

Abstract
The function of the lectin pathway of complement activation in vivo against West Nile virus (WNV) or many other pathogenic viruses has not been defined. Mice deficient in lectin pathway recognition molecules (mannose binding lectin-A (MBL-A) and mannose binding lectin-C (MBL-C)) or the effector enzyme mannan-binding lectin-associated serine protease-2 (MASP-2), were more vulnerable to WNV infection than wild type mice. Compared with studies of mice deficient in factors of the classical or alternative pathway, MBL-A-/- × MBL-C-/- or MASP-2-/- mice showed a less severe course of WNV infection. Indeed, a deficiency in lectin pathway activation did not significantly affect the kinetics of viral spread to the central nervous system (CNS) nor did it profoundly alter generation of adaptive B and T cell immune responses. We conclude that MBL-mediated recognition and lectin pathway activation has important yet subordinate functions in protecting against WNV infection and disease.
Keywords: flavivirus, complement, pathogenesis, lectin pathway, immunity
Introduction
Mannose-binding lectin (MBL) is a pattern recognition component of the complement system that binds carbohydrate groups on the surface of microbial pathogens and triggers the lectin activation pathway of complement. In humans, one MBL gene is expressed, whereas mice produce two closely related proteins, MBL-A and MBL-C. MBL proteins are expressed primarily in the liver and circulate in the blood, where they complex with mannan-binding lectin-associated serine proteases (MASP). In addition to MBL, there is a family of structurally related plasma proteins, called ficolins, which may serve as carbohydrate recognition molecules and drive lectin pathway mediated complement activation upon binding to a pathogen surface. MBL and ficolin engagement activates the associated MASP-2, which cleaves the downstream complement components C4 and C2 to form the C3 convertase complex C4b2a. The cleavage of C3, results in C3a release and C3b deposition. C3b marks pathogens for clearance by phagocytosis through one of several complement receptors. Additionally, complement deposition targets pathogens for destruction by forming a C5 convertase, C4b2a(C3b) that cleaves C5 to release the complement anaphylatoxin C5a and the larger cleavage fragment C5b, which promotes formation of the membrane-attack complex (MAC) by the terminal (C5-C9) complement components.
MBL and ficolins recognize many pathogens, including viruses, bacteria and fungi, and are believed to function both in direct innate immune restriction and induction of adaptive immunity. In humans, inherited MBL deficiencies are common, and are caused by frequently occurring polymorphisms in the MBL gene. MBL deficiencies are associated with more severe disease after Neisseria meningitides infection, and greater susceptibility to infection with HIV, hepatitis B virus, hepatitis C virus, and herpes simplex virus (HSV). Studies in MBL-A-/- × MBL-C-/- mice have established a role for MBL in limiting infections with Staphylococcus aureus, Pseudomonas aeruginosa, Candida albicans, and herpes simplex virus.
West Nile virus (WNV) is an enveloped, single-stranded positive polarity RNA virus in the Flaviviridae family. The virus cycles between mosquitoes and birds, but also infects humans, horses and other vertebrate animals. In humans, WNV infection is often asymptomatic, or manifests with a mild fever; however, in some cases, especially in elderly or immunosuppressed individuals, it can cause life-threatening encephalitis. The immunological basis of protection against WNV has been analyzed extensively in inbred mice, which are susceptible to WNV encephalitis. From these studies, it is evident that both innate and adaptive immune mechanisms are required for protection against neuroinvasive disease. Mice deficient in type I or II interferon, or T cell subsets such as γδ, regulatory, CD4+ or CD8+ T cells succumb to WNV disease with enhanced kinetics and frequency (reviewed in).
Protection against WNV encephalitis also requires an intact complement system as mice lacking the central complement component C3 uniformly succumbed to infection. Both the classical and alternative activation pathways are required, as mice deficient in C1q (classical pathway) or fB (alternative pathway) also showed greater susceptibility. As C4-/- mice showed a more severe phenotype after WNV infection than C1q-/- mice, we hypothesized that the lectin pathway independently protects against WNV infection. Consistent with this, mouse serum lacking MBL showed a decreased ability to directly neutralize insect cell-derived WNV in vitro.
Here, using mice that were genetically deficient in MBL-mediated pathway recognition and activation, we directly evaluated the contribution of MBL to immunity against WNV infection. MBL-A-/- × MBL-C-/- mice had relatively mild defects in WNV clearance, resulting in only moderately enhanced susceptibility. Contrary to our expectations, the in vivo studies revealed similar viral tissue burdens in several peripheral organs between wild type and MBL-A-/- × MBL-C-/- mice. Furthermore, a deficiency in MBL genes did not significantly impair induction of adaptive antiviral immune responses. Thus, while the lectin pathway contributes to protection against WNV, its effect is subordinate compared to the antiviral and priming functions of the classical and alternative pathways of complement activation.
