

Abstract
BACKGROUND AND PURPOSE
Vorinostat and romidepsin are histone deacetylase inhibitors (HDI), approved for the treatment of cutaneous T-cell lymphoma (CTCL). However, the mechanism(s) by which these drugs exert their anti-cancer effects are not fully understood. Since CTCL is associated with immune dysregulation, we investigated whether these HDI modulated cytokine expression in CTCL cells.
EXPERIMENTAL APPROACH
CTCL cell lines and primary CTCL cells were treated in vitro with vorinostat or romidepsin, or with STAT3 pathway inhibitors. Cell cycle parameters and apoptosis were analysed by propidium iodide and annexin V/propidium iodide staining respectively. Cytokine expression was analysed using QRT-PCR and elisa assays. STAT3 expression/phosphorylation and transcriptional activity were analysed using immunoblotting and transfection/reporter assays respectively.
KEY RESULTS
Vorinostat and romidepsin strongly down-regulated expression of the immunosuppressive cytokine, interleukin (IL)-10, frequently overexpressed in CTCL, at both the RNA and protein level in CTCL cell lines and at the RNA level in primary CTCL cells. Vorinostat and romidepsin also increased expression of IFNG RNA and decreased expression of IL-2 and IL-4 RNA, although to a lesser extent compared to IL-10. Transient exposure to vorinostat was sufficient to suppress IL-10 secretion but was not sufficient to irreversibly commit cells to undergo cell death. STAT3 pathway inhibitors decreased production of IL-10 and vorinostat/romidepsin partially decreased STAT3-dependent transcription without effects on STAT3 expression or phosphorylation.
CONCLUSIONS AND IMPLICATIONS
These results demonstrate that HDI modulate cytokine expression in CTCL cells, potentially via effects on STAT3. Immunomodulation may contribute to the clinical activity of HDI in this disease.
Keywords: cutaneous T-cell lymphoma, interleukin 10, romidepsin, vorinostat, cytokine, histone deacetylase, STAT3
Introduction
Histone deacetylase (HDAC) inhibitors (HDI) are emerging as an exciting new therapeutic option for lymphoid malignancies (Marks et al., 2004; Bolden et al., 2006; Richon et al., 2009). These drugs promote the acetylation of histones and, via effects on chromatin structure and transcription factor/cofactor binding, modulate expression of ∼2–10% of cellular genes. Although histone acetylation is generally associated with transcriptional activation, HDI can either increase or decrease expression of specific genes. HDI also increase the acetylation status and modulate the activity of a wide range of non-histone proteins, and effects on both histone and non-histone proteins may contribute to their anti-cancer effects (Choudhary et al., 2009). Vorinostat [suberoylanilide hydroxamic acid (SAHA); ChemBank ID 468] is a relatively low potency hydroxamate HDI whereas romidepsin (FK228; ChemBank ID 472) is a highly potent natural product HDI that acts via a pro-drug mechanism, requiring intracellular reduction for activity (Furumai et al., 2002). There are 11 Zn-dependent HDACs which are targeted by HDI in clinical development. Vorinostat appears to be relatively non-selective, inhibiting all Zn-dependent HDACs, whereas romidepsin is relatively selective for class I enzymes (Furumai et al., 2002; Khan et al., 2008).
Vorinostat and romidepsin have been approved by the US Food and Drug Administration, for the treatment of cutaneous T-cell lymphoma (CTCL) on the basis of positive phase II trial data (Olsen et al., 2007; Piekarz et al., 2009; Richon et al., 2009). CTCL are a heterogeneous group of lymphoproliferative disorders caused by clonal, skin-invasive T cells (Kim et al., 2005; 2006; Scarisbrick, 2006). Mycosis fungoides is the commonest form of CTCL and although typically indolent may progress towards or present in more advanced forms, most notably the leukaemic variant Sezary syndrome. Survival decreases with advanced stage with only ∼30% of Sezary syndrome patients surviving more than 5 years after diagnosis (Willemze et al., 2005). Phenotypic analyses indicate that Sezary syndrome is a malignancy of central memory T cells whereas mycosis fungoides is a malignancy of skin-resident, effector memory T cells (Campbell et al., 2010).
Although effective for the treatment of CTCL, the mechanism(s) of action of HDI in this disease are not fully understood. HDI induce apoptosis in CTCL cell lines and CTCL cells may be more sensitive than normal peripheral blood lymphocytes (Piekarz et al., 2004; Zhang et al., 2005; Chen et al., 2009). However, it is perhaps unlikely that induction of apoptosis alone can fully account for their pronounced clinical activity in CTCL since similar in vitro responses are observed in cells derived from solid tumours where clinical responses are much less impressive.
The development and progression of CTCL is associated with pronounced immune dysregulation (Kim et al., 2005). Numerous immunological abnormalities are observed, including decreased T-cell responses; decreased natural killer cell activity; decreased CD8+ T-cell, plasmacytoid and myeloid dendritic cell numbers; decreased lymphokine-activated killer cell activity; eosinophilia; and increased levels of IgE/IgA. In most cases, the malignant T cells exhibit a Th2-like phenotype characterized by secretion of interleukin (IL)-4, IL-5 and IL-10. Moreover, under some conditions, CTCL cells can acquire characteristics of regulatory T cells, with expression of FOXP3 and enhanced secretion of IL-10 and transforming growth factor (TGF)β (Berger et al., 2005; Kasprzycka et al., 2008; Krejsgaard et al., 2008). These alterations in cytokine secretion are thought to contribute to the systemic defects in immune function that characterize disease progression. Importantly, immunomodulating therapies have improved long-term prognosis in many individuals, whereas chemotherapy has not resulted in improved survival (Kim et al., 2005). Successful therapy is associated with ‘normalization’ of immune responses (Yoo et al., 2001). Thus, immunosuppression is considered to play a critical role in the development and progression of CTCL.
Given the key role of immunosuppression in CTCL, we hypothesized that HDI may have immunomodulatory activity in this disease and this may contribute to their clinical effectiveness. The aim of this study was to investigate the potential immunomodulatory effects of the two licensed HDI on CTCL cells with a particular emphasis on cytokine expression, as this is subject to tight regulation via effects on chromatin (Ansel et al., 2006). Vorinostat and romidepsin modulated cytokine expression in CTCL cells and effectively decreased expression of the key immunosuppressive cytokine IL-10. Immunomodulation may contribute to the clinical effects of vorinostat and romidepsin in CTCL.
Methods
Cells
The human CTCL cell lines HUT78 and SeAx were established from peripheral blood of patients with Sezary syndrome (Kaltoft et al., 1987; Bunn and Foss, 1996) and were from the American Type Culture Collection (Manassas, VA, USA) and a kind gift of Dr K Kaltoft respectively. Cells were maintained in RPMI1640 medium (Invitrogen, Paisley, UK) supplemented with 10% (v/v) heat-inactivated fetal calf serum (PAA, Pasching, Austria), 2 mM L-glutamine, 100 µg·mL−1 penicillin and 100 µg·mL−1 streptomycin (all Invitrogen). Culture medium for SeAx cells was additionally supplemented with IL-2 (200 U·mL−1; Peprotech, Rocky Hill, NJ, USA). Primary CTCL cells were isolated from the blood of two Sezary syndrome patients (CD4 : CD8 ratio > 10) following informed consent and with ethical approval from the Leiden University Medical Center review board. Peripheral blood mononuclear cells were isolated by Ficoll density centrifugation and CD4+ T cells were purified by negative selection (CD4+ T-cell isolation kit, Miltenyi Biotec, Bergisch Gladbach, Germany). Cells were cultured in RPMI1640 medium, supplemented with 10% (v/v) human AB serum (Greiner Bio-One, Alphen aan den Rijn, the Netherlands), 2 mM L-glutamine, 100 U·mL−1 penicillin and 100 µg·mL−1 streptomycin, and 200 U·mL−1 IL-2 and 5 ng·mL−1 IL-7 (PeproTech).
Cell growth inhibition assays and analysis of cell cycle progression and apoptosis
Cell growth inhibition assays were performed using the CellTiter 96 AQueous One Solution Assay (Promega, Southampton, UK) at 48 h after addition of drug. To determine the proportion of cells in different phases of the cell cycle, drug-treated cells were collected by centrifugation and fixed in 70% (v/v) ice cold ethanol and stored at 4°C. On the day of analysis, cells were collected by centrifugation and resuspended in 300 µL phosphate buffered saline containing 100 µg·mL−1 RNAse and 8.3 µg·mL−1 propidium iodide (Sigma Chemicals, Poole, UK) for 15 min. Cell fluorescence was analysed using a FACS Canto (Becton Dickinson, Oxford, UK). The proportion of cells in G1, S, or G2/M phases of the cell cycle was calculated as a proportion of all cells in cycle, and the proportion of cells with sub-G1 content was calculated as a proportion of all cells. Apoptosis was analysed by annexin V/propidium iodide staining (Pickering et al., 2007).
