

SUMMARY
Pore-forming toxins (PFTs) secreted by pathogenic bacteria are the most common bacterial protein toxins and are important virulence factors for infection. PFTs punch holes in host cell plasma membranes and although cells can counteract the resulting membrane damage, the underlying mechanisms at play remain unclear. Using Caenorhabditis elegans as a model, we demonstrate in vivo and in an intact epithelium that intestinal cells respond to PFTs by increasing levels of endocytosis, dependent upon RAB-5 and RAB-11, which are master regulators of endocytic and exocytic events. Furthermore, we find that RAB-5 and RAB-11 are required for protection against PFT, and to restore integrity to the plasma membrane. One physical mechanism involved is the RAB-11-dependent expulsion of microvilli from the apical side of the intestinal epithelial cells. Specific vesicle-trafficking pathways thus protect cells against an attack by PFTs on plasma membrane integrity, via altered plasma membrane dynamics.
INTRODUCTION
Pore-forming toxins (PFTs) are virulent bacterial proteins that perforate host cell plasma membranes to disable cellular viability, alter cytokine responses, and provide the pathogen nutrients (Aroian and van der Goot, 2007; Bischofberger et al., 2009). Many well known pathogenic bacteria such as Staphylococcus aureus, Group A and B streptococci, Vibrio cholerae, and Clostridium septicum employ PFTs (Aroian and van der Goot, 2007; Labandeira-Rey et al., 2007; Olivier et al., 2007). With 25–30% of all known virulent proteins, PFTs form the largest class of bacterial toxins (Alouf, 2003; Gonzalez et al., 2008).
It was recently shown that the large-pore PFT SLO, the small-pore PFT α-toxin, and medium-sized perforin pores trigger endocytic and/or exocytic events, which correlate with plasma membrane resealing (Husmann et al., 2009; Idone et al., 2008a; Idone et al., 2008b; Tam et al., 2010; Thiery et al., 2010). Correlation between these processes and cellular survival were not tested for SLO, but the data suggest that inhibition of endocytosis causes decreased survival when cells are exposed to α-toxin (Husmann et al., 2009), and perforin induced a shift from necrotic to apoptotic cell death (Thiery et al., 2010). These in vitro studies show that vesicle trafficking is involved in restoring plasma membrane integrity after an attack by PFT. However, conclusive demonstration that these events contribute to cellular survival following PFT attack and especially of their relevance towards productive responses in an intact tissue in vivo are currently lacking.
The roundworm Caenorhabditis elegans is extensively used as a model for mammalian pathogenesis and innate immune responses (Alegado et al., 2003; Schulenburg et al., 2004; Sifri et al., 2005). It has also been developed as a system to study cellular defenses against PFTs in vivo, and has proven instrumental in the identification of specific molecular PFT-defense pathways (Bellier et al., 2009; Bischof et al., 2008; Chen et al., 2010; Huffman et al., 2004). Where tested, these C. elegans findings always extended to mammalian systems (Bischof et al., 2008; Huffman et al., 2004). C. elegans has furthermore been developed into a relevant model to study vesicle trafficking (Grant and Donaldson, 2009). Using this system, we address a number of the uncertainties surrounding the physical mechanisms employed by cells to restore integrity to their membranes and survive an attack by PFTs. We show a clear correlation between vesicle trafficking, plasma membrane repair and survival in response to a bacterial PFT in vivo in an intact tissue context. We furthermore demonstrate a requirement of RAB-5 and RAB-11, master regulators of endocytic and exocytic events, in these processes, and involvement of microvilli expulsion from the cell surface.
RESULTS
PFTs Induce Intracellular Relocalization of Plasma Membrane Markers
To study the effects of PFTs on the plasma membrane in vivo, we used a C. elegans strain expressing PGP-1::GFP, a labeled ATP binding-cassette (ABC) transporter that, in fourth larval stage (L4) animals, localizes strictly to the apical plasma membrane of the intestinal cells (Sato et al., 2007) (Figure 1A). When L4 animals are exposed on agar plates to Cry5B PFT transgenically expressed by Escherichia coli (the normal laboratory food source for C. elegans), a striking relocalization of the GFP marker is observed, from the apical plasma membrane to intracellular vesicular structures (Figure 1B). Although most dramatic after at least two hours, this phenotype is observed as early as 5 minutes after exposure to PFT (Figure 1C). After two hours on E. coli-expressed Cry5B, 96.9% of the animals show the marker on intracellular vesicular structures, versus 0% in control-treated animals (P < 0.0001) (Table S1).
Figure 1. PFTs induce uptake of apical plasma membrane markers.

(A) Differential interference contrast (DIC) image of anterior half of C. elegans, with intestine false-colored in blue and pharynx in green; arrowhead indicates the transition from pharynx to intestine, and arrow indicates the posterior bulb of the pharynx (left). Fluorescence image (middle) and overlay over DIC image (right), showing PGP-1::GFP marks the apical plasma membrane of C. elegans intestinal cells. Scale bar: 50 μm. (B) Confocal images of PGP-1::GFP after 0 (left) or 2 hr (middle) exposure to E. coli-expressed Cry5B. Indicated area in the middle panel was magnified 3x (right), with arrows indicating intracellular PGP-1::GFP-positive vesicular structures. Scale bar: 25 μm left (same for middle); 10 μm right. (C) Confocal images showing intracellular PGP-1::GFP-positive vesicular structures after 5 minutes exposure to E. coli-expressed Cry5B, which are absent from untreated animals. Panels and scales as (B). (D) Deconvolved images showing V. cholerae expressing VCC induces PGP-1::GFP relocalization to intracellular vesicular structures after 2 hr exposure (middle and right), whereas V. cholera lacking VCC does not (left). Occasionally, vesicles are visible in control images; these were confirmed to be autofluorescent gut granules (see Experimental Procedures). Scale bar: 25 μm left (middle same scale); 10 μm right.
To confirm that the observed phenotype was not an artifact of PGP-1::GFP, another apical plasma-membrane marker was tested, OPT-2::GFP, a labeled peptide transporter (Nehrke, 2003). OPT-2::GFP similarly relocalizes to intracellular vesicular structures after treatment with Cry5B PFT (Figure S1A).
The results extend to other small-pore PFTs, as PGP-1::GFP also relocalizes to intracellular vesicles after exposure to Cry21A, another Bt crystal PFT that intoxicates nematodes (Wei et al., 2003) (92.4% of animals for Cry21A versus 0.0% for controls, P < 0.0001; Table S1, Figure S1B), or to a V. cholerae strain expressing VCC (48.4% with VCC versus 1.9% without VCC, P < 0.05; Table S1, Figure 1D). To examine the specificity of this response to PFTs, we exposed PGP-1::GFP animals for two hours to various other stressors, namely the pathogenic bacterium Pseudomonas aeruginosa (strain PA14, which kills C. elegans without apparently relying on a PFT (Bischof et al., 2008)), a high dose of the heavy metal copper (10mM CuSO4 (Bischof et al., 2008)), hyper osmotic stress (400mM NaCl (Lamitina et al., 2006)), and 35°C heat stress. None of these treatments induce PGP-1::GFP relocalization (Table S1, Figure S1C), indicating the observed response is not caused by cellular stress per se.
Relocalization of apical plasma membrane markers to intracellular punctate structures in C. elegans thus represents a response specific to membrane damage caused by small-pore PFTs.
