

Abstract
BACKGROUND AND PURPOSE
The uterotonins oxytocin and histamine, mediate contractile signals through specific G protein-coupled receptors, a process which is tightly controlled during gestation to prevent preterm labour. We previously identified G protein-coupled receptor kinase (GRK)2 and GRK6 as respective cardinal negative regulators of histamine H1 and oxytocin receptor signalling. GRK-mediated phosphorylation promotes arrestin recruitment, not only desensitizing receptors but activating an increasing number of diverse signalling pathways. Here we investigate potential roles that arrestins play in the regulation of myometrial oxytocin/histamine H1 receptor signalling.
EXPERIMENTAL APPROACH
Endogenous arrestins2 and 3 were specifically depleted using RNA-interference in a human myometrial cell line and the consequences of this for G protein-coupled receptor-mediated signalling were assessed using Ca2+/inositol 1,4,5-trisphophate imaging and standard mitogen-activated protein kinase (MAPK) assays.
KEY RESULTS
Depletion of arrestin3, but not arrestin2 enhanced and prolonged H1 receptor-stimulated Ca2+ responses, whilst depletion of either arrestin increased oxytocin receptor responses. Arrestin3 depletion decreased H1 receptor desensitization, whilst removal of either arrestin isoform was equally effective in preventing oxytocin receptor desensitization. Following arrestin3 depletion oxytocin-induced phospho-extracellular signal-regulated kinase1/2 signals were diminished and histamine-stimulated signals virtually absent, whereas depletion of arrestin2 augmented extracellular signal-regulated kinase1/2 responses to each agonist. Conversely, depletion of arrestin3 enhanced p38 signals to each agonist, whilst arrestin2 suppression increased oxytocin-, but not histamine-induced p38 MAPK responses.
CONCLUSIONS AND IMPLICATIONS
Arrestin proteins are key regulators of H1 and oxytocin receptor desensitization, and play integral roles mediating uterotonin-stimulated MAPK-signalling. These data provide insights into the in situ regulation of these receptor subtypes and may inform pathophysiological functioning in preterm labour.
Keywords: oxytocin receptor, histamine H1 receptor, arrestin, desensitization, MAPK, myometrium, contractile signalling
Introduction
Myometrial tone depends upon the dynamic balance between contractile [e.g. voltage-dependent and -independent Ca2+ channel activity, phospholipase C (PLC)-coupled receptor activity (Sanborn, 2007) ], and relaxatory inputs [e.g. plasmalemmal and sacroplasmic reticulum Ca2+ ATPases, and Na+/ Ca2+ exchangers, cyclic AMP concentrations and K+ channel activity (Sanborn, 2000; 2007; Yuan and Lopez Bernal, 2007)]. One important excitatory input is provided by the wide variety of PLC-coupled receptors (e.g. oxytocin, prostaglandin F2α, histamine H1) expressed on human myometrial cells, the activation of which stimulates the production of inositol 1,4,5-trisphosphate and diacylglycerol to initiate and prolong smooth muscle contraction through activation of Ca2+ mobilization and influx pathways respectively (Ku et al., 1995; Holda et al., 1996; Sanborn, 2001). During pregnancy the uterus is subjected to extensive remodelling and stretch due to the developing fetus. Despite these physical changes, it is essential that the uterine environment is maintained in a quiescent state throughout gestation to prevent preterm labour. At present, the exact mechanisms by which this process is achieved are not fully understood, but short-term [receptor desensitization and/or down-regulation (Engelhardt et al., 1997; Phaneuf et al., 1997; Willets et al., 2008; 2009;) ] as well as longer-term hormonal regulation of receptor expression levels are likely to be involved (Fuchs et al., 1984; Matsumoto et al., 1997; Helmer et al., 1998; Brodt-Eppley and Myatt, 1999).
Most G protein-coupled receptors are tightly regulated through the action of G protein-coupled receptor kinases (GRK) to prevent prolonged or inappropriate signalling (Willets et al., 2003). GRK proteins phosphorylate agonist-occupied G protein-coupled receptors (GPCRs) at key serine or threonine residues within the third intracellular loop or C-terminal tail, a process that usually leads to the recruitment of non-visual arrestin proteins (Willets et al., 2003; DeWire et al., 2007). Arrestin binding physically prevents GPCR/G protein interactions, whilst also promoting GPCR desensitization and internalization. Interestingly, accumulating evidence highlights that arrestin proteins have many more diverse roles than mediation of receptor desensitization and internalization (Luttrell and Lefkowitz, 2002), including acting as agonist-regulated adaptor scaffolds for extracellular signal-regulated kinases (ERK) and other mitogen-activated protein kinases (MAPK) (Luttrell and Lefkowitz, 2002; DeWire et al., 2007; Gesty-Palmer and Luttrell, 2008). We recently identified regulatory roles for GRK2 in H1 (Willets et al., 2008) and GRK6 in oxytocin receptor (Willets et al., 2009) signalling, respectively, in both ULTR (an immortalized human myometrial cell line) and primary myometrial cells. Because GRK-mediated GPCR phosphorylation promotes arrestin association, we have now gone on to investigate the potential roles of arrestin proteins in H1 and oxytocin receptor desensitization, as well as the ability of arrestins to affect receptor coupling to MAPK pathways.
Although little is presently known about the potential roles of arrestins in H1 receptor signalling, studies utilizing model cell systems recombinantly overexpressing either GPCR and/or arrestin constructs, highlight a potential role for arrestins in the regulation of oxytocin receptor signalling (Oakley et al., 2001; Smith et al., 2006). However, it should be noted that such overexpression studies are often not reflective of how GPCRs are regulated within their native environment (Tobin et al., 2008). Indeed, accumulating evidence indicates that the regulation of GPCRs is likely to be highly dependent on the cell background (Kong et al., 1994; Simon et al., 2003; Smith et al., 2006; Willets et al., 2009). Therefore, to assess the involvement of arrestins in different aspects of H1 and oxytocin receptor signalling/regulation, we have utilized small interfering (si)RNAs to specifically ablate endogenous arrestin2 and arrestin3 expression in the ULTR human myometrial cell line.
Methods
All drug and molecular target nomenclature conforms to the British Journal of Pharmacology's Guide to Receptors and Channels (Alexander et al., 2009).
Cell culture
An immortalized human myometrial cell line (ULTR) (Perez-Reyes et al., 1992) previously extensively characterized by us (Brighton et al., 2009; Willets et al., 2008; 2009;) and others (Olson et al., 2003; Ball et al., 2006), were maintained in Dulbecco's minimal essential medium, supplemented with 10% fetal calf serum, penicillin (100 IU·mL−1), streptomycin (100 µg·mL−1), and amphotericin B (2.5 µg·mL−1) and Glutamax-1. Cells were maintained under humidified conditions at 37°C, in air/5% CO2.
siRNA-targeted arrestin depletion
To deplete endogenous arrestin expression, cells were transfected with specific siRNAs designed to target either human arrestin2 (ARRB1) (5′-GGAGAUCUAUUACCAUGGtt-3′) or arrestin3 (ARRB2) siRNA (5′-CGAACAAGAUGACCAGGUAtt-3′). ULTR cells were seeded 24 h before transfection at a density of 150 000 cells per well of a six-well culture plate, and transfected with various concentrations (10 or 100 nM) of either negative control (non-targeting), anti-arrestin2, anti-arrestin3 or a combination of anti-arrestin2 and anti-arrestin3 siRNAs. After 48 h, cells were lysed and subjected to sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE) separation and transferred to nitrocellulose as described previously (Willets et al., 2001). Arrestin expression was detected using the A1CT polyclonal antibody (Ahn et al., 2004b) raised against arrestin2, which cross-reacts with arrestin3 allowing visualization of both proteins. The relative expression of individual arrestin proteins was determined using the GeneGnome image analysis system and software (Syngene, Cambridge, UK).
Assessment of oxytocin- and histamine-stimulated Ca2+ signalling
To assess oxytocin- and histamine-mediated intracellular calcium concentration ([Ca2+]i) changes, ULTR cells were seeded into 96-well plates and grown to 90% confluency. Cells were washed with Krebs buffer [composition (mM); HEPES 10, NaHCO3 1.3, D-Glucose 11.7, MgSO4 1.2, KH2PO4 1.2, KCl 4.7, NaCl 118 and CaCl2 1.3, pH 7.4] and loaded with 3 µM fluo4-AM (room temperature, 1 h). Cell monolayers were washed again with Krebs buffer prior to challenge with increasing concentrations of either oxytocin or histamine for various time periods. Agonist-stimulated fluorescence intensity changes were measured using a NovoStar imaging system (BMG Labtech, Aylesbury, UK), and plotted graphically as the maximum increase in fluorescence minus basal fluorescence. To assess whether arrestin depletion affected oxytocin- or histamine-stimulated Ca2+ signalling, ULTR cells were seeded 24 h before transfection at a density of 10 000 cells per well and transfected with either negative control (100 nM), anti-human arrestin2 (100 nM) or anti-human arrestin3 (10 nM) siRNAs. Transfection was achieved using Interferin™ transfection reagent (Polyplus, New York, NY, USA), as per manufacturers' instructions.