Immunoblotting
For analysis of STAT3, cells were lysed on ice using modified RIPA buffer plus protease and phosphatase inhibitor cocktails (Sigma Chemicals, Poole, UK). The protein concentration of the clarified supernatant was determined using the Bradford assay (Bio-Rad, Hemel Hempstead, UK) and immunoblotting was performed using an equal amount of protein per sample. Analysis of histones was performed by lysing equal cell numbers in sodium dodecyl sulphate-polyacrylamide gel electrophoresis sample buffer followed by sonication. The following primary antibodies were used: anti-acetylated histone H4 (K12) (Millipore, Watford, UK), anti-histone H4 (Millipore), anti-HSC70 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-STAT3 (New England Biolabs, Hitchin, UK), anti-phospho-Ser727 STAT3 (New England Biolabs), anti-phospho-Tyr705 STAT3 (New England Biolabs). Secondary antibodies (horseradish peroxidase conjugated anti-rabbit IgG and anti-mouse IgG) were from GE Healthcare (Chalfont St Giles, UK). Immunoblot signals were quantified using Quantity One image analysis software (Bio-Rad, Hemel Hempstead, UK).
Quantitative reverse transcription polymerase chain reaction (QRT-PCR)
Total RNA was isolated using the TRIzol reagent (Invitrogen) and converted to cDNA. QRT-PCR was performed using an ABI Prism 7900 Sequence Detection System (Applied Biosystems, Warrington, UK) with the following cycling variables: 94°C for 10 min, followed by 40 cycles of 94°C for 15 s and 60°C for 1 min. CTvalues were standardized to β-actin (for HUT78 cells) or glyceraldehyde-3-phosphate dehydrogenase (for SeAx and primary cells) to calculate ΔCT values. ΔΔCTvalues were calculated by subtracting the ΔCT values of control (DMSO-treated) cells for each time point. Fold change in expression was calculated using the formula 2T−(ΔΔC). All probes were from Applied Biosystems; Hs00174143-m1 (IFNγ); Hs99999043-m1 (TNF); Hs00174114-m1 (IL-2); Hs00929862-m1 (IL-4); Hs99999031-m1 (IL-5); Hs99999035-m1 (IL-10); Hs99999038-m1 (IL-13); Hs01106578-m1 (IL-12Rβ1); Hs00155486-m1 (IL-12Rβ2); human ACTB probe (β-actin); Hs99999905_m1 (GAPDH).
Enzyme-linked immunosorbent assays (elisa)
IL-10 secretion in cell culture supernatants was determined using elisa assays (R&D Systems) according to the manufacturer's instructions. To allow statistical comparison of the effects of HDI between different experiments, the IL-10 secretion from DMSO-treated cells was set to 100 for each experiment.
Transfections
HUT78 cells were transfected with the STAT3-luc reporter plasmid (Clontech, Oxford, UK) using electroporation (8 × 106 cells in a total volume of 0.25 mL serum free medium; 900 µF, 250 V in a 0.4 cm cuvette). Cells were co-transfected with the Renilla luciferase reporter plasmid pRLTK (Promega) as an internal control. Firefly and Renilla luciferase activity was determined using the Dual Luciferase reporter assay (Promega) according the manufacturer's instructions.
Data analysis
Data are shown as means ± SD. Means were compared using the repeated measures anova test and within-subject contrasts and Fisher LSD post hoc test (spss; SPSS (UK) Limited, Woking, UK).
Materials
Vorinostat was from Alexis Biochemicals (Nottingham, UK) and romidepsin was synthesized in-house (Yurek-George et al., 2007; Wen et al., 2008). Cucurbitacin I was from Tocris Bioscience (Bristol, UK). WP1066 and Stattic were from Calbiochem (Nottingham, UK).
Results
Histone acetylation, cell cycle arrest and induction of apoptosis
We first analysed the effects of vorinostat and romidepsin on the in vitro growth of Sezary syndrome-derived HUT78 cells, a well-validated cell line widely used for studies of CTCL. Both HDI inhibited HUT78 cell growth although, consistent with previous studies (Piekarz et al., 2004; Zhang et al., 2005), romidepsin was significantly more potent. IC50 values for growth inhibition by romidepsin and vorinostat were 1.22 ± 0.24 nM and 675 ± 214 nM respectively (mean ± SD derived from a minimum of three independent experiments).
We characterized the effects of vorinostat and romidepsin on histone H4K12 acetylation in HUT78 cells (Figure 1). For these experiments we used romidepsin at 10 and 25 nM (i.e. ∼10- and ∼25-fold the IC50 for growth inhibition). Vorinostat was used at up to 5 µM (∼10-fold the IC50 value for growth inhibition). We were unable to test higher concentrations of vorinostat due to poor aqueous solubility. Vorinostat induced rapid effects and histone acetylation reached a maximum level within 3 h. By contrast, induction of histone acetylation was relatively delayed in romidepsin-treated cells, especially at the lower concentration tested. The delayed induction of histone acetylation in romidepsin-treated cells may be due to the requirement for intracellular reduction for formation of the active compound (Furumai et al., 2002).
Figure 1.
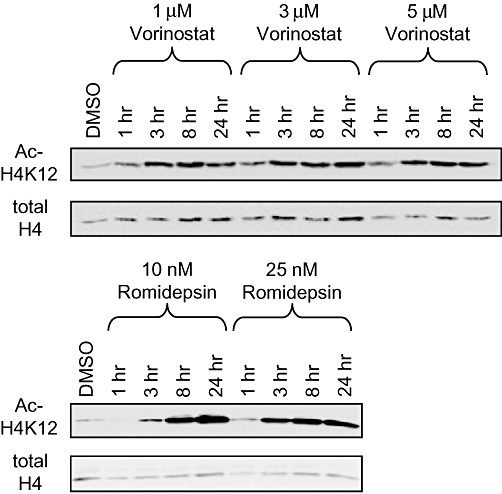
Histone deacetylase inhibitor-induced histone acetylation. HUT78 cells were treated with the indicated concentrations of vorinostat or romidepsin and the levels of acetylated histone H4 (K12) and total histone H4 were analysed by immunoblotting. Results shown are representative of at least three experiments.
Propidium iodide-staining experiments demonstrated that, overall, HDI had relatively modest effects on cell cycle parameters in HUT78 cells (Figure 2). Statistically significant increases in the proportion of cells in G0/G1 and decreases in the proportion of cells in G2/M were observed in cells treated with vorinostat at 1.0 or 2.5 µM. There was perhaps a trend towards accumulation of cells in G0/G1 and a decrease in cells in S phase in romidepsin-treated cells, but overall these changes were very modest and mostly not statistically significant. Changes in cell cycle profiles at higher concentrations appeared to be masked due to induction of cell death since HDI caused a significant increase in the proportion of cells with <G1 DNA content. To confirm that HDI-induced cell death was due to apoptosis, we performed annexin V staining (Figure 3). Vorinostat and romidepsin induced significant levels of apoptosis across a wide range of concentrations.
Figure 2.
