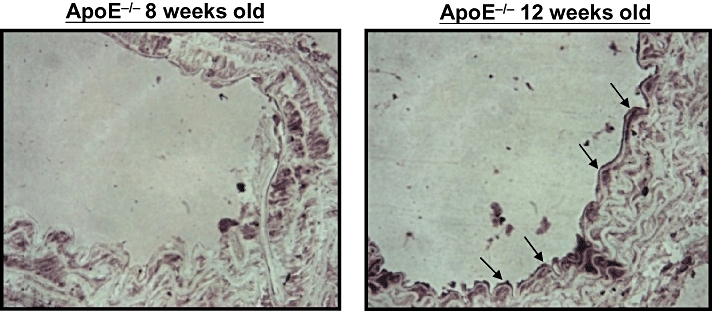
Immunohistochemical staining for adipocyte fatty acid-binding protein in the aortic endothelium of apolipoprotein E-deficient (ApoE−/−) mice at 8 and 12 weeks of age (×400).
Figure 3.
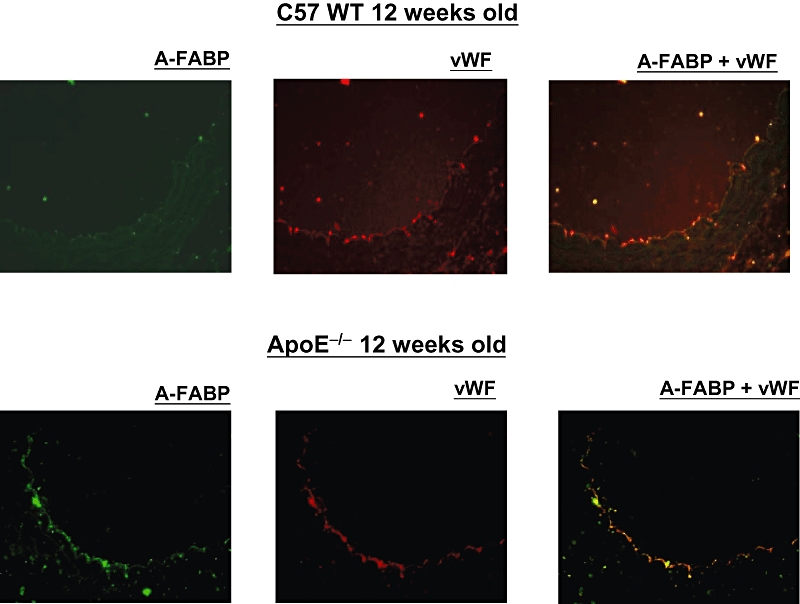
Immunofluorescent staining for adipocyte fatty acid-binding protein (A-FABP) (green) and von Willebrand factor (vWF; red) in the aortic endothelium of C57 wild-type mice and apolipoprotein E-deficient (ApoE−/−) mice at 12 weeks of age (×400).
Relaxations
Untreated mice
The sustained contractions to U46619 (10 to 50 nM) were not significantly different between aortae of wild-type and ApoE−/− mice from 8 to 18 weeks of age (data not shown).
There were no significant differences in relaxations to acetylcholine, UK14304 and sodium nitroprusside between aortae of wild-type mice at 8, 12 and 18 weeks of age. However, the relaxations to A23187 were significantly reduced in aortae of 12 weeks old wild-type mice compared with those of the younger group (Figure 4; Table 2).
Figure 4.
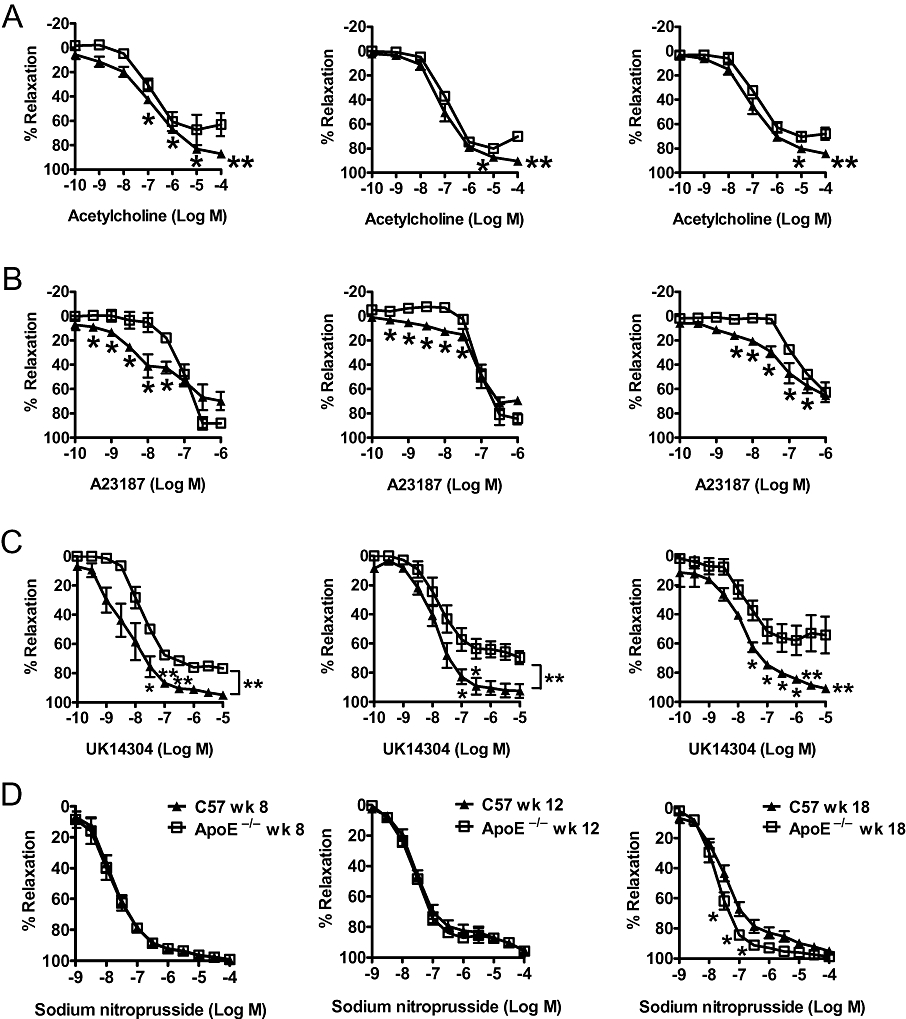
Endothelium-dependent relaxations induced by (A) acetylcholine, (B) A23187, (C) UK14304 (acting on α2-adrenoceptors on the endothelium) and endothelium-independent relaxations, (D) induced by sodium nitroprusside of U46619-contracted rings in aortae of age-matched wild-type (C57) and apolipoprotein E-deficient (ApoE−/−) mice at 8, 12 and 18 weeks old. Data are shown as means ± SEM *P < 0.05; **P < 0.01 compared with the control of age-matched C57 mice. n= 6–8.