PFTs Cause Increased Rates of Endocytosis
Most endocytosed cargo is thought to travel through early endosomes (Grant and Donaldson, 2009), which are labeled by mCherry::RAB-5 (Shi et al., 2009). When worms expressing both PGP-1::GFP and mCherry::RAB-5 are exposed to E. coli-expressed Cry5B, overlap is seen between PGP-1::GFP-labeled and RAB-5::mCherry-labeled vesicles (Figure 2A). The same is seen when these animals are exposed to V. cholerae that express VCC (Figure 2B). The colocalization of PGP-1::GFP and RAB-5::mCherry indicates that relocalization of PGP-1::GFP to punctate intracellular structures upon PFT attack marks an endocytic event.
Figure 2. PFTs induce plasma membrane uptake into early endosomes and increased rates of endocytosis.

(A) PGP-1::GFP-positive vesicles induced by 2 hr exposure to E. coli-expressed Cry5B PFT and vesicles positive for mCherry::RAB-5 overlap (indicated by arrows). Due to intensity differences, overlapping signals do not always appear yellow in merged image. Autofluorescence shown in blue in the merged image. Scale bar: 10 μm. (B) Two hr exposure to V. cholerae VCC induces PGP-1::GFP-positive vesicles that show similar overlap with RAB-5::mCherry. Scale bar: 10 μm. (C) After 2 hr exposure to TRITC-labeled BSA in absence of toxin, the dye is confined to the intestinal lumen. After simultaneous exposure to 1 μg/mL purified Cry5B PFT, TRITC-BSA is abundantly found inside intestinal cells. Scale bar: 25 μm. (D) Quantification of TRITC-BSA fluorescence, in absence or presence of 1 or 15 μg/mL purified Cry5B PFT, demonstrates PFT attack leads to increased signal intensity, consistent with increased endocytosis. Means of three experiments. Error bars are standard error of the mean. Statistics indicated here and elsewhere: ns: not significant, ***: P < 0.001, *: P < 0.05.
These data suggest that levels of endocytosis are increased in the presence of Cry5B PFT. When fed to C. elegans, tetramethylrhodamine isothiocyanate-labeled bovine serum albumin (TRITC-BSA) eventually localizes to autofluorescent gut granules in the intestinal cells, thought to be lysosomes, and can therefore be used to visualize fluid phase endocytosis (Hermann et al., 2005). We imaged animals after two hours exposure to 0, 1 or 15 μg/mL purified Cry5B in the presence of TRITC-BSA, and quantified fluorescence intensity in the eight anterior-most intestinal cells to measure the amount of endocytosis that had occurred. Images from the experiment (Figure 2C) as well as the quantification (Figure 2D, Table S1) show significantly increased TRITC-BSA fluorescence in the presence of Cry5B PFT. BSA has a diameter of 6.26 nm (Wu and Prausnitz, 1999) and is unlikely to passively diffuse into the cells through the 1–2 nm-sized Cry5B pores (Parker and Feil, 2005). Increased fluorescence was not due to altered autofluorescence of the intestine, as signals measured in the same experiments in the UV channel (indicating autofluorescence (Clokey and Jacobson, 1986)) showed no statistical difference between Cry5B - and control-treated animals (Figure S2A, B, Table S1). Thus, PFTs trigger endocytosis in C. elegans in vivo, consistent with observations in PFT-treated mammalian cells (Husmann et al., 2009; Idone et al., 2008b; Thiery et al., 2010).
RAB-5 and RAB-11 are required for cellular protection against PFT
RAB small GTPases are key regulators of vesicle-trafficking events (Grant and Donaldson, 2009), and we hypothesized that RABs would be involved in PFT defenses. We therefore did a provisional analysis of the involvement of various RABs (RAB-1, -5, -6.1, -6.2, -7, -11.1, -14, -21, 28, 37, 39 and UNC-108) in Cry5B defense in C. elegans, using RNA interference (RNAi) and subsequent exposure to E. coli-expressed Cry5B. This analysis yielded RAB-5 and RAB-11 as candidates, which led us to further investigate their roles in cellular responses to Cry5B. RAB-5, found on PFT-induced PGP-1::GFP-containing vesicles (Figure 2A, B), is the C. elegans homolog of Rab5, a master regulator of trafficking from the plasma membrane to early endosomes (Grant and Hirsh, 1999; Grant and Donaldson, 2009). Rab11 directs vesicles from the recycling endosome to the plasma membrane (Grant and Donaldson, 2009), and functions to regulate trafficking from the trans-Golgi network to the plasma membrane (Grant and Hirsh, 1999). The C. elegans genome encodes two closely related Rab11 homologues, rab-11.1 and rab-11.2, which differ mainly in the C-terminal region (Figure S3). By quantitative real time PCR, using primers that anneal to where rab-11.1 and rab-11.2 differ most, we found that rab-11.1 and rab-11.2 are both expressed in C. elegans, although rab-11.1 was expressed at higher levels than rab-11.2. Given the 85.2% identity between rab-11.1 and rab-11.2 nucleic acid sequences, it is likely our RNAi experiments below target both genes.
We wanted to determine whether RAB-5 and RAB-11 fulfill a role in protection against PFT attack. Because rab-5 and rab-11.1 are essential for development (Grant and Hirsh, 1999), no mutants are available that lend themselves for our assays, and we used feeding RNAi. We found that rab-5 and rab-11.1 RNAi also inhibit normal development, so resorted to diluting the rab-5 and rab-11.1 double-stranded (ds)RNA-expressing bacteria with empty vector control bacteria, effectively lowering the RNAi dose and allowing superficially normal development. The genetic background and sensitivity to RNAi of the different C. elegans strains used in this study varies, and for each strain we used the lowest dilution, or highest dose, of RNAi bacteria that still allowed normal development of the worms. The worm strains and RNAi dilutions used are discussed in Table S2. To confirm diluted RNAi still caused knockdown of gene expression, we exposed RAB-5::GFP or RAB-11.1::GFP animals to the highest dilutions (lowest concentrations) of respectively rab-5 and rab-11.1 RNAi used in this study. Under these conditions RNAi still results in efficient suppression of GFP expression (Figure S4A, S4B).
To determine a requirement of RAB-5 and RAB-11.1 in PFT defense, we exposed rrf-3(pk1426) RNAi-hypersensitive animals to diluted RNAi against rab-5 and rab-11.1, followed by 48 hours exposure to E. coli-expressed Cry5B on solid medium. Vector-RNAi control animals are only mildly affected by a low dose of PFT, but animals grown on rab-5 or rab-11.1 RNAi are significantly more intoxicated (Figure 3A). VPS-45 and RABX-5 are important factors for RAB-5 function (Gengyo-Ando et al., 2007; Sato et al., 2005). We found that mutation of vps-45 or rabx-5, or RNAi against vps-45 caused qualitative hypersensitivity to E. coli-expressed Cry5B (Figure S4C, S4D), which further validates the role of rab-5 and of endocytosis in PFT defenses.
Figure 3. RAB-5 and RAB-11 are required for defense against Cry5B PFT.

(A) rrf-3(pk1426) animals after rab-5 or rab-11.1 RNAi are qualitatively hypersensitive to a low (10%) dose of E. coli-expressed Cry5B PFT after 48 hr exposure. Scale bar: 0.5 mm. (B) VP303 animals grown in liquid on rab-5, rab-11.1, or sek-1 RNAi bacteria show significantly decreased survival rates on 15 μg/mL purified Cry5B, whereas pgp-1 and empty vector (L4440) controls do not. (C) In simultaneously performed assays, RNAi treatments did not cause significant hypersensitivity to CuSO4. (B) and (C) show means of at least three independent experiments. Statistics indicate difference between toxin treatment and its accompanying no-toxin control for each RNAi treatment. Error bars are standard error of the mean.
Since Cry5B PFT attacks the C. elegans intestine (Griffitts et al., 2001), we wanted to quantitatively determine the requirement of rab-5 and rab-11.1 for PFT defense in this tissue. We therefore performed RNAi against these genes in C. elegans strain VP303, which allows for RNAi to predominantly knock down gene expression in the intestine (see Table S2). Animals were grown in liquid on RNAi to the L4 larval stage at which point purified Cry5B PFT was added, and survival was scored six days later. RNAi against rab-5, rab-11.1 and sek-1 (p38 MAPKK, used as a hypersensitive control (Huffman et al., 2004)) caused significantly lower survival on Cry5B (Figure 3B, Table S1).
RNAi against pgp-1 was used as a negative control because activity of the clone could easily be confirmed by its ability to suppress PGP-1::GFP expression (not shown). pgp-1 RNAi causes significantly compromised survival in the absence of toxin, however does not cause hypersensitivity to Cry5B PFT (Figure 3B, Table S1), demonstrating that compromised health per se does not lead to increased PFT sensitivity. To directly confirm that the observed Cry5B-hypersensitivity caused by rab-5 or rab-11.1 RNAi was not due to general poor health, we tested survival on 2mM CuSO4 (simultaneously with the previous experiment and in identical setup). We found that at this dose, which causes a low fraction of lethality in C. elegans (Bischof et al., 2008), RNAi against rab-5, rab-11.1, pgp-1, or sek-1 does not cause significant hypersensitivity (Figure 3C, Table S1). Based on our data that (1) diluted rab-5 and rab-11.1 RNAi permit normal development and survival in absence of toxin, (2) decreased survival of pgp-1 RNAi in the absence of PFT does not lead to increased PFT sensitivity, and (3) rab-5 and rab-11.1 RNAi animals are not hypersensitive to CuSO4, we conclude that the PFT hypersensitivity caused by reduction of rab-5 and rab-11 function is not due to overall compromised health, but that there is a specific role for RAB-5 and RAB-11-mediated vesicle-trafficking pathways in the affected tissue in PFT defense.
RAB-5 and RAB-11 are required for PFT-induced vesicle formation
The formation of vesicles upon addition of PFT and the protective effect of RAB-5 and RAB-11 vesicle-trafficking pathways suggest that the vesicles themselves are involved in RAB-5 and RAB-11-mediated protection. This predicts the formation of these vesicles to be dependent upon RAB-5 and/or RAB-11. To test this, we exposed PGP-1::GFP-expressing animals to diluted rab-5 or rab-11.1 RNAi (see Table S2) and subsequently to Cry5B. Both RNAi treatments caused significant inhibition of the PFT-induced endocytic response, as measured by the fraction of animals showing PGP-1::GFP-containing intracellular vesicles (Figure 4A, B, Table S1). Furthermore, animals grown on rab-5 or rab-11.1 RNAi that still showed GFP relocalization in response to Cry5B generally contained less vesicles. Interestingly, RNAi against the p38 MAPKK sek-1 did not alter the endocytic response (Figure 4B, Table S1). No differences in PGP-1::GFP localization were noticed between controls and any of the RNAi-treated animals in the absence of toxin. These data indicate that there is a correlation between PFT protection and vesicle formation dependent upon RAB-5 and RAB-11, and that the p38 MAPK pathway is not likely to be upstream of this process in this time frame. The fact that RNAi of rab-11.1, which is involved in exocytosis, results in defects in endocytosis is likely a consequence of the fact that cells maintain a balance of endocytosis and exocytosis. Loss of exocytosis will therefore also result in rapid loss of endocytosis as cells naturally compensate to maintain homeostasis of the plasma membrane.
Figure 4. RAB-5 and RAB-11 are required for PFT-induced endocytosis.

(A) PGP-1::GFP animals show a diminished endocytic response to E. coli-expressed Cry5B PFT when grown on rab-5 or rab-11.1 RNAi. Images show anterior half of intestine. Scale bar: 25 μm. (B) Fractions of population showing PGP-1::GFP on endocytic vesicles after RNAi against rab-5, rab-11.1, or sek-1, and subsequent exposure to E. coli-expressed Cry5B. L4440 is empty vector control. Means of at least six independent experiments. Error bars are standard error of the mean.
Vesicle-Trafficking Pathways Remove Pores From the Plasma Membrane
Vesicle-trafficking pathways may provide protection against PFTs by dynamically removing pores from the membrane, perhaps by endocytosis of pore-contaminated membrane (Bischofberger et al., 2009; Idone et al., 2008a; Idone et al., 2008b). To test whether pores were being removed from the membrane, we developed an in vivo assay involving the small dye propidium iodide (PI), commonly used to study the presence of membrane pores in vitro (Idone et al., 2008b; Tam et al., 2010; Thiery et al., 2010; Walev et al., 2001), inspired by a published assay for membrane integrity in C. elegans intestinal cells (Luke et al., 2007). PI is membrane impermeable, and, when fed to live C. elegans, is restricted to the intestinal lumen (Figure 5A). The molecular diameter of PI is less than 0.96nm (Iwase et al., 1990), whereas the predicted pore size for Cry5B is 1 to 2nm (Parker and Feil, 2005). When fed to worms in liquid in the presence of purified Cry5B for two hours, PI stains the cytosol of the intestinal cells (Figure 5B). The same was observed when animals were exposed to E. coli-expressed Cry5B on solid medium and subsequently stained with PI (not shown). To confirm PI enters the cells via diffusion through pores and not via endocytosis like TRITC-BSA (see above), we studied uptake of the two dyes after a short exposure to PFT. We found that after a short (15 minutes) Cry5B exposure, small amounts of TRITC-BSA had entered the intestinal cells relative to what remained in the lumen, suggesting BSA uptake is rate-limited, consistent with an endocytic process (Figure S5A). However, large amounts of PI had entered the cells relative to what was still present in the lumen (Figure S5B), consistent with passive diffusion of PI through pores. This finding is further reinforced by experiments below.
Figure 5. RAB-5 and RAB-11 are required for removal of PFT from the plasma membrane.

(A) PI is restricted to the intestinal lumen when fed to C. elegans. PGP-1::GFP marks the apical boundary of the intestinal cells. Scale bar: 25 μm. (B) PI stains the cytosol of the intestinal cells when animals are simultaneously exposed to purified Cry5B PFT for 2 hr. Scale as (A). (C) Fractions of population showing cytosolic PI staining after 0.5 or 24 hr recovery after a 15 minute pulse with E. coli-expressed Cry5B. After 0.5 hr recovery all RNAi treatments resulted in statistically equal fractions of animals with compromised integrity (internalized PI) of the intestinal cells. The majority of control (L4440) animals and animals on pgp-1 RNAi have repaired their intestinal cells after 24 hr recovery. RNAi against rab-5 or rab-11.1 causes significantly impaired repair after 24 hr. Means of at least three independent experiments. Error bars are standard error of the mean.
To determine if cells are able to rectify membrane permeability to PI following PFT attack, we pulsed animals for 15 minutes with E. coli-expressed Cry5B, and allowed 30 minutes or 24 hours recovery prior to staining with PI. We found that 92.6% of the animals have PI-permeable intestinal cells 30 minutes after the Cry5B pulse, whereas only 18.3% had PI-permeable intestinal cells 24 hours after the pulse (Figure 5C, Table S1). Thus, as judged by PFT-mediated permeabilization of intestinal cells to PI, 74.3% (92.6% - 18.3%) of wild-type animals are able to eliminate all pores from their membrane during the 24 hours following a brief PFT pulse. During preliminary experiments we found intermediate fractions of animals showing membrane repair at time points between 0.5 – 24 h (not shown). Also, occasionally animals were found showing a mosaic repair pattern, i.e. some of the cells contained PI whereas others did not (Figure S5C), which was never observed after 30 minutes recovery.
To confirm membrane repair can occur in a single animal, wild-type animals were pulsed with E. coli-expressed Cry5B, immediately stained with propidium iodide and then recovered. After six hours to allow repair of pores, the same worms were fed SYTOX Green, a dye that behaves identical to PI in this assay except that it fluoresces at a different wavelength. We found animals that showed PI localized to the cytosol, but SYTOX Green localized to the lumen (Figure S5D), indicating membrane repair can occur within a single animal.
We next examined the requirement for RAB-5 and RAB-11 for pore elimination after a Cry5B pulse. While no differences are seen after 0.5 hours recovery, animals grown and recovered on diluted rab-5 or rab-11.1 RNAi bacteria show a significantly impaired restoration of membrane integrity compared to controls after 24 hours (Figure 5C, Table S1). pgp-1 RNAi did not cause statistically different fractions of animals displaying cytosolic PI at 0.5 or 24 hours recovery (Figure 5C, Table S1). These data indicate that RAB-5 and RAB-11- controlled vesicle-trafficking pathways are required for effective membrane repair after an attack by PFT. In addition, they further confirm that PI enters cells via pores, since RAB-5 or RAB-11 knock down would be predicted to decrease rather than increase PI uptake by the cells if it were entering via endocytosis.
Repair after physical damage to the plasma membrane requires Ca2+ influx through the lesions, which was also found to be true for repair of the large pores formed by SLO (Idone et al., 2008b). Extracellular Ca2+ is not likely required for repair of small pores formed by S. aureus α-toxin or V. cholerae VCC, as these are Ca2+-impermeable (Walev et al., 1993; Zitzer et al., 1997), although intracellular Ca2+-stores may still play a role. Based on analogy to Cry1Aa and Cry1Ac, Cry5B pores may however be permeable to Ca2+ (Kirouac et al., 2002). To determine whether Ca2+ was required for defense against Cry5B, we exposed wild type animals to purified Cry5B in liquid media with or without Ca2+, and quantified survival. We found that absence of Ca2+ from the media did not significantly alter Cry5B sensitivity (Figure S5E).
RAB-11 is linked to PFT-induced expulsion of plasma membrane into the intestinal lumen
To ascertain directly the consequences of PFT attack on the intestinal cells, C. elegans animals treated with Cry5B PFT for three hours were fixed, sectioned, and examined by transmission electron microscopy (EM). As a negative control, we fed Cry5B PFT to bre-5(ye17) mutant animals, which lack the receptor required for pore formation by Cry5B and hence are unaffected by the toxin (Griffitts et al., 2003). Dramatic changes at the cell surface are evident in Cry5B-intoxicated intestinal cells, which show gaps in the regular arrangement of microvilli and a concomitant increase in membranes present in the lumen of the intestine relative to the unaffected bre-5(ye17) control animals (Figure 6A). These data strongly suggest that one response of the animals to PFT attack is to dislodge or expel microvilli from the cell surface, probably as a way to rapidly remove pores from the cell surface. To confirm that the bre-5 mutation did not alter our findings, we performed a similar EM experiment where we used wild-type animals fed empty-vector bacteria as a negative control instead. The results were consistent, as empty-vector control-treated wild-type animals looked comparable to bre-5(ye17) (non-intoxicated) animals treated with Cry5B, and wild-type animals treated with Cry5B looked similar in both experiments (Figure S6).
Figure 6. Cry5B PFT induces expulsion of plasma membrane into the intestinal lumen.

(A) EM images showing extensive damage to the microvilli of the intestinal cells following 3 hr E. coli-expressed Cry5B treatment. Unintoxicated controls (receptor-negative bre-5(ye17) mutant) have healthy microvilli (left). Middle and right panels: intoxicated wild type animals. Intoxicated wild-type animals show microvilli deficiency (middle, arrow), and dislodged microvilli in intestinal lumen (right, arrows). Each panel shows a single focal plane from a different animal. All focal planes were analyzed to confirm the lack of microvilli or disconnection of membranous material. Scale bars: 0.5 μM. (B) Deconvolved images showing PGP-1::GFP positive material in the intestinal lumen (arrows) after 2 hr exposure to E. coli-expressed Cry5B. Scale bar: 10 μm. (C) Confocal images showing debris in the lumen after 5 minutes exposure to E. coli-expressed Cry5B. Scale bar: 10 μm. (D) Fluorescence image showing PGP-1::GFP-labeled material in the posterior bulb of the pharynx (indicated by arrow) after exposure to E. coli-expressed Cry5B. Scale bar: 10 μm. (E) Fractions of animals containing luminal PGP-1::GFP-positive material after E. coli-expressed Cry5B PFT treatment. Error bars are standard error of the mean.
To determine if microvilli expulsion is detectable in live animals, we exposed PGP-1::GFP-expressing animals to Cry5B PFT. We found an accumulation of detached, GFP-positive material in the lumen of the intestine of PFT-exposed animals (Figure 6B), consistent with our EM results that apical membrane is being dislodged into the intestinal lumen. Like the endocytosis phenotype, this is most obvious after two hours exposure to E. coli-expressed Cry5B (Figure 6B) but already visible after five minutes (Figure 6C). The membranous material appears to be detached from the intestinal cells, as it is sometimes encountered in the pharynx (Figure 6D) where pgp-1::gfp is not expressed (Figure 1A), likely resulting from backwards movement of luminal contents from the intestine into the pharynx.
Since the expulsion or dislodging of plasma membrane into the lumen would necessitate a replenishment of plasma membrane at the cell surface via fusion of exocytic vesicles, we hypothesized that perturbation of vesicle-trafficking pathways would perturb this ejection. We performed RNAi of rab-5 or rab-11.1 on PGP-1::GFP animals and then exposed them to E. coli-expressed Cry5B as above, after which we examined the presence of unattached PGP-1::GFP in the intestinal lumen. We found that RNAi of rab-11.1, but not rab-5, leads to a statistically significant decrease in animals displaying PGP-1::GFP-positive membrane structures in the intestinal lumen (Figure 6E, Table S1). The lack of phenotype after rab-5 RNAi in this assay might be explained by lack of penetrance with diluted RNAi for this particular phenomenon. Thus, the expulsion or dislodging of material into the lumen of the intestine requires RAB-11, likely via fusion of vesicles with the plasma membrane.
DISCUSSION
The data presented here directly correlate vesicle trafficking, plasma membrane repair, and survival after attack by a small-pore bacterial PFT and to do so in an intact tissue in vivo. We show that (1) PFT intoxication triggers an increase in endocytosis in vivo, (2) loss of either of two key RAB proteins, RAB-5 and RAB-11, master regulators of early and recycling endosome functions, results in strong hypersensitivity to PFT and loss of cellular protection against PFT, (3) loss of RAB-5 and RAB-11 results in significant decreases in PFT-induced endocytosis, showing a correlation between RAB-5, RAB-11, PFT-induced endocytosis of the plasma membrane, and cellular protection, (4) RAB-5 and RAB-11 are required for normal recovery of plasma membrane integrity (membrane resealing) in vivo following PFT attack, and (5) RAB-11 is required for PFT-induced expulsion of microvilli.
Taken together, these data demonstrate that an attack on the plasma membrane by small-pore PFTs leads to a rapid (within 5 minutes) and dramatic increase in plasma membrane dynamics, i.e. an increase in endocytosis and expulsion of microvilli, that relies on both endocytic (RAB-5) and exocytic (RAB-11) machinery and that functions to remove pores from the plasma membrane. Although we have not directly shown the presence of PFT on internalized or externalized membranes, we do demonstrate the removal of pores from the plasma membrane via PI permeability. Presence of PFTs on endosomes and externalized membranes has been seen or inferred in two studies in mammalian cells (Husmann et al., 2009; Idone et al., 2008b).
Our data appears consistent with a molecular-physical model that can explain how cells use vesicular trafficking pathways to repair membrane damage, the vertex fusion model (McNeil and Kirchhausen, 2005). Here, a large vesicle forms a patch by docking at the damaged section of plasma membrane via docking sites along the circumference of the patch, the vertex ring. Fusion between the two membranes then occurs along this vertex ring, excising a piece of damaged plasma membrane and releasing it (Wang et al., 2002). (This is opposed to fusion via the expanding pore model, where all boundary membrane is converted to outside membrane (Wang et al., 2002).) In the case of membrane damaged by PFT, we hypothesize RAB-11’s role is to control assembly and localization of the patch and/or to directly initiate vertex fusion, and that the dislodged microvilli represent the excised membrane, in this case released extracellularly (Figure 7). The role of RAB-5 in this model would be to use endocytosis to maintain homeostasis of the plasma membrane (Figure 7), as well as to contribute to pore removal via internalization (see below). The vertex fusion model for membrane repair was however proposed in the context of Ca2+-dependent repair (McNeil and Kirchhausen, 2005), whereas we did not observe a requirement for Ca2+ (Figure S5E).
Figure 7. Model for PFT pore removal by RAB-5 and RAB-11-dependent endo- and exocytosis.
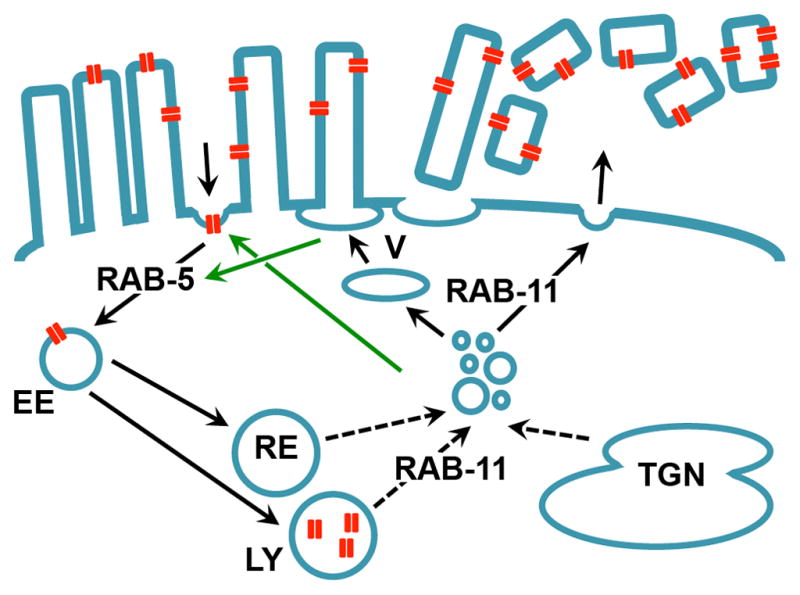
Microvilli containing PFT pores (red) are expelled from the apical cell surface through RAB-11-dependent vertex fusion (indicated with “V”) or some other mechanism. RAB-5-dependent endocytosis supports this process by maintaining membrane homeostasis through increased endocytosis (indicated by green arrow). Alternatively or in addition, pores are taken up from the plasma membrane into the cells by RAB-5-controlled endocytosis, and potentially transported to lysosomes. RAB-11-controlled exocytosis may balance increased endocytosis (indicated by green arrow).
Alternatively, PFT pores in the apical plasma membrane could be quickly detected, and taken up into the cells by RAB-5-controlled endocytosis. Pores are then potentially transported to the lysosomes for destruction. Simultaneous RAB-11-dependent exocytic events provide the new plasma membrane required to counterbalance the increased rates of endocytosis. These exocytosed vesicles could be derived from recycling endosomes or the trans-golgi network (with which Rab11 is known to interact (Grant and Hirsh, 1999; Grant and Donaldson, 2009)), or lysosomes (which are known to fuse with the plasma membrane in response to membrane damage (Idone et al., 2008a)). Simultaneously, microvilli are expelled from the cell surface in a RAB-11-dependent fashion, via vertex fusion (see above), or through some other mechanism (Figure 7), thus also playing a significant role in the removal of pore-contaminated membrane. In this way, RAB-5-mediated endocytosis and RAB-11-mediated exocytosis together play a role in removing pores from the membrane, while also balancing each other out to maintain plasma membrane homeostasis. The roles for RAB-5 and RAB-11 in PFT defense could be analogous to their functions during cytokinesis to reshape the plasma membrane (Furthauer and Gonzalez-Gaitan, 2009). In addition to removing pores, the dislodged microvilli might function to serve as decoys for unbound PFT, and to dramatically reduce the cell’s surface area so that the amount of pores relative to cell volume drops considerably and so that fewer PFTs insert de novo.
This study is consistent with previous studies in mammalian cells, and closes important gaps in our understanding of cellular defenses against PFTs. Consistent with our findings, others have demonstrated that both large- and small-pore PFTs trigger endocytic (Husmann et al., 2009; Idone et al., 2008b; Tam et al., 2010; Thiery et al., 2010) and exocytic (Husmann et al., 2009; Shaik et al., 2009; Tam et al., 2010) responses to PFTs in mammalian cells. Loss of microvilli has also been observed before in mammalian cells in response to small-pore and large-pore PFTs, such as the thermostable direct hemolysin produced by Vibrio parahaemolyticus (Sakurai et al., 1976) and streptolysin O (SLO) produced by Streptococcus pyogenes (Engel et al., 1995). SLO also triggered shedding of microparticles from cells in vitro (Babiychuk et al., 2009), and perforin was found to induce membrane blebbing (Keefe et al., 2005). What is demonstrated here, is a direct correlation between these events and survival following PFT attack, and the central role played in this process by conserved RAB-5 (endocytic) and RAB-11 (exocytic) vesicle-trafficking pathways. The striking parallel between our results and those in mammalian cells indicate that what we characterize here likely has significant relevance to cellular responses of mammalian cells to PFTs. The C. elegans system therefore provides an important genetically tractable, in-vivo model for future studies that detail vesicle trafficking and other fundamental cell biological process important for survival following the most common mode of bacterial toxin attack.
Some of the most problematic multi-drug resistant bacteria, e.g. MRSA (methicillin-resistant S. aureus), E. faecalis and S. pneumoniae (Woodford and Livermore, 2009) use PFTs as critical virulence factors. Elucidating host cell immunity towards PFTs can lead to the development of drugs that fortify cellular defenses against PFTs by promoting protective molecular-physical pathways or by mimicking the way cells disarm PFTs, e.g., by introducing decoy membranes. Such therapeutics would neutralize a broad and important class of virulence factors and hence could provide continued protection against many problematical bacterial infections. In summary, we demonstrate that RAB-5 and RAB-11 direct vesicle-trafficking pathways that remove PFTs from the plasma membrane and provide cellular protection against the single largest class of bacterial protein toxins.
EXPERIMENTAL PROCEDURES
Strains, culture conditions, PFT use
Worm strains used are outlined in Table S2 and were maintained as described (Brenner, 1974). Bacterial strains and culture conditions are detailed in Supplemental Experimental Procedures. E. coli-expressed Cry5B was used for figures 1B, 1C, 2A, 3A, 4A, 4B, 5C, 6A, 6B, 6C, 6D, 6E, S1A, S4C, S4D S5C, S5D and S6. Purified Cry5B (prepared as described (Cappello et al., 2006), but with omission of the sucrose gradient) was used for figures 2C, 2D, 3B, 5B, S2A, S5A, S5B and S5E. Cry21A and VCC were always administered expressed from E. coli or V. cholerae.
Microscopy and image editing
Endocytosis assays
Exposure of L4 stage PGP-1::GFP, OPT-2::GFP and PGP-1::GFP; mCherry::RAB-5 animals to OP50-Cry5B, OP50-Cry21A (OP50-pQE9 empty vector used as no-toxin control), P. aeruginosa (E. coli OP50 used as control), or V. cholerae was done as described, except 5-fluoro-2′-deoxy-uridine (FUdR) was omitted for P. aeruginosa (Bellier et al., 2009; Bischof et al., 2006; Bischof et al., 2008; Vaitkevicius et al., 2006). L4 PGP-1::GFP animals were exposed in 48-wells plates to M9 with 10mM CuSO4, 400mM NaCl or water control. 35°C heat shock was delivered on NGM plates without bacteria. Animals were scored as showing an endocytic response when the GFP marker was robustly found on more than 10 intracellular vesicles in the anterior half of the intestine. Signals in the UV channel were used to distinguish between GFP-positive vesicles and autofluorescent gut granules (Clokey and Jacobson, 1986). At least three independent repeats were performed for each experiment with at least 30 animals per treatment.
To determine endocytosis after RNAi treatment, PGP-1::GFP animals were synchronized to the L1 stage by hypochlorite treatment and overnight starvation in M9 media at room temperature, and then seeded onto RNAi plates (see Supplemental Experimental Procedures) and grown to the L4 stage at 20°C, before being exposed to OP50-Cry5B for two hours at 20°C. Animals were scored as above, and at least six independent repeats were done, with at least 24 animals per treatment.
Tetramethylrhodamine isothiocyanate-labeled albumin (TRITC-BSA) staining was based on published protocols (Grant et al., 2001; Hermann et al., 2005). TRITC-BSA was cleaned on a 10,000 MW centricon (Millipore) before use to remove any unbound TRITC. Synchronized L4 animals were incubated 15 minutes at 20°C in S-media (Bischof et al., 2006) with 5 mg/mL serotonin (5-HT) (Sigma) (explained below) and 80 μg/mL TRITC-BSA (Invitrogen) in a 96-wells plate. Then, purified Cry5B to 1 or 15 μg/mL final concentration, or Hepes pH8.0 to 1mM (no-toxin control) was added. After two hours incubation at 20°C, worms were transferred to presiliconized 1.6 mL microcentrifuge tubes (National Scientific Supply Co), washed three times, and imaged (see Microscopy and image editing). Three independent experiments were performed, with 10–17 animals per treatment.
When exposed to Cry5B, C. elegans rapidly ceases feeding (Wei et al., 2003). We found that feeding on Cry5B could be forced to continue by administration of exogenous serotonin (5-HT), a known regulator of feeding behavior (Horvitz et al., 1982). Hence 5-HT was added to equalize the amount of dye ingested by animals treated with different Cry5B doses.
Toxicity assays
Qualitative toxicity assays after RNAi treatment were based on a published protocol (Bischof et al., 2006). Synchronized L1 rrf-3(pk1426) animals were grown to L4 stage on RNAi at 20°C, after which they were transferred to OP50-Cry5B plates. Worms were then incubated at 20°C, and observed and imaged after 48 hours. At least three repeats were performed for each condition, with at least 15 animals per treatment.
Cry5B survival assays with RNAi in liquid were based on a published protocol (Chen et al., 2010). RNAi-bacteria were grown overnight at 37°C in LB media with 1 μg/mL carbenicillin. dsRNA expression was induced by incubating with 1mM IPTG for one hour at 37°C while shaking. Bacteria were pelleted and resuspended in an equal volume of S-media, and rab-5 and rab-11.1 clones were then diluted 1:6 with pL4440. Synchronized L1 VP303 animals were grown to the L4 larval stage in 48 wells plates on RNAi bacteria in S-media, in ~40 hours at 25°C while gently shaking. Then, FUdR to 200μM final (to inhibit development of the F1 generation), and either purified Cry5B to 15 μg/mL, Hepes pH 8.0 to 0.5 mM (no-Cry5B control), CuSO4 to 2 mM, or ddH2O (no-CuSO4 control) were added. This was incubated six days at 25°C while gently shaking, after which worms were scored for survival. Animals were scored as dead if no activity was observed after prodding an animal three times with a worm picker. At least three independent assays were performed for each treatment, with per assay three identical wells with ~20 animals.
Pore repair assay
Synchronized L1 rrf-3(pk1426) animals were grown on RNAi at 20°C. L4 stage animals were transferred from RNAi to an OP50-Cry5B plate, and incubated for 15 minutes at 20°C. After this Cry5B pulse, worms were transferred to fresh RNAi plates and allowed to recover at 20°C for 0.5 or 24 hours. They were then transferred to presiliconized microcentrifuge tubes with M9 with 5 mg/mL serotonin and incubated 15 minutes at 20°C while shaking, after which propidium iodide (Sigma) was added to 6.7 μg/mL. This was incubated 45 minutes at 20°C while shaking, after which worms were washed twice with M9 media and mounted on slides. Animals were scored positive for cytosolic PI staining if at least one of the enterocytes in the anterior half of the animal was filled with propidium iodide. At least three independent repeats were performed, with ~30 animals per treatment. For Figure 5A, B, animals were treated in wells containing S-media, Hepes (1 mM pH 8.0) or Cry5B (33.5 μg/mL), and PI, for 2 hr at 20° C.
Statistical analyses
Statistical analyses were performed using JMP 8 software (SAS Institute). Fractions of the population showing endocytosis in the PGP-1::GFP assay were compared by one-way ANOVA (CuSO4 and NaCl assay) or student’s t-test (remaining data sets in Table S1). Fractions of animals alive in the Cry5B and CuSO4 liquid assays (Figure 3B, C, S5E), and fractions of animals showing cytosolic staining in the pore repair assays (Figure 5C) were compared using two-way ANOVA with Tukey’s post test. Relative levels of TRITC-BSA fluorescence or autofluorescence intensity (Fig. 2D, S2B) were compared with one way ANOVA with Tukey’s post test. See Table S1 for mean values and standard error of the mean found for all experiments, and confidence limits and P-values calculated with JMP 8. Graphs were prepared using GraphPad Prism 5 software (GraphPad, San Diego, CA).
Supplementary Material
Acknowledgments
We thank the Caenorhabditis Genetics Stock Center, Ken Sato, Keith Nehrke, Kevin Strange, and Barth Grant for C. elegans strains, Larry Bischof for help with the provisional analysis of RAB proteins, Audrey Bellier for OP50-Cry21A, Barth Grant for the rab-11.1 RNAi clone, Anjon Audhya and Anita Hermann for advice on microscopy, Frans Los for advice on statistics, and Emily Troemel for critical reading of the manuscript. Funding: NIH/NIGMS grant R01GM071603 and NIH/NIAID 2R01AI056189.
Footnotes
Author Contributions: Conceived and designed the experiments: FCOL, CYK, RVA. Performed the experiments: FCOL, CYK, JES, CH, KLM, CAP. Analyzed the data: FCOL, KLM, CAP, RVA. Wrote the paper: FCOL, CYK, RVA.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alegado RA, Campbell MC, Chen WC, Slutz SS, Tan MW. Characterization of mediators of microbial virulence and innate immunity using the Caenorhabditis elegans host-pathogen model. Cell Microbiol. 2003;5:435–444. doi: 10.1046/j.1462-5822.2003.00287.x. [DOI] [PubMed] [Google Scholar]
- Alouf JE. Molecular features of the cytolytic pore-forming bacterial protein toxins. Folia Microbiol (Praha) 2003;48:5–16. doi: 10.1007/BF02931271. [DOI] [PubMed] [Google Scholar]
- Aroian R, van der Goot FG. Pore-forming toxins and cellular non-immune defenses (CNIDs) Curr Opin Microbiol. 2007;10:57–61. doi: 10.1016/j.mib.2006.12.008. [DOI] [PubMed] [Google Scholar]
- Babiychuk EB, Monastyrskaya K, Potez S, Draeger A. Intracellular Ca(2+) operates a switch between repair and lysis of streptolysin O-perforated cells. Cell Death Differ. 2009;16:1126–1134. doi: 10.1038/cdd.2009.30. [DOI] [PubMed] [Google Scholar]
- Bellier A, Chen CS, Kao CY, Cinar HN, Aroian RV. Hypoxia and the Hypoxic Response Pathway Protect against Pore-Forming Toxins in C. elegans. PLoS Pathog. 2009;5:e1000689. doi: 10.1371/journal.ppat.1000689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischof LJ, Huffman DL, Aroian RV. Assays for toxicity studies in C. elegans with Bt crystal proteins. Methods Mol Biol. 2006;351:139–154. doi: 10.1385/1-59745-151-7:139. [DOI] [PubMed] [Google Scholar]
- Bischof LJ, Kao CY, Los FC, Gonzalez MR, Shen Z, Briggs SP, van der Goot FG, Aroian RV. Activation of the unfolded protein response is required for defenses against bacterial pore-forming toxin in vivo. PLoS Pathog. 2008;4:e1000176. doi: 10.1371/journal.ppat.1000176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischofberger M, Gonzalez MR, van der Goot FG. Membrane injury by pore-forming proteins. Curr Opin Cell Biol. 2009;21:589–595. doi: 10.1016/j.ceb.2009.04.003. [DOI] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappello M, Bungiro RD, Harrison LM, Bischof LJ, Griffitts JS, Barrows BD, Aroian RV. A purified Bacillus thuringiensis crystal protein with therapeutic activity against the hookworm parasite Ancylostoma ceylanicum. Proc Natl Acad Sci U S A. 2006;103:15154–15159. doi: 10.1073/pnas.0607002103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CS, Bellier A, Kao CY, Yang YL, Chen HD, Los FC, Aroian RV. WWP-1 is a novel modulator of the DAF-2 insulin-like signaling network involved in pore-forming toxin cellular defenses in Caenorhabditis elegans. PLoS One. 2010;5:e9494. doi: 10.1371/journal.pone.0009494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clokey GV, Jacobson LA. The autofluorescent “lipofuscin granules” in the intestinal cells of Caenorhabditis elegans are secondary lysosomes. Mech Ageing Dev. 1986;35:79–94. doi: 10.1016/0047-6374(86)90068-0. [DOI] [PubMed] [Google Scholar]
- Engel F, Blatz R, Kellner J, Palmer M, Weller U, Bhadki S. Breakdown of the round window membrane permeability barrier evoked by streptolysin O: possible etiologic role in development of sensorineural hearing loss in acute otitis media. Infect Immun. 1995;63:1305–1310. doi: 10.1128/iai.63.4.1305-1310.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furthauer M, Gonzalez-Gaitan M. Endocytosis and mitosis: a two-way relationship. Cell Cycle. 2009;8:3311–3318. doi: 10.4161/cc.8.20.9700. [DOI] [PubMed] [Google Scholar]
- Gengyo-Ando K, Kuroyanagi H, Kobayashi T, Murate M, Fujimoto K, Okabe S, Mitani S. The SM protein VPS-45 is required for RAB-5-dependent endocytic transport in Caenorhabditis elegans. EMBO Rep. 2007;8:152–157. doi: 10.1038/sj.embor.7400882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez MR, Bischofberger M, Pernot L, van der Goot FG, Freche B. Bacterial pore-forming toxins: the (w)hole story? Cell Mol Life Sci. 2008;65:493–507. doi: 10.1007/s00018-007-7434-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant B, Hirsh D. Receptor-mediated endocytosis in the Caenorhabditis elegans oocyte. Mol Biol Cell. 1999;10:4311–4326. doi: 10.1091/mbc.10.12.4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant B, Zhang Y, Paupard MC, Lin SX, Hall DH, Hirsh D. Evidence that RME-1, a conserved C. elegans EH-domain protein, functions in endocytic recycling. Nat Cell Biol. 2001;3:573–579. doi: 10.1038/35078549. [DOI] [PubMed] [Google Scholar]
- Grant BD, Donaldson JG. Pathways and mechanisms of endocytic recycling. Nat Rev Mol Cell Biol. 2009;10:597–608. doi: 10.1038/nrm2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffitts JS, Huffman DL, Whitacre JL, Barrows BD, Marroquin LD, Muller R, Brown JR, Hennet T, Esko JD, Aroian RV. Resistance to a bacterial toxin is mediated by removal of a conserved glycosylation pathway required for toxin-host interactions. J Biol Chem. 2003;278:45594–45602. doi: 10.1074/jbc.M308142200. [DOI] [PubMed] [Google Scholar]
- Griffitts JS, Whitacre JL, Stevens DE, Aroian RV. Bt toxin resistance from loss of a putative carbohydrate-modifying enzyme. Science. 2001;293:860–864. doi: 10.1126/science.1062441. [DOI] [PubMed] [Google Scholar]
- Hermann GJ, Schroeder LK, Hieb CA, Kershner AM, Rabbitts BM, Fonarev P, Grant BD, Priess JR. Genetic analysis of lysosomal trafficking in Caenorhabditis elegans. Mol Biol Cell. 2005;16:3273–3288. doi: 10.1091/mbc.E05-01-0060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvitz HR, Chalfie M, Trent C, Sulston JE, Evans PD. Serotonin and octopamine in the nematode Caenorhabditis elegans. Science. 1982;216:1012–1014. doi: 10.1126/science.6805073. [DOI] [PubMed] [Google Scholar]
- Huffman DL, Abrami L, Sasik R, Corbeil J, van der Goot FG, Aroian RV. Mitogen-activated protein kinase pathways defend against bacterial pore-forming toxins. Proc Natl Acad Sci U S A. 2004;101:10995–11000. doi: 10.1073/pnas.0404073101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husmann M, Beckmann E, Boller K, Kloft N, Tenzer S, Bobkiewicz W, Neukirch C, Bayley H, Bhakdi S. Elimination of a bacterial pore-forming toxin by sequential endocytosis and exocytosis. FEBS Lett. 2009;583:337–344. doi: 10.1016/j.febslet.2008.12.028. [DOI] [PubMed] [Google Scholar]
- Idone V, Tam C, Andrews NW. Two-way traffic on the road to plasma membrane repair. Trends Cell Biol. 2008a;18:552–559. doi: 10.1016/j.tcb.2008.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Idone V, Tam C, Goss JW, Toomre D, Pypaert M, Andrews NW. Repair of injured plasma membrane by rapid Ca2+-dependent endocytosis. J Cell Biol. 2008b;180:905–914. doi: 10.1083/jcb.200708010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwase M, Lally ET, Berthold P, Korchak HM, Taichman NS. Effects of cations and osmotic protectants on cytolytic activity of Actinobacillus actinomycetemcomitans leukotoxin. Infect Immun. 1990;58:1782–1788. doi: 10.1128/iai.58.6.1782-1788.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keefe D, Shi L, Feske S, Massol R, Navarro F, Kirchhausen T, Lieberman J. Perforin triggers a plasma membrane-repair response that facilitates CTL induction of apoptosis. Immunity. 2005;23:249–262. doi: 10.1016/j.immuni.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Kirouac M, Vachon V, Noel JF, Girard F, Schwartz JL, Laprade R. Amino acid and divalent ion permeability of the pores formed by the Bacillus thuringiensis toxins Cry1Aa and Cry1Ac in insect midgut brush border membrane vesicles. Biochim Biophys Acta. 2002;1561:171–179. doi: 10.1016/s0005-2736(02)00342-5. [DOI] [PubMed] [Google Scholar]
- Labandeira-Rey M, Couzon F, Boisset S, Brown EL, Bes M, Benito Y, Barbu EM, Vazquez V, Hook M, Etienne J, et al. Staphylococcus aureus Panton-Valentine leukocidin causes necrotizing pneumonia. Science. 2007;315:1130–1133. doi: 10.1126/science.1137165. [DOI] [PubMed] [Google Scholar]
- Lamitina T, Huang CG, Strange K. Genome-wide RNAi screening identifies protein damage as a regulator of osmoprotective gene expression. Proc Natl Acad Sci U S A. 2006;103:12173–12178. doi: 10.1073/pnas.0602987103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luke CJ, Pak SC, Askew YS, Naviglia TL, Askew DJ, Nobar SM, Vetica AC, Long OS, Watkins SC, Stolz DB, et al. An intracellular serpin regulates necrosis by inhibiting the induction and sequelae of lysosomal injury. Cell. 2007;130:1108–1119. doi: 10.1016/j.cell.2007.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeil PL, Kirchhausen T. An emergency response team for membrane repair. Nat Rev Mol Cell Biol. 2005;6:499–505. doi: 10.1038/nrm1665. [DOI] [PubMed] [Google Scholar]
- Nehrke K. A reduction in intestinal cell pHi due to loss of the Caenorhabditis elegans Na+/H+ exchanger NHX-2 increases life span. J Biol Chem. 2003;278:44657–44666. doi: 10.1074/jbc.M307351200. [DOI] [PubMed] [Google Scholar]
- Olivier V, Haines GK, 3rd, Tan Y, Satchell KJ. Hemolysin and the multifunctional autoprocessing RTX toxin are virulence factors during intestinal infection of mice with Vibrio cholerae El Tor O1 strains. Infect Immun. 2007;75:5035–5042. doi: 10.1128/IAI.00506-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker MW, Feil SC. Pore-forming protein toxins: from structure to function. Prog Biophys Mol Biol. 2005;88:91–142. doi: 10.1016/j.pbiomolbio.2004.01.009. [DOI] [PubMed] [Google Scholar]
- Sakurai J, Honda T, Jinguji Y, Arita M, Miwatani T. Cytotoxic effect of the thermostable direct hemolysin produced by Vibrio parahaemolyticus on FL cells. Infect Immun. 1976;13:876–883. doi: 10.1128/iai.13.3.876-883.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M, Sato K, Fonarev P, Huang CJ, Liou W, Grant BD. Caenorhabditis elegans RME-6 is a novel regulator of RAB-5 at the clathrin-coated pit. Nat Cell Biol. 2005;7:559–569. doi: 10.1038/ncb1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T, Mushiake S, Kato Y, Sato K, Sato M, Takeda N, Ozono K, Miki K, Kubo Y, Tsuji A, et al. The Rab8 GTPase regulates apical protein localization in intestinal cells. Nature. 2007;448:366–369. doi: 10.1038/nature05929. [DOI] [PubMed] [Google Scholar]
- Schulenburg H, Kurz CL, Ewbank JJ. Evolution of the innate immune system: the worm perspective. Immunol Rev. 2004;198:36–58. doi: 10.1111/j.0105-2896.2004.0125.x. [DOI] [PubMed] [Google Scholar]
- Shaik GM, Draberova L, Heneberg P, Draber P. Vacuolin-1-modulated exocytosis and cell resealing in mast cells. Cell Signal. 2009;21:1337–1345. doi: 10.1016/j.cellsig.2009.04.001. [DOI] [PubMed] [Google Scholar]
- Shi A, Sun L, Banerjee R, Tobin M, Zhang Y, Grant BD. Regulation of endosomal clathrin and retromer-mediated endosome to Golgi retrograde transport by the J-domain protein RME-8. EMBO J. 2009;28:3290–3302. doi: 10.1038/emboj.2009.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sifri CD, Begun J, Ausubel FM. The worm has turned--microbial virulence modeled in Caenorhabditis elegans. Trends Microbiol. 2005;13:119–127. doi: 10.1016/j.tim.2005.01.003. [DOI] [PubMed] [Google Scholar]
- Tam C, Idone V, Devlin C, Fernandes MC, Flannery A, He X, Schuchman E, Tabas I, Andrews NW. Exocytosis of acid sphingomyelinase by wounded cells promotes endocytosis and plasma membrane repair. J Cell Biol. 2010;189:1027–1038. doi: 10.1083/jcb.201003053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiery J, Keefe D, Saffarian S, Martinvalet D, Walch M, Boucrot E, Kirchhausen T, Lieberman J. Perforin activates clathrin- and dynamin-dependent endocytosis, which is required for plasma membrane repair and delivery of granzyme B for granzyme-mediated apoptosis. Blood. 2010;115:1582–1593. doi: 10.1182/blood-2009-10-246116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaitkevicius K, Lindmark B, Ou G, Song T, Toma C, Iwanaga M, Zhu J, Andersson A, Hammarstrom ML, Tuck S, et al. A Vibrio cholerae protease needed for killing of Caenorhabditis elegans has a role in protection from natural predator grazing. Proc Natl Acad Sci U S A. 2006;103:9280–9285. doi: 10.1073/pnas.0601754103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walev I, Bhakdi SC, Hofmann F, Djonder N, Valeva A, Aktories K, