

Abstract
Alzheimer's disease (AD) is a well-studied process characterized by the presence of amyloid plaques and neurofibrillary tangles. In this study, a series of protein kinase C (PKC) activators were investigated, some of which also exhibit histone deacetylases (HDACs) inhibitory activity, under the hypothesis that such compounds might provide a new path forward in the discovery of drugs for the treatment of AD. The PKC activating properties of these drugs were expected to enhance the α-secretase pathway in the processing of amyloid precursor protein (APP), while their HDAC inhibition was anticipated to confer neuroprotective activity. We found that the benzolactams compounds 9 and 11-14 caused a concentration-dependent increase in sAPPα and decrease in β-amyloid (Aβ) production using concentrations of these drugs in the range of 0.1∼10 μM, consistent with a shift of APP metabolism towards the α-secretase-processing pathway. Moreover, 9-14 showed neuroprotective effects in the 10 to 20 μM range in the homocysteate (HCA) cortical neuron model of oxidative stress. In parallel, we found that the most neuroprotective compounds caused increased levels of histone acetylation (H4), thus indicating their likely ability to inhibit histone deacetylase activity. As the majority of the compounds studied also show nanomolar binding affinities for PKC, we conclude that it is possible to design, de novo, agents that combine both PKC activating properties along with HDAC inhibitory properties, thereby resulting in agents capable of modulating amyloid processing while showing neuroprotection. These findings may offer a new approach to therapies that exhibit disease-modifying effects, as opposed to symptomatic relief, in the treatment of AD.
Keywords: Alzheimer's Disease, amyloid β, HDAC inhibition, neuroprotection, PKC activation
Introduction
Alzheimer's disease (AD) is a devastating neurological disorder that is characterized by deteriorating cognition and memory, progressive impairment in the ability to carry out daily living activities, and a number of neuropsychiatric symptoms. Associated with this progressive deterioration is the pathological generation and accumulation of Aβ40-42 plaques, giving rise to the amyloid hypothesis: that the pathological accumulation of Aβ40-42 in the brain leads to oxidative stress, neuronal destruction and finally the clinical syndrome of AD. Following this hypothesis, strategies to decrease the production of Aβ40-42, stimulate the clearance of Aβ formed or prevent the aggregation of Aβ into amyloid plaques are being pursued. However, these strategies fail to recognize and treat other factors that are concomitant with AD pathology, for example oxidative stress, neuronal degeneration, and impaired memory and learning. Thus strategies that aim to target multiple pathological processes in AD need to be considered. One strategy to achieve these aims is combination therapy. Combination therapy has been used successfully in the treatment of cancer, HIV infection, and tuberculosis. An alternative to combinatorial therapy is to identify a single biological target that will act on or interdict multiple relevant processes.
In light of identifying single biological targets that will act on multiple processes relevant to AD, there has been a growing interest in protein kinase C (PKC), which has been shown to play a critical role in memory,1 in APP processing,2-4 and its activity has been found to be altered or defective in brains5-10 and peripheral tissues of AD patients.11, 12 PKC plays a role in amyloid precursor protein (APP) processing through its ability to modulate the activity of α-secretase, a metalloprotease neither completely identified nor characterized.2-4 Activation of α-secretase shifts formation of the amyloidogenic Aβ40-42, generated by β- and γ-secretases, to generation of the non-amyloidogenic sAPPα. Since Aβ plaque formation in the brain is considered to be a key player in the etiology of Alzheimer's pathophysiology,13, 14 suppression of neurotoxic Aβ40 or Aβ42 formation by PKC activation represents an alternative non-protease based approach to controlling Aβ levels.
In previous studies, we have demonstrated that the benzolactam analog, 8-(1-decynyl)benzolactam (BL), a novel PKC activator, can reverse K+ channels defects and enhance secretion of sAPPα in AD cells.15,16 We have also shown previously that another PKC activator, bryostatin 1, at subnanomolar concentrations, can dramatically enhance the secretion of the α-secretase product sAPPα in fibroblasts from AD patients. Moreover, when tested in vivo, BL was found to significantly increase the amount of sAPPα and reduce Aβ40 in the brains of APP[V717I] transgenic mice.17 In a more recently developed AD double-transgenic mouse, bryostatin 1 was found to be effective in reducing both brain Aβ40 and Aβ42, ameliorate the rate of premature death and improve behavioral outcome.17 Collectively, these data corroborate PKC and its activation as a potentially important target for ameliorating AD pathophysiology and cognitive impairment, and thus offers a promising target for drug development.
Another emerging approach could affect multiple processes to ameliorate AD pathophysiology and cognitive impairment while targeting a single biological molecule is epigenetic remodeling via histone deacetylase inhibition. Acetylation and deacetylation of histone proteins play critical roles in transcriptional regulation in eukaryotic cells. The acetylation status is determined by the activities of two families of enzymes, histone acetyltransferases (HATs), which add acetyl groups to conserved lysine residues within the amino-terminal tails of histone proteins, and HDACs, which remove these acetyl groups. In general, the acetylation of histone promotes a more relaxed chromatin structure, allowing for transcriptional activation. The HDACs are able to act as transcriptional repressors, due to histone deacetylation, that consequently promotes chromatin condensation. HDAC inhibitors selectively alter gene transcription, in part, by permitting chromatin remodeling by HAT activity and changes to the composition of multiprotein complexes bound to proximal region of specific gene promoters.18 Furthermore, HDACs interact with many non-histone protein substrates such as the hormone receptors, chaperone proteins, and cytoskeletal proteins, which regulate cell proliferation and cell death.19, 20
Impressively, HDAC inhibitors have been found to be neuroprotective in a broad array of cellular and animal models of acute and chronic neurodegenerative injury and disease, including AD, Huntington's disease (HD), spinal muscular atrophy (SMA) and spinal and bulbar muscular atrophy (SBMA), amyotrophic lateral sclerosis (ALS), ischemic stroke, and experimental autoimmune encephalomyelitis (EAE).21, 22 At least part of this broad neuroprotective capacity centers on the ability of HDAC inhibitors to increase neuronal resistance to oxidative stress; a pathological process that underlies many neurodegenerative disease pathologies and is particularly relevant to AD. However, in addition to neuroprotection, HDAC inhibition has also been shown to enhance synaptic plasticity, learning and memory in rodents. Furthermore, in a transgenic mouse model (CK-p25Tg) that allows the temporal and spatial induction of neuronal degeneration and displays many of the pathologic hallmarks of AD, HDAC inhibition was found to significantly improve associative and spatial learning after degeneration.23 Perhaps the most striking of these studies, though, was that HDAC inhibition could facilitate recovery of inaccessible long-term memories in this model.23
Based upon the foregoing body of research on PKC activators and HDAC inhibitors in AD and other neurodegenerative diseases, we believed that it would be valuable to design and create single chemical entities capable of targeting both PKC and HDAC enzymes simultaneously. From our previous work on PKC activators comprised of a benzolactam scaffold,24, 25 we believed that we could attach a modified side chain appendage to the phenyl ring that would allow either capable of interacting with the cell membrane or that would engage the catalytic site of the HDACs. This is perhaps best illustrated by Figure 1, which shows one of the benzolactams of the present paper docked to the crystal structure of the C1b domain of PKCδ as well as to the crystal structure of HDAC7. In the benzolactam-PKC complex, the side chain is located outside of the ligand binding domain while in the HDAC complex the benzolactam nucleus resides at the enzyme surface (Figure 1). Thus we hypothesized that such a chemical entity would bring together the disparate but synergistic capabilities of each pharmacological approach and offer a drug with enhanced therapeutic potential for use in the treatment of AD. Herein we detail the chemistry and biology of a novel series of small molecules possessing such dual pharmacological activity.
Figure 1.
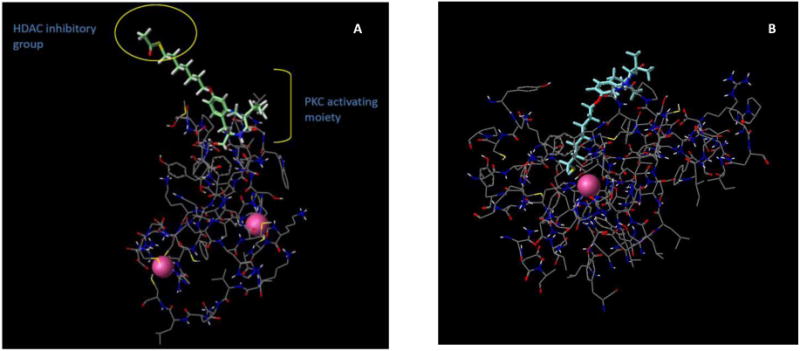
A. This figure was generated using PKCδ C1B (pdb: 1PTR) and illustrates how the HDAC inhibitory group resides outside of the ligand binding domain of PKC, thus allowing the benzolactam scaffold to engage the diacylglycerol binding site of PKC. B. Compound 13 in its more active free thiol form docked to HDAC7 (pdb: 3C10) which illustrates how the benzolactam moiety resides on the surface of the enzyme while the thiol group is coordinated to the catalytic zinc atom.