Table 2.
EC50 values and maximal relaxations of aortic rings
| Max. relaxation EC50 | WT 8 weeks | ApoE−/− 8 weeks | WT 12 weeks | ApoE−/− 12 weeks | WT 18 weeks | ApoE−/− 18 weeks | ApoE−/− 18 weeks + BMS |
|---|---|---|---|---|---|---|---|
| UK14304 | 95.07 ± 1.46 | 75.56 ± 0.00*** | 92.55 ± 4.74 | 69.61 ± 4.40*** | 90.80 ± 0.97 | 66.22 ± 4.66** | 87.82 ± 2.99† |
| −8.27 ± 0.27 | −7.17 ± 0.0* | −7.77 ± 0.19 | −6.75 ± 0.37* | −7.77 ± 0.09 | −6.76 ± 0.25* | −7.76 ± 0.18† | |
| Ach | 87.01 ± 0.97 | 63.06 ± 9.57*** | 90.53 ± 2.76 | 78.12 ± 3.35*** | 84.39 ± 1.89 | 68.01 ± 4.91*** | 90.49 ± 2.57† |
| −6.73 ± 0.12 | −5.71 ± 0.51*** | −6.86 ± 0.16 | −6.45 ± 0.08* | −6.64 ± 0.18 | −5.95 ± 0.22* | −7.01 ± 0.11† | |
| A23187 | 69.88 ± 7.50 | 88.15 ± 3.59 ns | 69.42 ± 3.41 | 84.54 ± 4.48* | 65.45 ± 3.26 | 62.73 ± 8.16 ns | 68.19 ± 7.67 ns |
| −7.14 ± 0.34 | −7.00 ± 0.09 ns | −6.85 ± 0.11 | −6.90 ± 0.04 ns | −6.87 ± 0.13 | −6.34 ± 0.11* | −6.87 ± 0.19† | |
| SNP | 99.51 ± 1.43 | 99.23 ± 1.76 ns | 95.29 ± 0.81 | 95.77 ± 0.53 ns | 95.13 ± 1.79 | 76.94 ± 17.47 ns | 96.98 ± 1.34 ns |
| −7.72 ± 0.12 | −7.77 ± 0.17 ns | −7.28 ± 0.14 | −7.41 ± 0.07 ns | −7.27 ± 0.17 | −7.66 ± 0.09 ns | −7.88 ± 0.15 n.s. |
The EC50 values (log M) and maximal relaxation (%) in response to various endothelium-dependent vasodilators [UK14304, acetylcholine (ACh), A23187 and sodium nitroprusside (SNP)] in wild-type mice (8 to 18 weeks of age), ApoE−/− (8 to 18weeks of age) and ApoE−/− mice with chronic treatment (18 weeks of age; ApoE−/− 18 wk + BMS) for 6 weeks. Data are shown in parallel for age-matched C57 mice. Data are shown as means ± SEM
P < 0.05;
P < 0.01;
P < 0.000 compared with the wild-type control.
P < 0.01; compared with the ApoE−/− mice without BMS309403 treatment. ns not significant. n= 5–7.
The relaxations to acetylcholine, the α2-adrenoceptor agonist UK 14304 and the calcium ionophore A23187 were impaired in aortae of ApoE−/− of all age groups. The maximal relaxation to UK14304 and acetylcholine was significantly reduced while the EC50 was significantly increased in aortae of ApoE−/− of all age groups. However, the maximal relaxation to A23187 was significantly increased in aorta of ApoE−/− at 12 weeks of age while the EC50 was significantly increased in aorta of ApoE−/− at 18 weeks of age (Figure 4; Table 2).
Except for an enhanced relaxation in aortae of 18 weeks old ApoE-/-, there were no statistically significant differences in the relaxations, maximal relaxation and EC50 to sodium nitroprusside between the different experimental groups (Figure 4; Table 2).
Chronic treatment with BMS309403
BMS309403 is an aromatic biphenyl azol compound that competes with fatty acids for the binding pocket of A-FABP with high specificity (Furuhashi et al., 2007). Chronic administration of BMS309403 (15 mg·kg−1·day−1; from 12 to 18 weeks of age) in ApoE−/− mice significantly improved the relaxations, maximal relaxation and EC50 to UK14304 (Figure 5; Table 2), acetylcholine (Figure 6; Table 2) and A23187 (except for the maximal relaxation; Figure 6; Table 2), compared with those obtained in aortae from age-matched, vehicle-treated ApoE−/− mice, without significantly affecting responses to sodium nitroprusside (Figure 5; Table 2).