Results
A deficiency of MBL or MASP-2 results in increased susceptibility to WNV infection
We recently observed that MBL in serum can bind to insect cell-derived WNV, leading to complement activation and neutralization of viral infectivity in vitro. To investigate whether MBL contributes to immune protection in vivo, we infected MBL-A-/- × MBL-C-/- mice on a C57BL/6 background with 102 PFU of WNV via footpad injection (Fig 1A). As seen previously, ∼30% of wild type C57BL/6 mice succumbed to WNV infection. Congenic MBL-A-/- × MBL-C-/- mice reproducibly showed a small yet statistically significant increased mortality rate (∼55%, P < 0.005). However, MBL-A-/- × MBL-C-/- mice showed similar kinetics of survival with a mean time to death that was not substantially different compared to wild type mice (mean time to death of 11.6 versus 11.9 days, P > 0.6). For both wild type and MBL-A-/- × MBL-C-/- mice, animals that succumbed to WNV infection showed similar clinical signs of encephalitis and limb paralysis. As our previous studies suggested that MBL preferentially bound to insect cell-derived WNV, we assessed whether infection with mammalian cell-derived WNV altered the susceptibility in MBL-A-/- × MBL-C-/- mice. However, similar to insect-cell derived virus, WNV propagated in Vero African green monkey cells also showed greater virulence in MBL-A-/- × MBL-C-/- mice with increased mortality observed (66% versus 24%, P = 0.006) (data not shown).
Figure 1.
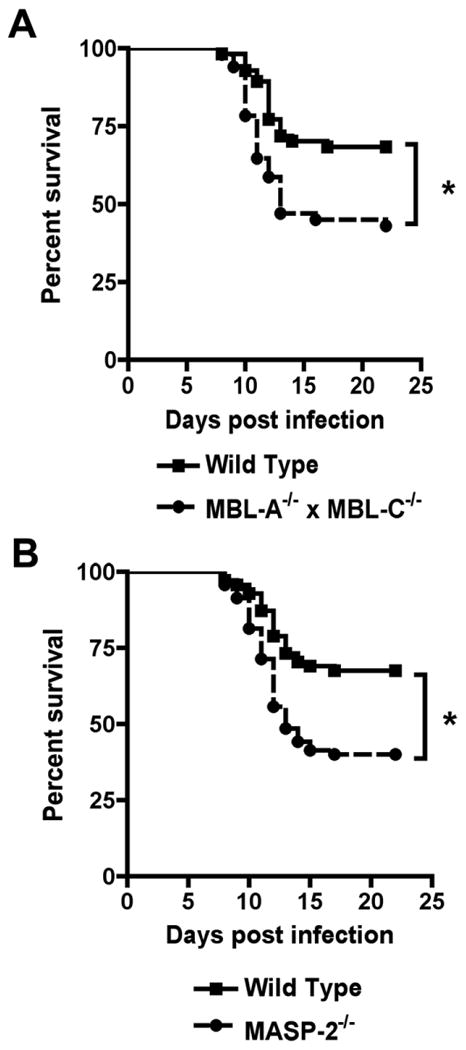
Mice deficient in components of the lectin pathway are more susceptible to lethal WNV infection. Wild type (A, n = 78; B, n = 71), MBL-A-/- × MBL-C-/- (n = 65) (A), or MASP-2-/- (n = 70) (B) C57BL/6 mice were infected subcutaneously with 102 PFU of WNV. Survival was monitored over the course of 22 days. Asterisks denote differences that were statistically significant (P < 0.05) as judged by the log rank test.
MBL proteins associate with MASP molecules to trigger downstream complement activation, with MASP-2 believed to be the functionally dominant serine protease. Infection of congenic MASP-2-/- mice with WNV showed a similar phenotype of modestly increased susceptibility to lethal infection (Fig 1B, P < 0.0005). Overall, an absence of key components of the lectin activation pathway resulted in increased susceptibility to WNV infection, although the difference was less than that observed with deficiencies in either the classical or alternative pathways.
Viral burden in wild type and MBL-A-/- × MBL-C-/- mice after subcutaneous infection
To begin to determine the cause for the higher mortality, we measured viral burden in tissues from WNV-infected wild type and MBL-A-/- × MBL-C-/- mice at several time points after infection. Because of its ability to neutralize insect cell-derived WNV, we hypothesized that MBL would limit viral spread at early time points in lymphoid tissues or the intravascular compartment. Surprisingly, no statistical difference in magnitude or kinetics of WNV infection was observed in the draining inguinal lymph node, serum, or spleen of wild type mice and MBL-A-/- × MBL-C-/- mice (Fig 2A, B, and C, P > 0.1). At day one after infection, we found slightly lower levels of WNV RNA that did not achieve statistical significance in the draining lymph node of MBL-A-/- × MBL-C-/- mice, as half of the samples had undetectable levels of viral RNA. However, by day 2, no difference in WNV RNA was observed, confirming that viral dissemination in the draining lymph nodes was not significantly affected by the absence of MBL-A and MBL-C.
Figure 2.
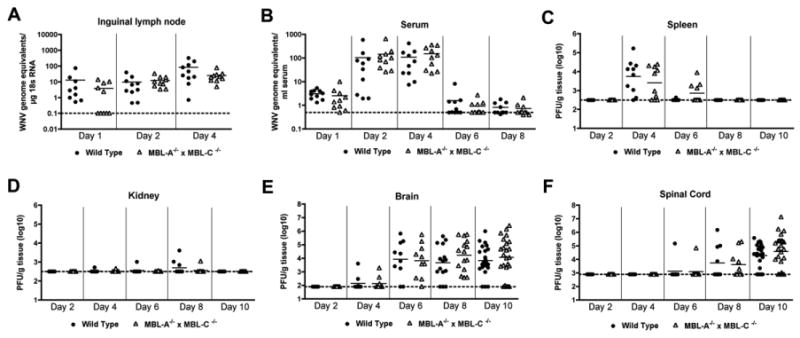
WNV tissue burden and spread in mice after subcutaneous infection. MBL-A-/- × MBL-C-/- and wild type mice were infected subcutaneously with WNV. At the indicated time points, tissues were harvested and analyzed for viral titers by quantitative RT-PCR ((A-B) inguinal lymph node and serum) or plaque assay ((C-F) spleen, kidney, brain, spinal cord). Data from 10 to 24 mice per time point are shown as WNV genome equivalents per μg 18S RNA (A, B) or as PFU per gram of tissue (C-F). Dotted lines indicate the limit of detection of the assay.
