

Abstract
Environmental estrogen mimics, including metalloestrogens that can activate estrogen receptor-alpha (ERα), may contribute to breast cancer risk. However, the underlying mechanisms through which these molecular mimics activate the estrogen receptor-alpha are generally, poorly understood. With concern to this important question, we investigated whether intracellular calcium may mediate the crosstalk between signaling pathways that activate ERα and the ligand binding domain of ERα. MCF-7 cells treated with EGF, ATP, extracellular calcium, or caffeine to increase intracellular calcium triggered a rapid recruitment of ERα to estrogen responsive promoters and stimulated expression of estrogen responsive genes including pS2, complement C3, and progesterone receptor. Induction was blocked by an antiestrogen but also by the chelation of intracellular calcium. Treatment with extracellular calcium also increased the growth of MCF-7 cells through an ER dependent mechanism. We found that EGF and extracellular calcium activated the C-terminus of ERα and the activation was blocked by the antiestrogen. Mechanistic investigations identified four potential sites on the solvent accessible surface of the ERα ligand binding domain as important for calcium activation of the receptor. Taken together, our results suggest that calcium mediates the crosstalk between ERα–activating signaling pathways and the ligand binding domain of ERα providing a potential explanation for the ability of certain environmental metalloestrogens to activate the receptor.
Keywords: estrogen receptor-alpha, cross talk, calcium
Introduction
Estrogen receptor-α (ERα) is a ligand inducible transcription factor that belongs to the superfamily of nuclear receptors (1). Upon hormone binding, ERα is activated and regulates the transcription of target genes. Similar to other nuclear receptors, ERα is divided into regions A through F (2). The N-terminal A/B region harbors transactivation function-1 (AF-1) that plays a role in ligand dependent and independent activation of the receptor. Region C is the DNA binding domain and region D is the hinge region. Region E is the hormone, or ligand, binding domain (LBD). It is responsible for ligand dependent activation of transactivation function-2 (AF-2). It is also important for dimerization and binds coactivators and corepressors (3). In the absence of hormone, ERα is associated with a complex containing heat shock proteins that render the receptor inactive. Upon hormone binding, a conformational change occurs in the LBD that results in the dissociation of the heat shock complex and the formation of the AF-2 domain (4). Hormone binding also results in the phosphorylation of serines in the A/B region that increases the activity of AF-1 (5,6).
Activation of signal transduction pathways also activates ERα (7–9), yet, the mechanisms are not fully understood. Growth factors, such as epidermal growth factor (EGF) and insulin like growth factor-1, activate the Ras-Raf-MEK-MAPK kinase and PI3K-AKT pathways resulting in the phosphorylation of serines in the N-terminal A/B region of the receptor (10). Although phosphorylation of the A/B region is thought to be responsible for the activation of the AF-1 function, it does not account for the conformational changes necessary for the activation of the AF-2 function in the C-terminal LBD suggesting that additional intracellular events are required for the cross talk between signal transduction pathways and ERα.
Metalloestrogens are metals that activate ERα in the absence of estradiol. Metalloestrogens fall into two subclasses, anions and bivalent cations that include cadmium (11–17). The ability of metalloestrogens to activate ERα is due to their ability to form a high affinity complex with the LBD of the receptor (13–16). Cadmium and calcium have similar properties (18) and cadmium replaces calcium in biological assays/systems (18–20) suggesting that metalloestrogens may mimic calcium and that calcium is an intracellular ligand of the LBD. The results of this study suggest that calcium mediates the cross talk between signal transduction pathways and the LBD of ERα.
Materials and Methods
Tissue culture
MCF-7 and MCF-7/LCC cells were obtained from the Tissue Culture Shared Resource, Georgetown University and authenticated by the resource using short tandem repeat profiling (PowerPlex 1.2, Promega) in March and October 2010. MCF-7 cells were grown as previously described (21). Two days prior to treatment, the medium was changed to phenol red-free IMEM supplemented with fibronectin, glutamine, HEPES, and transferrin. For calcium treatments, the media was changed to calcium- and phenol red-free DMEM supplemented with 5% charcoal stripped calf serum (CCS). MCF-7/LCC2 cells were grown in phenol red-free IMEM with 5%CCS and treated in phenol red-free IMEM.
Real time RT-polymerase chain reaction
The real time RT-PCR assay was performed as described (21). The reaction contained 1ug cDNA, Universal Master Mix, and Assay on Demand. The samples were run on the 7900HT and the data were analyzed by the 2−∆∆Ct method using the SDS 2.1 software (Applied Biosystems). Statistical analyses were performed using two-tailed and paired t-tests.
Chromatin immunoprecipitation (ChIP)
The ChIP assay was performed as previously described (21). Briefly, the cells were treated with formaldehyde, washed with PBS, and disrupted by sonication. The ERα was immunoprecipitated (H-184, Santa Cruz Biotech), the crosslink was reversed, and the amount of DNA was measured with a PCR assay.
Transient transfection
COS-1 cells (75,000/well) were plated in phenol red-free IMEM supplemented with 5%CCS and transfected with 2μg DNA containing 0.15μg ERα or ERα mutants (2,22-25), 1.5μg ERE-chloramphenicol acetyl transferase (CAT) construct, 0.07μg β-galactosidase, 7.5μl Superfect (Quigen), and 75μl phenol red-free IMEM media and 400μl IMEM with 5%CCS. As a control, cells were transfected with glucocorticoid receptor (GR) and MMTV-CAT. The media was replaced 3–4 hours later and changed to phenol red-free IMEM with 5%CCS 18–20 hours after transfection. Cells were treated in phenol red-free IMEM for 24 hours. The CAT and β-galactosidase assays were conducted as described (21). Statistical analyses were performed using paired and unpaired t-tests for the wild type and mutants, respectively.
Site-directed mutagenesis
To generate the mutants D351A, E419A and E423A, QuikChangeRXL Site Directed Mutagenesis (Stratagene) was used. Briefly, the reaction contained 10ng of pRER (22), 100ng/μl primers, 5μl 10x reaction buffer, 1μl dNTP mix, and 1μl PfuTurboR DNA polymerase (2.5U/μl). For the D351A mutant, the forward and reverse primers w e r e5’-GACCAACCTGGCAGCCAGGGAGCTGGTTC-3’ and 5’-GAACCAGCTCCCTGGCTGCCAGGTTGGTC-3’. For E419A, the forward and reverse primers were 5’-CAGGGAAAATGTGTAGCGGGCATGGTGGAGATC-3’ and 5’-GATCTCCACCATGCCCGCTACACATTTTCCCTG-3’. For E423A, the forward primer was 5’-GTAGAGGGCATGGTGGCGATCTTCGACATGCTG-3’ and the reverse primer was 5’-CAGCATGTCGAAGATCGCCACCATGCCCTCTAC-3’. The mutations were confirmed by sequencing. The sequencing primers were 5’-AAGAACAGCCTGGCCTTGT-3’ and 5’-TCCAGAGACTTCAGGGTGCT-3’.
Binding assay
Binding assays were performed using purified recombinant full length (375.8×10−15 moles) and LBD (557×10−15 moles) of human ERα (Invitrogen) in buffer containing 10mM Tris, 20% glycerol, and 2mM DTT. Briefly, ERα was incubated overnight with 45Ca (0.25×10−8M, 0.5×10−8M, 0.75×10−8M, 1×10−8M, 0.25×10−7M, 0.5×10−7M, 0.75×10−7M, 1×10−7M , 1×10−6M, 1.5×10−6M, 2×10−6M; specific activity 20.72mCi/mg; Amersham; Perkin Elmer) in the presence or absence of 100-fold molar excess of CaCl2 (1×10−6-2.5×10−4M). Unbound calcium was removed using DCC. Specific binding was calculated by subtracting non-specific binding from total binding and the binding affinity was determined by nonlinear regression analysis (Prism, GraphPad Software Inc).
Molecular modeling of calcium into potential binding sites
Model construction, visualization, analysis, and molecular dynamics were performed as previously described (21). The calcium motif and the histidine, glutamate, aspartate, asparagine, and cysteine side chain residues were used in the identification of the cation binding sites. Models were constructed with calcium docked manually into different binding sites. Final model building was carried out using the restrained molecular dynamics simulations where the optimum van der Waals and H-bond distance constraints were set between the calcium cation and the potential binding site residues. The quality of the models was checked by comparisonof coordination geometry with the representative models of calcium-binding proteins from the PDB database. Binding models were energy minimized followed by 25 ps molecular dynamics simulations using DISCOVER. In vacuo molecular dynamics simulations followed by energy minimizations on such dimolecular complex produced trigonal coordination geometries. The hydrogen bond distances between calcium and the side chainsranged from 2.5-3.0Å.
Results
Effects of intracellular calcium on ERα activity in MCF-7 cells
To ask whether calcium mediates the cross talk between signal transduction pathways and ERα, several approaches were taken to increase intracellular calcium including treatment with EGF, ATP, extracellular calcium, and caffeine. EGF increases intracellular calcium through the phospholipase C-gamma (PLCγ) pathway, extracellular ATP increases intracellular calcium by activating the P2 purinoreceptors (26), high concentrations of extracellular calcium increase intracellular calcium by activating the calcium sensing receptor and calcium channels (27), and caffeine increases calcium through the release of calcium from intracellular stores and by calcium induced calcium release (28). To determine whether EGF activates ERα through an increase in intracellular calcium, MCF-7 cells were treated with estradiol or EGF in the presence or absence of the antiestrogen ICI-182,780, the EGF receptor inhibitor AG1478, the PLCγ inhibitor U73122, the intracellular calcium chelator BAPTA-AM, and the extracellular calcium chelator BAPTA. As expected, treatment with estradiol resulted in an approximately 4- to 5-fold increase in progesterone receptor (PgR) mRNA that was blocked by the antiestrogen but not by the inhibitor of PLCγ or by the chelation of intracellular calcium (Figs.1A/B). Treatment with EGF resulted in an approximately 2- to 3-fold induction of PgR mRNA that was blocked by the antiestrogen, by the inhibitors of EGF receptor and PLCγ, and by the chelation of intracellular calcium but not by the chelation of extracellular calcium (Figs.1A/B). To determine whether EGF induces other estrogen responsive genes through the calcium pathway, the ability of the intracellular calcium chelator to block the induction of pS2 mRNA was measured (Fig.1B). Treatment with estradiol resulted in an approximately 4.6-fold induction of pS2 mRNA. Treatment with EGF resulted in an approximately 3-fold induction of pS2 mRNA that was blocked by the antiestrogen and the chelation of intracellular calcium.
Figure 1. Effects of EGF and calcium on ERα target genes.

MCF-7 cells were treated for 24 hours with estradiol (1nM), EGF (150ng/ml; A, B), ATP (100uM; C), and calcium (5mM; D) in the presence and absence of the EGF receptor inhibitor AG1478 (1uM), the calcium chelator BAPTA (1uM), the antiestrogen ICI-182,780 (500nM), the PLC inhibitor U73122 (2 μM), or the intracellular calcium chelator BAPTA-AM (1uM). The amount of PgR and pS2 mRNA was measured using a real time RT-PCR assay and normalized to the amount of 18S ribosomal RNA for EGF and ATP treatments or GAPDH mRNA for calcium treatments. Data are expressed as percent control (mean±SD; three independent experiments). *, p < 0.05; **; p < 0.005; ***, p < 0.0005. a, compared to control; b, compared to treatment.
To determine whether increasing intracellular calcium through alternate pathways also activates ERα, MCF-7 cells were treated with ATP, calcium, or caffeine in the presence or absence of the antiestrogen or the intracellular calcium chelator. Treatment with ATP or extracellular calcium resulted in an approximately 1.8- and 5-fold induction of PgR and a 2.1- and 3-fold induction of pS2 mRNA, respectively, that were blocked by the antiestrogen and the intracellular calcium chelator (Figs.1C/D). Treatment with caffeine resulted in an approximately 4-fold induction of PgR mRNA that was blocked by the antiestrogen, the chelation of intracellular calcium, but not by a PKA inhibitor (Fig.S1). Simultaneous treatment with estradiol and calcium did not result in a further increase in the expression of PgR or pS2 mRNA (Fig.S2). To determine whether increasing intracellular calcium recruits ERα to estrogen responsive promoters, the occupancy of the pS2 and complement C3 promoters was examined using a chromatin immunoprecipitation assay. Treatment with estradiol, calcium, or EGF induced a significant increase in ERα occupancy of the pS2 (Fig.2A) and complement C3 (data not shown) promoters.
Figure 2. Effects of EGF and calcium on ERα activity.

A. Effects of EGF and calcium on ERα activity in MCF-7 cells. Cells were treated for 1 hour with estradiol (1nM), calcium (5mM), or EGF (150ng/ml) and the occupancy of ERα on the pS2 promoter was examined using a chromatin immunoprecipitation assay.
B. Effects of calcium on growth. Cells treated with estradiol (1nM) and calcium (0.25, 0.5, 1.0, and 2.0mM) on day 0 or calcium (0.25, 0.5, and 1.0mM) on days 0, 1, 2 and 3 in the presence and absence of the antiestrogen ICI-182,780 (500nM). Cell number was measured on day 4 (mean±SD; three independent experiments). *, p < 0.05; **; p < 0.005; ***, p < 0.0005. a, compared to control; b, compared to treatment.
C. Effects of EGF and calcium on ERα target genes in MCF-7/LCC2 cells. MCF-7/LCC2 cells were treated for 24 hours with estradiol (1nM), EGF (150ng/ml), or calcium (5mM) in the presence and absence of the antiestrogen ICI-182,780 (500nM) or the intracellular calcium chelator BAPTA-AM (1uM). The amount of PgR mRNA was measured using a real time RT-PCR assay and normalized to the amount of 18S ribosomal RNA. Data are expressed as percent control (mean±SD; two independent experiments done in duplicate). *, p < 0.05; **; p < 0.005; ***, p < 0.0005. a, compared to control; b, compared to treatment
To ask whether calcium mimics the effects of estrogen on growth, MCF-7 cells were treated with estradiol or calcium on day 0 in the presence and absence of the antiestrogen and counted on day 4 (Fig.2B). As expected, treatment with estradiol resulted in an approximately 4.3-fold increase in cell number. Treatment with calcium (1.0 and 2.0mM) resulted in an approximately 2.3- and 2.2-fold increase, respectively, that was blocked by the antiestrogen. Cells were also treated with calcium on days 0, 1, 2 and 3 and counted on day 4 (Fig.2B). Daily treatment with calcium (0.25, 0.5, and 1.0mM) resulted in an approximately 2.7-, 3.5-, and 3.1-fold increase in cell number, respectively, that was blocked by the antiestrogen.
To ask whether intracellular calcium mediates the activation of ERα in hormone independent cells, MCF-7/LCC2 cells were treated with estradiol, EGF, or calcium in the presence and absence of an antiestrogen or the intracellular calcium chelator (Fig.2C). Treatment with estradiol resulted in an approximately 8-fold increase in PgR mRNA. Treatment with EGF or calcium resulted in an approximately 2-fold increase in PgR mRNA that was blocked by the antiestrogen and the chelation of calcium demonstrating that similar to estradiol, EGF and calcium activate ERα in hormone independent cells.
To measure intracellular calcium, MCF-7 cells were preincubated with the calcium dye FLUO-4-AM, treated, and the increase in intracellular calcium was detected as an increase in fluorescence (Fig.S3A). Treatment with EGF, extracellular calcium, and ATP increased intracellular calcium from approximately 200.9-224nM to 379nM (+/−100 nM), 550nM (+/−100nM), and 400nM (+/−50nM), respectively, demonstrating an increase in intracellular calcium. Taken together, the data suggest that an increase in intracellular calcium activates ERα.
Effects of EGF and calcium on the activation of ERα
To ask whether EGF and calcium activate the N-terminal AF-1 and/or C-terminal AF-2 domains of ERα, transient transfections were conducted with the HE15 construct that contains the AF-1 and DNA binding domains and the HE19 construct that contains the DNA and ligand binding domains (Fig.3C). COS-1 cells were transiently cotransfected with the mutants, an estrogen responsive-CAT reporter construct, and the β-galactosidase gene and treated with estradiol, EGF, or calcium in the presence and absence of the antiestrogen or the PLCγ inhibitor and the amount of CAT activity was measured (Figs.3A/B). In cells transfected with wtERα, treatment with estradiol resulted in an approximately 3-fold increase in CAT activity that was blocked by the antiestrogen. Treatment with EGF resulted in approximately 3.2-fold increase in CAT activity that was blocked by both the antiestrogen and the PLCγ inhibitor while treatment with calcium resulted in approximately in 3.2-fold induction of CAT activity that was blocked by the antiestrogen. Calcium also increased CAT activity with a time course similar to estradiol (Fig.S4). The GR is not activated by signal transduction pathways (9). As a negative control, cells were transiently cotransfected with GR and MMTV-CAT. Treatment with dexamethsone resulted in an approximately 7.3-fold increase in CAT activity but treatment with estradiol, EGF, or calcium did not induce CAT activity. In cells transfected with the N-terminus of ERα, treatment with estradiol did not increase CAT activity but, as expected, treatment with EGF resulted in a 2-fold increase in CAT. Treatment with calcium resulted in an approximately 1.5-fold increase in CAT activity. In cells transfected with the C-terminus of ERα, treatment with estradiol, EGF, or calcium resulted in an approximately 2.7-, 2-, and 1.8-fold induction of CAT activity that was blocked by the antiestrogen suggesting that EGF and calcium activate the AF-2 function in the LBD. To show an increase in intracellular calcium, cells were preincubated with FLUO-4-AM dye, treated, and intracellular calcium was measured (Fig.S3B). Treatment with EGF, extracellular calcium, or ATP resulted in an increase in intracellular calcium from 150nM to 329nM (+/−100), 400nM (+/−75), and 274nM (+/75), respectively.
Figure 3. Effects of EGF and calcium on the activity of ERα in COS-1 cells.

A,B. COS-1 cells were transiently transfected with wt ERα, truncated ERα mutants HE15 and HEG19, an ERE-CAT reporter or GR and an MMTV-CAT reporter andβ-gal and treated for 24 hours with estradiol (1nM), EGF (150ng/ml), and calcium (1mM) in the presence and absence of the antiestrogen ICI-182,780 (500 nM) and the PLC inhibitor U73122 (2μM). The amount of CAT activity was measured and normalized to the amount of β-galactosidase activity. The results are expressed as percent control (mean±SD; three independent experiments). *, p < 0.05; **; p < 0.005; ***, p < 0.0005. a, compared to control; b, compared to treatment.
C. Schematic representation of ERα mutants.
Interaction of calcium with ERα LBD
To ask whether calcium binds to ERα, purified recombinant human ERα was incubated with varying concentrations of 45Ca in the absence and presence of a 100-fold molar excess of CaCl2 to determine total and non-specific binding, respectively. The binding affinity was determined by nonlinear regression analysis (Fig.4A). The dissociation constant for calcium was approximately 1.0×10−6M (±0.4×10−6M, n=2). To ask whether calcium binds to the LBD, the binding assay was repeated with purified recombinant LBD (Fig.4B). Similar to the full length receptor, the dissociationconstant for calcium was approximately 0.5×10−6M (±0.16×10−6M, n=3) and the binding capacity was approximately 4 moles of 45Ca per mole LBD. Consistent with other calcium binding proteins, the low affinity of calcium for ERα suggests that at normal resting levels, intracellular calcium does not bind to ERα but at activated levels, calcium binds to the receptor.
Figure 4. Calcium interaction with Estrogen Receptor-α.

A,B. Calcium binding to ERα. Purified recombinant ERα (A) or the purified recombinant LBD of ERα (B) was incubated overnight at 4°C with radioactive 45Ca2+ in the presence and absence of a 100-fold excess of calcium. Unbound 45Ca2+ was removed and specific binding was determined as total binding minus non-specific binding. The results were analyzed using nonlinear regression analysis. Representative graph of two or three independent experiments, respectively.
C. Calcium activation of ERα ligand binding domain mutants. COS-1 cells were transiently cotransfected with wtERα or C381A, E423A, H516A, E523A, N532A, D538A, D545N, H547A, H547D, and E542A/D545A, an estrogen responsive CAT reporter gene, andβ-gal and treated for 24 hours with estradiol (1nM), or calcium (1mM) with or without the antiestrogen ICI-182,780 (500nM). The amount of CAT activity was measured, normalized to the amount of β-galactosidase activity, and expressed as percent control. (mean±SD; three independent experiments). *, p < 0.05, **; p < 0.005; ***, p < 0.0005.
Several bivalent metals including cadmium bind with high affinity to specific amino acids in the LBD of ERα (13, 16). To determine whether cadmium blocks the binding of calcium to the receptor, the LBD was incubated with 45Ca in the absence and presence of CdCl2 (Fig.S5). Cadmium blocked the binding of calcium with a Ki of approximately 10−6 M suggesting that cadmium competes with calcium for binding to the LBD.
Effects of calcium on the activation of ERα LBD
Several major structural changes are thought to occur upon activation of the LBD including the repositioning of helix H12 under helix H4 and over the ligand binding pocket, the rotation of helix H11 around its axis and movement towards helix H7 to form a continuous helix with helix H10, and the rotation of N-terminal end of helix H3 around its axis and movement toward helix H4. Amino acids on helices H4, H7, H10, H11, and H12 and in loop 11–12 that can interact with calcium and potentially facilitate these movements were tested in transient transfection assays. The ability of calcium to activate C381A (helix H4); E423A (helix H7); H516A (helix H10); E523A (helix H11); N532A (loop 11-12); and D538A, D545N, H547A, H547D, and E542A/D545A (helix H12) was measured (Figs.4C/D). Although all of the mutants were activated by the estradiol, not all of the mutants were activated by calcium. Treatment with calcium activated E423A, H547A, H547D, and D545N; there was an approximately 2- to 5-fold increase in CAT activity. However, calcium failed to activate C381A, H516A, E523A, N532A, D538A, and E542A/D545A suggesting that specific amino acids on helices H4, H10, H11, and H12 and in loop11-12 are necessary for calcium activation of ERα.
Molecular modeling of potential calcium binding sites in ERα LBD
Based on the mutational analysis, the crystal structure of the LBD (PDB:3ERD) (29), and the chemical properties of calcium, molecular modeling identified four potential binding sites for calcium on the solvent accessible surface of the LBD. Site A, located at the interface of helices H10 and H11, is formed by a direct interaction of calcium with the side chain imidazole group of his516 on helix H10 and with the side chain carboxyl groups of asn519 at the interface of helices H10 and H11 and glu523 on helix H11 (Fig.5A). Site B, located at the C-terminal end of helix H11, is formed by a direct interaction of the cation with the back bone carboxyl groups of lys529 on helix H11 and val534 in loop 11-12 and with the side chain carboxyl group of asn532 also in loop 11-12 (Fig5B). Site C, located at the N-terminal end of helix H12, is formed by a direct interaction of calcium with the side chain carboxyl groups of asp538, glu542, and asp 545 on helix H12 (Fig5C). The fourth site, site D, is located at the bend between helices H4 and H5 and is formed by the direct interaction of the cation with the side chain imidazole group of his377, the carboxyl group of glu380, and the thiol group of cys381 on helix H4 (Fig5D). The identification of four potential calcium binding sites is consistent with the results of the binding assay.
Figure 5. Molecular model of the interaction of calcium with ERα.
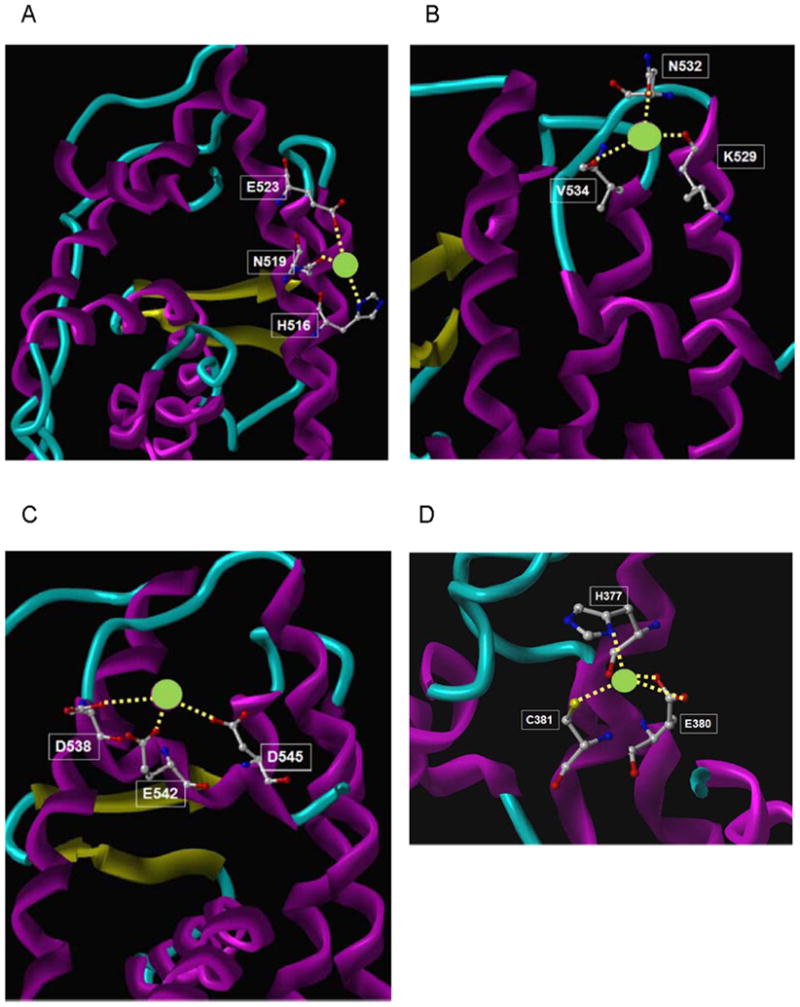
Four potential calcium binding sites on the solvent accessible surface of the ligand binding domain of ERα were identified by computational analysis. Calcium and amino acid side chains are presented as ball and stick model.
Effects of EGF on the activation of ERα LBD
To determine whether the sites that are necessary for calcium activation of ERα are required for EGF activation of the receptor, COS-1 cells were again transiently cotransfected with ERα wild type or LBD mutants, an estrogen responsive-CAT reporter construct, and the β-galactosidase gene (Figs.6A/B). As expected, treatment with estradiol resulted in an approximately 3- to 5-fold increase in CAT activity. Treatment with EGF activated C381A, E419A, E423A, N532A, E542A, D545N, H547A, and H547D; there was an approximately 1.9- to 3-fold increase in CAT activity (Fig.6A and data not shown). Treatment with EGF partially activated D538A and E542A/D545A; there was an approximately 1.5-fold increase in CAT activity. However, EGF failed to activate H516A and E523A providing additional evidence that calcium mediates the effects of the growth factor and on the LBD.
Figure 6. Effects of EGF on ERα LBD mutants.

A,B Effects of EGF on ERα LBD mutants. COS-1 cells were transiently cotransfected with wtERα or C381A, E419A, E423A, H516A, E523A, N532A, D538A, D545N, H547A, H547D, and E542A/D545A, an estrogen responsive CAT reporter gene, and β-gal and treated for 24 hours with estradiol (1nM) or EGF (150ng/ml) in the presence and absence of the antiestrogen ICI-182,780 (500nM). The amount of CAT activity was measured, normalized to the amount of β-galactosidase activity, and expressed as the percent control. (mean±SD; three independent experiments). *, p < 0.05, **; p < 0.005; ***, p < 0.0005.
C. Proposed model of calcium activation of ERα. Model compares helices H3, H4, H5, H7, H10, H11, and H12 in inactive and active state bound to calcium.
Discussion
At the time of diagnosis, approximately 70% of breast tumors are ER positive but one-third of these tumors fail to respond to endocrine therapy or become resistant (30). Several mechanisms are thought to be responsible for endocrine resistance including hormone independent activation of ERα due to overexpression, mutation, or activation of growth factor signal transduction pathways. Growth factors and cytokines are thought to activate nuclear receptors, in part, through the posttranslational modification of coactivators (31) and the phosphorylation of the N-terminal AF-1 domain (32). The results of this study show that calcium mediates the effects of EGF on the C-terminal AF-2 domain.
The mammary gland is a unique organ that, in addition to utilizing calcium as a second messenger, transports and concentrates the cation for milk production. There is increasing evidence that deregulation of calcium metabolism in the gland is linked to breast cancer (33). Women diagnosed with hypercalcemia, hypercalcemia associated with hyperparathyroidism, and idiopathic hypercalciurea have an increased risk of developing breast cancer (34–40). In patients, metastases overexpress the calcium sensing receptor (41,42). In breast cancer cells, the plasma membrane calcium-ATPase is overexpressed (43) and inhibition of the plasma membrane calcium-ATPase (44) and the T-type voltage operated calcium channel (45) inhibits proliferation. The association between the loss of calcium homeostasis and breast cancer may be due, in part, to the ability of calcium to activate ERα and provides a potential therapeutic target for the treatment of the disease.
Similar to other nuclear receptors (4,29,46–48), the LBD of ERα contains 11α-helices (H1, H3-H12) folded into a three layered antiparallel α-helical sandwich. The central core layer contains the ligand binding pocket and is formed by three α-helices (H5/6, H9, and H10) sandwiched between two additional layers of helices composed of H1-4, H7, H8, and H11. The central core of the ligand binding pocket is flanked by H12. Based upon the crystal structure of apo- and holo-RXR-α (47,49), it has been proposed that upon hormone binding, the N-terminal end of helix H3 rotates around its axis and bends towards helix H4 and the core of the pocket (47,49) which, in turn, causes helix H11 to rotate around its axis and tilt away from the pocket to form a continuous bent helix with helix H10 (Fig.6B). Together with helices H8 and H9, helix H10/H11 forms the dimerization domain. The repositioning of helix H11 also results in the repositioning of helix H12 over the binding pocket. The repositioning of helices H12 and H3 ultimately results in the formation of a shallow hydrophobic groove that constitutes the AF-2 domain, the binding site for coactivators. In addition to inducing major structural changes, estradiol and the agonist diethylstilbestrol (DES) form a cooperative network of van der Waals interactions with hydrophobic amino acids on helices H3, H5, H7, and H11 and in the turn between the β sheets (4,29,48) resulting in one to two additional turns at the ends of helices H3, H8, H11 and H12 (29).
In proteins, metals such as calcium have different functions including the stabilization of protein structure through interactions with different amino acids. In the case of calcium and ERα, the identification of potential binding sites at the ends of helices H10, H11, and H12 suggests a model (Fig.6C) that, similar to estradiol and DES, calcium alters and/or stabilizesα-helices in the LBD (32). At Site A, the interaction of calcium with his516 (C-terminal end of helix H10), asn519 (between helices H10 and H11), and glu523 (N-terminal end of helix H11) would promote the rotation of helix H11 around its axis and the formation of a continuous helix with helix H10. Calcium facilitates a similar movement of helices in the activation of I.3 lipase in Pseudomonas (50). In the inactive conformation, a lid covers the active site of the enzyme. The lid is formed by a bent helix-turn-helix-like structure that is similar to the structure of helices H10 and H11 in the apo-RXR-α. Binding of calcium to an aspartic acid on the N-terminal helix and to an aspartic acid on the C-terminal helix stabilizes the formation of a continuous helix between the two helices thereby opening the lid to the active site of lipase. At Sites B and C in ERα, the interaction of calcium with lys529, asn532, and val534, (C-terminal end of helix H11 and in loop 11-12; Site B) and with asp538, glu542, and asp 545 (N-terminal end of helix H12; Site C) would stabilize additional turns in helices H11 and H12 that would shorten and/or induce a conformational change in loop 11-12 similar to the effects of estradiol and DES resulting in the repositioning of helix H12 over the binding pocket. At Site D, the interaction of calcium with his377, glu380, and cys381 (helix H4) would induce a kink in the helix formed by helices H4 and H5 to facilitate the binding of coactivators to glu380 (29). Although the ability of EGF to activate the LBD appears to be mediated by calcium, not all of the amino acids that are required for calcium activation are necessary for EGF activation of the receptor. The amino acids located at the interface of helices H10 and H11 (Site A) are necessary; while the amino acids located at the N-terminal end of helix H12 (Site C) are partially required and the amino acids located at the C-terminal end of helix H11 (Site B) and the interface of helices H4 and H5 (Site D) are not required. The flexibility of loop 11–12 and helix H12 (48) as well as the posttranslational modification of coactivators by EGF may compensate for the loss of calcium at Sites B, C, and D. In EGF activation of ERα, the binding of calcium at Site A would reposition helix H11 causing loop 11–12 and helix H12 to pack against helices H3 and H11, while the interaction of coactivators with glu542 at Site C and glu380 at Site D (29) would further stabilize the active conformation of the AF-2 domain. Although the models propose that binding of calcium to amino acids in Sites A-D directly activates ERα, it is also possible that the binding of calcium indirectly activates the receptor, e.g., by the recruitment of coactivators. The mechanism by which calcium activates the LBD of ERα remains to be established.
The ability of calcium to mediate the effects of EGF and other activators of signal transduction pathways on the AF-2 domain provides a rationale basis for the ability of metalloestrogens to activate ERα. Bivalent metals such as cadmium, chromium, cobalt, copper, lead, mercury, nickel, and tin activate ERα due to their ability to form a high affinity complex with the LBD (13–16). Similar to calcium, these metals require glu523, asp538, and cys381 to activate ERα suggesting that metalloestrogens mimic a physiological activator of the receptor.
Supplementary Material
Acknowledgments
We thank Prof. Chambon and Dr. B. Katzenellenbogen for ERα mutants; Dr. Wolfe for helpful discussions; Drs. Byers, Fornace and Jordan for critical reading of the manuscript; NCI for computing time; and Advanced Biomedical Computing Center for support. This work was supported, in part, by grants from Komen, AICR, NIH-ES11745, NIH-P30-CA51008, U54-CA0100970, and the undergraduate program of HHMI.
References
- 1.Yamamoto KR. Steroid receptor regulated transcription of specific genes and gene networks. Annu Rev Genet. 1985;19:209–52. doi: 10.1146/annurev.ge.19.120185.001233. [DOI] [PubMed] [Google Scholar]
- 2.Kumar V, Green S, Stack G, Berry M, Jin JR, Chambon P. Functional domains of the human estrogen receptor. Cell. 1987;51:941–51. doi: 10.1016/0092-8674(87)90581-2. [DOI] [PubMed] [Google Scholar]
- 3.Bourguet W, Germain P, Gronemeyer H. Nuclear receptor ligand-binding domains: three-dimensional structures, molecular interactions and pharmacological implications. Trends Pharmacol Sci. 2000;21:381–8. doi: 10.1016/s0165-6147(00)01548-0. [DOI] [PubMed] [Google Scholar]
- 4.Brzozowski AM, Pike ACW, Dauter Z, et al. Molecular basis of agonism and antagonism in the estrogen receptor. Nature. 1997;389:753–8. doi: 10.1038/39645. [DOI] [PubMed] [Google Scholar]
- 5.Ali S, Metzger D, Bornert JM, Chambon P. Modulation of transcriptional activation by ligand-dependent phosphorylation of the human oestrogen receptor A/B region. EMBO J. 1993;12:1153–60. doi: 10.1002/j.1460-2075.1993.tb05756.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Le Goff P, Montano MM, Schodin DJ, Katzenellenbogen BS. Phosphorylation of the human estrogen receptor. Identification of hormone-regulated sites and examination of their influence on transcriptional activity. J Biol Chem. 1994;269:4458–66. [PubMed] [Google Scholar]
- 7.Ignar-Trowbridge DM, Nelson KG, Bidwell MC, et al. Coupling of dual signaling pathways: epidermal growth factor action involves the estrogen receptor. Proc Natl Acad Sci USA. 1992;89:4658–62. doi: 10.1073/pnas.89.10.4658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kato S, Endoh H, Masuhiro Y, et al. Activation of the estrogen receptor through phosphorylation by MAPK. Science. 1995;270:1491–4. doi: 10.1126/science.270.5241.1491. [DOI] [PubMed] [Google Scholar]
- 9.Power RF, Mani SK, Codina J, Conneely OM, O'Malley BW. Dopaminergic and ligand-independent activation of steroid hormone receptors. Science. 1991;254:1636–9. doi: 10.1126/science.1749936. [DOI] [PubMed] [Google Scholar]
- 10.Shupnik MA. Crosstalk between steroid receptors and the c-Src-receptor tyrosine kinase pathways: implications for cell proliferation. Oncogene. 2004;23:7979–89. doi: 10.1038/sj.onc.1208076. [DOI] [PubMed] [Google Scholar]
- 11.Johnson MD, Kenney N, Stoica A, et al. Cadmium mimics the in vivo effects of estrogen in the uterus and mammary gland. Nature Med. 2003;9:1081–4. doi: 10.1038/nm902. [DOI] [PubMed] [Google Scholar]
- 12.Garcia-Morales P, Saceda M, Kenney N, et al. Effect of cadmium on estrogen receptor levels and estrogen-induced responses in human breast cancer cells. J Biol Chem. 1994;269:16896–901. [PubMed] [Google Scholar]
- 13.Stoica A, Katzenellenbogen BS, Martin MB. Activation of estrogen receptor-alpha by the heavy metal cadmium. Mol Endocrinol. 2000;14:545–53. doi: 10.1210/mend.14.4.0441. [DOI] [PubMed] [Google Scholar]
- 14.Stoica A, Pentecost E, Martin MB. Effects of selenite on estrogen receptor-a expression and activity in MCF-7 breast cancer cells. J Cell Biochem. 2000;79:282–92. doi: 10.1002/1097-4644(20001101)79:2<282::aid-jcb110>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 15.Stoica A, Pentecost E, Martin MB. Effect of arsenite on estrogen receptor-a expression and activity in MCF-7 breast cancer cells. Endocrinol. 2000;141:3595–602. doi: 10.1210/endo.141.10.7704. [DOI] [PubMed] [Google Scholar]
- 16.Martin MB, Reiter R, Pham T, et al. Estrogen like activity of metals in MCF-7 breast cancer cells. Endocrinol. 2003;144:2425–36. doi: 10.1210/en.2002-221054. [DOI] [PubMed] [Google Scholar]
- 17.Choe SY, Kim SJ, Kim HG, et al. Evaluation of estrogenicity of major heavy metals. Sci Total Environ. 2003;312:15–21. doi: 10.1016/S0048-9697(03)00190-6. [DOI] [PubMed] [Google Scholar]
- 18.Evenas J, Forsen S, Malmendal A, Akke M. Backbone dynamics and energetics of a calmodulin domain mutant exchanging between closed and open conformations. J Mol Biol. 1999;289:603–17. doi: 10.1006/jmbi.1999.2770. [DOI] [PubMed] [Google Scholar]
- 19.Wimberly B, Thulin E, Chazin WJ. Characterization of the N-terminal half-saturated state of calbindin D9k: NMR studies of the N56A mutant. Protein Sci. 1995;4:1045–55. doi: 10.1002/pro.5560040603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Akke M, Forsen S, Chazin WJ. Solution structure of (Cd2+)1-calbindin D9k reveals details of the stepwise structural changes along the Apo-->(Ca2+)II1-->(Ca2+)I,II2 binding pathway. J Mol Biol. 1995;252:102–21. doi: 10.1006/jmbi.1995.0478. [DOI] [PubMed] [Google Scholar]
- 21.Veselik DJ, Divekar S, Dakshanamurthy S, et al. Activation of estrogen receptor- alpha by the anion nitrite. Cancer Res. 2008;68:3950–58. doi: 10.1158/0008-5472.CAN-07-2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reese JC, Katzenellenbogen BS. Mutagensis of cysteines in the hormone binding domain of the human estrogen receptor. J Biol Chem. 1991;266:10880–7. [PubMed] [Google Scholar]
- 23.Wrenn CK, Katzenellenbogen BS. Structure-function analysis of the hormone binding domain of the human estrogen receptor by region-specific mutagenesis and phenotypic screening in yeast. J Biol Chem. 1993;268:24089–98. [PubMed] [Google Scholar]
- 24.Pakdel F, Reese JC, Katzenellenbogen BS. Identification of charged residues in an N-terminal portion of the hormone-binidng domain of the human estrogen receptor important in transcriptional activity of the receptor. Mol Endocrinol. 1993;7:1408–17. doi: 10.1210/mend.7.11.8114756. [DOI] [PubMed] [Google Scholar]
- 25.Pakdel F, Katzenellenbogen BS. Human estrogen receptor mutants with altered estrogen and antiestrogen ligand discrimination. J Biol Chem. 1992;267:3429–37. [PubMed] [Google Scholar]
- 26.Dixon CJ, Bowler WB, Fleetwood P, Ginty AF, Gallagher JA, Carron JA. Extracellular nucleotides stimulate proliferation in MCF-7 breast cancer cells via P2-purinoceptors. Br J Cancer. 1997;75:34–9. doi: 10.1038/bjc.1997.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Journe F, Dumon JC, Kheddoumi N, et al. Extracellular calcium downregulates estrogen receptor alpha and increases its transcriptional activity through calcium-sensing receptor in breast cancer cells. Bone. 2004;35:479–88. doi: 10.1016/j.bone.2004.03.021. [DOI] [PubMed] [Google Scholar]
- 28.Tong J, McCarthy TV, MacLennan DH. Measurement of resting cytosolic Ca2+ concentrations and Ca2+ store size in HEK-293 cells transfected with malignant hyperthermia or central core disease mutant Ca2+ release channels. J Biol Chem. 1999;274:693–702. doi: 10.1074/jbc.274.2.693. [DOI] [PubMed] [Google Scholar]
- 29.Shiau AK, Barstad D, Loria PM, et al. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell. 1998;95:927–37. doi: 10.1016/s0092-8674(00)81717-1. [DOI] [PubMed] [Google Scholar]
- 30.Musgrove EA, Sutherland RL. Biological determinants of endocrine resistance in breast cancer. Nat Rev Cancer. 2009;9:631–43. doi: 10.1038/nrc2713. [DOI] [PubMed] [Google Scholar]
- 31.Han SJ, Lonard DM, O'Malley BW. Multi-modulation of nuclear receptor coactivators through posttranslational modifications. Trends Endocrinol Metab. 2009;20:8–15. doi: 10.1016/j.tem.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ordonez E, Thiyagarajan S, Cook JD, et al. Evolution of metal(loid) binding sites in transcriptional regulators. J Biol Chem. 2008;283:25706–14. doi: 10.1074/jbc.M803209200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee WJ, Monteith GR, Roberts-Thomson SJ. Calcium transport and signaling in the mammary gland: targets for breast cancer. Biochim Biophys Acta. 2006;1765:235–55. doi: 10.1016/j.bbcan.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 34.Almquist M, Manjer J, Bondeson L, Bondeson AG. Serum calcium and breast cancer risk: results from a prospective cohort study of 7,847 women. Cancer Causes Control. 2007;18:595–602. doi: 10.1007/s10552-007-9001-0. [DOI] [PubMed] [Google Scholar]
- 35.Garner CN, Ganetzky R, Brainard J, et al. Increased prevalence of breast cancer among patients with thyroid and parathyroid disease. Surgery. 2007;142:806–13. doi: 10.1016/j.surg.2007.07.024. [DOI] [PubMed] [Google Scholar]
- 36.Martin E, Miller M, Krebsbach L, Beal JR, Schwartz GG, Sahmoun AE. Serum calcium levels are elevated among women with untreated postmenopausal breast cancer. Cancer Causes Control. 2010;21:251–7. doi: 10.1007/s10552-009-9456-2. [DOI] [PubMed] [Google Scholar]
- 37.Michels KB, Xue F, Brandt L, Ekbom A. Hyperparathyroidism and subsequent incidence of breast cancer. Int J Cancer. 2004;110:449–51. doi: 10.1002/ijc.20155. [DOI] [PubMed] [Google Scholar]
- 38.Nilsson IL, Zedenius J, Yin L, Ekbom A. The association between primary hyperparathyroidism and malignancy: nationwide cohort analysis on cancer incidence after parathyroidectomy. Endocr Relat Cancer. 2007;14:135–40. doi: 10.1677/erc.1.01261. [DOI] [PubMed] [Google Scholar]
- 39.Palmer M, Adami HO, Krusemo UB, Ljunghall S. Increased risk of malignant diseases after surgery for primary hyperparathyroidism. A nationwide cohort study. Am J Epidemiol. 1988;127:1031–40. doi: 10.1093/oxfordjournals.aje.a114879. [DOI] [PubMed] [Google Scholar]
- 40.Pal SK, Blazer K, Weitzel J, Somlo G. An association between invasive breast cancer and familial idiopathic hyperparathyroidism: a case series and review of the literature. Breast Cancer Res Treat. 2009;115:1–5. doi: 10.1007/s10549-008-0056-8. [DOI] [PubMed] [Google Scholar]
- 41.Cheng I, Klingensmith ME, Chattopadhyay N, et al. Identification and localization of the extracellular calcium-sensing receptor in human breast. J Clin Endocrinol Metab. 1998;83:703–7. doi: 10.1210/jcem.83.2.4558. [DOI] [PubMed] [Google Scholar]
- 42.Mihai R, Stevens J, McKinney C, Ibrahim NB. Expression of the calcium receptor in human breast cancer--a potential new marker predicting the risk of bone metastases. Eur J Surg Oncol. 2006;32:511–5. doi: 10.1016/j.ejso.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 43.Lee WJ, Roberts-Thomson SJ, Holman NA, May FJ, Lehrbach GM, Monteith GR. Expression of plasma membrane calcium pump isoform mRNAs in breast cancer cell lines. Cell Signal. 2002;14:1015–22. doi: 10.1016/s0898-6568(02)00049-9. [DOI] [PubMed] [Google Scholar]
- 44.Lee WJ, Robinson JA, Holman NA, McCall MN, Roberts-Thomson SJ, Monteith GR. Antisense-mediated Inhibition of the plasma membrane calcium-ATPase suppresses proliferation of MCF-7 cells. J Biol Chem. 2005;280:27076–084. doi: 10.1074/jbc.M414142200. [DOI] [PubMed] [Google Scholar]
- 45.Bertolesi GE, Shi C, Elbaum L, et al. The Ca(2+) channel antagonists mibefradil and pimozide inhibit cell growth via different cytotoxic mechanisms. Mol Pharmacol. 2000;62:210–9. doi: 10.1124/mol.62.2.210. [DOI] [PubMed] [Google Scholar]
- 46.Renaud JP, Rochel N, Ruff M, et al. Crystal structure of the RAR-gamma ligand- binding domain bound to all-trans retinoic acid. Nature. 1996;378:681–9. doi: 10.1038/378681a0. [DOI] [PubMed] [Google Scholar]
- 47.Bourguet W, Ruff M, Chambon P, Gronemeyer H, Moras D. Crystal structure of the ligand-binding domain of the human nuclear receptor RXR-alpha. Nature. 1995;375:377–82. doi: 10.1038/375377a0. [DOI] [PubMed] [Google Scholar]
- 48.Tanenbaum DM, Wang Y, Williams SP, Sigler PB. Crystallographic comparison of the estrogen and progesterone receptor's ligand binding domain. Proc Natl Acad Sci USA. 1998;95:5998–6003. doi: 10.1073/pnas.95.11.5998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Egea PF, Mitschler A, Rochel N, Ruff M, Chambon P, Moras D. Crystal structure of the human RXRalpha ligand-binding domain bound to its natural ligand: 9-cis retinoic acid. EMBO J. 2000;19:2592–601. doi: 10.1093/emboj/19.11.2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kuwahara K, Angkawidjaja C, Matsumura H, Koga Y, Takano K, Kanaya S. Importance of the Ca2+-binding sites in the N-catalytic domain of a family I.3 lipase for activity and stability. Protein Eng Des Sel. 2008;21:737–44. doi: 10.1093/protein/gzn057. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.