Figure 5.
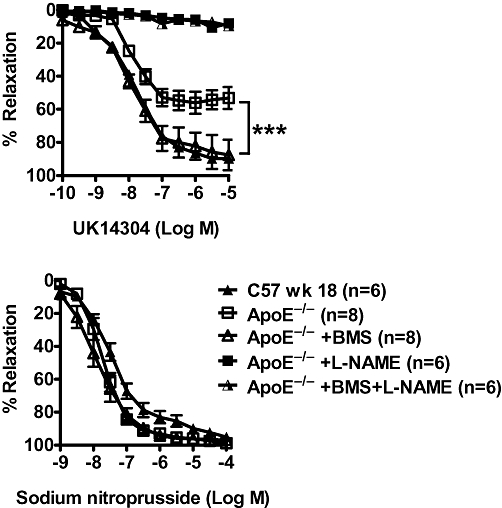
Relaxations of U46619-contracted rings induced by UK14304 (upper), and sodium nitroprusside (SNP) (lower) in aortae of 18 weeks old apolipoprotein E-deficient (ApoE−/−) mice treated with BMS309403 (15 mg·kg−1·day−1; ApoE−/−+BMS) or vehicle (4% Tween 80; ApoE−/−) for 6 weeks. In one set of experiments, the in vitro effect of NG-nitro-L-arginine methyl ester (L-NAME) (45 min of incubation at 10 mM) was determined (upper). Responses in wild-type mice (C57 wk 18) without any treatment are shown for comparison. Data are shown as means ± SEM ***P < 0.000 compared with the respective control. n= 6–8.
Figure 6.
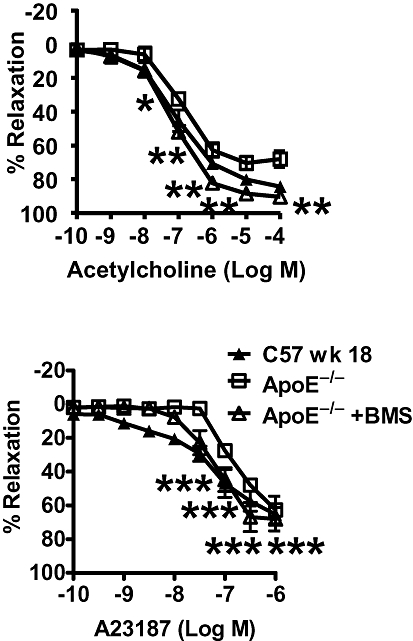
Endothelium-dependent relaxations of U46619-contracted rings induced by acetylcholine (upper) and the calcium ionophore A23187 (lower) in aortae of apolipoprotein E-deficient (ApoE−/−) mice after chronic treatment [BMS309403 (15 mg·kg−1·day−1; ApoE−/−+BMS) or vehicle (4% Tween 80; ApoE−/−)] for 6 weeks. Responses in wild-type mice (C57 wk 18) without any treatment are shown for comparison. Data are shown as means ± SEM *P < 0.05; **P < 0.01; ***P < 0.000 compared with the respective control. n= 6–10.
Incubation with PTX (400 ng·mL−1) before obtaining the UK14304-induced relaxation abolished the potentiating effect of the chronic treatment with BMS309403 on the response to the α2-adrenoceptor agonist. No impairment in UK14304-induced relaxation was observed with PTX in the aortae of vehicle-treated mice (Figure 7).
Figure 7.

Endothelium-dependent relaxations of U46619-contracted rings induced by UK14304 in aortae of apolipoprotein E-deficient (ApoE−/−) mice after chronic treatment with BMS309403 (15 mg·kg−1·day−1; ApoE−/−+BMS). The preparations were treated with Pertussis toxin (PTX; 400 ng·mL−1) or vehicle control (0.1 mM sodium phosphate) 1.5 h before obtaining the UK14304-induced relaxation in aortae of treated and untreated ApoE−/− mice. Data are shown as means ± SEM *P < 0.05 compared with the respective control. n= 6–8.
In aortae isolated from both BMS309403-treated and vehicle-treated ApoE−/− mice, the relaxation evoked by UK14304 was abolished by incubation with L-NAME (10 mM) (Figure 5). L-NAME abolished also the relaxations to acetylcholine and A23187 (data not shown).
eNOS levels
The levels of both the total and phosphorylated eNOS (Ser1177) were significantly reduced in aortae of 18 week-old ApoE−/− compared with the wild-type controls of the same age. The phosphorylated to total eNOS ratio was not changed significantly (Figure 8A).
Figure 8.