Results
Design of PKC-agonist/HDAC-inhibitor compounds
In order to create ligands that would retain α-secretase activating properties while also exhibiting a neuroprotective action through HDAC inhibition, a small library of benzolactam analogs was generated. This chemical scaffold has been shown previously by us to have high binding affinity for PKC and to function as an agonist, especially when it is adorned with lipophilic side chains capable of interacting with the cell membrane.26 High structural diversity is possible for this side chain, since it does not interact with the diacylglycerol (DAG) recognition site of PKC. Thus, we imagined that the side chain could incorporate functional groups capable of interacting with the zinc atom present within the catalytic site of the HDACs. From our earlier work, we found that certain thiol-based HDAC inhibitors were less toxic and therefore more effective than their hydroxamate-based counterparts at protecting cortical neurons from oxidative stress,27 so thioacetate group and lipoic acid group were selected as zinc binding group (ZBG) to append to benzolactam scaffold.
All compounds were synthesized starting from the common intermediate 1,28 which was subjected to benzyl deprotection with hydrogen to obtain the phenol 2 (Scheme 1). Next, 2 was treated with 7-bromo-1-heptene in the presence of potassium carbonate as a base and potassium iodide at 80 °C for 7 h to afford the final compound 3 in 31% yield. Compound 5 was synthesized in 16% yield from 2 following two steps: treatment with Boc-protected 2-bromoethanolamine in the presence of cesium carbonate to give the intermediate phenolic ether in 20% yield followed by acetylation of the free hydroxyl group using acetic anhydride and pyridine as a base. The synthesis of compound 6 was carried out starting from the Boc-protected amino compound 5 by coupling it with (±)-α-lipoic acid using EDCI and HOBT followed by deacetylation under basic conditions in 80% yield. Compound 7 was prepared from 1 by protection of the free hydroxyl group with TBDMSCl, followed by debenzylation in the usual way to afford 4. Compound 4 was reacted with (±)-α-lipoic acid using DCC as the coupling reagent followed by silyl deprotection to give the final product in 36% yield. Compounds 9–14 were also derived from 2, which was coupled with the appropriate disubstitued alkanes to provide the intermediate 8 (Scheme 2). Compound 8 was then reacted with potassium thioacetate at 50 °C for 2 h to afford the products 9–14 in 12-67% yield as shown in Scheme 2.
Scheme 1.

Reagents and Conditions (a) H2, 10% Pd/C, EtOAc; (b) TBDMS-Cl, Et3N, DMAP, CH2Cl2, 0 °C to rt, 12 h; (c) H2, 20% Pd(OH)2/C, EtOAc, rt, 1 h; (d) 7-bromo-1-heptene, K2CO3, KI, DMF, 80 °C;. (e) BocNHCH2CH2Br, Cs2CO3, 100 °C, 24 h; (f) Ac2O, pyridine, CH2Cl2, 0 °C to rt, 20 h; (g) TFA, CH2Cl2, 0 °C to rt, 2 h; (h) (±)-α-Lipoic acid, EDCI, HOBT, DIEA, DMF, 0 °C to rt, 12 h; (i) K2CO3, MeOH, 0 °C, 30 min; (j) (±)-α-Lipoic acid, DCC, DMAP, CH2Cl2, rt, 16 h;. (k) PTSA, MeOH, rt, 3 h.
Scheme 2.

Reagents and Conditions (a) Cs2CO3, DMF, 80 °C, 8 h; (b) AcSK, DMF, 50 °C, 1 h.
Clog P and tPSA values were calculated for the hybrid ligands (Table 1). Clog P values for these final compounds ranged from 0.92 to 4.67. The tPSA values were 61.80 to 90.90. These values combined with our previous in vivo studies of other benzolactam analogs predicted good cellular permeability and blood brain barrier penetration for the synthesized compounds, designed to possess PKC activating and HDAC inhibitory activity.17 The binding affinities of the compounds for mouse PKCα were determined as described previously.26 The Ki[nM] values for each of the compounds 2, 3, 6, 7 and 9–14 are listed in Table 1. Four of these compounds, 6, 7, 12 and 13, were also examined for possible PKC isoform selectivity at α, β, γ, δ and ε, however, only minor differences were observed (data not shown).
Table 1. PKC binding affinity of the benzolactams together with their calculated LogP values and topological polar surface areas.
| Code | Clog Pa,c | tPSA b,c | Ki [nM] ± SEM |
|---|---|---|---|
| 2 | 0.92 | 72.79 | 4840 ± 320 |
| 3 | 4.65 | 61.80 | 5.4 ± 0.1 |
| 6 | 4.11 | 78.87 | 148 ± 18 |
| 7 | 4.66 | 90.90 | 5.7 ± 1.0 |
| 9 | 2.21 | 78.87 | 21.0 ± 2.8 |
| 10 | 2.70 | 78.87 | 15.8 ± 0.8 |
| 11 | 3.19 | 78.87 | 6.6 ± 0.2 |
| 12 | 3.68 | 78.87 | 8.7 ± 0.7 |
| 13 | 4.17 | 78.87 | 4.5 ± 0.4 |
| 14 | 4.67 | 78.87 | 2.8 ± 0.5 |
| Bryostatin 1 | 3.80 | 240.14 | 1.4 ± 0.253 |
| SAHA | 1.44 | 78.42 | --- |
Clog P (KOWWIN) were calculated from http://146.107.217.178/lab/alogps/start.html.
tPSA were calculated from http://www.molinspiration.com/cgi-bin/properties?textMode=1
Clogp and tPSA values of the free thiol analogs of compound 9–14 are available in supporting information
Compounds designed to possess PKC activating and HDAC inhibitory activities increase sAPPα production and reduce Aβ production
Activation of PKC by DAG, benzolactam, or other activators in fibroblasts has been shown to increase α-secretase-mediated cleavage of the amyloid precursor protein and thus increase production of the non-amyloidogenic sAPPα.17 To examine the effect of the benzolactam analogs on altering sAPPα production and release HEK293 stably transfected with human APP were treated with compounds 9–14 at concentrations ranging from 0.001 to 10 μM and sAPPα levels measured by sAPPα ELISA. Treatment with compounds 12 and 14 resulted in an increase of sAPPα levels at concentrations of 1 to 10 μM (Figure 2). Treatment with compounds 9, 11 and 13 resulted in an increase of sAPPα levels at a concentration of 10 μM. In contrast, compound 10 showed a statistically significant elevation in sAPPα at a concentration of 1 nM.
Figure 2.

The effect of 6 HDAC-PKC hybrid ligands on sAPPα levels. Data are presented as percent of control ± SEM. *Significantly higher compared to control without drug p < 0.01; ** Significantly lower compared to control without drug p < 0.01.
Theoretically, if APP synthesis is held relatively constant, the production of Aβ should be reduced if more of the APP is processed through the α-secretase pathway. In order to confirm that treatment with compounds 9-14 were indeed enhancing the α-secretase APP processing pathway we examined whether the increased levels of sAPPα corresponded to a decrease in the production of Aβ40. Cells were treated with compounds 9–14 for 24 h at concentrations ranging from 0.001 μM to 10 μM and Aβ40 levels measured by Aβ40 ELISA. Treatment with compounds 13 and 14 resulted in a significant decrease of Aβ40 levels at concentrations of 1 to 10 μM (Figure 3). Treatment with compounds 9, 10, 11 and 12 resulted in significant decrease of Aβ40 levels at concentrations of 10 μM. In general, there was good agreement with a compound's ability increase sAPPα and its ability to decrease Aβ40.
Figure 3.

The effect of 6 HDAC-PKC hybrid ligands in reducing Aβ production. Data are presented as the percent of control ± SEM. *Significantly higher compared to control without drug p < 0.01; ** Significantly lower compared to control without drug p < 0.01.
Compounds designed to possess PKC activating and HDAC inhibitory activities induce histone H4 acetylation and protect cortical neurons from oxidative stress-induced death
Oxidative stress has been implicated to play a crucial role in the pathogenesis of AD and has been observed in the AD brain. Indeed, oxidative stress occurs early in the progression of Alzheimer disease, even before the development of the pathologic hallmarks, neurofibrillary tangles and senile plaques. The rationale for designing and synthesizing compounds with dual activity – to activate PKC and inhibit HDAC activity – stems from our prior work where we have demonstrated that HDAC inhibition can protect against neuronal oxidative stress in vitro.