Determination of oxytocin- and histamine-stimulated phosphoinositide turnover and receptor desensitization in single cells
Oxytocin- and H1 receptor-stimulated PLC activity was assessed in ‘real time’ using the e-GFP-tagged pleckstrin homology domain of phospholipase Cδ (eGFP-PH) as a biosensor of receptor-mediated PLC activity (Willets et al., 2005; 2008; 2009;). Cells were seeded onto glass coverslips and transfected with eGFP-PH (0.5 µg) using Lipofectamine2000 (Invitrogen, Paisley, UK) as per manufacturers' instructions. After 48 h, agonist-stimulated fluorescent eGFP-PH membrane-cytoplasmic translocations were assessed using an Olympus FV500 scanning laser confocal microscope as described previously (Willets et al., 2005; 2008;). Cells were maintained at 37°C using a temperature controller and micro-incubator (PDMI-2 and TC202A; Burleigh, Digitimer, Cambridge, UK), and perfused with Krebs–Henseleit buffer (composition: NaCl 134 mM, KCl 6 mM, MgCl2 1 mM, glucose 10 mM, HEPES 10 mM and CaCl2 1.3 mM, pH 7.4) at 5 mL·min−1. Images were captured using an oil immersion 60× objective, with inositol 1,4,5-trisphosphate levels determined by increases in cytosolic fluorescence in a defined area of interest exactly as described previously (Willets et al., 2005; 2008;). Oxytocin and H1 receptor desensitization was determined using our previously validated protocols. Briefly, to assess oxytocin receptor desensitization, cells were stimulated with a maximal oxytocin concentration (100 nM) for 30 s (termed R1), followed by a 5 min wash period before a second 30 s oxytocin challenge (100 nM, termed R2). R2 responses were significantly attenuated compared with R1 and the resulting reduction in the R2/R1 ratio is interpreted as an indication of oxytocin receptor desensitization (Willets et al., 2009). Due to the presence of a significant receptor reserve, a slightly different protocol was applied to observe H1 receptor desensitization (Willets et al., 2008). Here, cells were challenged with an approximate EC50 histamine concentration (10 µM) for 30 s, 5 min before (R1) and 5 min after (R2) a desensitizing histamine pulse (100 µM, for 1 min). Again reduced R2/R1 ratios were interpreted as an indication of receptor desensitization (Willets et al., 2008).
Detection of MAPK activation
Agonist-driven ERK1/2 (MAPK3/MAPK1) activity was detected using Western blotting techniques as described previously (Brighton et al., 2009). Agonist-stimulated p38 MAPK (MAPK11-14) phosphorylation was also detected through a similar Western blotting approach. Briefly, ULTR cells were seeded into six-well plates and grown to confluency. Cells were then deprived of serum for 24 h before agonist addition. Next, signalling was terminated by the addition of lysis buffer [composition: 20 mM Tris-HCl (pH 7.4), 1% (v·v−1) Triton X-100, 2 mM EDTA, 25 mM β-glycerophosphate, 1 mM sodium orthovanidate, 500 µM phenylmethanesulphonylfluoride, 0.1 mg·mL−1 leupeptin, 0.2 mg·mL−1 benzamidine, and 0.1 mg·mL−1 pepstatin]. Insoluble material was cleared by centrifugation and an equal volume of 2× sample buffer [composition: 250 mM Tris-HCl, pH 6.8, 0.01% (w·v−1) bromophenol blue, 2% (w·v−1) sodium dodecyl sulphate, 40% (v·v−1) glycerol and 50 mM ditiothreitol] and added before heating (5 min, 100°C) and gel loading. Samples were separated by SDS-PAGE and transferred to nitrocellulose using Western blotting techniques. Detection of threonine- and tyrosine-phosphorylated extracellular signal-regulated kinase (pERK)1/2 was via a specific anti-pERK1/2 antibody (Promega, Southampton, UK), which detects the dual phosphorylated P-loop pTEpY motif of ERK1 and ERK2; phospho-p38 MAPK (pTGpY) was detected using a specific anti-phospho-p38 antibody (Cell Signaling, Madison, WI, USA). Immune-reactive bands were visualized using HRP-conjugated anti-rabbit secondary antibody (Sigma, Poole, UK), ECL reagent and Hyperfilm (GE Healthcare, Little Chalfont, UK). Densitometric analysis of the resultant autoradiographs was undertaken using the GeneGnome image analysis system and software (Syngene, Cambridge, UK). For data relating to ERK1/2, the densities of p42 and p44 ERK proteins (ERK 1 and 2) were averaged and related to basal (unstimulated) levels. To ensure that all samples contained the same levels of protein, total ERK and p38 levels were determined by running additional gels in parallel with the detection of pERK and phospho-p38. For ERK1/2 samples, uniform protein loading was confirmed by detection of total-ERK1/2 proteins using an anti-total ERK1/2 antibody (Santa Cruz, CA, USA) and for p38 samples, an antibody against total p38 (Cell Signaling, Madison, WI, USA).
Data analysis
All concentration–response curves were generated and EC50 values determined using non-linear regression analysis software Prism, version 5.0 (GraphPad Software Inc., San Diego, CA, USA). Data were analysed using one-way or two-way anova, followed by appropriate post hoc testing (Excel 5.0, Microsoft, Redmond, WA, USA). Significance was accepted when P < 0.05.
Results
Oxytocin- and histamine-mediated elevation of [Ca2+]i
Stimulation of fluo-4-loaded cells with either oxytocin or histamine resulted in concentration-dependent increases in fluorescence indicating elevation of [Ca2+]i with typical peak and plateau profiles (Figure 1A). Concentration–response analysis revealed EC50 values of 1.1 nM [pEC50 (M) = 8.95 ± 0.20] and 105 nM [pEC50 (M) = 6.98 ± 0.16] for oxytocin and histamine respectively (Figure 1B).
Figure 1.
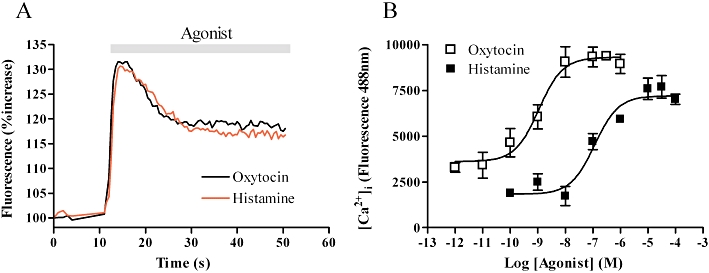
Characterization of histamine and oxytocin induced intracellular calcium concentration ([Ca2+]i) changes in myometrial cells. ULTR cells were loaded with Fluo4AM (3 µM, for 1 h) and agonist-induced [Ca2+]i changes monitored using a NovoStar imaging system (see Methods). (A) Representative traces showing typical temporal profiles of [Ca2+]i changes after stimulation with maximal concentrations of oxytocin (100 nM) or histamine (100 µM). (B) Concentration–response curves generated after stimulation with varying concentrations of oxytocin or histamine. Data are shown as means ± SEM from six separate experiments.
Depletion of arrestin isoform expression in ULTR cells
To optimize endogenous arrestin protein depletion, ULTR cells were transfected with either 10 or 100 nM of siRNA targeting arrestin2, arrestin3 or a negative control siRNA. Initial experiments revealed that maximal arrestin depletion could be achieved 48 h after transfection (data not shown). Optimal arrestin2 depletion was attained following application of 100 nM anti-arrestin2 siRNA (Figure 2A and C). The A1CT antibody also detects arrestin3, albeit with lower affinity, enabling visualization of both arrestins on one blot. Increased exposure of the same blot (Figure 2B and C) highlighted the successful and selective depletion of arrestin3 immunoreactivity with concentrations of anti-arrestin3 siRNA of >10 nM. We routinely observed a >70% reduction in the expression of the targeted arrestin isoform when compared with cell lysates transfected with negative control siRNA. Importantly, each anti-arrestin siRNA appeared selective for the isoform targeted (Figure 2A–C). In all subsequent experiments 100 nM of anti-arrestin2 and 10 nM of anti-arrestin3 siRNA were used to maximally deplete targeted endogenous arrestin isoform.
Figure 2.
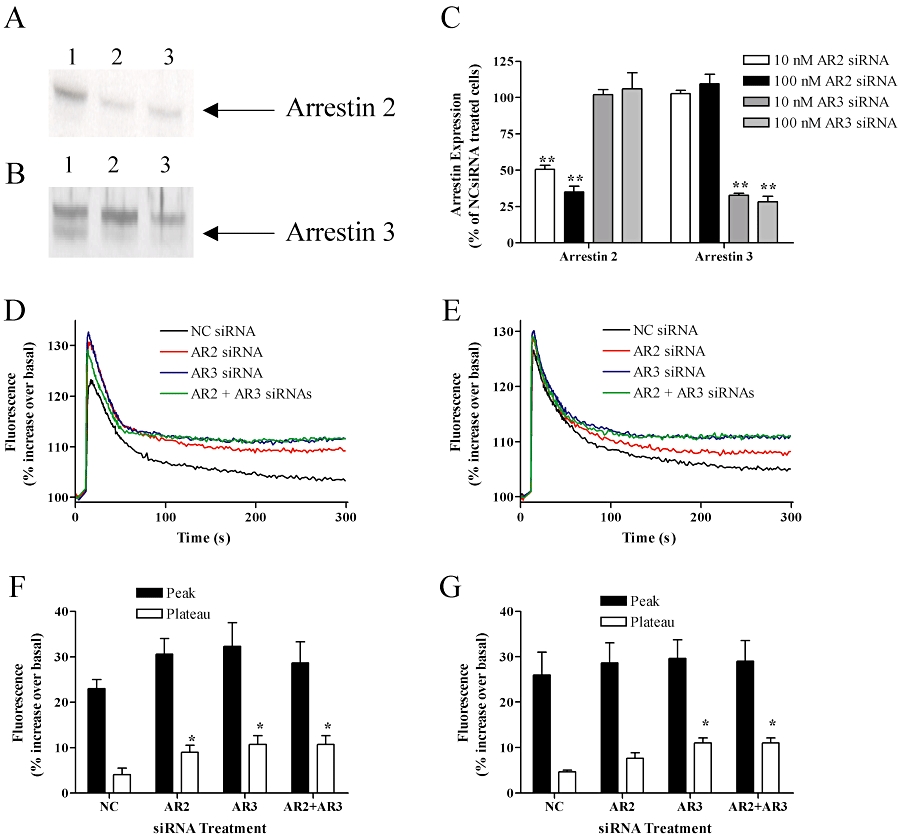
Arrestin depletion prolongs oxytocin- and histamine-induced Ca2+ signalling. ULTR cells were transfected with siRNAs against arrestin2 (AR2, 100 nM), arrestin3 (AR3, 10 nM) or negative control (NC, 100 nM) siRNA. After 48 h cells were lysed and arrestin expression levels determined via Western blotting (see Methods). (A) Representative immunblot showing arrestin2 expression following treatment with negative control (lane 1), anti-arrestin2 (lane 2) or both anti-arrestin2 and anti-arrestin3 (lane 3) siRNAs respectively. (B) Representative immunblot showing arrestin3 expression (the lower band) following transfection with negative control (lane 1), anti-arrestin3 (lane 2) or anti-arrestin2 and anti-arrestin3 (lane 3) siRNAs respectively. Cumulative data are also shown quantifying the extent of siRNA-mediated arrestin suppression in ULTR cells (C). Data are expressed as means ± SEM from four separate experiments. Significant differences seen following knockdown relative to negative control transfected cells are indicated as **P < 0.01. Oxytocin (100 nM) (D) and histamine (100 µM)-stimulated (E) intracellular calcium concentration ([Ca2+]i) signals were examined in the presence or absence of arrestin2 or arrestin3 or both arrestins. Mean trace data are shown from n= 3 experiments and SEM are omitted for clarity. Cumulative data show peak and plateau [Ca2+]i changes (at 200 s) following oxytocin (F) and histamine (G) stimulation taken from the temporal traces shown in (D) and (E). Oxytocin-stimulated plateau [Ca2+]i levels were significantly enhanced (*P < 0.05, one-way anova, Bonferroni's post hoc test) following knockdown of either arrestin isoform, while histamine-stimulated plateau [Ca2+]i levels were significantly enhanced (*P < 0.05, one-way anova, Bonferroni's post hoc test) only in the absence of arrestin3. Mean basal fluorescent [Ca2+]i-values for non-transfected cells (25564 ± 357) were unaltered following transfection with negative control (24685 ± 1151), anti-arrestin2 (24931 ± 399), anti-arrestin3 24338 ± 764) or both anti-arrestin3 and 3 (25185 ± 862) siRNAs (data are mean arbitrary fluorescent units ± SEM, for n= 3 experiments for each condition).