In several immunodeficient mice lacking key proteins that function in innate and adaptive immunity, WNV replicates to high levels in visceral tissues such as the kidney or liver. However, MBL-A-/- × MBL-C-/- mice showed little capacity to sustain WNV infection in these tissues (Fig 2D). Consistent with these findings, dissemination to the brain and spinal cord occurred with the same kinetics in wild type and MBL-A-/- × MBL-C-/- mice. Beginning at days 6 and 8, WNV was detected in the brain and spinal cord of wild type and MBL-A-/- × MBL-C-/- mice (Fig 2E and F). While we observed an overall trend towards increased WNV replication in the CNS of MBL-A-/- × MBL-C-/- mice, this did not reach statistical significance despite the inclusion of large numbers of mice (P > 0.2, n = 15 (day 8); n = 24 (day 10)).
As seen previously in adult C57BL/6 mice, the viral burden data in the brain and spinal cord did not fit a Gaussian distribution; while most mice sustained high titers, a subset were below the limit of detection (∼102 to 103 PFU/g) of the plaque assay. The latter data points may reflect a relatively low or complete absence of CNS infection in this 8 to 12 week-old age cohort. As the few negative data points could create sufficient variability to limit detection of differences in the cohort of mice that showed definitive CNS infection, we stratified the viral burden data and compared samples with documented spread to the brain and spinal cord. Using this analysis, differences in viral burden at day 10 between wild type and MBL-A-/- × MBL-C-/- mice while still small, became more pronounced in the brain (1.3 × 104 versus 4.3 × 104 PFU/g, P = 0.07) and spinal cord (4.5 × 104 versus 1.4 × 105 PFU/g, P = 0.05). Overall, an absence of MBL resulted in a small increase in WNV lethality that was associated with relatively modest differences in viral replication at late times after infection in the CNS.
WNV replication in wild type and MBL-A-/- × MBL-C-/- mice after intracranial infection
The modest increase in viral infection in the CNS of MBL-A-/- × MBL-C-/- mice at only late time points suggested a possible direct innate immune effect of MBL that restricted replication in neuronal tissues. Prior studies have established that MBL and MASP are deposited in brain tissue during inflammatory or ischemic insults. To assess whether the lectin pathway could have a more direct effect in the CNS, we infected wild type and MBL-A-/- × MBL-C-/- mice with WNV by an intracranial route and measured viral titers in different regions of the CNS at days 2, 4 and 6 after infection. WNV replicated rapidly in neuronal tissues, reaching high (1 to 5 × 107 PFU/g) titers at days 4 and 6 after infection in multiple regions including the cortex, brainstem, cerebellum, and spinal cord. However, no difference in the kinetics of spread or magnitude of replication was observed in CNS tissues from MBL-A-/- × MBL-C-/- mice (Fig 3, P > 0.6). These results suggest that the increased viral burden in CNS tissues at late times after subcutaneous infection was not due to a direct effect of MBL.
Figure 3.
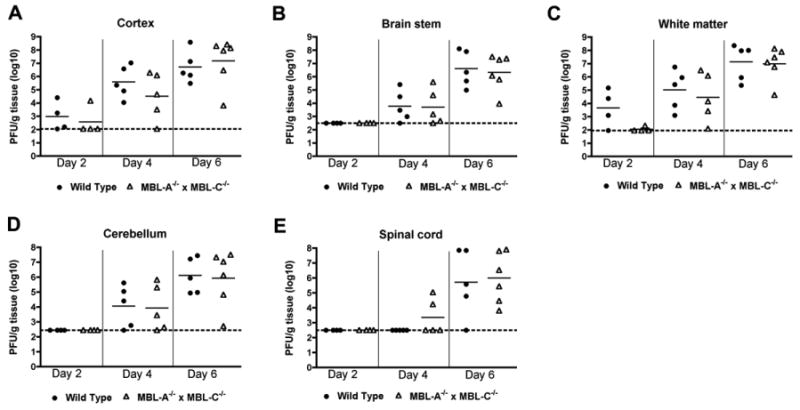
WNV tissue burden and spread in the CNS after direct intracranial inoculation. MBL-A-/- × MBL-C-/- and wild type mice (n = 4 to 6 mice per time point) were infected intracranially with 101 PFU of WNV, and CNS regions (A, cerebral cortex; B, brain stem; C, white matter; and D, cerebellum) and (E) spinal cord were harvested at indicated time points. Viral titers were determined by plaque assay. Data are shown as PFU per gram of tissue. Dotted lines indicate the limit of detection of the assay.
The effect of an MBL deficiency on T and B cell responses to WNV
Given the intracranial challenge results, we hypothesized that the differences in viral burden and mortality after subcutaneous WNV infection could be due to altered adaptive immunity. Complement activation is known to augment adaptive immune responses by facilitating antigen uptake and presentation and lowering the threshold for B cell activation. Moreover, prior studies have shown defects in adaptive B and T cell responses in mice lacking components of the classical and alternative pathways of complement activation. However, detailed studies on the development of antigen-specific immune responses against pathogens in MBL-A-/- × MBL-C-/- mice have not been performed. To investigate the contribution of MBL to adaptive immune responses, we examined WNV-specific T cell and antibody responses.