Dynamic equilibria exist between the acetylation and deacetylation status of lysine residues within histone proteins, which are regulated in turn by both the HATs and the HDACs. By inhibiting the deacetylation reaction through the application of HDAC inhibitors, the global acetylation level of the histones in neurons can be increased, and profound changes in gene expression can occur. From our earlier work, we found that certain thiol-based HDAC inhibitors were less toxic and therefore more effective than their hydroxamate-based counterparts at protecting cortical neurons from oxidative stress.27
To directly gauge the HDAC inhibitory of this new class of compounds we investigated their ability to alter histone acetylation levels. Because of the instability of the free mercaptans, we were unable to isolate these compounds in sufficient purity to allow for direct assay of their inhibitory action using the purified enzymes. Therefore, cultures of primary cortical neurons were treated with the various benzolactam analogs (10 μM) for a period of 8 h, and Western blot analysis then performed using acetyl-histone H4-specific antibodies (Figure 4A). Compounds 11–14 all induced an increase in histone H4 acetylation at this concentration, consistent with inhibiting HDAC activity. For compounds 12 and 13, histone H4 acetylation was comparable to that induced by the prototypical HDAC inhibitors, trichostatin A (TSA) and suberoylanilide hydroxamic acid (SAHA). Compounds 9 and 10 were less effective at inducing histone H4 acetylation at 10 μM, but modest acetylation could be seen when treated at 20 μM (Figure 4B). As expected, treatment of primary cortical neurons with compound 3 did not increase histone H4 acetylation, even at the 20 μM level, in spite of the fact that it has high affinity for PKC, thus further underscoring the importance of the sulfur bearing tail for HDAC inhibition. Interestingly, compounds 6 and 7 containing a lipoic acid side chain, also failed to induce any histone H4 acetylation in these cells. While lipoic acid has been shown to act as an HDAC inhibitor,29 this requires initial reduction to the di-mercaptan, which may not be taking place appropriately in this model. Thus the degree of histone H4 acetylation observed appears to correlate with the ability of the free mercapto group, derived from its acetylthio pro-drug form by the action of estereases, to engage the catalytic zinc atom present in the bottom of the gorge region of the HDAC enzyme.30
Figure 4.
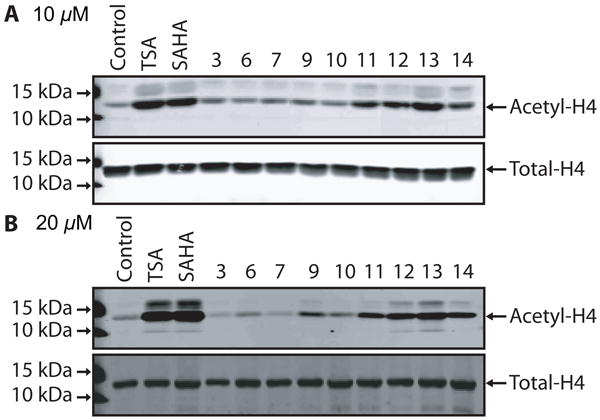
Effect of different PKC-HDAC hybrid ligands (A: 10 μM and B: 20 μM) on the acetylation levels of histone H4.
Having determined that compounds 9–14 do indeed display HDAC inhibitory activity, we investigated their ability to protect cultured primary cortical neurons from oxidative stress-induced death. In this model of neurodegeneration oxidative stress is induced by the presence of a 5 mM concentration of the glutamate analog, HCA, which depletes the cellular antioxidant glutathione by the competitive inhibition of cysteine uptake at the level of the plasma membrane cystine/glutamate antiporter system xc-. Because cysteine is required for the synthesis of glutathione, the inhibition of its uptake results in glutathione depletion. Cellular redox homeostasis, therefore, becomes disrupted with the accumulation of endogenously produced and unopposed oxidants resulting in neuronal degeneration over about a 24 h period of time. Importantly, this model utilizes embryonic primary neurons (E17), which, at this early developmental stage, lack ionotropic and metabotropic receptors and are not susceptible to excitotoxicity. Rather death is induced by accumulation of unopposed free radicals and the neurons exhibit a number of apoptotic features.31
Primary cortical neurons were treated with compounds 3, 6, 7, and 9–14 at concentrations ranging from 1 μM to 20 μM in the presence or absence of HCA for 48 h – well beyond the time for degeneration to occur in this model. As is apparent in Figure 5, in the absence of HCA, none of the compounds show any toxicity when neuron viability was measured by MTT assay, even at the highest concentrations tested (20 μM). For comparison purposes, the effects of the commercially available, prototypical HDAC inhibitor, TSA, was again carried out in the same studies. In contrast to the compounds being tested and as previously published,32 TSA showed considerable dose-dependent toxicity the absence of HCA. In the presence of HCA, compounds 9 through 14, with a sulphur atom in the side chain appendage showed 100 percent neuroprotection from HCA-induced oxidative toxicity at 20 μM, correlating with the ability of these compounds to induce histone acetylation. Indeed, within this set, compounds 11–13, showed complete protection at a dose of 10 μM, corresponding to their ability to induce histone H4 acetylation at the same concentration.
Figure 5.

Dose dependent survival of cortical neurons upon exposure to PKC-HDAC hybrid ligands, TSA and SAHA in the absence (left bar) or presence (right bar) of HCA. Data are presented as the percent of control ± SEM. Neuron survival was quantified by the MTT assay, a colorimetric assay in which the amount of yellow MTT is reduced to purple formazan by active mitochondrial reductase enzymes in viable cells.
In contrast to the protection seen with compounds 9–14, compounds 3, 6 and 7 showed no neuroprotection even at a 20 μM level. In the case of compound 3, which has high affinity for PKC, this again underscores the importance of the sulfur bearing tail for HDAC inhibition and the capacity for HDAC inhibition to protect neurons from oxidative stress-induced death. Indeed, the lack of protection in this system with the lipoic acid side chain containing compounds 6 and 7, which also failed to induce any histone H4 acetylation, adds further support for this argument. Moreover, the neuroprotective action of the acetylated thiols is not due to some general radical trapping by the sulfur atom, as previously published experiments by us failed to show any improvement in survival until millimolar concentrations were reached.27
Discussion
This article elaborates on the chemistry and biology of compounds that offer a different therapeutic approach to the treatment of AD. By coupling the idea of generating neuroprotective agents that work through the inhibition of histone deacetylases together with the desire to activate PKC and limit the production of one of the key culprits in this disease, namely Aβ, we have investigated the design, synthesis, and testing of appropriately substituted benzolactam analogs.
PKC is a multienzyme protein complex that phosphorylates serine and threonine residues in a host of proteins. It plays important roles in intracellular signal transduction related to various cellular events, including proliferation, differentiation, and apoptosis.33, 34 The N-terminal regulatory domain of the classical PKCs contains two cysteine-rich membrane-targeting C1 (C1A and C1B) domains that bind to the natural PKC activator DAG as well as to certain natural products such as phorbol esters and indolactam-V analogs, and the Ca2+-dependent membrane binding C2 domain. In its inactive state PKCα resides in the cytosol, where the pseudosubstrate sequence of the regulatory domain of PKC interacts with the catalytic domain and prevents access of substrate to the catalytic site. However, in response to elevated Ca2+ ion levels, PKCα translocates to the membrane where it interacts with natural phospholipids and DAG (or DAG mimics) to become fully active.35-37 In order to engineer compounds that can interact with and to activate the enzyme PKC and thus increase α-secretase processing of APP, we have utilized the indolactam-V analog, benzolactam, as a scaffold. Benzolactam has been shown previously by us to have high binding affinity for PKC and to function as a PKC agonist.26 Indeed, as presented in Table 1, PKC binding affinity of the different benzolactams compounds, 3, 6, 7 and 9–14, were all in the low nanomolar range. Furthermore, the treatment of HEK293 cells expressing human APP with compounds 9–14 all resulted in an increase in sAPPα and a concomitant decrease in Aβ40 levels, consistent with greater PKC activation and enhanced α-secretase activity.
These benzolactams bear side chain appendages potentially capable of interacting with and inhibiting the activity of HDAC enzymes, thereby causing the hyperacetylation of histone proteins and, inter alia, neuroprotection. In mammalian cells, the HDACs are divided into four classes that depend on their sequence/structural homology to yeast deacetylases, expression patterns and catalytic mechanisms.38, 39 Eighteen HDAC enzymes have been identified and classified19, 40 based on homology to these yeast HDACs: Class I HDACs include HDAC1, 2, 3, and 8; Class II HDACs include HDAC4, 5, 6, 7, 9, and 10; Class III include SIRT1-7; and Class IV is represented by HDAC11. All class I, II and IV HDACs are zinc-dependent enzymes. The active site of these HDACs is found within a highly conserved catalytic domain containing a divalent zinc cation that is coordinated by both histidine and aspartate residues. Deacetylation of the HDAC substrates occurs through attack by a water molecule that is activated through interaction with this zinc cation coupled with deprotonation through a histidine-aspartate charge-relay system.41 By contrast, the Class III sirtuins require NAD+ for their enzymatic activity.42
Acetylation and deacetylation of histones play an important role in transcriptional regulation of eukaryotic cells and HDAC inhibitors selectively alter gene transcription, in part, by permitting chromatin remodeling and by changing the composition of multiprotein complexes bound to proximal region of specific gene promoters.18 Further, the HDACs interact with many non-histone protein substrates such as the hormone receptors, chaperone proteins, and cytoskeletal proteins.19, 20 Thus an important question that still needs to be adequately addressed concerns identification of the individual HDAC or subset of HDACs that one might best inhibit in order to achieve a desirable therapeutic outcome. Genetic studies, knockout studies in yeast, and the use of siRNAs in mammalian cells suggest that the Class I HDACs are essential to cell proliferation and survival.43, 44 Indeed, recent evidence from the Tsai laboratory suggests that HDAC1 activity is inhibited by p25/Cdk5 leading to aberrant cell cycle activity, double-strand DNA breaks, and neuronal death.45 Interestingly, CDK5 hyperactivity, cell cycle reentry and DNA damage are all seen in AD pathophysiology downstream of Aβ.46 These results suggest HDAC1 is critical for neuronal survival and should be avoided as one thinks about HDAC isoform selectivity. Despite this necessity for HDAC1 in neurons, the Tsai laboratory has also found that global HDAC inhibition can induce sprouting of dendrites, increase the number of synapses, and reinstate learning behavior and access to long-term memories in an inducible p25/Cdk5 transgenic mouse model of Alzheimer's disease.47 Based upon our earlier work HDAC6 might be considered an important target for achieving excellent levels of neuroprotection.48 Supporting this assertion, the inactivation of HDAC6 has been shown to induce the acetylation and increased activity of antioxidant proteins peroxiredoxin 1 and 2, which are important for redox homeostasis.49 HDAC6 has also been shown to interact with tau, a microtubule-associated protein that forms neurofibrillary tangles in Alzheimer's disease. In these studies, HDAC6-selective inhibition or knockdown was found to attenuate site-specific tau phosphorylation.50 In contrast to these findings Pandey et al. have found that HDAC6 is important for mediating macroautophagy-lysosome pathways that can compensate for ubiquitin-proteasome system dysfunction, and that HDAC6 expression can suppress degeneration in a fly model expressing pathological Aβ fragments 51. While HDAC6 expression can promote autophagy and neuroprotection, it is important to note that HDAC6 knockdown alone in this model does not induce neurodegeneration, which is consistent with the findings that HDAC6-null mice are viable, relatively normal, and fertile.52 How these findings will impact HDAC6 as a target for neuroprotection remains to be evaluated. The development of inhibitors selective for the individual HDAC isoforms and/or conditional knockout mice may resolve some of the issues in this area.