Arrestin depletion enhances oxytocin- and histamine-stimulated Ca2+ signals
To assess whether arrestin depletion affected oxytocin- or histamine-stimulated [Ca2+]i signalling, cells were transfected with either negative control (100 nM), anti-arrestin2 (100 nM), anti-arrestin3 (10 nM) or both anti-arrestin2 and anti-arrestin3 siRNAs. Agonist-stimulated changes in [Ca2+]i were monitored 48 h after transfection after addition of a single maximal oxytocin (100 nM) or histamine (100 µM) concentration. For both oxytocin and histamine, transfection with negative control siRNA had no effects on basal fluorescence ([Ca2+]i) values (Figure 2), or the magnitude or peak-plateau profiles of oxytocin- or histamine-stimulated [Ca2+]i signals (data not shown). However, following siRNA depletion of either arrestin2 or arrestin3, peak-plateau oxytocin-stimulated [Ca2+]i changes were significantly enhanced when compared with negative control transfected cells (P < 0.01 two-way anova; Bonferroni's post hoc test). Combined depletion of both arrestin isoforms by co-transfection of anti-arrestin2 and anti-arrestin3 siRNAs failed to enhance oxytocin-induced [Ca2+]i changes to any greater extent than by depletion of individual arrestin isoforms. While arrestin depletion (either individually or in combination) did not affect histamine-stimulated peak [Ca2+]i changes (Figure 2E), the plateau phase was significantly enhanced siRNA-mediated arrestin3 suppression (P < 0.01 two-way anova; Bonferroni's post hoc test). Knockdown of arrestin2 did not significantly affect the plateau phase of the histamine-stimulated Ca2+ response and failed to increase the effect seen on arrestin3 depletion (Figure 2E). These data suggest that arrestin2 and 3 regulate oxytocin receptor signalling, whilst arrestin3 alone is responsible for the regulation of H1 receptor signalling.
Effects of arrestin isoform depletion on oxytocin and H1 receptor desensitization
We have previously determined that GRK2 (Willets et al., 2008) and GRK6 (Willets et al., 2009) are the respective key regulators of H1 and oxytocin receptor desensitization in ULTR and primary human myometrial cell. These findings prompt the question of whether GRK-mediated phosphorylation leads to arrestin binding and what the consequences of this for downstream signalling are? To answer this question, myometrial cells were co-transfected with eGFP-PH and either negative control (100 nM), anti-arrestin2 (100 nM) or anti-arrestin3 (10 nM) siRNAs. As expected application of our previously validated desensitization protocols highlighted markedly reduced R2 values compared with R1 for both histamine and oxytocin challenge. Reduction in the R2/R1 ratio is indicative of receptor desensitization and was comparable to that previously observed in the presence or absence of negative control siRNA (i.e. 50% for H1 and 75% for oxytocin receptor responses (Willets et al., 2008; 2009;) (Figure 3A, D and G). Suppression of arrestin3, but not arrestin2 expression markedly reversed H1 receptor desensitization (Figure 3B, C and G). In contrast, depletion of either arrestin isoform was equally efficient at reversing oxytocin receptor desensitization (Figure 3E–G). These findings highlight a key role for arrestin3 in the regulation of H1 receptor desensitization whilst oxytocin receptor desensitization is mediated equally by both arrestin2 and arrestin3.
Figure 3.
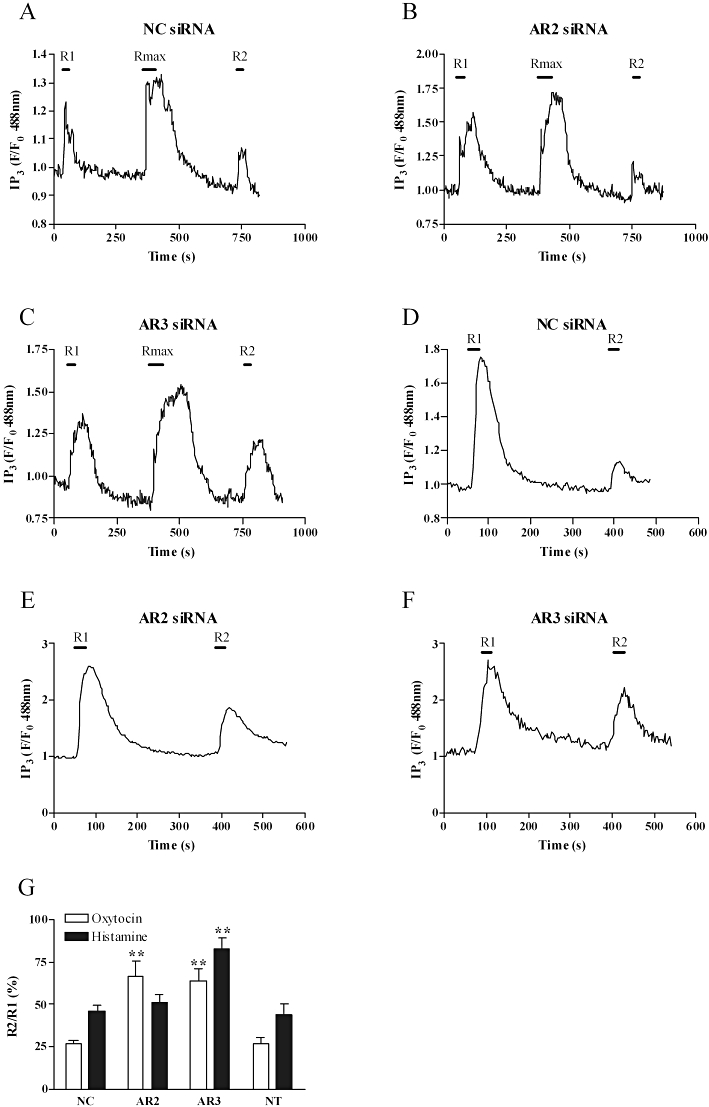
Suppression of arrestin expression prevents oxytocin and H1 receptor desensitization. Cells were co-transfected with e-GFP-tagged pleckstrin homology domain of phospholipase Cδ (0.5 µg) and either anti-arrestin2 (AR2, 100 nM), anti-arrestin3 (AR3, 10 nM) or negative control (NC, 100 nM) as described in the Methods section. After 48 h, H1 receptor desensitization was assessed using the standard desensitization protocol (R1, R2 = 10 µM histamine for 30 s; Rmax= 100 µM histamine for 60 s) and oxytocin receptor desensitization assessed using an alternative desensitization protocol whereby 100 nM oxytocin was applied for 30 s during R1 and R2 with 5 min washout period. Representative traces show the effects of negative control (A) anti-arrestin2 (B) and anti-arrestin3 (C) siRNA treatment on H1 receptor during the R1/Rmax/R2 protocol. Representative traces also display the effects of negative control (D) anti-arrestin2 (E) and anti-arrestin3 (F) siRNA treatment on oxytocin receptor responses during the alternative R1/R2 protocol. Cumulative data (G) show a significant (**P < 0.01, one-way anova, Dunnett's post hoc test) decrease in the extent of H1 receptor desensitization after depletion of endogenous arrestin3 and oxytocin receptor desensitization after suppression of arrestin2 or arrestin3 expression. Data are presented as means ± SEM for the % change in R2/R1 ratio for between 7 and 17 cells from at least six separate experiments. IP3, inositol 1,4,5-trisphosphate.