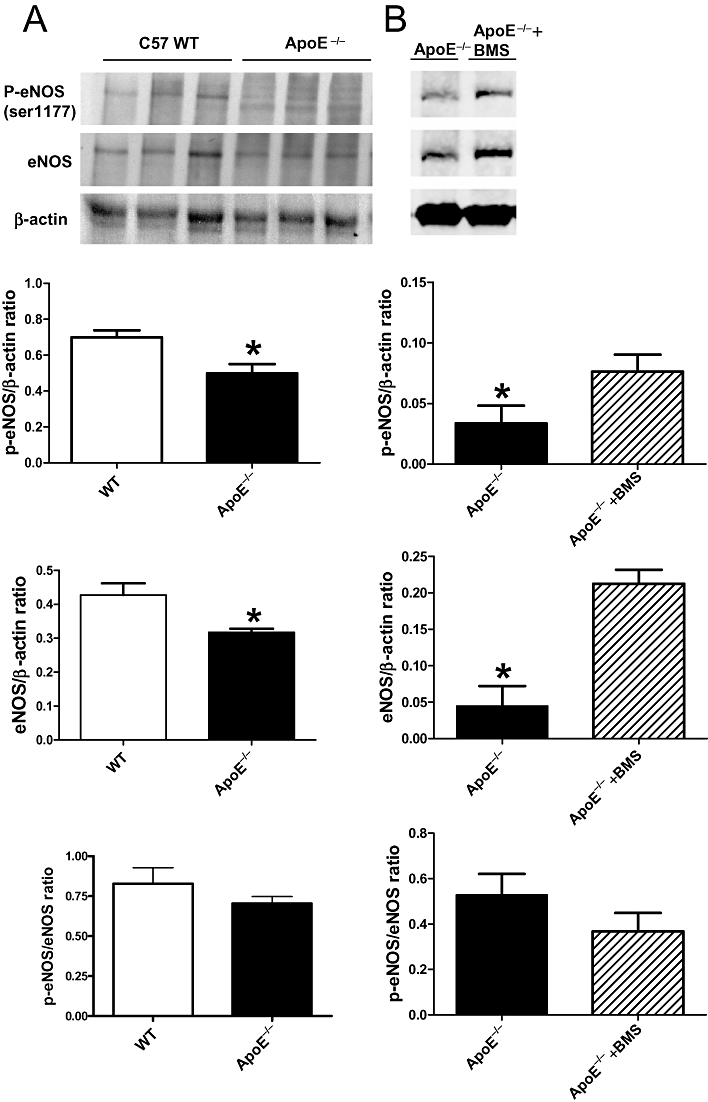
Western blot analysis of phosphorylated eNOS (Ser1177) (shown as p-eNOS) and total eNOS in (A) apolipoprotein E-deficient (ApoE−/−) and C57 wild-type (WT) mice at 18 weeks of age without drug treatment. (B) ApoE−/− mice with (ApoE−/−+BMS) or without (ApoE−/−, vehicle-treated group) chronic treatment of BMS309403 for 6 weeks (n= 3–4). The bar graphs show the data as means ± SEM *P < 0.05 compared with control. n= 3.
Chronic treatment with BMS309403 (15 mg·kg−1·day−1) for 6 weeks significantly increased the phosphorylated eNOS (Ser1177) and total eNOS but not the phosphorylated to total eNOS ratio in aortae of 18 weeks old ApoE−/−mice, compared with values in aortae from the vehicle-treated mice (Figure 8B).
HMECs
Real-time PCR analysis showed that palmitate increased the A-FABP mRNA expression in a dose-dependent manner in HMECs (Figure 9A). The protein abundance of A-FABP was elevated significantly following incubation with the fatty acid for 24 h (Figure 9B). Palmitate decreased eNOS phosphorylation under both basal and insulin-stimulated conditions (Figure 9C), but did not affect the expression of total eNOS. The change in eNOS phosphorylation was accompanied by a decreased level of cGMP (Figure 9D). The impaired eNOS phosphorylation and cGMP production by palmitate were prevented by simultaneous incubation with BMS309403 (Figure 9E, F).
Figure 9.
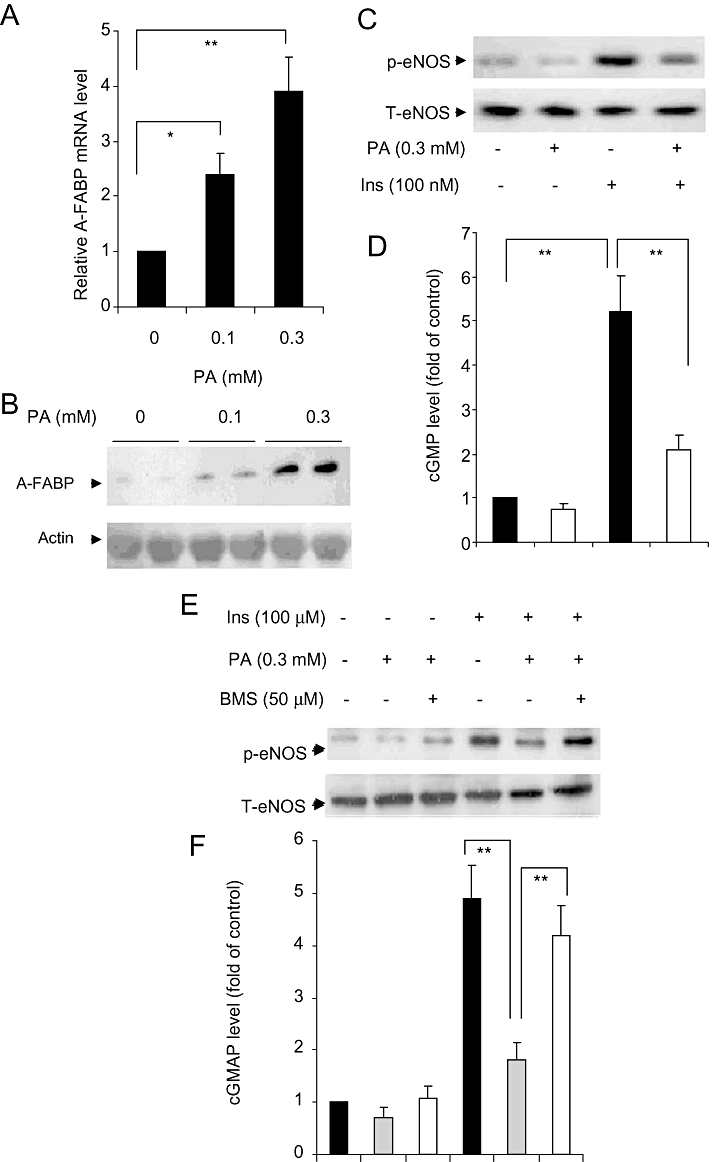
(A) The relative abundance of mRNA for the adipocyte fatty acid-binding protein (A-FABP) gene in human microvascular endothelial cells (HMECs) was quantified by real-time PCR analysis and normalized against 18S RNA. (B, C) The protein levels of A-FABP, phosphorylated (p-eNOS) and total eNOS (T-eNOS) with or without stimulation with palmitate (PA) and insulin (Ins) was analyzed by Western blotting. (D) NO production measured by cGMP assay using the same condition as in panel C (n= 5–6). (E) Protein levels of p-eNOS (ser1177) and total eNOS with or without A-FABP inhibitor treatment in the presence of palmitate (0.1 to 0.3 mM; PA) or/and insulin (100 nM; Ins) in HMECs. (F) NO production measured by cGMP assay using the same conditions as in panel E (n= 4–5). The bar graphs show the data as means ± SEM *P < 0.05; **P < 0.01.