Conclusion
With regard to the compounds prepared and tested herein, compounds 9-14 all showed the capacity to induce histone H4 acetylation in neurons at 10 and/or 20 μM concentration and protect in a cortical neuron oxidative toxicity model. In fact, a very good agreement is observed between the ability of a compound to induce histone H4 acetylation and its ability to protect. Compounds 12 and 13 are particularly intriguing, as, in addition to enhancing the acetylation of histone H4 and being neuroprotective, they contain high affinity for PKC, increase the production of sAPPα, and decrease Aβ40. These compounds also, on their own, show little toxicity toward the cortical neurons at concentrations as high as 20 μM. A critical aspect when designing single compounds with multiple targets is matching potency for the individual targets. Importantly, we found that the concentration where best efficacy for increasing sAPPα production, decreasing Aβ40, inducing histone acetylation, and neuroprotection for compounds 12 and 13 was in the same range; 10 μM. In light of the present findings, efforts are being made to explore these novel agents in other AD cell models. More importantly, these compounds will be studied in vivo, and results of the testing of these compounds in transgenic mouse models of AD are underway, and will be reported in due course.
Experimental Section
Chemistry
1H NMR and 13C NMR spectra were recorded on Bruker spectrometer at 300/400 MHz and 75/100 MHz, respectively, with TMS as an internal standard. HRMS experiment was performed on Q-TOF-2TM (Micromass). TLC was performed with Merck 60F254 silica gel plates. Preparative TLC was performed with analtech 1000 mm silica gel GF plates. Column chromatography was performed using Merck silica gel (40-60 mesh). HPLC was carried out on an ACE AQ column (100 × 4.6 mm and 250 ×10 mm), with detection at 210, 240, 254, 280, and 300 nm on a Shimadzu SPD-10A VP detector; flow rate = 2.0 – 3.5 mL/min; from 10% acetonitrile in water to 100% acetonitrile with 0.05% TFA.
9-Hydroxy-5-hydroxymethyl-2-isopropyl-1-methyl-(2S,5S)-1,4,5,6-tetrahydro-2H-benzo[e][1,4] diazocin-3-one (2)
Compound 1 (500 mg, 1.36 mmol) was dissolved in MeOH (20 mL) and subjected to a debenzylation reaction with H2 in presence of 10% Pd/C as catalyst. After 30 min, the reaction mixture was filtered, the filtrate was concentrated, and purified by column chromatography to afford the title compound (288 mg, 76%); [α]D20 = −331.18 (c = 0.10 in MeOH); 1H NMR (300 MHz, CD3OD): δ= 6.86 (d, J = 8.3 Hz, 1H), 6.55 (d, J = 2.2 Hz, 1H), 6.37 (dd, J = 2,3, 8.2 Hz, 1H), 4.28 (m, 1H), 3.59 (dd, J = 4.8, 11.0 Hz, 1H), 3.47 (m, 2H), 2.86 (m, 2H), 2.73 (s, 3H), 2.38 (m, 1H), 1.08 (d, J = 6.7 Hz, 3H), 0.93 (d, J = 6.7 Hz, 3H); 13C NMR (100 MHz, CD3OD): δ= 175.9, 158.3, 154.4, 133.6, 124.8, 111.2, 108.9, 65.9, 55.6, 37.9, 37.0, 29.8, 20.9, 20.1; ESI-HRMS calculated for [C15H23N2O3 + H]+: 279.1703, found: 279.1693; HPLC purity: 96.2%.
9-Hept-6-enyloxy-5-hydroxymethyl-2-isopropyl-1-methyl-1,4,5,6-tetrahydro-2H-benzo[e][1,4] diazocin-3-one (3)
To a solution of compound 2 (40 mg, 0.14 mmol) in anhydrous DMF (2 mL) was added 7-bromo-1-heptene (44 μL, 0.29 mmol), KI (72 mg, 0.43 mmol) and K2CO3 (80 mg, 0.58 mmol), then heated to 80 °C for 8 h. The mixture was cooled and extracted with ethyl acetate (50 mL), washed with water (30 mL) and brine (30 mL), dried over anhydrous Na2SO4, concentrated, and purified by column chromatography to afford the title compound (17 mg, 31%); [α]D20 = −221.23 (c = 0.09 in MeOH); 1H NMR (400 MHz, CDCl3): δ= 6.93 (br s, 1H), 6.92 (d, J = 8.0 Hz, 1H), 6.52 (d, J = 4.0 Hz, 1H), 6.41 (dd, J = 4.0, 8.0 Hz, 1H), 5.87–5.77 (m, 1H), 5.03–4.94 (m, 2H), 3.93–3.89 (m, 3H), 3.72–3.66 (m, 2H), 3.50–3.48 (m, 2H), 3.03 (dd, J = 8.0, 16.0 Hz, 1H), 2.77 (s, 3H), 2.77–2.69 (m, 1H), 2.45–2.37 (m, 1H), 2.09–2.08 (m, 2H), 1.79– 1.76 (m, 2H), 1.47–1.46 (m, 4H), 1.05 (d, J = 8.0 Hz, 3H), 0.87 (d, J = 8.0 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ= 174.4, 159.0, 152.8, 139.0, 132.5, 123.2, 114.7, 106.8, 106.6, 70.0, 68.1, 66.1, 54.8, 36.7, 35.3, 33.9, 29.4, 28.9, 28.5, 25.8, 20.7, 20.1; ESI-HRMS calculated for [C22H34N2O3 + H]+: 375.2648, found: 375.2642; HPLC purity: 95.1%.
5-(O-tert-Butyldimethylsilanyloxymethyl)9-hydroxy-2-isopropyl-1-methyl-(2S,5S)-1,4,5,6-tetra-hydrodro-2H-benzo[e][1,4]diazocin-3-one (4)
To a solution of compound 1 (1.00 g, 2.71 mmol) in anhydrous CH2Cl2 (20 mL) was added Et3N (2.30 mL, 16.50 mmol) and tert-butyldimethylsilyl chloride (1.60 g, 10.62 mmol) at 0 °C, the mixture was stirred for another 5 min followed by the addition of N,N-dimethyl-4-aminopyridine (66 mg, 0.54 mmol), and then stirred for an additional 3 h. The reaction mixture was quenched by a saturated aqueous NH4Cl solution, and then extracted with CH2Cl2 (80 mL), washed with aqueous HCl solution (0.5 N, 10 mL), water (40 mL) and brine (40 mL), dried over anhydrous Na2SO4, and concentrated to afford the crude residue, which was dissolved in ethyl acetate (20 mL) and subjected to a debenzylation reaction with hydrogen under balloon pressure in the presence of 20% Pd(OH)2/C as catalyst. The reaction mixture was filtered after 30 min, and the filtrate was concentrated and purified by column chromatography to afford the title compound (806 mg, 76%); [α]D20 = −232.51 (c = 0.11 in MeOH); 1H NMR (400 MHz, CDCl3): δ= 6.82 (d, J = 8.1 Hz, 1H), 6.50 (s, 1H), 6.30 (dd, J = 2,4, 8.2 Hz, 1H), 6.12 (d, J = 3.2 Hz, 1H), 6.06 (s, 1H), 4.06 (m, 1H), 3.64 (dd, J = 3.9, 9.8 Hz, 1H), 3.50 (dd, J = 2.4, 15.3 Hz, 2H), 2.98 (dd, J = 8.0, 16.4 Hz, 1H), 2.75 (s, 3H), 2.68 (dd, J = 8.0, 16.4 Hz, 1H), 2.42 (m, 1H), 1.06 (d, J = 7.0 Hz, 3H), 0.91 (s, 9H), 0.89 (d, J = 7.0 Hz, 3H), 0.09 (s, 3H), 0.07 (s, 3H); 13C NMR (100 MHz, CDCl3): δ= 173.3, 155.8, 152.8, 132.4, 122.7, 108.9, 107.0, 71.3, 65.9, 53.8, 36.7, 35.5, 28.3, 25.9, 19.9, 18.3, −5.3, −5.4; ESI-HRMS calculated for [C21H36N2O3Si + H]+: 393.2568, found: 393.2572.
2-Isopropyl-1-methyl-9-(2-tert-butyloxycarboxamidoethoxy)-3-oxo-(2S,5S)-1,2,3,4,5,6-hexahydrobenzo[e][1,4]diazocin-5-ylmethyl acetate (5)
To a solution of compound 2 (500 mg, 1.81 mmol) in anhydrous DMF (10 mL) was added 2-tert-Boc-aminoethylbromide (805 mg, 3.61 mmol) and Cs2CO3 (1.80 g, 5.52 mmol), the mixture was heated to 100 °C for 24 h. The mixture was extracted with ethyl acetate (200 mL), washed with water (100 mL) and brine (100 mL), dried over anhydrous Na2SO4, concentrated, and purified by column chromatography to afford the intermediate alcohol (152 mg, 20%); [α]D20 = −189.10 (c = 0.10 in MeOH); 1H NMR (400 MHz, CDCl3): δ= 6.94 (d, J = 9.0 Hz, 1H), 6.68 (br s, 1H), 6.51 (d, J = 2.0 Hz, 1H), 6.39 (dd, J = 2.0, 9.0 Hz, 1H), 5.01 (br s, 1H), 3.97 (dd, J = 5.0, 10.0 Hz, 2H), 3.88 (br s, 1H), 3.68 (m, 2H), 3.49 (m, 4H), 3.04 (dd, J = 7.8, 16.5 Hz, 1H), 2.77 (s, 3H), 2.69 (dd, J = 7.8, 16.5 Hz, 1H), 2.41 (m, 1H), 1.45 (s, 9H), 1.05 (d, J = 7.0 Hz, 3H), 0.86 (d, J = 7.0 Hz, 3H); 13C NMR (75 MHz, CDCl3): δ= 173.7, 158.5, 156.2, 152.9, 132.6, 123.7, 106.7, 106.4, 79.8, 67.3, 66.0, 54.8, 40.4, 36.8, 35.2, 29.9, 28.6, 28.5, 20.7, 20.1; ESI-HRMS calculated for [C22H36N3O5 + H]+: 422.2649, found: 422.2638.
The alcohol (200 mg, 0.47 mmol) was dissolved in anhydrous CH2Cl2 (3 mL) and cooled to 0 °C, pyridine (0.23 mL, 2.82 mmol) and Ac2O (0.20 mL, 2.12 mmol) were added sequentially and the reaction mixture was stirred at the room temperature for 12 h. The reaction mixture was extracted with ethyl acetate (60 mL), washed with aqueous HCl solution (1N, 10 mL), water (20 mL) and brine (20 mL), dried over anhydrous Na2SO4, concentrated, and purified by column chromatography to afford 5 (170 mg, 78%); 1H NMR (400 MHz, CDCl3): δ= 6.89 (d, J = 8.1 Hz, 1H), 6.54 (s, 1H), 6.40 (d, J = 8.1 Hz, 1H), 5.93 (s, 1H), 4.99 (br s, 1H), 4.32 (m, 1H), 4.15 (dd, J = 3.4, 11.0 Hz, 1H), 3.92 (m, 3H), 3.44 (m, 3H), 2.89 (m, 2H), 2.73 (s, 3H), 2.37 (m, 1H), 2.06 (s, 3H), 1.41 (s, 9H), 1.02 (d, J = 6.4 Hz, 3H), 0.87 (d, J = 6.4 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ= 173.1, 170.8, 158.6, 156.0, 152.9, 132.6, 123.4, 107.7, 107.1, 79.7, 71.7, 67.3, 67.0, 51.7, 40.3, 37.3, 35.9, 28.5, 21.0, 20.2, 19.9; ESI-HRMS calculated for [C24H37N3O6 + Na]+: 486.2575, found: 486.2567.
1N-{2-[5-Hydroxymethyl-2-isopropyl-1-methyl-3oxo-(2S,5S)-1,2,3,4,5,6-hexahydrobenzo[e][1,4] diazocin-9-yloxy]ethyl}-5-(1,2-dithiolan-3-yl)pentanamide (6)
To a solution of compound 5 (63 mg, 0.14 mmol) in anhydrous CH2Cl2 (1.6 mL) was added TFA (0.4 mL) at 0 °C, and the reaction mixture was stirred at room temperature for 3 h. Solvent and excess TFA was removed under reduced pressure and the crude residue was dried under high vacuo and subjected to next reaction without further purification.
To a solution of (±)-α-Lipoic acid (25 mg, 0.12 mmol) in anhydrous CH2Cl2 (2 mL) was added HOBT (16 mg, 0.12 mmol) and EDCI (24 mg, 0.12 mmol) at 0 °C and stirred for 15 min. The crude amine dissolved in anhydrous CH2Cl2 (2 mL, free amine was generated from TFA amine-salt by adding N,N-diisopropylethylamine (0.1 mL) to CH2Cl2 solution of TFA salt) was added to the active ester solution and the resultant mixture was stirred at room temperature for 12 h. The reaction was quenched by adding saturated aqueous NH4Cl solution (10 mL) and extracted with ethyl acetate (40 mL), washed with water (20 mL) and brine (20 mL), dried over anhydrous Na2SO4, and concentrated to give crude residue. The residue was dissolved in anhydrous MeOH (2 mL) and cooled to 0 °C, K2CO3 (16 mg, 0.12 mmol) was added and stirred for another 30 min at same temperature. The mixture was extracted with ethyl acetate (50 mL), washed with water (25 mL) and brine (25 mL), dried over anhydrous Na2SO4, concentrated, and purified by column chromatography to afford the title compound (49 mg, 80%); [α]D20 = −136.44 (c = 0.11 in MeOH); 1H NMR (400 MHz, CDCl3,): δ= 6.93 (d, J = 8.0 Hz, 1H), 6.84 (s, 1H), 6.52 (s, 1H), 6.40 (d, J = 8.0 Hz, 1H), 6.11 (t, J = 6.0 Hz, 1H), 4.0 (m, 2H), 3.86 (m, 2H), 3.59 (m, 3H), 3.48 (m, 3H), 3.12 (m, 3H), 2.76 (s, 3H), 2.75 (m, 1H), 2.42 (m, 2H), 2.22 (t, J = 8.0 Hz, 2H), 1.84 (m, 1H), 1.66 (m, 4H), 1.46 (m, 2H), 1.05 (t, J = 7.0 Hz, 3H), 0.87 (t, J = 7.0 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ= 173.8, 173.0, 158.1, 152.7, 132.5, 123.8, 106.7, 106.2, 70.2, 66.7, 65.7, 56.3, 54.4, 40.2, 38.9, 38.4, 36.5, 36.3, 35.2, 34.5, 28.8, 28.2, 22.6, 20.3, 19.9; ESI-HRMS calculated for [C25H39N3O4S2 + Na]+: 532.2275, found: 532.2262. HPLC purity: 98.6%.
(2S,5S)-1,2,3,4,5,6-hexahydro-5-(hydroxymethyl)-2-isopropyl-1-methyl-3-oxobenzo[e][1,4] diazocin-9-yl 5-(1,2-dithiolan-3-yl)pentanoate (7)
To a solution of (±)-α-Lipoic acid (30 mg, 0.15 mmol) in CH2Cl2 (2 mL) was added DCC (33 mg, 0.16 mmol) and N,N-dimethyl-4-aminopyridine (9 mg, 0.07 mmol) at 0 °C, then added compound 4 solution (68 mg, 0.17 mmol, dissolved in CH2Cl2 (1 mL)). The reaction mixture was warmed to room temperature and stirred for another 3 h, then extracted with ethyl acetate (40 mL), washed with saturated aqueous NaHCO3 solution (10 mL), water (20 mL) and brine (20 mL), dried over anhydrous Na2SO4, concentrated, and purified by column chromatography to yield the ester intermediate. The ester was subjected to TBDMS deprotection with p-toluenesulphonic acid (28 mg, 0.15 mmol) in MeOH for 3 h at room temperature, then extracted with ethyl acetate (40 mL), washed with water (20 mL) and brine (20 mL), dried over anhydrous Na2SO4, concentrated and purified by column chromatography to afford the title compound (25 mg, 36%); [α]D20 = −235.88 (c = 0.17 in MeOH); 1H NMR (400 MHz, CDCl3): δ= 7.03 (d, J = 8.3 Hz, 1H), 6.78 (s, 1H), 6.68 (d, J = 2.2 Hz, 1H), 6.60 (dd, J = 2.2, 8.3 Hz, 1H), 3.81 (m, 1H), 3.66 (m, 1H), 3.47 (m, 3H), 3.12 (m, 3H), 2.77 (s, 3H), 2.74 (m, 1H), 2.53 (m, 2H), 2.43 (m, 2H), 1.76 (m, 1H), 1.72 (m, 4H), 1.53 (m, 3H), 1.03 (d, J = 6.4 Hz, 3H), 0.83 (d, J = 6.4 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ= 173.9, 172.2, 152.6, 150.4, 132.6, 128.2, 114.3, 112.7, 69.7, 66.2, 56.6, 54.4, 40.5, 38.8, 37.0, 35.1, 34.8, 34.4, 28.9, 28.4, 24.8, 20.8, 20.0; ESI-HRMS calculated for [C23H34N2O4S2 + H]+: 467.2033, found: 467.2032; HPLC purity: 96.3%.
Thioacetic acid S-[3-(5-hydroxymethyl-2-isopropyl-1-methyl-3-oxo-1,2,3,4,5,6-hexahydro-benzo[e] [1,4]diazocin-9-yloxy)-propyl] ester (9)
To a solution of compound 2 (50 mg, 0.18 mmol) in anhydrous DMF (2 mL) was added 1,3-Diiodopropane (207 μL, 1.80 mmol) and Cs2CO3 (234 mg, 0.72 mmol), then heated to 80 °C for 8 h. The mixture was cooled and then extracted with ethyl acetate (30 mL), washed with water (15 mL) and brine (15 mL), dried over anhydrous Na2SO4, concentrated, and purified by column chromatography to afford iodo-substituted intermediate (20 mg, 0.037 mmol). The intermediate was dissolved in anhydrous DMF (0.5 mL), potassium thioacetate (20 mg, 0.19 mmol) was added to the solution, and then the mixture was heated to 50 °C for 2 h. The mixture was extracted with ethyl acetate (30 mL), washed with water (15 mL) and brine (15 mL), dried over anhydrous Na2SO4, concentrated, and purified by column chromatography to afford 9 (6 mg, 12%); [α]D20 = −232.12 (c = 0.075 in MeOH); 1H NMR (400 MHz, CDCl3): δ= 6.94 (d, J = 8.0 Hz, 1H), 6.85 (br s, 1H), 6.54 (d, J = 2.0 Hz, 1H), 6.43 (dd, J = 2.0, 8.0 Hz, 1H), 4.01 (br s, 1H), 3.98 (t, J = 8.0 Hz, 2H), 3.74 (dd, J = 4.0, 12.0 Hz, 1H), 3.59–3.51 (m, 2H), 3.07–3.01 (m, 3H), 2.80 (s, 3H), 2.80–2.76 (m, 1H), 2.48–2.41 (m, 1H), 2.34 (s, 3H), 2.08 –2.02 (m, 2H), 1.06 (d, J = 8.0 Hz, 3H), 0.89 (d, J = 8.0 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ= 195.8, 174.4, 158.6, 152.6, 132.4, 123.2, 107.0, 106.7, 70.3, 66.2, 65.9, 54.6, 36.5, 35.3, 30.7, 29.4, 28.3, 25.9, 20.3, 19.9; ESI-HRMS calculated for [C20H30N2O4S + H]+: 395.2005, found 395.2006; HPLC purity: 96.4%.
Thioacetic acid S-[4-(5-hydroxymethyl-2-isopropyl-1-methyl-3-oxo-1,2,3,4,5,6-hexahydro-benzo [e][1,4]diazocin-9-yloxy)-butyl] ester (10)
The title compound (yield 56%) was obtained according to compound 9 except using 1,4-diiodobutane: [α]D20 = −186.50 (c = 0.25 in MeOH); 1H NMR (400 MHz, CDCl3,): δ= 6.92 (d, J = 8.0 Hz, 1H), 6.62 (br s, 1H), 6.51 (d, J = 2.0 Hz, 1H), 6.40 (dd, J = 2.0, 8.0 Hz, 1H), 3.93 (t, J = 8.0 Hz, 2H), 3.89 (br s, 1H), 3.70 (dd, J = 4.0, 12.0 Hz, 1H), 3.54–3.49 (m, 2H), 3.05–3.01 (m, 1H), 2.95 (t, J = 8.0 Hz, 2H), 2.79 (s, 3H), 2.79–2.76 (m, 1H), 2.48–2.41 (m, 1H), 2.34 (s, 3H), 1.86–1.74 (m, 4H), 1.05 (d, J = 8.0 Hz, 3H), 0.87 (d, J = 8.0 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ= 195.9, 173.7, 158.7, 152.6, 133.3, 123.0, 106.5, 106.3, 69.7, 67.2, 66.0, 54.5, 36.6, 34.9, 30.6, 28.8, 28.4, 28.2, 26.3, 20.5, 19.8; ESI-HRMS calculated for [C21H32N2O4S + H]+: 409.2161, found: 409.2164; HPLC purity: 95.1%.
Thioacetic acid S-[5-(5-hydroxymethyl-2-isopropyl-1-methyl-3-oxo-1,2,3,4,5,6-hexahydro-benzo[e] [1,4]diazocin-9-yloxy)-pentyl] ester (11)
The title compound (yield 38%) was obtained according to compound 9 except using 1,5-diiodopentane: [α]D20 = −261.70 (c = 0.45 in MeOH); 1H NMR (400 MHz, CDCl3,): δ= 6.92 (d, J = 8.0 Hz, 1H), 6.74 (br s, 1H), 6.51 (d, J = 2.0 Hz, 1H), 6.40 (dd, J = 2.0, 8.0 Hz, 1H), 3.91(t, J = 8.0 Hz, 2H), 3.89 (br s, 1H), 3.70 (dd, J = 4.0, 8.0 Hz, 1H), 3.54–3.49 (m, 2H), 3.05–3.01 (m, 1H), 2.90 (t, J = 8.0 Hz, 2H), 2.78 (s, 3H), 2.78–2.70 (m, 1H), 2.48–2.41 (m, 1H), 2.33 (s, 3H), 1.86–1.76 (m, 2H), 1.67–1.61 (m, 2H), 1.55–1.49 (m, 2H), 1.05 (d, J = 8.0 Hz, 3H), 0.87 (d, J = 8.0 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ= 195.9, 174.0, 158.7, 152.6, 132.3, 123.0, 106.6, 106.3, 69.8, 67.5, 65.8, 54.5, 36.5, 35.0, 30.6, 29.3, 28.9, 28.8, 28.2, 25.3, 20.5, 19.9; ESI-HRMS calculated for [C22H34N2O4S + H]+: 423.2318, found: 423.2317; HPLC purity: 99.5%.
Thioacetic acid S-[6-(5-hydroxymethyl-2-isopropyl-1-methyl-3-oxo-1,2,3,4,5,6-hexahydro-benzo[e] [1,4]diazocin-9-yloxy)-hexyl] ester (12)
The title compound (yield 67%) was obtained according to compound 9 except using 1,6-diiodohexane: [α]D20 = −197.20 (c = 0.55 in MeOH); 1H NMR (400 MHz, CDCl3): δ= 6.92 (d, J = 8.4 Hz, 1H), 6.52 (d, J = 2.0 Hz, 1H), 6.39 (dd, J = 2.0, 8.0 Hz, 1H), 6.20 (br s, 1H), 3.92–3.89 (m, 3H), 3.76–3.70 (m, 1H), 3.52–3.50 (m, 2H), 3.06 (dd, J = 6.8, 16.4 Hz, 1H), 2.88 (t, J = 7.2 Hz, 2H), 2.80 (s, 3H), 2.80–2.70 (m, 1H), 2.45–2.43 (m, 1H), 2.33 (s, 3H), 1.78–1.74 (m, 2H), 1.62–1.56 (m, 2H), 1.47–1.43 (m, 4H), 1.05 (d, J = 6.4 Hz, 3H), 0.87 (d, J = 6.4 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ= 196.1, 174.1, 158.8, 152.6, 132.3, 122.9, 106.7, 106.4, 69.9, 67.8, 65.9, 54.6, 36.6, 35.1, 30.7, 29.5, 29.2, 29.0, 28.6, 28.3, 25.6, 20.5, 19.9; ESI-HRMS calculated for [C23H36N2O4S + H]+: 437.2474, found: 437.2478; HPLC purity: 99.9%.
Thioacetic acid S-[7-(5-hydroxymethyl-2-isopropyl-1-methyl-3-oxo-1,2,3,4,5,6-hexahydro benzo[e] [1,4]diazocin-9-yloxy)-heptyl] ester (13)
The title compound (yield 49%) was obtained according to compound 9 except using 1,7-ditosylheptane: [α]D20 = −183.43 (c = 0.18 in MeOH); 1H NMR (400 MHz, CDCl3): δ= 6.93 (d, J = 8.0 Hz, 1H), 6.88 (br s, 1H), 6.54 (d, J = 2.0 Hz, 1H), 6.43 (dd, J = 2.0, 8.0 Hz, 1H), 4.01 (br s, 1H), 3.91 (t, J = 8.0 Hz, 2H), 3.74 (dd, J = 4.0 Hz, 1H), 3.59–3.51 (m, 2H), 3.03 (dd, J = 8.0, 16.0 Hz, 1H), 2.87 (t, J = 8.0 Hz, 2H), 2.79 (s, 3H), 2.79–2.76 (m, 1H), 2.45–2.43 (m, 1H), 2.32 (s, 3H), 1.78–1.72 (m, 2H), 1.60–1.57 (m, 2H), 1.47–1.38 (m, 6H), 1.06 (d, J = 8.0 Hz, 3H), 0.89 (d, J = 8.0 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ= 196.1, 174.3, 158.9, 152.6, 132.3, 122.9, 107.0, 106.6, 70.3, 67.9, 65.9, 54.6, 36.5, 36.3, 30.7, 29.4, 29.2, 29.1, 28.9, 28.7, 28.3, 25.9, 20.3, 19.9; ESI-HRMS calculated for [C24H38N2O4S + Na]+: 473.2450, found: 473.2443; HPLC purity: 98.5%.