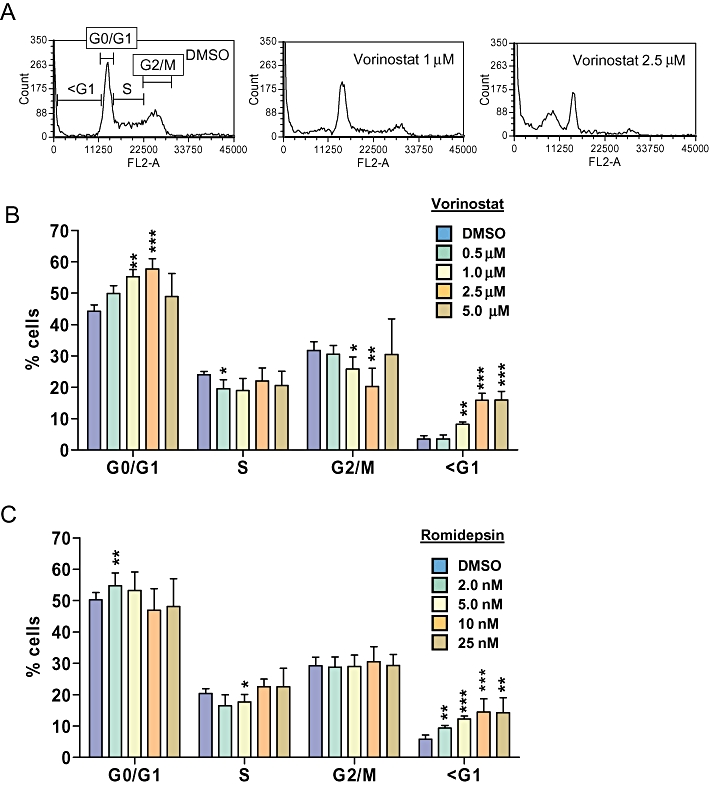
Effect of histone deacetylase inhibitors on cell cycle parameters in HUT78 cells. A. Representative cell cycle profiles showing HUT78 cells treated with DMSO or vorinostat (1 µM or 2.5 µM). B,C. HUT78 cells were treated with the indicated concentrations of vorinostat (B) or romidepsin (C) for 24 h. Cells were treated with DMSO as control. The graphs show the proportion of cells in different phases of the cell cycle determined by flow cytometric analysis of propidium iodide-stained cells. Graphs show mean (±SD) values derived from at least three independent experiments, each performed in duplicate. Statistically significant differences compared to DMSO-treated cells are shown (*P≤ 0.05, **P≤ 0.01, ***P≤ 0.001).
Figure 3.
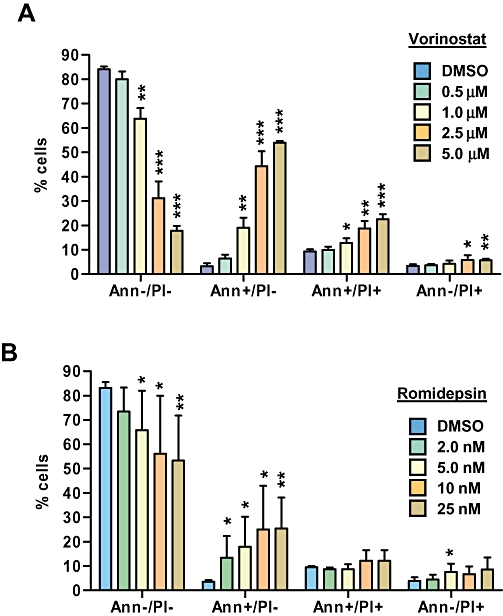
Effect of histone deacetylase inhibitors on apoptosis in HUT78 cells. HUT78 cells were treated with the indicated concentrations of vorinostat (A) or romidepsin (B). After 24 h, induction of apoptosis was analysed by flow cytometric analysis of propidium iodide (PI) and annexin (Ann) V-stained cells. Graphs show mean (±SD) values derived from at least three independent experiments, each performed in duplicate. Statistically significant differences compared to DMSO-treated cells are shown (*P≤ 0.05, **P≤ 0.01, ***P≤ 0.001).
Effect of HDI on cytokine, IL-12RB1 and IL-12RB2 gene expression in CTCL cells
We analysed the effects of vorinostat and romidepsin on cytokine expression in HUT78 cells. Cells were treated with vorinostat for up to 8 h whereas cells were treated with romidepsin for up to 24 h since the onset of action of romidepsin is relatively delayed compared to vorinostat (Figure 1). The expression of IFNG and TNF (Th1 cytokines), IL-4, IL-5, IL-10, IL-13 (Th2/regulatory cytokines) and IL-2 (a T-cell growth-stimulating cytokine) were analysed by QRT-PCR. Both HDI induced statistically significant increases in the expression of IFNG and decreases in the expression of IL-2, IL-4 and IL-10 (Figure 4). The effects of romidepsin were delayed compared to vorinostat. In contrast to vorinostat, romidepsin induced the expression of IL-5, particularly at 24 h. IL-13 was down-regulated by vorinostat at 8 h, but was not consistently regulated following vorinostat treatment. Overall, there were clear effects of HDI on cytokine expression in HUT78 cells. IL-10 was the most dramatically regulated cytokine and its expression was maximally repressed by vorinostat and romidepsin by 95% and 99% respectively.
Figure 4.
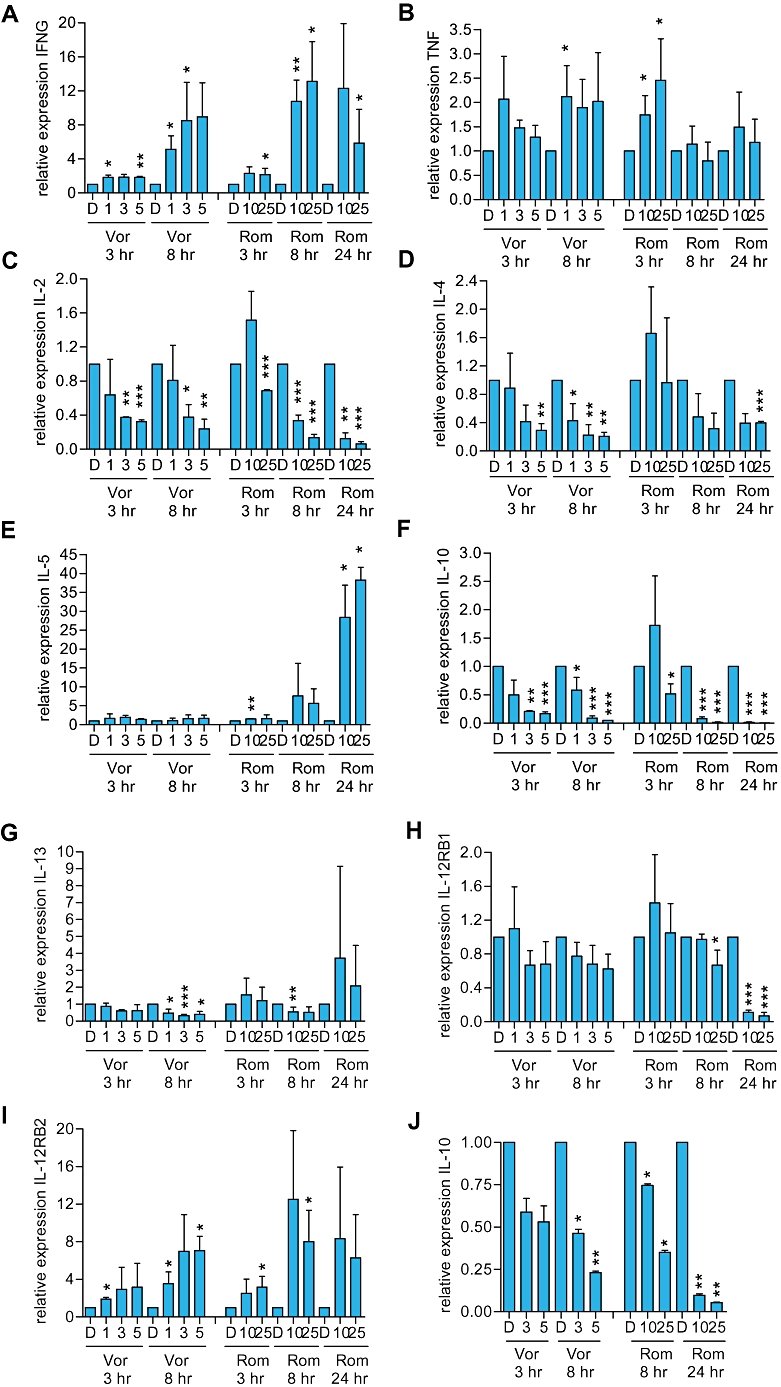
Effect of histone deacetylase inhibitors on cytokine and IL-12RB1/B2 RNA expression in cutaneous T-cell lymphoma cells. A–I. HUT78 or (J) SeAx cells were treated with the indicated concentrations of vorinostat (Vor; µM), romidepsin (Rom; nM) or DMSO (D) as a control. After the indicated times (A) IFNG (B) TNF (C) IL-2 (D) IL-4 (E) IL-5 (F, J) IL-10 (G) IL-13 (H) IL-12RB1 and (I) IL-12RB2 RNA expression was analysed by QRT-PCR. Data are the means (±SD) derived from two to five separate experiments. Statistically significant differences compared to DMSO-treated cells are shown (*P≤ 0.05, **P≤ 0.01, ***P≤ 0.001).