Characteristics of oxytocin- and histamine-mediated ERK1/2 phosphorylation
The effects of oxytocin and H1 receptor activation on the ERK1/2 signalling pathway in ULTR cells were investigated by immunuoblotting techniques using antibodies for total and phospho-forms (p) of ERK1/2. Oxytocin induced a 200% increase over basal in pERK1/2, which peaked at 3 min and rapidly returned towards basal after 5 min. In contrast, histamine-stimulated pERK1/2 responses, whilst peaking at 3 min, were more prolonged, declining over the succeeding 15 min. Concentration–response analysis for pERK1/2 immunoreactivity at 3 min after agonist addition generated EC50 values of 1.3 nM for oxytocin [pEC50 (M) = 8.84 ± 0.18] and 7.1 µM for histamine [pEC50 (M) = 5.14 ± 0.21] (Figure 4C, D and F). In agreement with previous findings in ULTR cells (Willets et al., 2008), histamine-stimulated pERK1/2 and phospho-p38 signals were blocked by the H1 antagonist diphenhydramine (10 µM), but not by the H2 receptor antagonist, cimetidine (10 µM, data not shown).
Figure 4.
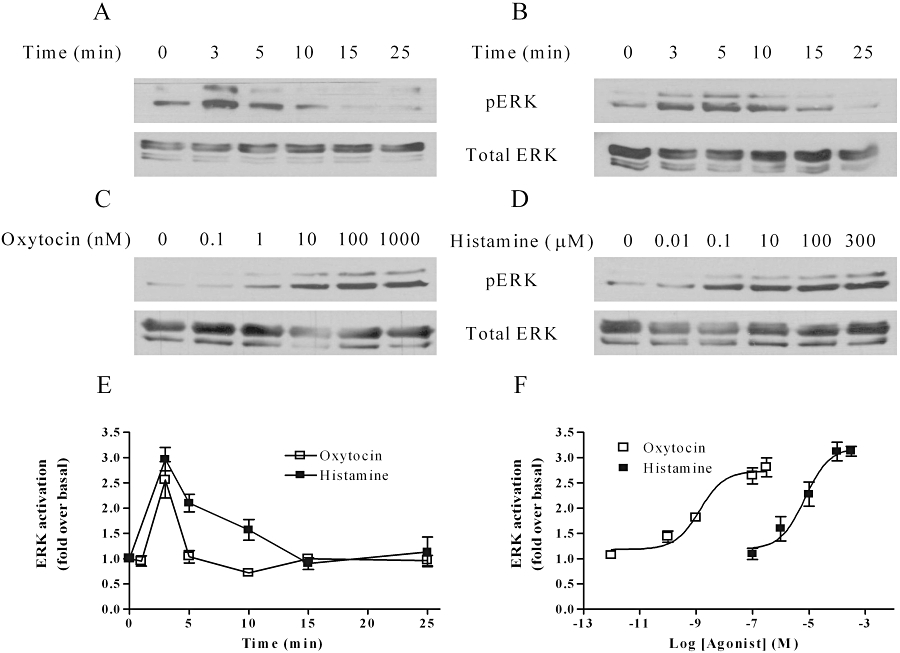
Time courses and concentration-dependencies for oxytocin- and histamine-stimulated extracellular signal-regulated kinase (ERK)1/2 phosphorylation. ULTR cells were deprived of serum for 24 h prior to agonist stimulation, and threonine- and tyrosine-phosphorylated extracellular signal-regulated kinase (pERK)1/2 levels determined by standard immunoblotting techniques (A–D, upper panels). To ensure that all samples contained the same levels of protein, total ERK levels were determined by running additional gels in parallel with the detection of pERK using an anti-ERK1 antibody (A–D, lower panels). Representative immunoblots show time courses for responses to oxytocin (100 nM) (A) or histamine (100 µM) (B), and concentration-dependencies for oxytocin (C) and histamine (D). Concentration-dependencies were determined at the peak ERK1/2 phosphorylation time point (3 min). Mean data (± SEM) for time courses (E) and concentration-dependencies (F) are shown for n= 3 experiments.
Differential effects of arrestin isoform depletion on oxytocin- and histamine-stimulated ERK1/2 signalling
For at least some GPCRs, arrestin proteins have well-documented roles as agonist-regulated adaptor scaffolds for cell surface receptor-to-ERK1/2 signalling (Ahn et al., 2004b; DeWire et al., 2007), although their potential role in myometrium has not yet been investigated. Here, we examined the effects of specific siRNA-targeted arrestin suppression on oxytocin- and histamine-stimulated ERK1/2 phosphorylation (Figure 4). Transfection of ULTR cells with negative control siRNA had no effect on basal pERK1/2 levels, or the magnitude or time course of oxytocin- or histamine-stimulated ERK1/2 responses compared with non-transfected cells (Figures 4 and 5). However, following siRNA-mediated depletion of arrestin2, a marked increase in oxytocin-mediated ERK1/2 activation was observed, with a doubling of the peak response, which was maintained over a more sustained time course (Figure 5A and C). Conversely, depletion of arrestin3 attenuated peak pERK1/2 levels, and virtually abolished any sustained signal (Figure 5A and C). As observed with oxytocin challenge, depletion of arrestin2 also enhanced both the peak and sustained phases of histamine-stimulated ERK1/2 activation (Figure 5B and D), although this effect was not as great as for oxytocin-stimulated responses. Strikingly, the histamine-evoked pERK1/2 response was completely absent after depletion of arrestin3 (Figure 5B and D). Previous reports highlight a role for PKC in mediating Gαq/11 coupled receptor-ERK1/2 activation (Kim et al., 2005; DeWire et al., 2007). To assess whether PKC was involved in uterotonic-ERK1/2 responses in ULTR cells, either oxytocin (100 nM) or histamine (100 µM) was applied for 3 min, in the presence or absence of the PKC inhibitor Go6976 (1 µM, 15 min pre-incubation). PKC inhibition had no effect upon histamine-stimulated ERK signals; however, oxytocin-induced ERK1/2 phosphorylation was inhibited by a third (Figure 6A, B and E). Interestingly, both oxytocin- and histamine-evoked pERK1/2 responses appear heavily reliant on extracellular Ca2+, because agonist-stimulated pERK1/2 signals were virtually undetectable when extracellular Ca2+ was removed from the assay buffer (Figure 6C–E). These data indicate that histamine-induced ERK1/2 phosphorylation is entirely reliant on the presence of arrestin3, and is enhanced following depletion of arrestin2. In contrast, the peak phase of oxytocin-induced ERK1/2 phosphorylation requires both PKC activity and the presence of arrestin3, whilst again oxytocin-induced ERK1/2 activation was enhanced in the absence of arrestin2.
Figure 5.
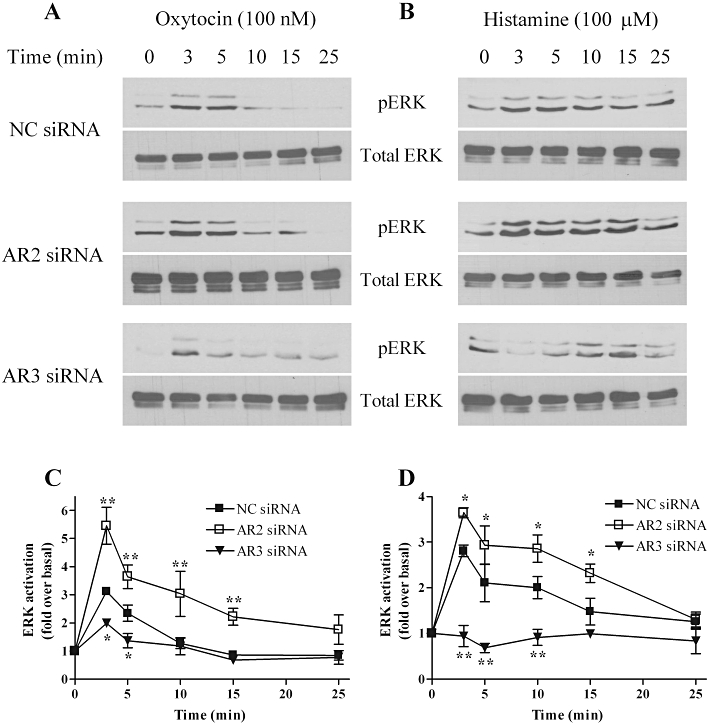
Arrestins differentially regulate oxytocin- and histamine-stimulated extracellular signal-regulated kinase (ERK)1/2 signalling. ULTR cells were transfected with negative control (NC, 100 nM), anti-arrestin2 (AR2, 100 nM) or anti-arrestin3 (AR3, 10 nM) siRNA for 48 h. Cells were serum-starved for the last 24 h prior to agonist stimulation for the times stated. ULTR cells were lysed and threonine- and tyrosine-phosphorylated extracellular signal-regulated kinase (pERK)1/2 levels determined by standard immunoblotting techniques (upper panels). To ensure that all samples contained the same levels of protein, total ERK levels were determined by running additional gels in parallel with the detection of pERK using an anti-ERK1 antibody (A–D, lower panels). Representative immunoblots show the effects of arrestin depletion on oxytocin (A) or histamine (B)-stimulated pERK1/2 responses. Cumulative densitometric analysis of oxytocin (C) or histamine (D)-stimulated ERK1/2 phosphorylation. Data are shown means ± SEM of n= 4 experiments. Depletion of arrestin3 significantly attenuated both oxytocin and histamine-stimulated pERK1/2 responses, whilst arrestin2 depletion significantly enhanced oxytocin and histamine-stimulated ERK1/2 signals (*P < 0.05, **P < 0.01, two-way anova, Bonferroni's post hoc test) when compared with negative control-treated cells. Mean basal pERK1/2 levels were similar in non-transfected (11722 ± 1592), to negative control (12937 ± 2970), anti-arrestin2 (11837 ± 1961) and anti-arrestin3 (12743 ± 2792) transfected cells (data are mean absorbance units·mm−2± SEM, n= 4).
Figure 6.