Discussion
The present study demonstrates the role of endothelial A-FABP in endothelial dysfunction.
Endothelial dysfunction is characterized by impairment of endothelium-dependent relaxations/dilatations (Vanhoutte, 2004; 2009;). Endothelial regeneration leads to dysfunction in pigs and humans (Shimokawa et al., 1989; Vanhoutte, 2004; Lee et al., 2007; Rodríguez-Mañas et al., 2009). Endothelial dysfunction is further impaired and the atherosclerotic process is accelerated when endothelial regeneration occurs under hypercholesterolemic conditions (Shimokawa and Vanhoutte, 1988; 1989; Williams et al., 2000; Crauwels et al., 2003; Kawashima, 2004; Hans et al., 2009). In the present study, relaxations to the endothelium-dependent vasodilators acetylcholine (muscarinic agonist), UK14304 (α2-adrenoceptor agonist) and A23187 (calcium ionophore) were impaired in the aorta of ApoE−/− mice from 8 to 18 weeks of age compared with age-matched wild-type controls. Since in the present study, L-NAME, an inhibitor of eNOS, abolished the endothelium-dependent relaxations irrespective of the chronic treatment imposed, they can be attributed to the release of endothelium-derived NO (Lloréns et al., 2007). By contrast to the response to the endothelium-dependent agonists, the relaxation induced by the endothelium-independent vasodilator sodium nitroprusside was not different between the ApoE−/− mice and wild-type mice at the age of either 8 or 12 weeks. However, such endothelium-independent relaxation was enhanced at 18 weeks of age in ApoE−/− mice, making the endothelial dysfunction even more obvious. The amounts of both phosphorylated eNOS (Ser1177) and total eNOS were down-regulated in aortae of 18 weeks old ApoE−/− mice although the ratio of Ser1177 phosphorylated to total eNOS was not changed significantly. Taken in conjunction, these findings suggest that impaired endothelial NO production and endothelial dysfunction occur early in life in the ApoE−/− mice and are due to a reduced production of NO rather than to a diminished sensitivity of the vascular smooth muscle to the endothelial mediator.
A-FABP is a cytoplasmic small lipid binding protein abundantly expressed in adipocytes, which can be released into the blood stream (Yeung et al., 2009). As a lipid-binding chaperone, it facilitates the intracellular transport of fatty acids. A-FABP also reversibly binds fatty acids or cholesterol and hence modulates many lipid-signalling cascades for metabolic and inflammatory responses, which have been linked to atherosclerosis (Wellen and Hotamisligil, 2005). Its expression can be induced in macrophages by lipopolysaccharide/Toll receptor activation (Kazemi et al., 2005), oxidized LDL (Fu et al., 2000), peroxisome proliferator-activated receptor (PPAR)-γ agonists, and decreased by treatment with cholesterol-lowering statins (Llaverias et al., 2004). The modulation of inflammatory responses and the activation of the NFκB pathway by A-FABP significantly up-regulates the transcriptional activity of various inflammatory components including TNF-α and cyclooxygenase-2 (Makowski et al., 2005), which play an important role in the formation of foam cells and atherosclerotic plaques (Makowski et al., 2001). This involvement of A-FABP in inflammatory reactions by activation of the NFκB-dependent pathway (Makowski et al., 2005), would result in increased oxidative stress (Pierce et al., 2009), which in turn could lead to a reduced NO bioavailability (Rodríguez-Mañas et al., 2009), explaining the reduced endothelium-dependent relaxations observed in the present studies.
High circulating levels of A-FABP are also associated with an increased risk of developing atherosclerosis (Yeung et al., 2007). In addition, in the pig coronary artery, the genomic expression of A-FABP is induced preferentially in endothelial cells after regeneration (Lee et al., 2007). In the present study, A-FABP was detected by real-time PCR, as well as by immunohistochemical and immunofluorescent staining in the aortic endothelium of ApoE−/− mice at 12 weeks of age and onwards but not in younger ApoE−/− or in wild-type mice up to 18 weeks of age. The co-localization of endothelial A-FABP and von Willebrand factor indicate the presence of A-FABP in the endothelial layer. These observations suggest that the appearance of A-FABP parallels that of endothelial dysfunction.