Thioacetic acid S-[8-(5-hydroxymethyl-2-isopropyl-1-methyl-3-oxo-1,2,3,4,5,6-hexahydro-benzo[e] [1,4]diazocin-9-yloxy)-octyl] ester (14)
The title compound (yield 39%) was obtained according to compound 9 except using 1,8-dibromooctane: [α]D20 = −154.37 (c = 0.07 in MeOH); 1H NMR (400 MHz, CDCl3): δ= 6.94 (d, J = 8.0 Hz, 1H), 6.93 (br s, 1H), 6.55 (d, J = 2.4 Hz, 1H), 6.44 (dd, J = 2.4, 8.0 Hz, 1H), 4.00 (br s, 1H), 3.92 (t, J = 6.4 Hz, 2H), 3.73 (dd, J = 4.0, 11.2 Hz, 1H), 3.58–3.51 (m, 2H), 3.03–2.99 (m, 1H), 2.88 (t, J = 7.2 Hz, 2H), 2.80 (s, 3H), 2.80 –2.76 (m, 1H), 2.48–2.41 (m, 1H), 2.34 (s, 3H), 1.79–1.75 (m, 2H), 1.60–1.57 (m, 2H), 1.46–1.36 (m, 8H), 1.07 (d, J = 6.8 Hz, 3H), 0.90 (d, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ= 196.1, 174.4, 158.9, 152.6, 132.3, 122.9, 106.9, 106.6, 70.1, 67.9, 65.8, 54.6, 36.5, 35.2, 30.6, 29.5, 29.3, 29.2, 29.1, 29.0, 28.7, 28.3, 26.0, 20.3, 19.9; ESI-HRMS calculated for [C25H40N2O4S + H]+: 465.2787, found: 465.2787; HPLC purity: 98.4%.
sAPPα and Aβ40 detection
Human APP stably transfected HEK293 cells lines were maintained in DMEM with 10% FBS. Cells were seeded onto 6-well plate, and incubated at 37 °C overnight. Compounds 9-14 were first dissolved in DMSO as a 10 mM stock and diluted into the respective concentration with fresh DMEM with 10% FBS. After exposure overnight, the cell medium was replaced by fresh medium containing the PKC agonist. After 48 h incubation, the medium was collected and both the soluble APP (sAPPα) and Aβ40 levels in the medium were examined by using the sAPPα and Aβ40 ELISA kits obtained from Biosource according to the manufacturer's instructions.
Primary Neurons and Cell Culture
Cell cultures were obtained from the cerebral cortex of fetal Sprague-Dawley rats (embryonic day 17), as described previously.31 All experiments were initiated 24 h after plating. Under these conditions, the cells are not susceptible to glutamate-mediated excitotoxicity.
Oxidative Toxicity and Neuron Viability Assays
For cytotoxicity studies, cells were rinsed with warm PBS and then placed in minimum essential medium (Invitrogen) containing glucose (5.5 g L-1), 10% fetal calf serum, L-glutamine (2 mM), and cystine (100 μM). Oxidative stress was induced by the addition of the glutamate analog HCA (5 mM) to the media. HCA was diluted from 100-fold concentrated solutions that were adjusted to pH 7.5. In combination with HCA, TSA, SAHA, or the novel PKC-HDAC compounds were added at different concentrations. Viability was assessed after 48 h by calceinacetoxymethyl ester (AM)/ethidium homodimer-1 staining (live/dead assay; Molecular Probes, Eugene, OR) under fluorescence microscopy and the MTT assay method. Each bar represents the mean ± SE of four replicates.
Histone Precipitation and Western Blot Analysis
Approximately one million treated neurons were incubated in 1 mL hypotonic lysis buffer containing Tris-HCl (pH 8.0, 10 mM), KCl (1 mM), MgCl2(1.5 mM), DTT(1 mM), aprotinin (1 mM), pepstatin (1 mM), and PMSF (0.4 mM) for 30 min rotating at 4 °C. Nuclei were pelleted by centrifugation for 10 min at 10,000 rpm, resuspended in H2SO4 (0.4 N, 200 μL), and rotated for 12 h at 4 °C. Following centrifugation at 13,000 rpm for 10 min the supernatant was transferred to a new tube, and the histone proteins were precipitated by adding 100% TCA (66 μL) dropwise followed by 30 min incubation on ice. Histone proteins were pelleted by centrifugation at 13000 rpm for 10 min, washed twice with ice-cold acetone, dried at room temperature for 20-40 min, and resuspended in H2O (50 μL). Twenty microliters of total histone proteins were boiled in Laemmli buffer and electrophoresed under reducing conditions on 15% polyacrylamide gel. Histone proteins were transferred to a nitrocellulose membrane (Bio-Rad). Nonspecific binding was inhibited by incubation in Tris-buffered saline with Tween 20 (TBST: 50 mM Tris-HCl, pH 8.0, 0.9% NaCl, and 0.1% Tween 20) containing 5% nonfat dried milk for at least 1.5 h. Primary antibodies against acetylated histone H4 or total histone H4 (Upstate) were diluted 1:1000 or 1:4000, respectively, in TBST containing 5% milk and incubated with the membrane for 3 h at room temperature, followed by incubation with antirabbit horseradish peroxidase-conjugated secondary antibodies for 2 h at room temperature. Acetyl histone H4 and total histone H4 immunoreactivity was detected according to the enhanced chemiluminescent protocol (Amersham Biosciences).
Docking Studies
The 3D structures of compound 13 was generated and optimized by using Sybyl program according to minimal energy evaluation, and the crystal structure of PKCδ C1b (PDB: 1PTR) and HDAC7 (PDB: 3C10) were used as template receptor structures. Docking simulations were performed via Autodocking method in FlexX program.
The program system used for the docking simulations was “Sybyl”, developed by Tripos Inc.
Supplementary Material
Acknowledgments
This work is supported by the National Institutes of Health (Grant RO1AG022941, APK and RO1AG025888, YS) and in part by the Intramural Research Program of the National Institutes of Health, Center for Cancer Research. Support also comes from the Adelson Medical Research Foundation (BL) and Burke Foundation (BL). We thank Dr. Rong He and Ms Kathryn McLaughlin for assistance in the preparation of the manuscript.
References
- 1.Pascale A, Amadio M, Govoni S, Battaini F. Pharmacol Res. 2007;55:560. doi: 10.1016/j.phrs.2007.04.013. [DOI] [PubMed] [Google Scholar]
- 2.Gandy S, Greengard P. Int Rev Neurobiol. 1994;36:29. doi: 10.1016/s0074-7742(08)60302-5. [DOI] [PubMed] [Google Scholar]
- 3.Buxbaum JD, Thinakaran G, Koliatsos V, O'Callahan J, Slunt HH, Price DL, Sisodia SS. J Neurosci. 1998;18:9629. doi: 10.1523/JNEUROSCI.18-23-09629.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buxbaum JD, Liu KN, Luo Y, Slack JL, Stocking KL, Peschon JJ, Johnson RS, Castner BJ, Cerretti DP, Black RA. J Biol Chem. 1998;273:27765. doi: 10.1074/jbc.273.43.27765. [DOI] [PubMed] [Google Scholar]
- 5.Cole G, Dobkins KR, Hansen LA, Terry RD, Saitoh T. Brain Res. 1988;452:165. doi: 10.1016/0006-8993(88)90021-2. [DOI] [PubMed] [Google Scholar]
- 6.Masliah E, Cole G, Shimohama S, Hansen L, DeTeresa R, Terry RD, Saitoh T. J Neurosci. 1990;10:2113. doi: 10.1523/JNEUROSCI.10-07-02113.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shimohama S, Narita M, Matsushima H, Kimura J, Kameyama M, Hagiwara M, Hidaka H, Taniguchi T. Neurology. 1993;43:1407. doi: 10.1212/wnl.43.7.1407. [DOI] [PubMed] [Google Scholar]
- 8.Wang HY, Pisano MR, Friedman E. Neurobiol Aging. 1994;15:293. doi: 10.1016/0197-4580(94)90023-x. [DOI] [PubMed] [Google Scholar]
- 9.Masliah E, Cole GM, Hansen LA, Mallory M, Albright T, Terry RD, Saitoh T. J Neurosci. 1991;11:2759. doi: 10.1523/JNEUROSCI.11-09-02759.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chachin M, Shimohama S, Kunugi YU, Taniguchi T. Jpn J Pharmacol. 1996;71:175. doi: 10.1254/jjp.71.175. [DOI] [PubMed] [Google Scholar]