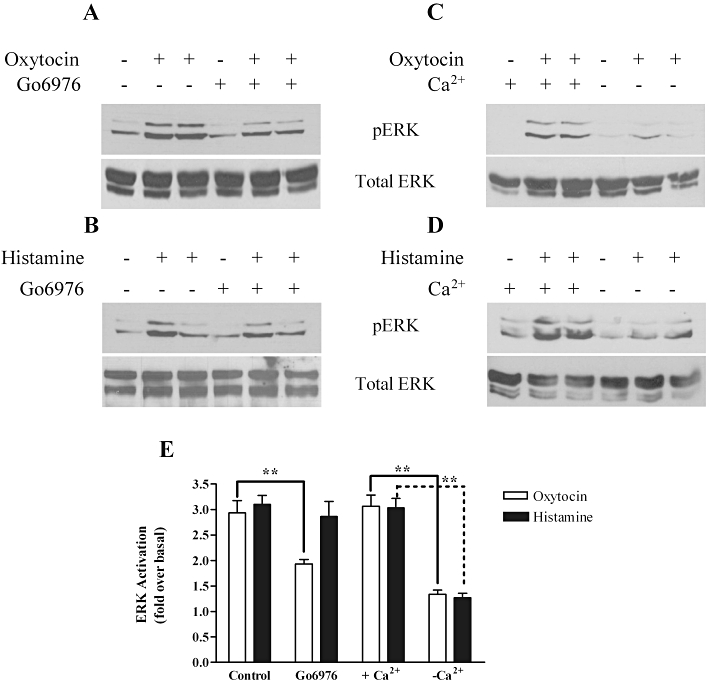
Ca2+ and protein kinase C (PKC) dependence of oxytocin- and histamine-stimulated threonine- and tyrosine-phosphorylated extracellular signal-regulated kinase (pERK)1/2 responses. Representative immunoblots are displayed showing (A) oxytocin (100 nM) and (B) histamine (100 µM)-stimulated pERK1/2 signals in the presence or absence of the PKC inhibitor Go6976 (1 µM, for 15 min prior to agonist challenge). Representative immunoblots (upper panels) are shown for (C) oxytocin and (D) histamine-stimulated pERK1/2 responses generated in the presence or nominal absence of extracellular Ca2+ (i.e. in nominally Ca2+-free Krebs). To ensure that all samples contained the same levels of protein, total extracellular signal-regulated kinase (ERK) levels were determined by running additional gels in parallel with the detection of pERK using an anti-ERK1 antibody (A–D, lower panels). Cumulative densitometric analysis pERK1/2 responses (E) show that removal of extracellular Ca2+ dramatically inhibits oxytocin- and histamine-evoked pERK1/2 immunonoreactivity (**P < 0.01, one-way anova, Dunnett's post hoc test). PKC inhibition partially inhibited oxytocin- (**P < 0.01, one-way anova, Dunnett's post hoc test), but not histamine-induced pERK1/2 responses. Data are shown as means ± SEM of n= 4 experiments.
Oxytocin- and histamine-stimulated phosphorylation of p38 MAPK
Oxytocin and histamine induced time- and concentration-dependent activation of p38 MAPK signalling (Figure 7). Oxytocin induced a maximal increase in phospho-p38 immunoreactivity after 5 min, which gradually declined to basal levels within 25 min (Figure 7A and E), whilst histamine induced maximal p38 phosphorylation after 10 min, declining to basal by 25 min (Figure 7B and E). Concentration–response analysis at the respective peak time points for the two agonists revealed EC50 values of 0.23 nM [pEC50 (M) = 9.65 ± 0.2] for oxytocin (Figure 7C and F) and 89 nM [pEC50 (M) = 7.05 ± 0.3] for histamine (Figure 7D and F) respectively.
Figure 7.
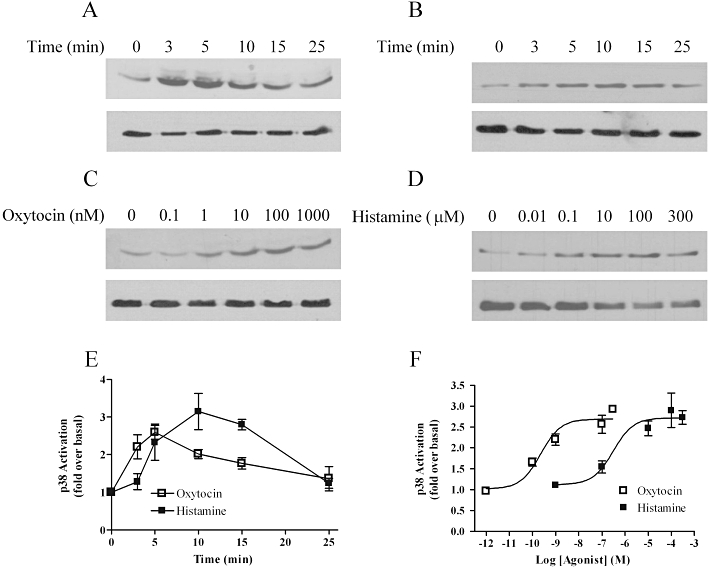
Time course and concentration-dependence of oxytocin- and histamine-stimulated p38 mitogen-activated protein kinase phosphorylation. Cells were deprived of serum for 24 h prior to agonist stimulation, and phospho-p38 (pp38) levels determined by standard immunoblotting techniques (A–D upper panels). To ensure that all samples contained the same levels of protein, total p38 levels were determined by running additional gels in parallel with the detection of p38 using an anti-p38 antibody (A–D, lower panels). Representative immunoblots show the time courses of oxytocin (100 nM) (A), and histamine (100 µM) (B) and concentration-dependence of oxytocin- (C) and histamine- (D) stimulated p38 activation. Concentration-dependencies were determined at the peak p38 phosphorlyation time point (oxytocin 5 min; histamine 10 min). Mean data (± SEM) for time courses (E) and concentration-dependencies (F) are shown for n= 3–5 separate experiments.
Differential effects of arrestin isoform depletion on oxytocin- and histamine-stimulated p38 MAPK signalling
Although less widely investigated, there is accruing evidence of an intermediary role for arrestins linking GPCRs to p38 MAPK (Sun et al., 2002; Bruchas et al., 2006; Gong et al., 2008). To investigate the role that arrestins might play in the oxytocin- and/or histamine-mediated p38 MAPK signalling, we examined the time course profiles of p38 phosphorylation stimulated by both receptors following siRNA-mediated depletion of arrestin2 or 3. When compared with negative control siRNA-transfected cells, the peak and sustained phases of oxytocin-induced p38 signalling were significantly enhanced in ULTR cells transfected with anti-arrestin2 or anti-arrestin3 siRNAs (Figure 8A and C). In the presence of negative control or arrestin2 siRNAs, the profile of histamine-induced p38 activation was similar to that observed in non-transfected cells, whereas arrestin3 depletion markedly increased both the peak and sustained phases of histamine-stimulated p38 signalling (Figure 8B and D). Furthermore, mean basal p38 phosphorylation levels were similar in non-transfected and siRNA-transfected cells (Figure 8). Our findings suggest that arrestin proteins play a key role in negatively regulating uterotonic p38 MAPK signalling in myometrial cells. Moreover, both arrestin isoforms are equally adept at limiting oxytocin-induced p38 signals, whereas arrestin3 appears to be selectively involved in modulating histamine-evoked signals.
Figure 8.
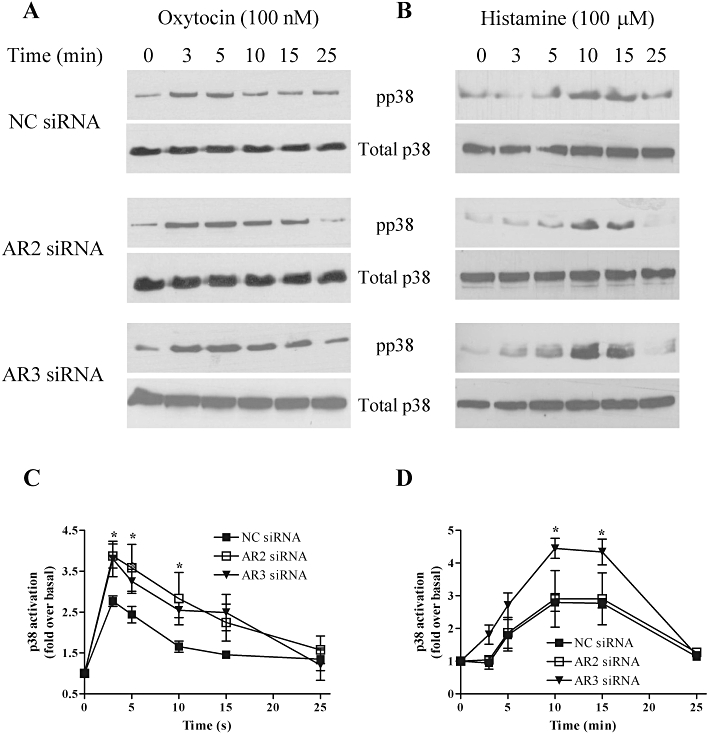
Arrestins differentially regulate oxytocin and histamine-stimulated extracellular signal-regulated kinase (ERK) signalling. ULTR cells were transfected with negative control (NC 100 nM), anti-arrestin2 (AR2, 100 nM) or anti-arrestin3 (AR3, 10 nM) siRNA for 48 h. Cells were deprived of serum for the last 24 h prior to agonist stimulation for the times stated. ULTR cells were lysed and phospho-p38 immunoreactivity determined by standard immunoblotting techniques (upper panels). To ensure that all samples contained the same levels of protein, total p38 levels were determined by running additional gels in parallel with the detection of phospho-p38 using an anti-p38 antibody (A–D, lower panels). Representative immunoblots show the effects of arrestin depletion on oxytocin (100 nM) (A) or histamine (100 µM) (B) stimulated p38 responses. Cumulative densitometric analysis of oxytocin (C) or histamine (D) stimulated p38 phosphorylation. Data are shown as means ± SEM of n= 4 experiments. Depletion of arrestin3 significantly enhanced both oxytocin- and histamine-stimulated p38 responses, whereas arrestin 2 depletion significantly enhanced oxytocin- but not histamine-stimulated p38 responses (*P < 0.05, **P < 0.01, two-way anova, Bonferroni's post hoc test), when compared with negative control-treated cells. Mean basal p38 phosphorylation levels were similar in non-transfected (9358 ± 1203), to negative control (8245 ± 2233), anti-arrestin2 (8677 ± 2023) and anti-arrestin3 (9546 ± 2683) transfected cells (data are mean absorbance units·mm−2± SEM, n= 4).
Discussion and conclusions
Here we investigated how the uterotonins, oxytocin and histamine cause their respective GPCRs (oxytocin and H1) to recruit arrestin isoforms and how this contributes to the desensitization of G protein-mediated signalling, whilst also contributing to the recruitment of alternate receptor-driven signalling mechanisms within myometrial smooth muscle cells. Oxytocin-mediated myometrial contraction has an established role in the progression of labour (Blanks and Thornton, 2003; Zingg and Laporte, 2003). Histamine, most likely released from infiltrating or resident uterine mast cells (Massey et al., 1991; Bytautiene et al., 2004b), can also induce myometrial contractions via H1 receptor activation (Rudolph et al., 1993; Bytautiene et al., 2003), a process implicated in preterm labour (Bytautiene et al., 2003; 2004a,b;). Regulation of GPCR signalling is complex, involving many different regulatory processes; however, GPCR desensitization, mediated by GRK and arrestin proteins, is fundamental in preventing prolonged or inappropriate signalling (Willets et al., 2003; Premont and Gainetdinov, 2007). We previously identified GRK6 and GRK2 as the respective primary initiators of myometrial oxytocin and H1 receptor desensitization (Willets et al., 2008; 2009;), findings that suggest these receptors are also likely to recruit arrestin proteins. Although H1 receptor–arrestin interactions are presently underexplored, a number of studies have demonstrated oxytocin receptor–arrestin interactions (Oakley et al., 2001; Hasbi et al., 2004; Smith et al., 2006; Conti et al., 2009). However, all previous studies to date have been performed in model cell (HEK293/COS-7) systems with recombinantly expressed oxytocin receptors. Therefore, our study is the first to assess the roles of arrestin proteins in oxytocin and H1 receptor regulation within the myometrium.
Initial studies assessed the effects of siRNA-mediated selective depletion of arrestin2 and 3 isoforms on oxytocin and H1 receptor signalling and desensitization. By examining a simple signalling readout ([Ca2+]i) and by using previously validated protocols to assess how prior agonist exposure affects subsequent responses to rechallenge with agonist [assessed at a single cell level using the eGFP-PH biosensor of phosphoinositide turnover; (Nelson et al., 2008; Willets et al., 2008)] we have been able to demonstrate different arrestin isoform dependencies of the two receptors. Thus, while knockdown of either arrestin2 or 3 augmented oxytocin-mediated Ca2+ responses and attenuated the extent of agonist-mediated oxytocin receptor desensitization, only arrestin3 knockdown had similar effects on H1 receptor signalling and desensitization. Previous studies also demonstrate an agonist- and GRK-dependent interaction of recombinant oxytocin receptors and arrestin3 in HEK293/COS-7 cell backgrounds (Oakley et al., 2001; Hasbi et al., 2004; Smith et al., 2006), with one study pinpointing two C-terminal Ser-Ser-Ser (368–370/377–379) clusters as the oxytocin receptor domains undergoing GRK phosphorylation and arrestin3 binding (Oakley et al., 2001). However, these studies all focused on oxytocin receptor–arrestin3 interactions and did not investigate the possibility of an oxytocin receptor–arrestin2 interaction. Here, we provide novel data on oxytocin receptor regulation by both arrestin2 and 3 in myometrial cells endogenously expressing this GPCR subtype, as well as the first evidence that H1 receptor signalling/desensitization is modulated by arrestin3, but not arrestin2.
An emerging theme over the past 10 years has been a realization that whilst arrestin binding results in GPCR uncoupling from G proteins and desensitization/internalization, it can also allow the receptor-arrestin complex to fulfil alternate signalling functions within the cell (Luttrell and Lefkowitz, 2002; DeWire et al., 2007; Gesty-Palmer and Luttrell, 2008). To investigate (some of) the signalling consequences of oxytocin and H1 receptor-arrestin binding, we assessed how the selective depletion of arrestin2 or 3 affects the time courses of agonist-stimulated ERK and p38 MAPK phosphorylation in human myometrial ULTR cells. Initial experiments established that oxytocin and histamine stimulated both ERK and p38 MAPK responses with similar peak increases, but somewhat different time courses of response. Similar findings have previously been obtained for oxytocin in myometrial preparations (Robinson and Dickenson, 2001; Zhong et al., 2003; Devost et al., 2008) and H1 receptor-mediated ERK1/2 and p38 MAPK activation has been reported in non-myometrial preparations (Robinson and Dickenson, 2001). Our data confirm a role for PKC in oxytocin-induced activation of ERK1/2 signals (Zhong et al., 2003); however, PKC appears to play a lesser role in mediating oxytocin ERK1/2 signalling in non-pregnant ULTR, than immortalized pregnant PHM1 myometrial cells (Zhong et al., 2003). These differences in oxytocin ERK1/2 signalling may reflect physiological changes in oxytocin receptor regulation induced by pregnancy. Indeed, removal of extracellular Ca2+ induced a far more pronounced inhibition of oxytocin-stimulated ERK1/2 signalling in non-pregnant myometrial (ULTR) cells. Moreover, histamine-stimulated ERK1/2 signals were abolished in the absence of extracellular Ca2+, and were PKC-independent. Collectively, these data infer that Ca2+ entry is the main driving force behind the peak phase of oxytocin and histamine ERK1/2 signalling in ULTR cells, which mirrors previous findings mediated by the gonadotropin-releasing hormone receptor in LβT-2 cells (Bonfil et al., 2004).
Existing work on GPCR-bound arrestins acting as signalling scaffolds has often focused on how this mechanism complements others utilized by GPCRs to activate MAPK signalling (e.g. those involving small GTPases, transactivation, growth-factor-shedding, etc.). For example, arrestins have been shown to scaffold ERK1/2 to the GPCR, leading to retention of active ERK1/2 within the cytoplasm and prolongation of signalling (DeWire et al., 2007). Here we demonstrate that selective arrestin3 knockdown in ULTR cells markedly attenuates ERK1/2 activation by either oxytocin or histamine. Previous studies have often reported a different time course of arrestin-dependency, with initial peak responses being generally less dependent on GPCR-arrestin-ERK1/2 signalling than the sustained phase (Ahn et al., 2004a; DeWire et al., 2007). Our oxytocin data appear to concur with this scenario because both PKC and arrestin3 appear to contribute equally to peak ERK1/2 activation, while sustained ERK phosphorylation is arrestin3-dependent. By contrast, we show that arrestin3 depletion completely ablates histamine-mediated ERK1/2 signalling and markedly attenuates the oxytocin-stimulated response at the earliest time point investigated. For both oxytocin- and H1 receptor-stimulated ERK1/2 responses arrestin2 knockdown resulted in enhanced initial responses that were evident across the first 15 min of the time course studied here. These data echo those reported in the earliest arrestin/siRNA studies, where knockdown of arrestin3 decreased, and arrestin2 enhanced, AT1A and V2 vasopressin receptor-ERK1/2 signalling in a HEK293 cell background (Ahn et al., 2004b; Ren et al., 2005). The enhancement of oxytocin receptor-ERK1/2 signalling seen when arrestin2 is selectively depleted correlates with the observed decrease in receptor desensitization, and desensitization/internalization of this receptor subtype certainly occurs very rapidly in myometrial derived cells (Conti et al., 2009). However, a similar explanation cannot be elaborated for the qualitatively similar enhancement of H1 receptor-ERK1/2 signalling as desensitization of this GPCR was not significantly affected by arrestin2 knockdown. Therefore, the precise mechanism(s) whereby arrestin2 knockdown facilitates both oxytocin and H1 receptor-ERK1/2 signalling have yet to be established.
Although less studied, GPCR coupling to another MAPK signalling pathway has also been reported to occur via arrestin-dependent mechanisms. As for the GPCR-ERK1/2 signalling pathway, it is likely that arrestins will be one of a number of different mechanisms that can link activated GPCRs to p38 MAPK signalling. However, preliminary experiments demonstrated that both oxytocin and H1 receptor subtypes can increase p38 MAPK phosphorylation in ULTR cells, albeit with somewhat different time courses of activation. Selective arrestin2 or 3 depletion had similar effects on the oxytocin receptor-p38 MAPK signalling, enhancing p38 MAPK phosphorylation, suggesting that each isoform normally suppresses rather than facilitates oxytocin receptor coupling to p38 MAPK. For H1 receptor-p38 MAPK signalling arrestin3 knockdown enhanced the response, whilst arrestin2 knockdown was without effect. Interestingly, none of the manipulations of arrestin expression altered the overall time courses of p38 phosphorylation stimulated by oxytocin or histamine (see Figure 8C and D).
Previous studies that have manipulated cellular arrestin levels have generally reached different conclusions with respect to the roles of arrestins in p38 MAPK activation. Indeed, Sun et al. (2002) showed that arrestin-3 overexpression enhanced, whilst antisense or siRNA knockdown of arrestin3 attenuated, both CXCR4 and CCR5 chemokine receptor signalling to p38 MAPK in HEK293 cells. Within a native cell background, Bruchas et al. (2006) showed that arrestin3 can act as a scaffold for p38 MAPK signalling and demonstrated that siRNA-knockdown of arrestin3 attenuated κ-opioid receptor signalling to this readout. These studies focused on manipulating cellular arrestin3 levels and no conclusions can be drawn with respect to isoform-selective arrestin effects within these pathways. More recently, a β2-adrenoceptor–arrestin2 (but not arrestin3) interaction was proposed to act as a proximal scaffold eventually leading (via Rac1/NADPH oxidase-dependent intermediate steps) to p38 MAPK activation (Gong et al., 2008). Collectively, these studies highlight a positive intermediary role for arrestin2 or 3 in GPCR coupling to p38 MAPK. Conversely, ablation of arrestin2 and 3 isoforms in double-knockout mouse embryonic fibroblasts dramatically enhanced CXCR2-stimulated p38 MAPK signalling (Zhao et al., 2004), again with an intermediary role of Rac1/NADPH oxidase being implicated. Our data are mostly in agreement with the latter study and suggest that the failure of arrestin isoform depletions to suppress p38 MAPK signalling argues against arrestins acting as p38 scaffolds for oxytocin or histamine H1 receptors in myometrium. Instead, enhanced p38 MAPK signalling following selective arrestin2 or 3 knockdown most likely reflects the decreased ability to desensitize oxytocin and histamine signalling in the absence of these proteins, suggesting that oxytocin and H1 receptor coupling to p38 MAPK requires the receptor to remain at the cell surface.
In summary, this study builds on our previous work defining the GRK isoenzymic specificity of receptor desensitization by now identifying an important role for arrestin protein isoforms in the acute desensitization of the contractile myometrial oxytocin and H1 receptors. Acute regulation of GPCR-mediated contractile signalling may add an additional ‘brake’ preventing inappropriate myometrial contraction, which in terms of H1 receptor activation may be brought about through infection-induced mast cell degranulation (Massey et al., 1991; Bytautiene et al., 2004b). Furthermore, acute desensitization of oxytocin receptor signalling is likely to play an important role in the timing of myometrial contractions during labour, especially because uterine contractions cease despite the presence of circulating oxytocin. In addition, we have identified arrestin isoforms as key regulators of oxytocin- and histamine-stimulated MAPK signalling, which, considering the plethora of physiological processes mediated by MAPK signalling pathways, indicates that arrestins potentially play a significant role influencing myometrial sensitivity to external stimuli and fine tuning myometrial contractility. Indeed, MAPK signalling is reported to play a key role in the up-regulation of cyclooxygenase-2 expression (Bartlett et al., 1999; Molnar et al., 1999), highlighting a potential role for arrestins in the control of prostaglandin production and labour induction.
Acknowledgments
We thank Tobias Meyer (Stanford University, USA) for generously providing the eGFP-PH biosensor and Robert J. Lefkowitz (Duke University, USA) for kindly providing the arrestin (A1CT) antibody.
Glossary
Abbreviations
- [Ca2+]i
intracellular calcium concentration
- eGFP-PH
e-GFP-tagged pleckstrin homology domain of phospholipase Cδ
- ERK
extracellular signal-regulated kinase
- Go6976
12-(2-Cyanoethyl)-6,7,12,13-tetrahydro-13-methyl-5-oxo-5H-indolo(2,3-a)pyrrolo(3,4-c)-carbazole
- GPCR
G protein-coupled receptor
- GRK
G protein-coupled receptor kinase
- MAPK
mitogen-activated protein kinase
- pERK
threonine- and tyrosine-phosphorylated extracellular signal-regulated kinase
Conflict of interest
None.
Supplementary material
Supporting Information: Teaching Materials; Figs 1–8 as PowerPoint slide.
References
- Ahn S, Shenoy SK, Wei H, Lefkowitz RJ. Differential kinetic and spatial patterns of β-arrestin and G protein-mediated ERK activation by the angiotensin II receptor. J Biol Chem. 2004a;279:35518–35525. doi: 10.1074/jbc.M405878200. [DOI] [PubMed] [Google Scholar]
- Ahn S, Wei H, Garrison TR, Lefkowitz RJ. Reciprocal regulation of angiotensin receptor-activated extracellular signal-regulated kinases by β-arrestins 1 and 2. J Biol Chem. 2004b;279:7807–7811. doi: 10.1074/jbc.C300443200. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th edition. Br J Pharmacol. 2009;158:S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball A, Wang JW, Wong S, Zielnik B, Mitchell J, Wang N, et al. Phorbol ester treatment of human myometrial cells suppresses expression of oxytocin receptor through a mechanism that does not involve activator protein-1. Am J Physiol Endocrinol Metab. 2006;291:E922–E928. doi: 10.1152/ajpendo.00602.2005. [DOI] [PubMed] [Google Scholar]
- Bartlett SR, Sawdy R, Mann GE. Induction of cyclooxygenase-2 expression in human myometrial smooth muscle cells by interleukin-1β: involvement of p38 mitogen-activated protein kinase. J Physiol. 1999;520:399–406. doi: 10.1111/j.1469-7793.1999.00399.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanks AM, Thornton S. The role of oxytocin in parturition. BJOG. 2003;110(Suppl 20):46–51. doi: 10.1016/s1470-0328(03)00024-7. [DOI] [PubMed] [Google Scholar]
- Bonfil D, Chuderland D, Kraus S, Shahbazian D, Friedberg I, Seger R, et al. Extracellular signal-regulated kinase, Jun N-terminal kinase, p38, and c-Src are involved in gonadotropin-releasing hormone-stimulated activity of the glycoprotein hormone follicle-stimulating hormone beta-subunit promoter. Endocrinology. 2004;145:2228–2244. doi: 10.1210/en.2003-1418. [DOI] [PubMed] [Google Scholar]
- Brighton PJ, McDonald J, Taylor AH, Challiss RA, Lambert DG, Konje JC, et al. Characterization of anandamide-stimulated cannabinoid receptor signaling in human ULTR myometrial smooth muscle cells. Mol Endocrinol. 2009;23:1415–1427. doi: 10.1210/me.2009-0097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodt-Eppley J, Myatt L. Prostaglandin receptors in lower segment myometrium during gestation and labor. Obstet Gynecol. 1999;93:89–93. doi: 10.1016/s0029-7844(98)00378-0. [DOI] [PubMed] [Google Scholar]
- Bruchas MR, Macey TA, Lowe JD, Chavkin C. κ-opioid receptor activation of p38 MAPK is GRK3- and arrestin-dependent in neurons and astrocytes. J Biol Chem. 2006;281:18081–18089. doi: 10.1074/jbc.M513640200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bytautiene E, Vedernikov YP, Saade GR, Romero R, Garfield RE. Effect of histamine on phasic and tonic contractions of isolated uterine tissue from pregnant women. Am J Obstet Gynecol. 2003;188:774–778. doi: 10.1067/mob.2003.162. [DOI] [PubMed] [Google Scholar]
- Bytautiene E, Romero R, Vedernikov YP, El-Zeky F, Saade GR, Garfield RE. Induction of premature labor and delivery by allergic reaction and prevention by histamine H1 receptor antagonist. Am J Obstet Gynecol. 2004a;191:1356–1361. doi: 10.1016/j.ajog.2004.06.092. [DOI] [PubMed] [Google Scholar]
- Bytautiene E, Vedernikov YP, Saade GR, Romero R, Garfield RE. Degranulation of uterine mast cell modifies contractility of isolated myometrium from pregnant women. Am J Obstet Gynecol. 2004b;191:1705–1710. doi: 10.1016/j.ajog.2004.04.008. [DOI] [PubMed] [Google Scholar]
- Conti F, Sertic S, Reversi A, Chini B. Intracellular trafficking of the human oxytocin receptor: evidence of receptor recycling via a Rab4/Rab5 ‘short cycle’. Am J Physiol Endocrinol Metab. 2009;296:E532–E542. doi: 10.1152/ajpendo.90590.2008. [DOI] [PubMed] [Google Scholar]
- Devost D, Carrier ME, Zingg HH. Oxytocin-induced activation of eukaryotic elongation factor 2 in myometrial cells is mediated by protein kinase C. Endocrinology. 2008;149:131–138. doi: 10.1210/en.2007-0548. [DOI] [PubMed] [Google Scholar]
- DeWire SM, Ahn S, Lefkowitz RJ, Shenoy SK. β-arrestins and cell signaling. Annu Rev Physiol. 2007;69:483–510. doi: 10.1146/annurev.physiol.69.022405.154749. [DOI] [PubMed] [Google Scholar]
- Engelhardt S, Zieger W, Kassubek J, Michel MC, Lohse MJ, Brodde OE. Tocolytic therapy with fenoterol induces selective down-regulation of β-adrenergic receptors in human myometrium. J Clin Endocrinol Metab. 1997;82:1235–1242. doi: 10.1210/jcem.82.4.3885. [DOI] [PubMed] [Google Scholar]
- Fuchs AR, Fuchs F, Husslein P, Soloff MS. Oxytocin receptors in the human uterus during pregnancy and parturition. Am J Obstet Gynecol. 1984;150:734–741. doi: 10.1016/0002-9378(84)90677-x. [DOI] [PubMed] [Google Scholar]
- Gesty-Palmer D, Luttrell LM. Heptahelical terpsichory. Who calls the tune? J Recept Signal Transduct Res. 2008;28:39–58. doi: 10.1080/10799890801941921. [DOI] [PubMed] [Google Scholar]
- Gong K, Li Z, Xu M, Du J, Lv Z, Zhang Y. A novel protein kinase A-independent, β-arrestin-1-dependent signaling pathway for p38 mitogen-activated protein kinase activation by β2-adrenergic receptors. J Biol Chem. 2008;283:29028–29036. doi: 10.1074/jbc.M801313200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasbi A, Devost D, Laporte SA, Zingg HH. Real-time detection of interactions between the human oxytocin receptor and G protein-coupled receptor kinase-2. Mol Endocrinol. 2004;18:1277–1286. doi: 10.1210/me.2003-0440. [DOI] [PubMed] [Google Scholar]
- Helmer H, Hackl T, Schneeberger C, Knofler M, Behrens O, Kaider A, et al. Oxytocin and vasopressin 1a receptor gene expression in the cycling or pregnant human uterus. Am J Obstet Gynecol. 1998;179:1572–1578. doi: 10.1016/s0002-9378(98)70027-4. [DOI] [PubMed] [Google Scholar]
- Holda JR, Oberti C, Perez-Reyes E, Blatter LA. Characterization of an oxytocin-induced rise in [Ca2+]i in single human myometrium smooth muscle cells. Cell Calcium. 1996;20:43–51. doi: 10.1016/s0143-4160(96)90049-4. [DOI] [PubMed] [Google Scholar]
- Kim J, Ahn S, Ren XR, Whalen EJ, Reiter E, Wei H, et al. Functional antagonism of different G protein-coupled receptor kinases for β-arrestin-mediated angiotensin II receptor signaling. Proc Natl Acad Sci U S A. 2005;102:1442–1447. doi: 10.1073/pnas.0409532102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong G, Penn R, Benovic JL. A β-adrenergic receptor kinase dominant negative mutant attenuates desensitization of the β2-adrenergic receptor. J Biol Chem. 1994;269:13084–13087. [PubMed] [Google Scholar]
- Ku CY, Qian A, Wen Y, Anwer K, Sanborn BM. Oxytocin stimulates myometrial guanosine triphosphatase and phospholipase-C activities via coupling to Gαq/11. Endocrinology. 1995;136:1509–1515. doi: 10.1210/endo.136.4.7895660. [DOI] [PubMed] [Google Scholar]
- Luttrell LM, Lefkowitz RJ. The role of β-arrestins in the termination and transduction of G-protein-coupled receptor signals. J Cell Sci. 2002;115:455–465. doi: 10.1242/jcs.115.3.455. [DOI] [PubMed] [Google Scholar]
- Massey WA, Guo CB, Dvorak AM, Hubbard WC, Bhagavan BS, Cohan VL, et al. Human uterine mast cells. Isolation, purification, characterization, ultrastructure, and pharmacology. J Immunol. 1991;147:1621–1627. [PubMed] [Google Scholar]
- Matsumoto T, Sagawa N, Yoshida M, Mori T, Tanaka I, Mukoyama M, et al. The prostaglandin E2 and F2α receptor genes are expressed in human myometrium and are down-regulated during pregnancy. Biochem Biophys Res Commun. 1997;238:838–841. doi: 10.1006/bbrc.1997.7397. [DOI] [PubMed] [Google Scholar]
- Molnar M, Rigo J, Jr, Romero R, Hertelendy F. Oxytocin activates mitogen-activated protein kinase and up-regulates cyclooxygenase-2 and prostaglandin production in human myometrial cells. Am J Obstet Gynecol. 1999;181:42–49. doi: 10.1016/s0002-9378(99)70434-5. [DOI] [PubMed] [Google Scholar]
- Nelson CP, Willets JM, Davies NW, Challiss RA, Standen NB. Visualizing the temporal effects of vasoconstrictors on PKC translocation and Ca2+ signaling in single resistance arterial smooth muscle cells. Am J Physiol Cell Physiol. 2008;295:C1590–C1601. doi: 10.1152/ajpcell.00365.2008. [DOI] [PubMed] [Google Scholar]
- Oakley RH, Laporte SA, Holt JA, Barak LS, Caron MG. Molecular determinants underlying the formation of stable intracellular G protein-coupled receptor-β-arrestin complexes after receptor endocytosis*. J Biol Chem. 2001;276:19452–19460. doi: 10.1074/jbc.M101450200. [DOI] [PubMed] [Google Scholar]
- Olson DM, Zaragoza DB, Shallow MC, Cook JL, Mitchell BF, Grigsby P, et al. Myometrial activation and preterm labour: evidence supporting a role for the prostaglandin F receptor – a review. Placenta. 2003;24:S47–S54. doi: 10.1053/plac.2002.0938. [DOI] [PubMed] [Google Scholar]
- Perez-Reyes N, Halbert CL, Smith PP, Benditt EP, McDougall JK. Immortalization of primary human smooth muscle cells. Proc Natl Acad Sci U S A. 1992;89:1224–1228. doi: 10.1073/pnas.89.4.1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phaneuf S, Asboth G, Carrasco MP, Europe-Finner GN, Saji F, Kimura T, et al. The desensitization of oxytocin receptors in human myometrial cells is accompanied by down-regulation of oxytocin receptor messenger RNA. J Endocrinol. 1997;154:7–18. doi: 10.1677/joe.0.1540007. [DOI] [PubMed] [Google Scholar]
- Premont RT, Gainetdinov RR. Physiological roles of G protein-coupled receptor kinases and arrestins. Annu Rev Physiol. 2007;69:511–534. doi: 10.1146/annurev.physiol.69.022405.154731. [DOI] [PubMed] [Google Scholar]
- Ren XR, Reiter E, Ahn S, Kim J, Chen W, Lefkowitz RJ. Different G protein-coupled receptor kinases govern G protein and β-arrestin-mediated signaling of V2 vasopressin receptor. Proc Natl Acad Sci U S A. 2005;102:1448–1453. doi: 10.1073/pnas.0409534102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson AJ, Dickenson JM. Activation of the p38 and p42/p44 mitogen-activated protein kinase families by the histamine H1 receptor in DDT1MF-2 cells. Br J Pharmacol. 2001;133:1378–1386. doi: 10.1038/sj.bjp.0704200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph MI, Reinicke K, Cruz MA, Gallardo V, Gonzalez C, Bardisa L. Distribution of mast cells and the effect of their mediators on contractility in human myometrium. Br J Obstet Gynaecol. 1993;100:1125–1130. doi: 10.1111/j.1471-0528.1993.tb15178.x. [DOI] [PubMed] [Google Scholar]
- Sanborn BM. Relationship of ion channel activity to control of myometrial calcium. J Soc Gynecol Investig. 2000;7:4–11. doi: 10.1016/s1071-5576(99)00051-9. [DOI] [PubMed] [Google Scholar]
- Sanborn BM. Hormones and calcium: mechanisms controlling uterine smooth muscle contractile activity. The Litchfield Lecture. Exp Physiol. 2001;86:223–237. doi: 10.1113/eph8602179. [DOI] [PubMed] [Google Scholar]
- Sanborn BM. Hormonal signaling and signal pathway crosstalk in the control of myometrial calcium dynamics. Semin Cell Dev Biol. 2007;18:305–314. doi: 10.1016/j.semcdb.2007.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon V, Robin MT, Legrand C, Cohen-Tannoudji J. Endogenous G protein-coupled receptor kinase 6 triggers homologous beta-adrenergic receptor desensitization in primary uterine smooth muscle cells. Endocrinology. 2003;144:3058–3066. doi: 10.1210/en.2002-0138. [DOI] [PubMed] [Google Scholar]
- Smith MP, Ayad VJ, Mundell SJ, McArdle CA, Kelly E, Lopez Bernal A. Internalization and desensitization of the oxytocin receptor is inhibited by Dynamin and clathrin mutants in human embryonic kidney 293 cells. Mol Endocrinol. 2006;20:379–388. doi: 10.1210/me.2005-0031. [DOI] [PubMed] [Google Scholar]
- Sun Y, Cheng Z, Ma L, Pei G. β-arrestin2 is critically involved in CXCR4-mediated chemotaxis, and this is mediated by its enhancement of p38 MAPK activation. J Biol Chem. 2002;277:49212–49219. doi: 10.1074/jbc.M207294200. [DOI] [PubMed] [Google Scholar]
- Tobin AB, Butcher AJ, Kong KC. Location, location, location . . . site-specific GPCR phosphorylation offers a mechanism for cell-type-specific signalling. Trends Pharmacol Sci. 2008;29:413–420. doi: 10.1016/j.tips.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willets JM, Challiss RA, Kelly E, Nahorski SR. G protein-coupled receptor kinases 3 and 6 use different pathways to desensitize the endogenous M3 muscarinic acetylcholine receptor in human SH-SY5Y cells. Mol Pharmacol. 2001;60:321–330. doi: 10.1124/mol.60.2.321. [DOI] [PubMed] [Google Scholar]
- Willets JM, Challiss RA, Nahorski SR. Non-visual GRKs: are we seeing the whole picture? Trends Pharmacol Sci. 2003;24:626–633. doi: 10.1016/j.tips.2003.10.003. [DOI] [PubMed] [Google Scholar]
- Willets JM, Nahorski SR, Challiss RA. Roles of phosphorylation-dependent and -independent mechanisms in the regulation of M1 muscarinic acetylcholine receptors by G protein-coupled receptor kinase 2 in hippocampal neurons. J Biol Chem. 2005;280:18950–18958. doi: 10.1074/jbc.M412682200. [DOI] [PubMed] [Google Scholar]
- Willets JM, Taylor AH, Shaw H, Konje JC, Challiss RA. Selective regulation of H1 histamine receptor signaling by G protein-coupled receptor kinase 2 in uterine smooth muscle cells. Mol Endocrinol. 2008;22:1893–1907. doi: 10.1210/me.2007-0463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willets JM, Brighton PJ, Mistry R, Morris GE, Konje JC, Challiss RA. Regulation of oxytocin receptor responsiveness by G protein-coupled receptor kinase 6 in human myometrial smooth muscle. Mol Endocrinol. 2009;23:1272–1280. doi: 10.1210/me.2009-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan W, Lopez Bernal A. Cyclic AMP signalling pathways in the regulation of uterine relaxation. BMC Pregnancy Childbirth. 2007;7:S10. doi: 10.1186/1471-2393-7-S1-S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao M, Wimmer A, Trieu K, Discipio RG, Schraufstatter IU. Arrestin regulates MAPK activation and prevents NADPH oxidase-dependent death of cells expressing CXCR2. J Biol Chem. 2004;279:49259–49267. doi: 10.1074/jbc.M405118200. [DOI] [PubMed] [Google Scholar]
- Zhong M, Yang M, Sanborn BM. Extracellular signal-regulated kinase 1/2 activation by myometrial oxytocin receptor involves GαqGβγ and epidermal growth factor receptor tyrosine kinase activation. Endocrinology. 2003;144:2947–2956. doi: 10.1210/en.2002-221039. [DOI] [PubMed] [Google Scholar]
- Zingg HH, Laporte SA. The oxytocin receptor. Trends Endocrinol Metab. 2003;14:222–227. doi: 10.1016/s1043-2760(03)00080-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.