To examine the potential beneficial role of inhibition of A-FABP in endothelial dysfunction, BMS309403, an orally active small-molecule inhibitor of A-FABP (Furuhashi et al., 2007), was used in both in vivo and in vitro experiments. The in vivo experiments reported here demonstrate that the inhibitor administered for 6 weeks in ApoE−/− mice (starting at the 12th week of age, a time at which A-FABP was detected first in the aortic endothelial cells of these mice) improved the endothelium-dependent relaxations induced by acetylcholine, UK14304 and A23187 without affecting responses in preparations of age-matched vehicle-treated animal. Such improvement was associated with an increase in phosphorylated (Ser1177) and total eNOS in the treated-group. By contrast, the endothelium-independent relaxation to sodium nitroprusside was not affected by the chronic treatment with BMS309403. This reveals that inhibition of A-FABP plays a beneficial effect specifically on the endothelium but not at the level of the vascular smooth muscle cells. This improvement is likely to be related, at least in part, to Gi proteins because incubation with PTX reduced the enhancement in UK14304-induced relaxation caused by the A-FABP inhibitor, a reduction that was not observed in vehicle-treated mice. Inhibition by PTXof UK14304-induced endothelium-dependent relaxations is also observed in porcine coronary arteries, illustrating the Gi protein dependency of the response to the α2-adrenoceptor agonist (Shimokawa et al., 1989; 1991;).
The plasma levels of triglycerides and LDL-C were augmented and those of HDL-C reduced, in ApoE−/− mice compared with the wild-type mice, in confirmation of earlier observations (Williams et al., 2000; Moghadasian et al., 2001). The chronic treatment with BMS309403 improved the lipid profile in terms of a reduced level of triglycerides while the plasma glucose and insulin levels remained unaffected. These observations are in line with the therapeutic effect of the orally active A-FABP inhibitor reported to reduce the formation of fatty streak lesions and macrophage-derived foam cells in ApoE−/− mouse models with severe atherosclerosis under the influence of a high fat diet (Furuhashi et al., 2007).
The experiments in cultured human endothelial cells demonstrated that A-FABP can be induced at both the mRNA and protein levels by fatty acids such as palmitate. Concomitantly, palmitate decreased eNOS phosphorylation, both under basal conditions and during activation by insulin, resulting in a decreased production of cGMP. The latter is an indicator of NO production (Fournet-Bourguignon et al., 2000) and its reduced accumulation would explain the impairment in endothelium-dependent relaxation observed in the present ex vivo studies with mouse arteries (Lee et al., 2007). This interpretation is strengthened by the observation that the selective inhibitor of A-FABP ameliorates the lipid-induced impairment in eNOS phosphorylation and cGMP production in the human endothelial cells.
In conclusion, the present findings demonstrate the age-dependent presence of A-FABP in the endothelium of ApoE−/− mice with endothelial dysfunction at 12 weeks of age and onwards. This phenomenon is likely to be related to endothelial dysfunction. Inhibition of A-FABP with BMS309403, both in vivo and in vitro, improved endothelial function in terms of eNOS phosphorylation, NO production or endothelium-dependent relaxations. These findings suggest that pharmacological inhibition of A-FABP represents a viable strategy for treating endothelial dysfunction and atherosclerosis.
Acknowledgments
This work was supported in part by grants from the Small Project Funding from the University of Hong Kong, the Research Grant Council of Hong Kong (HKU200707176115 and HKU7772/08 M), the Research Centre of Heart, Brain, Hormone & Healthy Aging of the University of Hong Kong, the Collaborative Research Fund from Research Grant Council of Hong Kong (HKU 2/07C), and the NSFC/GRC joint research scheme (Project No: N_HKU 735/08).
Glossary
Abbreviations
- A-FABP
adipocyte fatty acid-binding protein
- ApoE−/−
apolipoprotein E-knockout
- BMS309403
{2-[2′-(5-ethyl-3,4-diphenyl-1H-pyrazol-1-yl)biphenyl-3-yloxy] acetic acid}
- eNOS
endothelial nitric oxide synthase
Conflicts of interest
None.
Supplementary material
Supporting Information: Teaching Materials; Figs 1–9 as PowerPoint slide.
References
- Boord JB, Maeda K, Makowski L, Babaev VR, Fazio S, Linton MF, et al. Adipocyte fatty acid-binding protein, aP2, alters late atherosclerotic lesion formation in severe hypercholesterolemia. Arterioscler Thromb Vasc Biol. 2002;22:1686–1691. doi: 10.1161/01.atv.0000033090.81345.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crauwels HM, Van Hove CE, Holvoet P, Herman AG, Bult H. Plaque-associated endothelial dysfunction in apolipoprotein E-deficient mice on a regular diet. Effect of human apolipoprotein AI. Cardiovasc Res. 2003;59:189–199. doi: 10.1016/s0008-6363(03)00353-5. [DOI] [PubMed] [Google Scholar]