To evaluate WNV-specific T cell responses, wild type and MBL-A-/- × MBL-C-/- mice were infected subcutaneously with WNV. At day 7, the percentage and number of IFN-γ-secreting splenic CD4+ and CD8+ T cells after ex vivo restimulation with immunodominant WNV peptides or PMA and ionomycin was measured by flow cytometry (Fig 4A, B, and C). Notably, spleens from wild type and MBL-A-/- × MBL-C-/- mice contained similar percentages and numbers of stimulated and WNV-specific CD4+ and CD8+ T cells (Fig 4A, B and data not shown). To confirm that the WNV-specific CD8+ T cells were equivalently cytolytic in MBL-A-/- × MBL-C-/- mice, an in vivo killing assay was performed. At 8 days after WNV infection, CD45.2+ wild type or MBL-A-/- × MBL-C-/- mice were injected with a mixture of CFSE-labeled WNV peptide-pulsed CD45.1+ splenocytes and unlabeled, unpulsed CD45.1+ control splenocytes as target cells. Six hours later, spleens were harvested and the percentage of remaining WNV peptide-pulsed target cells was determined. Compared to wild type mice, MBL-A-/- × MBL-C-/- mice showed slightly lower (69 versus 83%) levels of antigen-specific cytotoxicity but this difference did not attain statistical significance (Fig 4D and E, P > 0.2). Overall, our analysis of WNV-specific T cell responses did not provide compelling evidence for an essential role of MBL in generating peripheral CD4+ and CD8+ responses against WNV.
Figure 4.
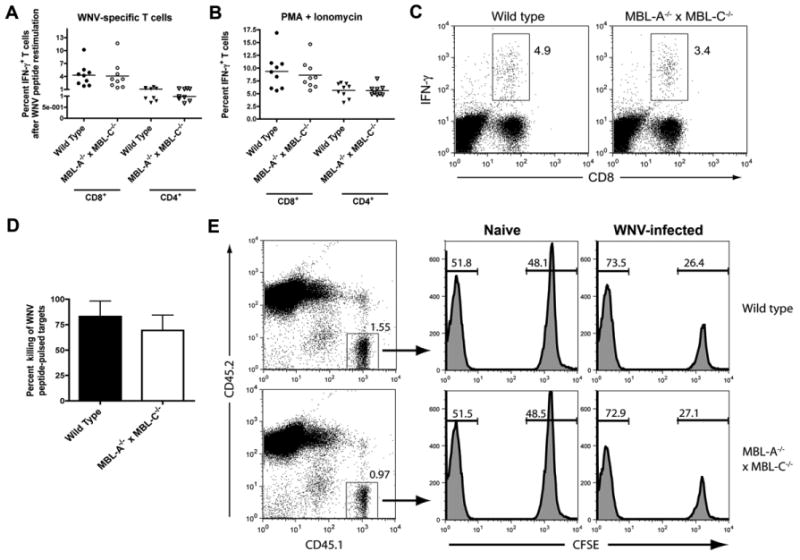
WNV-specific T cell responses remain intact in MBL-A-/- × MBL-C-/- mice. A-C. Wild type and MBL-A-/- × MBL-C-/- mice were infected subcutaneously with 102 PFU of WNV. Seven days later, spleens were harvested and splenocytes were (A) restimulated in vitro with Db-restricted WNV NS4b peptide or I-Ab-restricted WNV NS3 peptides or (B) activated with PMA and ionomycin. Six hours later, CD8+ and CD4+ T cells were analyzed for the expression of intracellular IFN-γ by flow cytometry. Data are combined from three independent experiments with a total n = 9 mice per group. C. A representative flow cytometry dot plot showing intracellular IFN-γ staining following Db-restricted WNV NS4B peptide re-stimulation comparing samples from wild type or MBL-A-/- × MBL-C-/- mice. Numbers in dot plots indicate the percentage of IFN-γ+ cells within the CD8+ T cell population. D-E. CD8+ T cell cytotoxicity function remains intact in MBL-A-/- × MBL-C-/- mice. Eight days after subcutaneous WNV infection, wild type or MBL-A-/- × MBL-C-/- CD45.2+ mice were given CD45.1+ target cells (CFSE+ WNV NS4b-pulsed naïve splenocytes or CSFE- control unpulsed splenocytes) by intravenous administration. Six hours later, mice were sacrificed and splenocytes were analyzed for the ratio of peptide pulsed to non-pulsed cells by interrogating cells in the CD45.1 gate. D. The percentage of in vivo killing was calculated by determining the ratio of peptide-pulsed versus unpulsed cells for each mouse, and by normalizing to that seen in naive mice. Data is combined from two independent experiments with a total of four WNV-infected mice per group. E. Representative flow cytometry plots showing differential killing of target cells in naive and WNV-infected mice.
Although peripheral T cell responses against WNV appeared normal in MBL-A-/- × MBL-C-/- mice, we assessed whether leukocyte migration to the inflamed brains might be impaired, as this also can result in enhanced viral replication in CNS tissues. We infected wild type and MBL-A-/- × MBL-C-/- mice subcutaneously with WNV and analyzed CD45+ leukocyte subsets within brains at day 8 by flow cytometric analysis. Notably, we observed no difference in accumulation of WNV-specific CD8+ T cells between wild type and MBL-A-/- × MBL-C-/- mice as judged after restimulation of brain leukocytes with a Db-restricted WNV immunodominant NS4B peptide (Fig 5A and B). Analogously, no significant difference in the leukocyte subset composition was detected between wild type and MBL-A-/- × MBL-C-/- mice, as the total number of CD45+ leukocytes, CD4+ T cells, CD8+ T cells, CD19+ B cells, activated macrophages and microglia was equivalent (Fig 5C and data not shown). Thus, an absence of MBL-A and MBL-C did not alter the trafficking or accumulation of antigen-specific or other inflammatory leukocytes in the brain after WNV infection.
Figure 5.
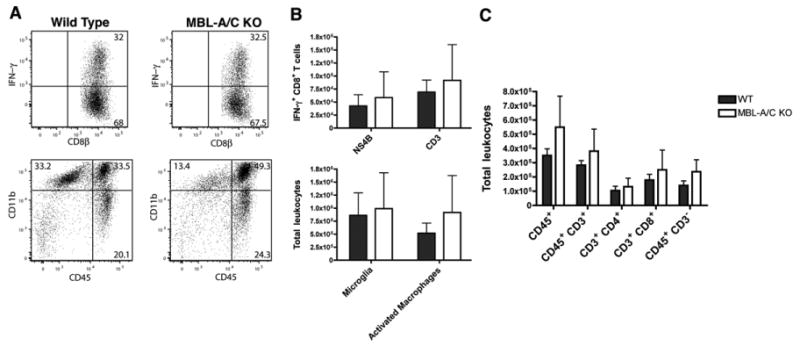
MBL-A-/- × MBL-C-/- mice show similar accumulation of leukocytes in the brain following WNV infection. Wild type and MBL-A-/- × MBL-C-/- mice were infected subcutaneously with WNV. At day 8 after infection, brains were harvested and the leukocyte compartment within brain tissue was analyzed by flow cytometry for the (C) percentage and numbers of total leukocytes, T cell subsets, CD11b+ CD45- microglia and CD11b+ CD45+ activated macrophages A-B. Leukocytes were restimulated in vitro with a Db-restricted NS4B peptide, and percentages and total numbers of WNV-specific IFN-γ-secreting CD8+ T cells (after NS4B peptide stimulation) or total activated IFN-γ+ CD8+ T cells (after anti-CD3 stimulation) was determined. Data was pooled from experiments with 5 to 8 individual mice.
We next measured WNV-specific antibody responses in wild type and MBL-A-/- × MBL-C-/- mice. Levels of WNV E protein-specific IgM and IgG in sera from infected mice were equivalent at days 6, 8 and 10 (Fig 6A and B, P > 0.5). Effective anti-WNV antibody responses in C57BL/6 mice are characterized by a dominance of complement-fixing IgG2b and IgG2c antibody isotypes, whereas WNV-specific IgG1 is typically observed at lower levels. This pattern was sustained in MBL-A-/- × MBL-C-/- mice, where antigen-specific IgG2b levels were equivalent to those in wild type sera (Fig 6C, P > 0.9). To compare the functional activity of WNV-specific antibodies, we performed a focus reduction neutralization assay in which virions were pre-incubated with serial dilutions of heat-inactivated serum from infected animals. We observed a small (1.4-fold) reduction in neutralizing activity of sera from MBL-A-/- × MBL-C-/- mice at day 8 after infection that attained statistical significance (Fig 6C, P < 0.05). Neutralizing antibody titers, however, were not significantly different at day 10 after infection. Overall, a deficiency of MBL had modest effects on antigen-specific antibody production, with only a transient impact on neutralizing antibody titers.
Figure 6.
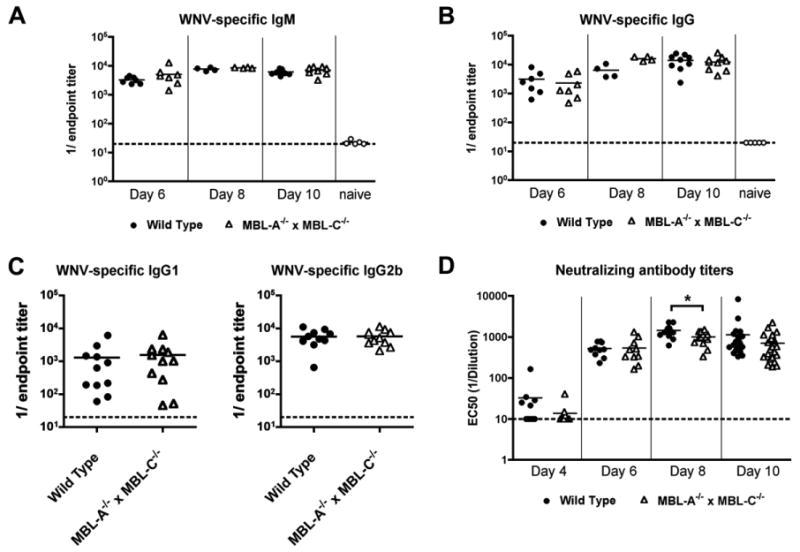
MBL-A-/- × MBL-C-/- mice generate efficient WNV-specific antibody responses during primary infection. Wild type and MBL-A-/- × MBL-C-/- mice were infected subcutaneously with WNV. At the indicated time points, serum was harvested, serially diluted, and WNV E protein-specific IgM (A) and IgG (B) were measured by ELISA using an endpoint titer analysis. C. At day 10, WNV-specific IgG1 and IgG2b were measured using isotype-specific secondary antibody reagents. Data are shown as reciprocal endpoint titers. D. Sera from the indicated days after infection also was tested for neutralizing activity using a focus reduction assay. Indicated data points represent serum dilutions that reduced focus formation by 50%. Asterisks denote differences that were statistically significant (P < 0.05) as judged by the Mann-Whitney test. Data are from 10 to 23 mice per group. Dotted lines indicate the limit of detection of the assay.