We also investigated the effects of HDI on expression of genes encoding IL-12RB1 and IL-12RB2, the low and high affinity subunits of the IL-12RB respectively. IL-12RB1 is expressed on both Th1 and Th2 cells, whereas IL-12RB2 is expressed more strongly on Th1 cells (Rogge et al., 1997; Szabo et al., 1997; Wu et al., 1997). IL-12RB2 expression was induced by both vorinostat and romidepsin (Figure 4). IL-12RB1 expression was not altered in vorinostat-treated cells but was consistently decreased in romidepsin-treated cells at 24 h.
We focused our subsequent mechanistic studies on IL-10 which was particularly strongly down-regulated. IL-10 is frequently expressed in CTCL and is considered to play a key immunosuppressive role in various malignancies (Mosser and Zhang, 2008). We first confirmed modulation of IL-10 RNA using SeAx cells which, like HUT78 cells, constitutively express IL-10 (Kasprzycka et al., 2008). Vorinostat and romidepsin both significantly decreased IL-10 RNA expression in SeAx cells, although the kinetics were somewhat slower than HUT78 cells (Figure 4J). Both drugs also down-regulated IL-10 RNA expression in two samples of primary CTCL cells, isolated from the blood of patients with Sezary syndrome (Figure 5A and B).
Figure 5.
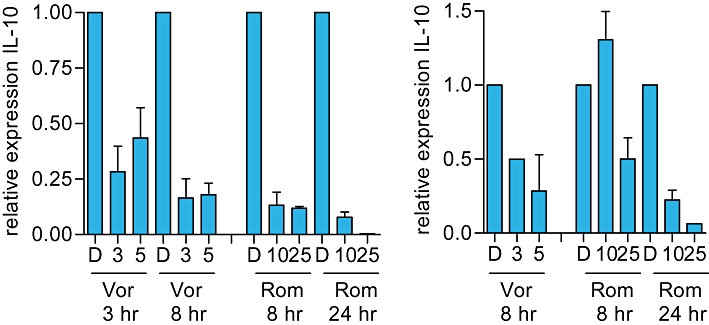
Effect of histone deacetylase inhibitors on IL-10 RNA expression in primary cutaneous T-cell lymphoma (CTCL) cells. A,B. Primary CTCL cells derived from two patients were treated with the indicated concentrations of vorinostat (Vor; µM), romidepsin (Rom; nM) or DMSO (D) as a control. After the indicated times, IL-10 RNA expression was analysed by QRT-PCR. Data are means (±SD) of duplicate determinations.
Effect of HDI on IL-10 secretion
We determined whether HDI inhibited the secretion of IL-10 from CTCL cells using elisa assays. Control (DMSO-treated) HUT78 and SeAx cells produced readily detectable levels of IL-10 in culture supernatants (34.5 ± 14.1 pg/h/1 × 106 cells and 42.7 ± 2.5 pg/h/1 × 106 cells respectively). Vorinostat and romidepsin significantly reduced IL-10 secretion from HUT78 cells (Figure 6A), and romidepsin significantly reduced IL-10 secretion from SeAx cells (Figure 6B). Since the effects of HDI on cytokine expression were rapid whereas effects on cell death occurred over a more protracted time course, we performed washout experiments to investigate in more detail the relationship between cytokine modulation and cell death. We selected vorinostat for these studies since, in contrast to romidepsin, histone acetylation is rapidly reversed following removal of vorinostat from cells (Crabb et al., 2008). When HUT78 cells were exposed continuously to vorinostat for 24 h, there was a significant increase in cell death (Figure 6C). However, when cells were transiently exposed to vorinostat for 6 h and then analysed 18 h after the drug had been removed, there was no increase in cell death. By contrast, vorinostat reduced IL-10 secretion from HUT78 cells when measured at 24 h, even when the drug was removed after 6 h (Figure 6D). The effect was not as dramatic compared to cells that were continuously exposed to drug for 24 h, but the reduction in IL-10 was still statistically significant compared to control cells. Therefore, 6 h exposure to vorinostat was sufficient to modulate cytokine gene expression but was not sufficient to irreversibly commit cells to undergo apoptosis.
Figure 6.
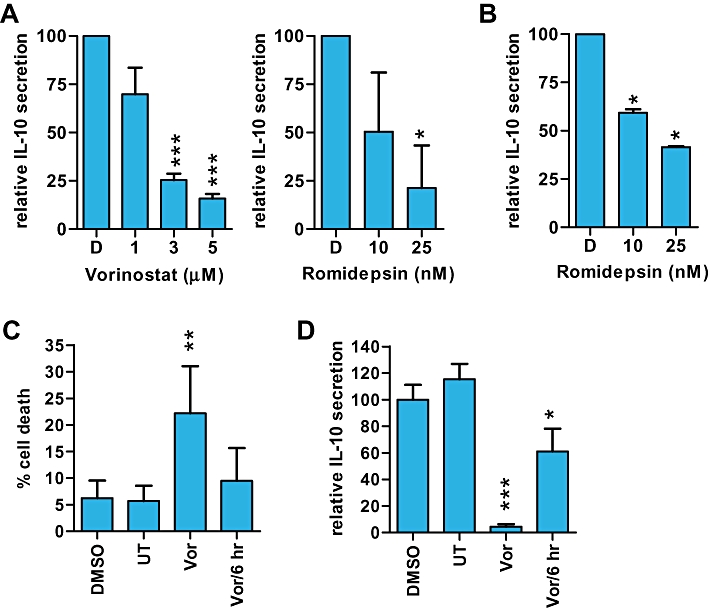
Effect of histone deacetylase inhibitors (HDI) on IL-10 secretion in cutaneous T-cell lymphoma cells. A. HUT78 cells were pretreated with vorinostat or romidepsin, washed to remove drug and resuspended in fresh HDI-supplemented media for a further 3.5 (vorinostat) or 3 h (romidepsin). IL-10 secretion was analysed by elisa. Data are means (±SD) of at least three separate experiments each performed in duplicate. B. IL-10 secretion from SeAx cells treated with the indicated concentrations of romidepsin or DMSO (D) for 24 h (elisa). Data are mean (±SD) of duplicate determinations. C,D. Washout experiments. HUT78 cells were treated with DMSO, vorinostat (Vor; 3 µM) or left untreated (UT) for 24 h. Cells were treated with vorinostat for 6 h (Vor/6 h), washed thoroughly and analysed at 24 h. C. Sub-G1 DNA content. D. IL-10 elisa. Data are means (±SD) derived from four experiments. Statistically significant differences compared to DMSO-treated cells are shown (*P≤ 0.05, **P≤ 0.01, ***P≤ 0.001).
Effect of HDI on STAT3 activity
The STAT3 transcription factor is commonly activated by tyrosine phosphorylation in CTCL and has been linked to growth and cytokine expression of the malignant cells (Mitchell and John, 2005). In particular, previous studies have implicated STAT3 as a key regulator of IL-10 in CTCL cells (Kasprzycka et al., 2008; Krejsgaard et al., 2008) suggesting that HDI might down-regulate IL-10 expression by interfering with STAT3 activity. We therefore first examined whether expression of IL-10 in HUT78 cells was also dependent on STAT3 activity. HUT78 cells were treated with three distinct STAT3 pathway inhibitors, Stattic (Schust et al., 2006), cucurbitacin I (Blaskovich et al., 2003) or WP1066 (Hussain et al., 2007).
Stattic inhibits STAT3 activation by preventing kinases from binding the STAT3 SH2 domain whereas cucurbitacin I and WP1066 have been shown to act as JAK inhibitors. Analysis of STAT3 expression confirmed that these compounds all interfered with STAT3 activity (Figure 7A and Table 1). Whereas WP1066 had no effect on total STAT3 expression, STAT3 expression was clearly decreased in stattic-treated cells. Although initially reported to act as a JAK inhibitor, cucurbitacin I caused a large decrease in the mobility of STAT3 suggesting accumulation of post-translation modifications, although this was variable between experiments. A similar effect of cucurbitacin I has been reported in SeAx cells (van Kester et al., 2008). Relative to HSC70, all inhibitors decreased tyrosine phosphorylation of STAT3, and stattic and cucurbitacin I (but not WP1066) also decreased serine phosphorylation. However, only WP1066 caused a decrease in tyrosine phosphorylation relative to total STAT3. None of the inhibitors decreased the amount of serine phosphorylation STAT3 when compared to total STAT3. Therefore, these STAT3 pathway inhibitors act via distinct mechanisms. WP1066 appears to act primarily as a JAK inhibitor, selectively down-regulating STAT3 tyrosine phosphorylation, whereas the decrease in the levels of tyrosine- and serine-phosphorylated STAT3 in stattic or cucurbitacin I-treated cells is associated with reduced total STAT3 expression and/or accumulation of an altered form. Despite acting via distinct mechanisms, all three STAT3 pathway inhibitors decreased expression of IL-10 RNA (data not shown) and secretion of IL-10 from HUT78 cells, although this failed to reach significance for cucurbitacin I (Figure 7B). Time course experiments using WP1066 demonstrated that IL-10 secretion was reduced by approximately 50% within 5 h (data not shown).