- Flavahan NA, Shimokawa H, Vanhoutte PM. Pertussis toxin inhibits endothelium-dependent relaxations to certain agonists in porcine coronary arteries. J Physiol. 1989;408:549–560. doi: 10.1113/jphysiol.1989.sp017475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fournet-Bourguignon MP, Castedo-Delrieu M, Bidouard JP, Leonce S, Saboureau D, Delescluse I, et al. Phenotypic and functional changes in regenerated porcine coronary endothelial cells: increased uptake of modified LDL and reduced production of NO. Circ Res. 2000;86:854–861. doi: 10.1161/01.res.86.8.854. [DOI] [PubMed] [Google Scholar]
- Fu Y, Luo N, Lopes-Virella MF. Oxidized LDL induces the expression of ALBP/aP2 mRNA and protein in human THP-1 macrophages. J Lipid Res. 2000;41:2017–2023. [PubMed] [Google Scholar]
- Furuhashi M, Tuncman G, Gorgun CZ, Makowski L, Atsumi G, Vaillancourt E, et al. Treatment of diabetes and atherosclerosis by inhibiting fatty-acid-binding protein aP2. Nature. 2007;447:959–965. doi: 10.1038/nature05844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuhashi M, Fucho R, Görgün CZ, Tuncman G, Cao H, Hotamisligil GS, et al. Adipocyte/macrophage fatty acid-binding proteins contribute to metabolic deterioration through actions in both macrophages and adipocytes in mice. J Clin Invest. 2008;118:2640–2650. doi: 10.1172/JCI34750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hans CP, Feng Y, Naura AS, Zerfaoui M, Rezk BM, Xia H, et al. Protective effects of PARP-1 knockout on dyslipidemia-induced autonomic and vascular dysfunction in ApoE mice: effects on eNOS and oxidative stress. Plos One. 2009;4:e7430. doi: 10.1371/journal.pone.0007430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoenig MR, Bianchi C, Rosenzweig A, Sellke FW. Decreased vascular repair and neovascularization with ageing: mechanisms and clinical relevance with an emphasis on hypoxia-inducible factor-1. Curr Mol Med. 2008;8:754–767. doi: 10.2174/156652408786733685. [DOI] [PubMed] [Google Scholar]
- Hui X, Li H, Zhou Z, Lam KS, Xiao Y, Wu D, et al. Adipocyte fatty acid binding protein mediates inflammatory responses in macrophages through a positive feedback loop involving c-Jun N-terminal kinases and activator protein-1. J Biol Chem. 2010;258:10273–10282. doi: 10.1074/jbc.M109.097907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawashima S. Malfunction of vascular control in lifestyle-related diseases: endothelial nitric oxide (NO) synthase/NO system in atherosclerosis. J Pharmacol Sci. 2004;96:411–419. doi: 10.1254/jphs.fmj04006x6. [DOI] [PubMed] [Google Scholar]
- Kazemi MR, McDonald CM, Shigenaga JK, Grunfeld C, Feingold KR. Adipocyte fatty acid-binding protein expression and lipid accumulation are increased during activation of murine macrophages by toll-like receptor agonists. Arterioscler Thromb Vasc Biol. 2005;25:1220–1224. doi: 10.1161/01.ATV.0000159163.52632.1b. [DOI] [PubMed] [Google Scholar]
- Kennedy S, Fournet-Bourguignon MP, Breugnot C, Castedo-Delrieu M, Lesage L, Reure H, et al. Cells derived from regenerated endothelium of the porcine coronary artery contain more oxidized forms of apolipoprotein-B-100 without a modification in the uptake of oxidized LDL. J Vasc Res. 2003;40:389–398. doi: 10.1159/000072817. [DOI] [PubMed] [Google Scholar]
- Krusinová E, Pelikánová T. Fatty acid binding proteins in adipose tissue: a promising link between metabolic syndrome and atherosclerosis? Diabetes Res Clin Pract. 2008;82(Suppl 2):S127–S134. doi: 10.1016/j.diabres.2008.09.023. [DOI] [PubMed] [Google Scholar]
- Lee MYK, Tse HF, Siu CW, Zhu SG, Man RYK, Vanhoutte PM. Genomic changes in regenerated porcine coronary arterial endothelial cells. Arterioscler Thromb Vasc Biol. 2007;27:2443–2449. doi: 10.1161/ATVBAHA.107.141705. [DOI] [PubMed] [Google Scholar]
- Libby P. Inflammatory mechanisms: the molecular basis of inflammation and disease. Nutr Rev. 2007;65:S140–S146. doi: 10.1111/j.1753-4887.2007.tb00352.x. [DOI] [PubMed] [Google Scholar]
- Llaverias G, Noé V, Peñuelas S, Vázquez-Carrera M, Sánchez RM, Laguna JC, et al. Atorvastatin reduces CD68, FABP4, and HBP expression in oxLDL-treated human macrophages. Biochem Biophys Res Commun. 2004;318:265–274. doi: 10.1016/j.bbrc.2004.04.021. [DOI] [PubMed] [Google Scholar]
- Lloréns S, de Mera RM, Pascual A, Prieto-Martín A, Mendizábal Y, de Cabo C, et al. The senescence-accelerated mouse (SAM-P8) as a model for the study of vascular functional alterations during aging. Biogerontology. 2007;8:663–672. doi: 10.1007/s10522-007-9108-4. [DOI] [PubMed] [Google Scholar]
- Makowski L, Hotamisligil GS. The role of fatty acid binding proteins in metabolic syndrome and atherosclerosis. Curr Opin Lipidol. 2005;16:543–548. doi: 10.1097/01.mol.0000180166.08196.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makowski L, Boord JB, Maeda K, Babaev VR, Uysal KT, Morgan MA, et al. Lack of macrophage fatty-acid-binding protein aP2 protects mice deficient in apolipoprotein E against atherosclerosis. Nature Medicine. 2001;7:699–705. doi: 10.1038/89076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makowski L, Brittingham KC, Reynolds JM, Suttles J, Hotamisligil GS. The fatty acid-binding protein, aP2, coordinates macrophage cholesterol trafficking and inflammatory activity. Macrophage expression of aP2 impacts peroxisome proliferator-activated receptor gamma and IkappaB kinase activities. J Biol Chem. 2005;280:12888–12895. doi: 10.1074/jbc.M413788200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moghadasian MH, McManus BM, Nguyen LB, Shefer S, Nadji M, Godin DV, et al. Pathophysiology of apolipoprotein E deficiency in mice: relevance to apo E-related disorders in humans. FASEB J. 2001;15:2623–2630. doi: 10.1096/fj.01-0463com. [DOI] [PubMed] [Google Scholar]
- Pierce GL, Lesniewski LA, Lawson BR, Beske SD, Seals DR. Nuclear factor-B activation contributes to vascular endothelial dysfunction via oxidative stress in overweight/obese middle-aged and older humans. Circulation. 2009;119:1284–1292. doi: 10.1161/CIRCULATIONAHA.108.804294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Mañas L, El-Assar M, Vallejo S, López-Dóriga P, Solís J, Petidier R, et al. Endothelial dysfunction in aged humans is related with oxidative stress and vascular inflammation. Aging Cell. 2009;8:226–238. doi: 10.1111/j.1474-9726.2009.00466.x. [DOI] [PubMed] [Google Scholar]
- Shimokawa H, Vanhoutte PM. Dietary cod-liver oil improves endothelium-dependent responses in hypercholesterolemic and atherosclerotic porcine coronary arteries. Circulation. 1988;78:1421–1430. doi: 10.1161/01.cir.78.6.1421. [DOI] [PubMed] [Google Scholar]
- Shimokawa H, Vanhoutte PM. Impaired endothelium-dependent relaxation to aggregating platelets and related vasoactive substances in porcine coronary arteries in hypercholesterolemia and atherosclerosis. Circ Res. 1989;64:900–914. doi: 10.1161/01.res.64.5.900. [DOI] [PubMed] [Google Scholar]
- Shimokawa H, Flavahan NA, Vanhoutte PM. Natural course of the impairment of endothelium-dependent relaxations after balloon endothelium removal in porcine coronary arteries. Possible dysfunction of a Pertussis toxin-sensitive G protein. Circ Res. 1989;65:740–753. doi: 10.1161/01.res.65.3.740. [DOI] [PubMed] [Google Scholar]
- Shimokawa H, Flavahan NA, Vanhoutte PM. Loss of endothelial Pertussis toxin-sensitive G protein function in atherosclerotic porcine coronary arteries. Circulation. 1991;83:652–660. doi: 10.1161/01.cir.83.2.652. [DOI] [PubMed] [Google Scholar]
- Taddei S, Virdis A, Ghiadoni L, Salvetti G, Bernini G, Magagna A, et al. Age-related reduction of NO availability and oxidative stress in humans. Hypertension. 2001;38:274–279. doi: 10.1161/01.hyp.38.2.274. [DOI] [PubMed] [Google Scholar]
- Tang EH, Vanhoutte PM. Prostanoids and reactive oxygen species: team players in endothelium-dependent contractions. Pharmacol Ther. 2009;122:140–149. doi: 10.1016/j.pharmthera.2009.02.006. [DOI] [PubMed] [Google Scholar]
- Tang EH, Ku DD, Tipoe GL, Feletou M, Man RY, Vanhoutte PM. Endothelium-dependent contractions occur in the aorta of wild-type and COX2-/- knockout but not COX1-/- knockout mice. J Cardiovasc Pharmacol. 2005;46:761–765. doi: 10.1097/01.fjc.0000187174.67661.67. [DOI] [PubMed] [Google Scholar]
- Vanhoutte PM, Shimokawa H. Endothelium-derived relaxing factor and coronary vasospasm. Circulation. 1989;80:1–9. doi: 10.1161/01.cir.80.1.1. [DOI] [PubMed] [Google Scholar]
- Vanhoutte PM. Endothelial function and dysfunction. Heart Metab. 2004;22:5–10. [Google Scholar]
- Vanhoutte PM. Endothelial dysfunction: the first step toward coronary arteriosclerosis. Circ J. 2009;73:595–601. doi: 10.1253/circj.cj-08-1169. [DOI] [PubMed] [Google Scholar]
- Vanhoutte PM, Tang EH. Endothelium-dependent contractions:when a good guy turns bad! J Physiol. 2008;586:5295–5304. doi: 10.1113/jphysiol.2008.161430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wamhoff BR, Lynch KR, Macdonald TL, Owens GK. Sphingosine-1-phosphate receptor subtypes differentially regulate smooth muscle cell phenotype. Arterioscler Thromb Vasc Biol. 2008;28:1454–1461. doi: 10.1161/ATVBAHA.107.159392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Huang Y, Lam KS, Li Y, Wong WT, Ye H, et al. Berberine prevents hyperglycemia-induced endothelial injury and enhances vasodilatation via adenosine monophosphate-activated protein kinase and endothelial nitric oxide synthase. Cardiovasc Res. 2009;82:484–492. doi: 10.1093/cvr/cvp078. [DOI] [PubMed] [Google Scholar]
- Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. J Clin Invest. 2005;115:1111–1119. doi: 10.1172/JCI25102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams KJ, Scalia R, Mazany KD, Rodrigueza WV, Lefer AM. Rapid restoration of normal endothelial functions in genetically hyperlipidemic mice by a synthetic mediator of reverse lipid transport. Arterioscler Thromb Vasc Biol. 2000;20:1033–1039. doi: 10.1161/01.atv.20.4.1033. [DOI] [PubMed] [Google Scholar]
- Yeung DC, Xu A, Cheung CW, Wat NM, Yau MH, Fong CH, et al. serum adipocyte fatty acid-binding protein levels were independently associated with carotid atherosclerosis. Arterioscler Thromb Vasc Biol. 2007;27:1796–1802. doi: 10.1161/ATVBAHA.107.146274. [DOI] [PubMed] [Google Scholar]
- Yeung DC, Xu A, Tso AW, Chow WS, Wat NM, Fong CH, et al. Circulating levels of adipocyte and epidermal fatty acid-binding proteins in relation to nephropathy staging and macrovascular complications in type 2 diabetic patients. Diabetes Care. 2009;32:132–134. doi: 10.2337/dc08-1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yildiz O. Vascular smooth muscle and endothelial functions in aging. Ann N Y Acad Sci. 2007;1100:353–360. doi: 10.1196/annals.1395.038. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.