Discussion
This report extends our understanding of how different complement activation pathways confer protection against WNV, a pathogenic enveloped RNA virus. We demonstrate that mice deficient in the lectin pathway recognition and activation (MBL-A-/- × MBL-C-/- or MASP-2-/-) show a small but statistically significant increased mortality rate after WNV infection. This finding indicates that MBL recognition of WNV contributes to protection against WNV infection. Somewhat unexpectedly, a deficiency of lectin pathway activation did not alter the kinetics or the magnitude of viral spread to different organs, nor did it profoundly affect development of adaptive immune responses.
Prior studies demonstrated that mice lacking the central complement component C3 were completely vulnerable to peripheral inoculation with the same low dose (102 PFU) of virulent WNV. In comparison, C1q-/- mice, which fail to activate the classical pathway efficiently, or factor B-/- mice, which are defective in alternative pathway activation, showed higher but incomplete (83% and 96%, respectively) mortality rates at the same viral dose compared to the ∼30% observed in congenic wild type mice. A role for the C4-dependent and C1q-independent lectin pathway of complement activation in restricting WNV infection was inferred because the phenotype of C4-/- mice was more severe than that observed with C1q-/- mice. Our results confirm that the lectin pathway is essential for efficient control of WNV infection. However, the rather subtle nature of the phenotype, at both the virological and immunological level, suggests that lectin pathway activation in vivo is less essential than classical or alternative pathway activation against WNV.
At first glance, these results appear to contrast with our recent study in which MBL bound to high- or pauci-mannose-containing carbohydrates on insect cell-derived WNV and neutralized viral infectivity in vitro. Based on cell culture experiments, we predicted that MBL served as an extracellular pattern recognition element that neutralized WNV directly. Surprisingly, we did not observe early or more rapid dissemination of WNV or a defect in adaptive immune priming in MBL-A-/- × MBL-C-/- mice. WNV infection spread with similar kinetics in blood and tissues, and the disease course was not accelerated compared to wild type mice. Rather, the increased mortality rate in MBL-A-/- × MBL-C-/- mice was associated with small differences in viral tissue burdens at late time points after infection in the CNS. However, in the original study, MBL had potent neutralizing functions only on WNV derived from insect cells, and not on virus generated in mammalian cells, because the N-linked glycans on the virion failed to bind MBL efficiently after processing to complex and hybrid carbohydrates. Thus, after subcutaneous infection, once WNV undergoes one round of replication in mouse cells, it becomes resistant to MBL-mediated neutralization. In comparison, mammalian cell-generated Dengue virus (DENV), a related flavivirus, remains susceptible to MBL-mediated neutralization (Fuchs 2010). This is due to the display of a second N-linked glycan on the viral envelope protein that retains a high mannose glycan even when produced in mammalian cells. We predict that MBL may have greater protective functions against viruses such as DENV, which display ligands for MBL after animal infection. Unfortunately, since inbred mice inherently are highly resistant to DENV infection with the exception of those lacking both type I and II IFN signaling, currently, it is not possible to study the function of MBL in vivo against DENV using the available deficient mice.
Complement deficiencies can impact adaptive immune responses, as seen in CR2-/- or C3-/- mice in which antibody and/or T cell responses are attenuated, respectively. A specific role for MBL in modulating antigen-specific IgM and IgG responses has been described. However, some of the observed differences in antibody production may be due to use of a mixed genetic background form of the MBL-A-/- × MBL-C-/-. Indeed, no difference in antibody response was observed by another group that used fully-backcrossed mice on the C57BL/6 background. Our experiments revealed only small differences in the quality of neutralization or quantity of antibody response or antigen-specific function of CD4+ or CD8+ T cells in the context of WNV infection in fully-backcrossed MBL-A-/- × MBL-C-/- mice.
In addition to MBL, the lectin pathway can be activated independently by ficolins, which recognize alternative carbohydrate ligands, such as acetylated or sulfated oligosaccharides. It is conceivable that ficolins could substitute and thus explain the relatively modest viral and immunological phenotypes in MBL-A-/- × MBL-C-/- mice. Two experiments suggest against this: (a) MBL-A-/- × MBL-C-/- mouse serum (which contains ficolins) was unable to deposit complement or neutralize WNV in vitro; and (b) although ficolin-mediated complement activation bypasses a requirement for MBL, it still requires MASP-2, the major serine protease that activates C4 and C2 after MBL or ficolin binding. The rather subtle phenotype of MBL-A-/- × MBL-C-/- mice in vivo after WNV infection independently was confirmed in MASP-2-/- mice, which also showed modestly increased mortality yet small differences in tissue viral burden (data not shown). Nonetheless, a definitive confirmation of the role of the lectin pathway in immunity to WNV may await infection studies with mice with multiple deficiencies (MBL-A-/- × MBL-C-/- × ficolin-/- or MASPnull (MASP-1/3-/- × MASP-2-/-)), which have not been generated or remain unavailable.
MBL has previously been suggested to contribute to protective immunity against several pathogens, including bacteria, viruses, fungi, and parasites. Analogous to our studies, direct MBL binding and modest neutralization of HSV-2 virions was demonstrated. Infection of MBL-A-/- × MBL-C-/- mice with HSV-2 resulted in higher viral loads in liver, but not in spleen or brain. In other studies, decreased survival of MBL-A-/- × MBL-C-/- mice after Staphylococcus aureus or Candida albicans infection was attributed to altered cytokine responses, reduced phagocytosis, or delayed leukocyte infiltration into affected tissues. MBL has been reported to interact with Toll-like receptors 2 and 6 to promote cytokine secretion in response to pathogen recognition. Thus, it remains possible that the modestly enhanced susceptibility of MBL-A-/- × MBL-C-/- mice during WNV infection is modulated in part, by altered cytokine responses, although this did not appear to affect viral titers in blood or peripheral tissue compartments.
This study expands our knowledge of the in vivo mechanisms of complement-mediated antiviral immunity. While MBL and the lectin activation pathway contribute to control of WNV infection in vivo, the phenotype was subtle compared to that observed with deficiencies in components of the classical or alternative activation pathways. For WNV, an arthropod-borne flavivirus, the modification of N-linked glycans on the virion structural proteins to complex carbohydrates, which occurs within one round of replication in animal cells, likely overcomes much of the direct inhibitory effect of MBL. Although we observed slightly higher WNV levels in the brain in MBL mice, which correlate with an enhanced mortality rate, the rather modest phenotype precluded further delineation of the specific mechanism. Based on extensive immunological analysis, a deficiency in MBL did not cause major defects in adaptive B or T cell responses. Although the studies with WNV suggest a relatively small effect, it remains intriguing to consider that severe infection of other flaviviruses (e.g., DENV) in humans could be associated with MBL deficiencies or polymorphisms that result in low expression or functionally impaired MBL.
Materials and Methods
Mice
MBL-A-/- × MBL-C-/- mice on a C57BL/6 background and congenic wild type mice were purchased from Jackson Laboratories. MASP-2-/- mice on a C57BL/6 background were obtained commercially (Omeros, Inc). All mice were housed in a pathogen-free mouse facility at Washington University School of Medicine. Studies were performed in compliance and with approval of the Washington University School of Medicine Animal Safety Committee. B6.SJL (CD45.1) mice were purchased from Taconic.
Cell lines and virus stocks
BHK21-15, Vero, and C6/36 Aedes albopictus cells were propagated as previously described (Diamond et al., 2003). The New York strain of WNV (WNV-NY) was generated from an infectious cDNA clone (strain 385-99) as described previously, and passaged once in C6/36 Aedes albopictus insect cells. BHK21-15 cells were used to measure viral titer of infected cells or tissues by plaque assay.
Infection of mice
For in vivo studies, age- and gender-matched 8 to 12 week old mice were infected with 102 PFU of WNV diluted in Hanks balanced salt solution (HBSS) supplemented with 1% heat-inactivated fetal bovine serum (FBS). Virus was administered subcutaneously (by footpad injection in a volume of 50 μl). Intracranial inoculation was performed by injecting 101 PFU of WNV in 10 μl HBSS/1% heat-inactived FBS. Virus preparations were tested for endotoxin (Associates of Cape Cod) and found to be less than 0.25 EU/ml.
Quantification of viral tissue burden and viremia
To monitor viral spread in vivo, mice were infected as described above, and sacrificed at specific time points after inoculation. After extensive cardiac perfusion with PBS, organs were harvested, weighed, homogenized, and virus titers were determined by standard plaque assay on BHK21 cells as described. Viral burden in serum and lymph nodes was determined by quantitative RT-PCR using WNV-specific primers and probe as described. Lymph node RNA was normalized by measuring 18S rRNA content in parallel using the 18S Taqman kit from Applied Biosystems.
Antigen-specific T cell responses
WNV-specific CD4+ and CD8+ T cell responses were measured after restimulation of splenic T cells ex vivo with WNV specific peptides followed by intracellular cytokine staining for IFN-γ as described previously. Briefly, seven days after infection mice were euthanized and splenocyte suspensions (2 × 107 cells/well) prepared and cultured in 96 well plates in the presence of Golgi Plug (BD Biosciences) and 1μM of the Db-restricted NS4b peptide or the IAb-restricted NS3 peptides. Six hours later, cells were washed and stained with fluorochrome-labeled antibodies to CD4 and CD8 (BD Biosciences). Cells were fixed in 2% paraformaldehyde, permeabilized in saponin buffer (0.5 % saponin, 0.1% BSA in PBS), and incubated with Alexa 647-conjugated anti-IFN-γ antibody (eBioscience). Cells were then washed and analyzed on a FACSCalibur cytometer (BD Biosciences). The frequency and number of WNV-specific T cells was determined based on the percentage of IFN-γ+ cells within the CD4+ and the CD8+ T cell gate following peptide stimulation. Staining on unstimulated or PMA (125 ng/ml) and ionomycin (5 μg/ml)-treated splenocytes was performed as negative and positive controls, respectively.