Figure 7.
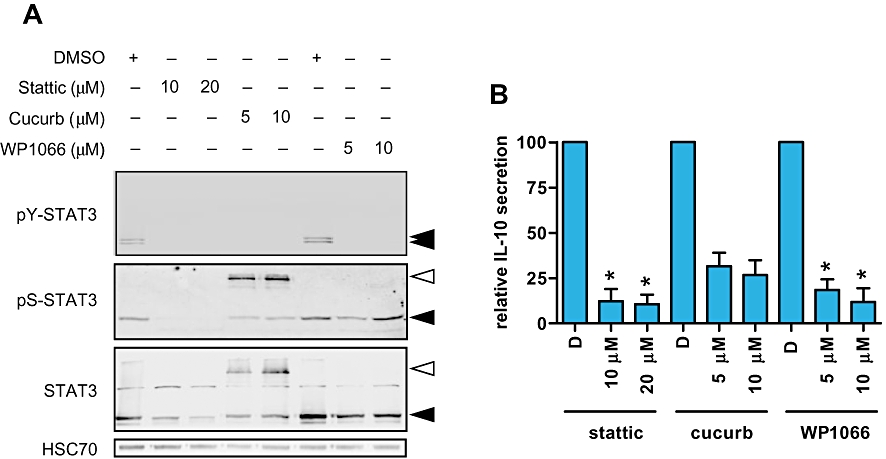
STAT3-dependent expression of IL-10 expression in HUT78 cells. HUT78 cells were treated with the indicated concentrations of the STAT3 pathway inhibitors [stattic, cucurbitacin I (cucurb) or WP1066] for 8 h. DMSO was used as a solvent control. In (A), expression of total, serine- and tyrosine-phosphorylated STAT3 was analysed by immunoblotting. HSC70 was analysed as a loading control. Results are representative of two independent experiments. Open arrowheads indicate a slower migrating isoform detected in cucurbitacin I-treated cells. In (B), supernatants were collected (D, DMSO) and levels of IL-10 analysed by elisa. Data are means (±SD) from two separate experiments each performed in duplicate. Statistically significant differences compared to DMSO-treated cells are shown (*P≤ 0.05).
Table 1.
Quantitation of STAT3 immunoblotting data
| Stattic | Cucurbitacin I | WP1066 | ||||
|---|---|---|---|---|---|---|
| 10 µM | 20 µM | 5 µM | 10 µM | 5 µM | 10 µM | |
| Total STAT3 versus HSC70 | 0.28 ± 0.13 | 0.13 ± 0.09 | 0.55 ± 0.67 | 0.53 ± 0.71 | 1.01 ± 0.39 | 1.56 ± 1.09 |
| PY-STAT3 versus HSC70 | 0.26 ± 0.33 | 0.07 ± 0.10 | 0.15 ± 0.22 | 0.10 ± 0.13 | 0.07 ± 0.10 | 0.04 ± 0.03 |
| PS-STAT3 versus HSC70 | 0.22 ± 0.06 | 0.11 ± 0.04 | 0.25 ± 0.12 | 0.31 ± 0.04 | 0.65 ± 0.04 | 1.29 ± 0.32 |
| PY-STAT3 versus total STAT3 | 1.49 ± 2.01 | 1.19 ± 1.69 | 2.11 ± 2.98 | 3.70 ± 5.23 | 0.06 ± 0.08 | 0.03 ± 0.00 |
| PS-STAT3 versus total STAT3 | 0.99 ± 0.77 | 1.35 ± 1.28 | 1.31 ± 1.39 | 6.69 ± 9.08 | 0.70 ± 0.31 | 1.19 ± 1.04 |
Data are means derived from two independent experiments (±SD) and are normalized to DMSO-treated cells (set to 1.0). Expression values are shown relative to the loading control (HSC70), or total STAT3 for tyrosine- or serine-phosphorylated STAT3. For cucurbitacin I, only the intensity of the ‘canonical’ STAT3 isoform was determined.
Since STAT3 was required for optimal expression of IL-10, it was possible that HDI downmodulated STAT3 activity. We investigated the effects of HDI on STAT3 levels and phosphorylation at 3 (vorinostat) or 8 h (romidepsin) following drug treatment, times at which IL-10 RNA levels are effectively repressed (Figure 4). HDI did not significantly alter STAT3 expression or reduce tyrosine or serine phosphorylation (Figure 8A). However, when HUT78 cells were transfected with a STAT3-specific reporter construct to monitor transcriptional activity, vorinostat and romidepsin rapidly reduced STAT3 activity (Figure 8B). In these experiments, HDI did not alter the expression of the co-transfected control Renilla luciferase plasmid demonstrating that the effects of HDI on STAT3-dependent transcription were selective.
Figure 8.
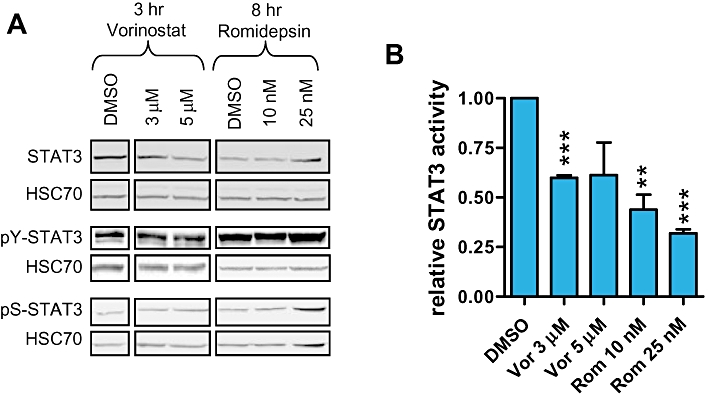
Histone deacetylase inhibitors (HDI) decrease STAT3 activity. A. HUT78 cells were treated with the indicated concentrations of vorinostat or romidepsin. After 3 (vorinostat) or 8 h (romidepsin), cells were collected and expression of total, tyrosine- and serine-phosphorylated STAT3 was analysed by immunoblotting. Data are from the same blots but with intervening lanes removed. HSC70, loading control. Results are representative of three separate experiments. B. HUT78 cells were transfected with STAT3-dependent firefly luciferase and control Renilla luciferase plasmids. After 24 h, cells were treated with the indicated concentrations of HDI for 3 (vorinostat) or 8 h (romidepsin) and luciferase assays were performed. The graph shows firefly luciferase activity normalized to Renilla luciferase activity and set to 1.0 for DMSO-treated cells. Data are means (±SD) derived from three separate experiments. Statistically significant differences compared to DMSO-treated cells are shown (**P≤ 0.01, ***P≤ 0.001).
Discussion
Immune dysregulation is a key feature of CTCL (Kim et al., 2005) and we therefore hypothesized that the clinical effectiveness of HDI in this disease may involve immunomodulation. Extensive data demonstrate that HDI can act as immunomodulators in non-malignant settings, for example via modulation of cytokine expression, dendritic cell maturation and T-cell function. However, there are relatively few studies of immunomodulation by HDI in cancers, and none in CTCL. Our results have confirmed previous studies demonstrating that clinically achievable concentrations of HDI exert a predominantly pro-apoptotic effect in CTCL cells, with little effect on the cell cycle (Piekarz et al., 2004; Zhang et al., 2005; Fantin et al., 2008; Chen et al., 2009). However, in addition to their ability to promote apoptosis, we show that HDI exert strong in vitro immunomodulatory activity in CTCL cells.