- 11.Govoni S, Bergamaschi S, Racchi M, Battaini F, Binetti G, Bianchetti A, Trabucchi M. Neurology. 1993;43:2581. doi: 10.1212/wnl.43.12.2581. [DOI] [PubMed] [Google Scholar]
- 12.Govoni S, Racchi M, Bergamaschi S, Trabucchi M, Battaini F, Bianchetti A, Binetti G. Ann N Y Acad Sci. 1996;777:332. doi: 10.1111/j.1749-6632.1996.tb34442.x. [DOI] [PubMed] [Google Scholar]
- 13.Cai H, Wang Y, McCarthy D, Wen H, Borchelt DR, Price DL, Wong PC. Nat Neurosci. 2001;4:233. doi: 10.1038/85064. [DOI] [PubMed] [Google Scholar]
- 14.Vassar R, Citron M. Neuron. 2000;27:419. doi: 10.1016/s0896-6273(00)00051-9. [DOI] [PubMed] [Google Scholar]
- 15.Bhagavan S, Ibarreta D, Ma D, Kozikowski AP, Etcheberrigaray R. Neurobiol Dis. 1998;5:177. doi: 10.1006/nbdi.1998.0195. [DOI] [PubMed] [Google Scholar]
- 16.Ibarreta D, Duchâen M, Ma D, Qiao L, Kozikowski AP, Etcheberrigaray R. Neuroreport. 1999;10:1035. doi: 10.1097/00001756-199904060-00026. [DOI] [PubMed] [Google Scholar]
- 17.Etcheberrigaray R, Tan M, Dewachter I, Kuiperi C, Van der Auwera I, Wera S, Qiao L, Bank B, Nelson TJ, Kozikowski AP, Van Leuven F, Alkon DL. Proc Natl Acad Sci U S A. 2004;101:11141. doi: 10.1073/pnas.0403921101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gui CY, Ngo L, Xu WS, Richon VM, Marks PA. Proc Natl Acad Sci U S A. 2004;101:1241. doi: 10.1073/pnas.0307708100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marks PA, Dokmanovic M. Expert Opin Investig Drugs. 2005;14:1497. doi: 10.1517/13543784.14.12.1497. [DOI] [PubMed] [Google Scholar]
- 20.Minucci S, Pelicci PG. Nat Rev Cancer. 2006;6:38. doi: 10.1038/nrc1779. [DOI] [PubMed] [Google Scholar]
- 21.Gray SG, Dangond F. Epigenetics. 2006;1:67. doi: 10.4161/epi.1.2.2678. [DOI] [PubMed] [Google Scholar]
- 22.Abel T, Zukin RS. Curr Opin Pharmacol. 2008;8:57. doi: 10.1016/j.coph.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Minamiyama M, Katsuno M, Adachi H, Waza M, Sang C, Kobayashi Y, Tanaka F, Doyu M, Inukai A, Sobue G. Hum Mol Genet. 2004;13:1183. doi: 10.1093/hmg/ddh131. [DOI] [PubMed] [Google Scholar]
- 24.Kozikowski AP, Ma D, Pang YP, Shum P, Likic V, Mishra PK, Macura S, Basu A, Lazo JS, Ball RG. J Am Chem Soc. 1993;115:3957. [Google Scholar]
- 25.Kozikowski AP, Wang S, Ma D, Yao J, Ahmad S, Glazer RI, Bogi K, Acs P, Modarres S, Lewin NE, Blumberg PM. J Med Chem. 1997;40:1316. doi: 10.1021/jm960875h. [DOI] [PubMed] [Google Scholar]
- 26.Kozikowski AP, Nowak I, Petukhov PA, Etcheberrigaray R, Mohamed A, Tan M, Lewin N, Hennings H, Pearce LL, Blumberg PM. J Med Chem. 2003;46:364. doi: 10.1021/jm020350r. [DOI] [PubMed] [Google Scholar]
- 27.Kozikowski AP, Chen Y, Gaysin A, Chen B, D'Annibale MA, Suto CM, Langley BC. J Med Chem. 2007;50:3054. doi: 10.1021/jm070178x. [DOI] [PubMed] [Google Scholar]
- 28.Clark DE. J Pharm Sci. 1999;88:815. doi: 10.1021/js980402t. [DOI] [PubMed] [Google Scholar]
- 29.Dashwood RH, Ho E. Semin Cancer Biol. 2007;17:363. doi: 10.1016/j.semcancer.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miller TA, Witter DJ, Belvedere S. J Med Chem. 2003;46:5097. doi: 10.1021/jm0303094. [DOI] [PubMed] [Google Scholar]
- 31.Ratan RR, Murphy TH, Baraban JMJ. Neurochem. 1994;62:376. doi: 10.1046/j.1471-4159.1994.62010376.x. [DOI] [PubMed] [Google Scholar]
- 32.Langley B, D'Annibale MA, Suh K, Ayoub I, Tolhurst A, Bastan B, Yang L, Ko B, Fisher M, Cho S, Beal MF, Ratan RR. J Neurosci. 2008;28:163. doi: 10.1523/JNEUROSCI.3200-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Newton AC. J Biol Chem. 1995;270:28495. doi: 10.1074/jbc.270.48.28495. [DOI] [PubMed] [Google Scholar]
- 34.Quest AF. Enzyme Protein. 1996;49:231. doi: 10.1159/000468635. [DOI] [PubMed] [Google Scholar]
- 35.Hennings H, Blumberg PM, Pettit GR, Herald CL, Shores R, Yuspa SH. Carcinogenesis. 1987;8:1343. doi: 10.1093/carcin/8.9.1343. [DOI] [PubMed] [Google Scholar]
- 36.Newton AC, Keranen LM. Biochemistry. 1994;33:6651. doi: 10.1021/bi00187a035. [DOI] [PubMed] [Google Scholar]
- 37.Edwards AS, Newton AC. Biochemistry. 1997;36:15615. doi: 10.1021/bi9718752. [DOI] [PubMed] [Google Scholar]
- 38.Rundlett SE, Carmen AA, Kobayashi R, Bavykin S, Turner BM, Grunstein M. Proc Natl Acad Sci U S A. 1996;93:14503. doi: 10.1073/pnas.93.25.14503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Taunton J, Hassig CA, Schreiber SL. Science (New York, N Y) 1996;272:408. doi: 10.1126/science.272.5260.408. [DOI] [PubMed] [Google Scholar]
- 40.Marks P, Rifkind RA, Richon VM, Breslow R, Miller T, Kelly WK. Nat Rev Cancer. 2001;1:194. doi: 10.1038/35106079. [DOI] [PubMed] [Google Scholar]
- 41.Finnin MS, Donigian JR, Cohen A, Richon VM, Rifkind RA, Marks PA, Breslow R, Pavletich NP. Nature. 1999;401:188. doi: 10.1038/43710. [DOI] [PubMed] [Google Scholar]
- 42.Frye RA. Biochem Biophys Res Commun. 2000;273:793. doi: 10.1006/bbrc.2000.3000. [DOI] [PubMed] [Google Scholar]
- 43.Bernstein BE, Tong JK, Schreiber SL. Proc Natl Acad Sci U S A. 2000;97:13708. doi: 10.1073/pnas.250477697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Glaser KB, Li J, Staver MJ, Wei RQ, Albert DH, Davidsen SK. Biochem Biophys Res Commun. 2003;310:529. doi: 10.1016/j.bbrc.2003.09.043. [DOI] [PubMed] [Google Scholar]
- 45.Kim D, Frank CL, Dobbin MM, Tsunemoto RK, Tu W, Peng PL, Guan JS, Lee BH, Moy LY, Giusti P, Broodie N, Mazitschek R, Delalle I, Haggarty SJ, Neve RL, Lu Y, Tsai LH. Neuron. 2008;60:803. doi: 10.1016/j.neuron.2008.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lopes JP, Oliveira CR, Agostinho P. Cell Cycle. 2009;8:97. doi: 10.4161/cc.8.1.7506. [DOI] [PubMed] [Google Scholar]
- 47.Fischer A, Sananbenesi F, Wang X, Dobbin M, Tsai LH. Nature. 2007;447:178. doi: 10.1038/nature05772. [DOI] [PubMed] [Google Scholar]
- 48.Kozikowski AP, Chen Y, Gaysin A, Chen B, D'Annibale MA, Suto CM, Langley BC. J Med Chem. 2007;50:3054. doi: 10.1021/jm070178x. [DOI] [PubMed] [Google Scholar]
- 49.Parmigiani RB, Xu WS, Venta-Perez G, Erdjument-Bromage H, Yaneva M, Tempst P, Marks PA. Proc Natl Acad Sci U S A. 2008;105:9633. doi: 10.1073/pnas.0803749105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ding H, Dolan PJ, Johnson GV. J Neurochem. 2008;106:2119. doi: 10.1111/j.1471-4159.2008.05564.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, Nedelsky NB, Schwartz SL, DiProspero NA, Knight MA, Schuldiner O, Padmanabhan R, Hild M, Berry DL, Garza D, Hubbert CC, Yao TP, Baehrecke EH, Taylor JP. Nature. 2007;447:859. doi: 10.1038/nature05853. [DOI] [PubMed] [Google Scholar]
- 52.Zhang Y, Kwon S, Yamaguchi T, Cubizolles F, Rousseaux S, Kneissel M, Cao C, Li N, Cheng HL, Chua K, Lombard D, Mizeracki A, Matthias G, Alt FW, Khochbin S, Matthias P. Mol Cell Biol. 2008;28:1688. doi: 10.1128/MCB.01154-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wender PA, DeBrabander J, Harran PG, Jimenez JM, Koehler MF, Lippa B, Park CM, Siedenbiedel C, Pettit GR. Proc Natl Acad Sci U S A. 1998;95:6624. doi: 10.1073/pnas.95.12.6624. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.