CD8+ T cell cytotoxicity was evaluated using an in vivo target cell killing assay. As targets, splenocytes from B6.SJL (CD45.1) mice were harvested and divided into two tubes. Half of the cells were labeled with 1 μM CFSE according to standard protocols (Molecular Probes). Labeled cells were incubated for 1 h at 37 °C with 1 μM of the Db-restricted NS4b peptide. Cells were washed, counted and mixed (1:1 ratio) with unlabeled cells that were not exposed to peptide. The cell mixture (107 total cells) was injected intravenously into naïve or WNV-infected (day 8 after inoculation) CD45.2 WT or MBL-A-/- × MBL-C-/- mice. After 6 hours, mice were sacrificed and splenocytes were incubated with conjugated antibodies to CD45.1 and CD45.2. Target cells were identified by gating on CD45.1+ splenocytes. The percentage of killing was calculated by determining the ratio of CFSE+ cells versus CFSE- cells for each WNV-infected mouse, and by normalizing for the ratio seen in naive mice: (1 (ratio WNV infected/ratio naive)) × 100.
Analysis of infiltrating leukocytes in the brain
Brain infiltrating CD4+ and CD8+ T cells, macrophages and microglia were isolated and quantified as previously described. Intracellular IFN-γ staining of brain lymphocytes was performed as described above. CD3 stimulation was performed with hamster anti-mouse CD3 (clone 145-2C11, BD Bioscience) for six hours in the presence of Golgi Plug prior to intracellular staining for IFN-γ. Lymphocytes were processed on an LSRII (BD Biosciences) using FACS Diva 6.1.1 (BD Biosciences) and analyzed with FlowJo software (Treestar). Additional antibodies for six-color analysis included: PerCP-Cy5.5 anti-CD8β (Biolegend), Pacific blue anti-CD4 (Invitrogen), Alexa 647 anti-IFN γ (Biolegend), Alexa-700 anti-CD45 (Biolegend), and PE anti-CD11b (BD Bioscience). The total numbers of specific cells was determined by multiplying the percentage of positive cells by the total numbers of brain lymphocytes harvested.
WNV-specific antibody responses
WNV-specific antibody titers were measured by ELISA as described previously. Briefly, individual wells of a microtiter plate were coated overnight at 4°C with 5 μg/ml of recombinant WNV E protein. Plates were washed and blocked with 1% bovine serum albumin in PBS/0.05% Tween 20 (blocking buffer). Serum samples from naive or WNV-infected mice were heat inactivated (56°C for 30 minutes), serially diluted in blocking buffer, and added to wells for 1 hour at 37 °C. Following several washes, biotin-conjugated goat anti-mouse IgG or IgM (Sigma Aldrich), IgG1 or IgG2b (BD Biosciences) (1 μg/ml in blocking buffer) were added for 1 hour at room temperature. Plates were then washed and incubated with streptavidin-conjugated horseradish peroxidase (2 μg/ml in blocking buffer; Vector Labs) for 1 hour at room temperature. After several washes, plates were developed with tetramethylbenzidine substrate (Pierce) and the reaction was stopped with 1N H2SO4. The OD450 was measured and adjusted by subtracting the OD450 of BSA-control wells. Best-fit curves were calculated using Prism (GraphPad Software) for each serum dilution series and endpoint titers were determined as three standard deviation units above background signal.
The neutralizing activity of serum antibodies against WNV was assessed using a focus reduction neutralization assay that was modified from a published protocol for Dengue virus. WNV (2.4 × 102 PFU in DMEM with 5% FBS) was pre-incubated with serial dilutions (in DMEM with 5% FBS) of heat-inactivated mouse serum at 37°C for 30 minutes. Virus-serum mixtures were added in duplicate to individual wells of tissue culture 96-well microtiter plates containing Vero cells at ∼90% confluency. Virus was incubated with Vero cells for 1 hour at 37°C, after which wells were overlaid with 1% carboxymethylcellulose (Sigma) in MEM with 4% FBS. After culture at 37°C for 24 hours, overlay media was removed and wells were washed with PBS. Cells were fixed with 1% paraformaldehyde in PBS (10 minutes at room temperature) and permeabilized with 0.1% saponin and 0.1% BSA in PBS (saponin buffer; 5 minutes at room temperature). Cells were stained with humanized E16 (50 μl at 200 ng/ml in saponin buffer) for 2 hours at room temperature. Following several washes, wells were incubated with horseradish peroxidase-conjugated anti-human IgG antibody (Sigma; 250 ng/ml in saponin buffer) for 1 hour at room temperature. Wells were washed and infectious foci were visualized with TrueBlue Substrate (KPL) after a 5 to 10 minute incubation at room temperature. Wells were rinsed with water and dried prior to analysis with a Biospot counter (Cellular Technology) using Immunocapture software. Neutralization curves (% reduction in spot numbers in samples pre-incubated with serum compared to wells with virus pre-incubation with medium alone) were graphed using Prism software, and EC50 values calculated.
Statistical analysis
In vitro experiments were analyzed with a two-tailed T-test. To analyze viral tissue burdens, differences in log titers were analyzed by the Mann-Whitney test. Kaplan-Meier survival data was analyzed by the log rank test. All statistical analyses were performed with Prism software.
Acknowledgments
We thank E. Mehlhop and M. Vogt for performing pilot studies with the MBL-A-/- × MBL-C-/- mice. We thank C. Tedford (Omeros, Inc) for shipment of the MASP-2-/- mice. This work was supported by a grant from the NIH (the Midwest Regional Center of Excellence for Biodefense and Emerging Infectious Diseases Research (U54 AI057160)).
Abbreviations
- WNV
West Nile virus
- DENV
Dengue virus
- CNS
central nervous system
- MBL
mannose binding lectin
- MASP
mannan-associated serine protease
- PFU
plaque forming units
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.