Overall, there was an increase in the expression of the pro-inflammatory cytokine interferon (IFN)γ and a decrease in the expression of the immunosuppressive cytokine IL-10. There was also a consistent decrease in the expression of IL-2, a T-cell growth-promoting cytokine. Importantly, at least some CTCL are dependent on IL-2 suggesting that HDI may be able to effectively interfere with this growth-promoting autocrine loop. Interestingly, of the ‘classic’ Th2 cytokines IL-4, IL-5 and IL-13 which are co-ordinately regulated via epigenetic mechanisms from a single genetic locus (Ansel et al., 2006), only IL-4 was consistently down-regulated in HDI-treated cells. Thus, HDI appears to trigger selective ‘reprogramming’ of cytokine expression, generally favouring Th1 cytokines at the expense of Th2 (IL-4)/regulatory (IL-10) cytokines, but not a general Th2 to Th1 ‘repolarization’. The effects of HDI were not restricted to cytokine gene expression and we detected induction of RNA encoding IL-12RB2, required for development of Th1 cells. We also detected decreased expression of the key Th2 transcription factor GATA3 in vorinostat and romidepsin-treated cells, but only at relatively late time points (data not shown). Overall, these results are consistent with the hypothesis that HDI do exert immunomodulatory activity in CTCL cells. A very recently published gene expression microarray study also reported modulation of cytokine RNA in HDI-treated CTCL cell lines, including IL-10, although modulation of RNA or protein expression was not confirmed by other techniques (Wozniak et al., 2010).
Our mechanistic studies focused on IL-10, a critical immunosuppressive cytokine expressed by various cells of the immune system, non-immune cells and tumour cells (Mosser and Zhang, 2008). Both IL-10 (Mosser and Zhang, 2008) and STAT3 (Yu et al., 2009) have been strongly linked to immunosuppression. IL-10 is highly expressed in skin lesions and peripheral blood mononuclear cells from patients with CTCL, especially in more advanced disease (Asadullah et al., 1996; Buhl and Sogaard, 1997; Shohat et al., 2001; Luftl et al., 2002; Krejsgaard et al., 2008). Ex vivo studies of CTCL samples treated have shown that IL-10 produced by CTCL cells has immunosuppressive activity since neutralizing antibodies enhanced differentiation of dendritic cells and expression of IL-12RB2 (Zaki et al., 2001; Berger et al., 2002). IL-10 is a key immunosuppressive factor produced by regulatory T cells and treatment of CTCL cells with specific cytokines induces regulatory T-cell activity and enhances secretion of IL-10 (Berger et al., 2005; Kasprzycka et al., 2008; Krejsgaard et al., 2008). Thus, IL-10 is likely to play a key role in immune dysregulation in CTCL and its inhibition may contribute to normalization of immune activity. Since activation of the IL-10 receptor leads to stimulation of STAT3 activity which in turn activates IL-10 expression, a positive feedback loop may operate to maintain high STAT3 activity and IL-10 expression. Importantly, transient exposure of cells to vorinostat for a time sufficient for cytokine modulation was not sufficient to irreversibly commit cells to undergo cell death. It is possible that, under clinical conditions, cytokine modulation may occur more readily following HDI administration than apoptosis induction since apoptosis induction requires drug levels to be maintained for an extended time.
Although HDAC11 has previously been implicated in repression of IL-10 (Villagra et al., 2009), this does not appear to play a major role in CTCL since both vorinostat and romidepsin reduce, rather than increase expression of IL-10. Instead, the mechanism of IL-10 modulation by HDI in CTCL cells appears to, at least partially, involve inhibition of STAT3 activity. We (this study) and others (Kasprzycka et al., 2008; Krejsgaard et al., 2008) have demonstrated that STAT3 plays a key role in mediating IL-10 expression in CTCL cells and we have now shown that vorinostat and romidepsin decrease STAT3-dependent transcription. Vorinostat and romidepsin did not alter STAT3 phosphorylation or decrease total STAT3 expression; similar results have been reported previously for vorinostat and panobinostat (LBH589) (Fantin et al., 2008; Chen et al., 2009). Since IL-10 signalling activates STAT3, it was possible that STAT3 inhibition might occur downstream of IL-10 downmodulation by HDI. However, the rapidity of the response and the fact that short-term incubation with HDI does not alter STAT3 tyrosine phosphorylation, indicate a more direct effect of HDI on STAT3. Interestingly, the HDI trichostatin A has also been shown to down-regulate IL-10 expression in T cells derived from patients with systemic lupus erythematosus (Mishra et al., 2001), also associated with STAT3 activation (Harada et al., 2007). By contrast, in normal murine CD4+CD25+ cells, trichostatin A induces expression of both IL-10 and FOXP3. In HUT78 cells, we demonstrated that vorinostat or romidepsin also increased FOXP3 RNA levels, but were unable to detect any changes in the expression of FOXP3 protein (J.E. Adams and G. Packham, unpubl. data).
STAT3 is an acetylated protein; however, previous studies have shown that direct acetylation of STAT3 enhances transcriptional activity (Ray et al., 2002; Yuan et al., 2005). STAT3 forms complexes with class I HDACs, including HDAC1 and HDAC3 (Ray et al., 2008; Togi et al., 2009), and we speculate that recruitment of this complex to the IL-10 promoter enhances transcription. Indeed, the transactivating functions of STAT1, 2 and 5 require HDAC activity (Mitchell and John, 2005). Consistent with a key role for nuclear HDACs, both the pan-HDI SAHA and the class I selective FK228 down-regulated IL-10 expression. Although it may seem counterintuitive that HDACs may play a role in transcriptional activation, active transcription may be associated with cyclical changes in chromatin modifications, including histone acetylation (Metivier et al., 2003). Alternatively, HDI may somehow interfere with the nuclear localization of phosphorylated STAT3, as previously demonstrated in vivo in a phase 2 trial of vorinostat, especially in clinically responsive patients (Duvic et al., 2007). However, it is important to note that the downmodulation of STAT3 transcriptional activity is not as dramatic as the reduction of endogenous IL-10 gene expression. This may reflect limitations of transient transfections to study chromatin-mediated effects. Alternatively, it is possible that HDI co-ordinately inhibit the activity of multiple transcription factors required for optimal IL-10 expression (Saraiva and O'Garra, 2010). Candidates would include NF-κB and Sp1 which are also modulated by acetylation.
The majority of CTCL appear to have activated STAT3 in vivo, although the extent of activation varies from case to case. Importantly, relatively high levels of nuclear, phosphorylated STAT3 predict poor responses to vorinostat (Fantin et al., 2008). This is consistent with the functional links between HDI and STAT3 that we have described, and we speculate that malignant cells with higher levels of STAT3 activity may require greater exposure to HDI to effectively inhibit this activity. Both cytokine-dependent and independent STAT3 activation has been described in CTCL (Mitchell and John, 2005). By blocking STAT3 activity downstream of its activation by JAK signalling, it is likely that HDI can counter STAT3 function independent of its mechanism of activation.
Interestingly, vorinostat has also been demonstrated to decrease secretion of IL-5 and increase secretion of the Th1 chemokine IP10 in cell lines derived from Hodgkin lymphoma (Buglio et al., 2008), a tumour that is characterized by a strong Th2 cell infiltrate and also appears to show clinical responses to HDI response (Stimson et al., 2009). Together, these and other findings suggest that it may be possible to exploit HDI as immunomodulatory rather than cytostatic/cytotoxic agents. This would have important implications for the selection of tumour types and individual patients for HDI therapy, as well as for monitoring responses. The development of HDI for cancer therapy has paid relatively little attention to potential immunomodulatory activity and it will be important to understand the role of specific HDACs in immune cells since this may lead to the development of HDI optimized for immune modulation. It will be important to extend these studies to further primary material representing various stages of this heterogeneous disease and to confirm our findings by performing detailed studies on cytokine production in individuals receiving HDI. It will also be important to investigate the consequences of HDI in terms of immune function, given that the effects of HDI on cytokine production are likely to be complex and are not restricted to IL-10. HDI are also likely to exert effects on non-malignant immune cells which may act to either counter or reinforce direct immunoregulatory effects of HDI on CTCL cells. However, our findings are consistent with the idea that HDI may exert clinical anti-cancer effects via immunomodulatory activity.
Acknowledgments
We thank Coby Out for excellent technical assistance and Dr K Kaltoft for the kind gift of SeAx cells. We are very grateful for the helpful discussion with Dr Sean Whittaker and Dr Tracey Mitchell. This work was supported by Leukaemia and Lymphoma Research, Cancer Research UK and the Netherlands Organisation for Scientific Research (NWO).
Glossary
Abbreviations
- CTCL
cutaneous T-cell lymphoma
- DMSO
dimethyl sulphoxide
- elisa
enzyme-linked immunosorbent assays
- HDAC
histone deacetylase
- HDI
HDAC inhibitor
- IFN
interferon
- IL
interleukin
- QRT-PCR
quantitative reverse transcription polymerase chain reaction
- SAHA
suberoylanilide hydroxamic acid
- TGF
transforming growth factor
Conflict of interest
The authors state no conflict of interest.
Supplementary material
Supporting Information: Teaching Materials; Fig 1 as PowerPoint slide.
References
- Ansel KM, Djuretic I, Tanasa B, Rao A. Regulation of Th2 differentiation and IL-4 locus accessibility. Annu Rev Immunol. 2006;24:607–656. doi: 10.1146/annurev.immunol.23.021704.115821. [DOI] [PubMed] [Google Scholar]
- Asadullah K, Docke WD, Haeussler A, Sterry W, Volk HD. Progression of mycosis fungoides is associated with increasing cutaneous expression of interleukin-10 mRNA. J Invest Dermatol. 1996;107:833–837. doi: 10.1111/1523-1747.ep12330869. [DOI] [PubMed] [Google Scholar]
- Berger CL, Hanlon D, Kanada D, Dhodapkar M, Lombillo V, Wang N, et al. The growth of cutaneous T-cell lymphoma is stimulated by immature dendritic cells. Blood. 2002;99:2929–2939. [PubMed] [Google Scholar]
- Berger CL, Tigelaar R, Cohen J, Mariwalla K, Trinh J, Wang N, et al. Cutaneous T-cell lymphoma: malignant proliferation of T-regulatory cells. Blood. 2005;105:1640–1647. doi: 10.1182/blood-2004-06-2181. [DOI] [PubMed] [Google Scholar]
- Blaskovich MA, Sun J, Cantor A, Turkson J, Jove R, Sebti SM. Discovery of JSI-124 (cucurbitacin I), a selective Janus kinase/signal transducer and activator of transcription 3 signaling pathway inhibitor with potent antitumor activity against human and murine cancer cells in mice. Cancer Res. 2003;63:1270–1279. [PubMed] [Google Scholar]
- Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5:769–784. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- Buglio D, Georgiakis GV, Hanabuchi S, Arima K, Khaskhely NM, Liu YJ, et al. Vorinostat inhibits STAT6-mediated TH2 cytokine and TARC production and induces cell death in Hodgkin lymphoma cell lines. Blood. 2008;112:1424–1433. doi: 10.1182/blood-2008-01-133769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buhl L, Sogaard H. Immunohistochemical expression of IL-10 in mycosis fungoides. Exp Dermatol. 1997;6:195–198. doi: 10.1111/j.1600-0625.1997.tb00205.x. [DOI] [PubMed] [Google Scholar]
- Bunn PA, Jr, Foss FM. T-cell lymphoma cell lines (HUT102 and HUT78) established at the National Cancer Institute: history and importance to understanding the biology, clinical features, and therapy of cutaneous T-cell lymphomas (CTCL) and adult T-cell leukemia-lymphomas (ATLL) J Cell Biochem Suppl. 1996;24:12–23. doi: 10.1002/jcb.240630503. [DOI] [PubMed] [Google Scholar]
- Campbell JJ, Clark RA, Watanabe R, Kupper TS. Sezary syndrome and mycosis fungoides arise from distinct T-cell subsets: a biologic rationale for their distinct clinical behaviors. Blood. 2010;116:767–771. doi: 10.1182/blood-2009-11-251926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Fiskus W, Eaton K, Fernandez P, Wang Y, Rao R, et al. Cotreatment with BCL-2 antagonist sensitizes cutaneous T-cell lymphoma to lethal action of HDAC7-Nur77-based mechanism. Blood. 2009;113:4038–4048. doi: 10.1182/blood-2008-08-176024. [DOI] [PubMed] [Google Scholar]
- Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- Crabb SJ, Howell M, Rogers H, Ishfaq M, Yurek-George A, Carey K, et al. Characterisation of the in vitro activity of the depsipeptide histone deacetylase inhibitor spiruchostatin A. Biochem Pharmacol. 2008;76:463–475. doi: 10.1016/j.bcp.2008.06.004. [DOI] [PubMed] [Google Scholar]
- Duvic M, Talpur R, Ni X, Zhang C, Hazarika P, Kelly C, et al. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL) Blood. 2007;109:31–39. doi: 10.1182/blood-2006-06-025999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fantin VR, Loboda A, Paweletz CP, Hendrickson RC, Pierce JW, Roth JA, et al. Constitutive activation of signal transducers and activators of transcription predicts vorinostat resistance in cutaneous T-cell lymphoma. Cancer Res. 2008;68:3785–3794. doi: 10.1158/0008-5472.CAN-07-6091. [DOI] [PubMed] [Google Scholar]
- Furumai R, Matsuyama A, Kobashi N, Lee KH, Nishiyama M, Nakajima H, et al. FK228 (depsipeptide) as a natural prodrug that inhibits class I histone deacetylases. Cancer Res. 2002;62:4916–4921. [PubMed] [Google Scholar]
- Harada T, Kyttaris V, Li Y, Juang YT, Wang Y, Tsokos GC. Increased expression of STAT3 in SLE T cells contributes to enhanced chemokine-mediated cell migration. Autoimmunity. 2007;40:1–8. doi: 10.1080/08916930601095148. [DOI] [PubMed] [Google Scholar]
- Hussain SF, Kong LY, Jordan J, Conrad C, Madden T, Fokt I, et al. A novel small molecule inhibitor of signal transducers and activators of transcription 3 reverses immune tolerance in malignant glioma patients. Cancer Res. 2007;67:9630–9636. doi: 10.1158/0008-5472.CAN-07-1243. [DOI] [PubMed] [Google Scholar]
- Kaltoft K, Bisballe S, Rasmussen HF, Thestrup-Pedersen K, Thomsen K, Sterry W. A continuous T-cell line from a patient with Sezary syndrome. Arch Dermatol Res. 1987;279:293–298. doi: 10.1007/BF00431220. [DOI] [PubMed] [Google Scholar]
- Kasprzycka M, Zhang Q, Witkiewicz A, Marzec M, Potoczek M, Liu X, et al. Gamma c-signaling cytokines induce a regulatory T cell phenotype in malignant CD4+ T lymphocytes. J Immunol. 2008;181:2506–2512. doi: 10.4049/jimmunol.181.4.2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Kester MS, Out-Luiting JJ, von dem Borne PA, Willemze R, Tensen CP, Vermeer MH. Cucurbitacin I inhibits Stat3 and induces apoptosis in Sezary cells. J Invest Dermatol. 2008;128:1691–1695. doi: 10.1038/sj.jid.5701246. [DOI] [PubMed] [Google Scholar]
- Khan N, Jeffers M, Kumar S, Hackett C, Boldog F, Khramtsov N, et al. Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors. Biochem J. 2008;409:581–589. doi: 10.1042/BJ20070779. [DOI] [PubMed] [Google Scholar]
- Kim EJ, Hess S, Richardson SK, Newton S, Showe LC, Benoit BM, et al. Immunopathogenesis and therapy of cutaneous T cell lymphoma. J Clin Invest. 2005;115:798–812. doi: 10.1172/JCI24826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EJ, Lin J, Junkins-Hopkins JM, Vittorio CC, Rook AH. Mycosis fungoides and sezary syndrome: an update. Curr Oncol Rep. 2006;8:376–386. doi: 10.1007/s11912-006-0061-1. [DOI] [PubMed] [Google Scholar]
- Krejsgaard T, Gjerdrum LM, Ralfkiaer E, Lauenborg B, Eriksen KW, Mathiesen AM, et al. Malignant Tregs express low molecular splice forms of FOXP3 in Sezary syndrome. Leukemia. 2008;22:2230–2239. doi: 10.1038/leu.2008.224. [DOI] [PubMed] [Google Scholar]
- Luftl M, Feng A, Licha E, Schuler G. Dendritic cells and apoptosis in mycosis fungoides. Br J Dermatol. 2002;147:1171–1179. doi: 10.1046/j.1365-2133.2002.04994.x. [DOI] [PubMed] [Google Scholar]
- Marks PA, Richon VM, Miller T, Kelly WK. Histone deacetylase inhibitors. Adv Cancer Res. 2004;91:137–168. doi: 10.1016/S0065-230X(04)91004-4. [DOI] [PubMed] [Google Scholar]
- Metivier R, Penot G, Hubner MR, Reid G, Brand H, Kos M, et al. Estrogen receptor-alpha directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell. 2003;115:751–763. doi: 10.1016/s0092-8674(03)00934-6. [DOI] [PubMed] [Google Scholar]
- Mishra N, Brown DR, Olorenshaw IM, Kammer GM. Trichostatin A reverses skewed expression of CD154, interleukin-10, and interferon-gamma gene and protein expression in lupus T cells. Proc Natl Acad Sci USA. 2001;98:2628–2633. doi: 10.1073/pnas.051507098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell TJ, John S. Signal transducer and activator of transcription (STAT) signalling and T-cell lymphomas. Immunology. 2005;114:301–312. doi: 10.1111/j.1365-2567.2005.02091.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosser DM, Zhang X. Interleukin-10: new perspectives on an old cytokine. Immunol Rev. 2008;226:205–218. doi: 10.1111/j.1600-065X.2008.00706.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen EA, Kim YH, Kuzel TM, Pacheco TR, Foss FM, Parker S, et al. Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J Clin Oncol. 2007;25:3109–3115. doi: 10.1200/JCO.2006.10.2434. [DOI] [PubMed] [Google Scholar]
- Pickering BM, de Mel S, Lee M, Howell M, Habens F, Dallman CL, et al. Pharmacological inhibitors of NF-kappaB accelerate apoptosis in chronic lymphocytic leukaemia cells. Oncogene. 2007;26:1166–1177. doi: 10.1038/sj.onc.1209897. [DOI] [PubMed] [Google Scholar]
- Piekarz RL, Robey RW, Zhan Z, Kayastha G, Sayah A, Abdeldaim AH, et al. T-cell lymphoma as a model for the use of histone deacetylase inhibitors in cancer therapy: impact of depsipeptide on molecular markers, therapeutic targets, and mechanisms of resistance. Blood. 2004;103:4636–4643. doi: 10.1182/blood-2003-09-3068. [DOI] [PubMed] [Google Scholar]
- Piekarz RL, Frye R, Turner M, Wright JJ, Allen SL, Kirschbaum MH, et al. Phase II multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T-cell lymphoma. J Clin Oncol. 2009;27:5410–5417. doi: 10.1200/JCO.2008.21.6150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray S, Sherman CT, Lu M, Brasier AR. Angiotensinogen gene expression is dependent on signal transducer and activator of transcription 3-mediated p300/cAMP response element binding protein-binding protein coactivator recruitment and histone acetyltransferase activity. Mol Endocrinol. 2002;16:824–836. doi: 10.1210/mend.16.4.0811. [DOI] [PubMed] [Google Scholar]
- Ray S, Lee C, Hou T, Boldogh I, Brasier AR. Requirement of histone deacetylase1 (HDAC1) in signal transducer and activator of transcription 3 (STAT3) nucleocytoplasmic distribution. Nucleic Acids Res. 2008;36:4510–4520. doi: 10.1093/nar/gkn419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richon VM, Garcia-Vargas J, Hardwick JS. Development of vorinostat: current applications and future perspectives for cancer therapy. Cancer Lett. 2009;280:201–210. doi: 10.1016/j.canlet.2009.01.002. [DOI] [PubMed] [Google Scholar]
- Rogge L, Barberis-Maino L, Biffi M, Passini N, Presky DH, Gubler U, et al. Selective expression of an interleukin-12 receptor component by human T helper 1 cells. J Exp Med. 1997;185:825–831. doi: 10.1084/jem.185.5.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saraiva M, O'Garra A. The regulation of IL-10 production by immune cells. Nat Rev Immunol. 2010;10:170–181. doi: 10.1038/nri2711. [DOI] [PubMed] [Google Scholar]
- Scarisbrick JJ. Staging and management of cutaneous T-cell lymphoma. Clin Exp Dermatol. 2006;31:181–186. doi: 10.1111/j.1365-2230.2005.02019.x. [DOI] [PubMed] [Google Scholar]
- Schust J, Sperl B, Hollis A, Mayer TU, Berg T. Stattic: a small-molecule inhibitor of STAT3 activation and dimerization. Chem Biol. 2006;13:1235–1242. doi: 10.1016/j.chembiol.2006.09.018. [DOI] [PubMed] [Google Scholar]
- Shohat M, Hodak E, Sredni B, Shohat B, Sredni D, David M. Cytokine profile of patients with mycosis fungoides and the immunomodulatory effect of AS101. Acta Derm Venereol. 2001;81:255–257. doi: 10.1080/00015550152572877. [DOI] [PubMed] [Google Scholar]
- Stimson L, Wood V, Khan O, Fotheringham S, La Thangue NB. HDAC inhibitor-based therapies and haematological malignancy. Ann Oncol. 2009;20:1293–1302. doi: 10.1093/annonc/mdn792. [DOI] [PubMed] [Google Scholar]
- Szabo SJ, Dighe AS, Gubler U, Murphy KM. Regulation of the interleukin (IL)-12R beta 2 subunit expression in developing T helper 1 (Th1) and Th2 cells. J Exp Med. 1997;185:817–824. doi: 10.1084/jem.185.5.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Togi S, Kamitani S, Kawakami S, Ikeda O, Muromoto R, Nanbo A, et al. HDAC3 influences phosphorylation of STAT3 at serine 727 by interacting with PP2A. Biochem Biophys Res Commun. 2009;379:616–620. doi: 10.1016/j.bbrc.2008.12.132. [DOI] [PubMed] [Google Scholar]
- Villagra A, Cheng F, Wang HW, Suarez I, Glozak M, Maurin M, et al. The histone deacetylase HDAC11 regulates the expression of interleukin 10 and immune tolerance. Nat Immunol. 2009;10:92–100. doi: 10.1038/ni.1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen S, Packham G, Ganesan A. Macrolactamization versus macrolactonization: total synthesis of FK228, the depsipeptide histone deacetylase inhibitor. J Org Chem. 2008;73:9353–9361. doi: 10.1021/jo801866z. [DOI] [PubMed] [Google Scholar]
- Willemze R, Jaffe ES, Burg G, Cerroni L, Berti E, Swerdlow SH, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768–3785. doi: 10.1182/blood-2004-09-3502. [DOI] [PubMed] [Google Scholar]
- Wozniak MB, Villuendas R, Bischoff JR, Blanco-Aparicio C, Martinez Leal JF, de La Cueva P, et al. Vorinostat interferes with the signaling transduction pathway of T cell receptor and synergizes with PI3K inhibitors in cutaneous T-cell lymphoma. Haematologica. 2010;95:613–621. doi: 10.3324/haematol.2009.013870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, Warrier RR, Wang X, Presky DH, Gately MK. Regulation of interleukin-12 receptor beta1 chain expression and interleukin-12 binding by human peripheral blood mononuclear cells. Eur J Immunol. 1997;27:147–154. doi: 10.1002/eji.1830270122. [DOI] [PubMed] [Google Scholar]
- Yoo EK, Cassin M, Lessin SR, Rook AH. Complete molecular remission during biologic response modifier therapy for Sezary syndrome is associated with enhanced helper T type 1 cytokine production and natural killer cell activity. J Am Acad Dermatol. 2001;45:208–216. doi: 10.1067/mjd.2001.116345. [DOI] [PubMed] [Google Scholar]
- Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9:798–809. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan ZL, Guan YJ, Chatterjee D, Chin YE. Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science. 2005;307:269–273. doi: 10.1126/science.1105166. [DOI] [PubMed] [Google Scholar]
- Yurek-George A, Cecil AR, Mo AH, Wen S, Rogers H, Habens F, et al. The first biologically active synthetic analogues of FK228, the depsipeptide histone deacetylase inhibitor. J Med Chem. 2007;50:5720–5726. doi: 10.1021/jm0703800. [DOI] [PubMed] [Google Scholar]
- Zaki MH, Shane RB, Geng Y, Showe LC, Everetts SE, Presky DH, et al. Dysregulation of lymphocyte interleukin-12 receptor expression in Sezary syndrome. J Invest Dermatol. 2001;117:119–127. doi: 10.1046/j.0022-202x.2001.01354.x. [DOI] [PubMed] [Google Scholar]
- Zhang C, Richon V, Ni X, Talpur R, Duvic M. Selective induction of apoptosis by histone deacetylase inhibitor SAHA in cutaneous T-cell lymphoma cells: relevance to mechanism of therapeutic action. J Invest Dermatol. 2005;125:1045–1052. doi: 10.1111/j.0022-202X.2005.23925.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.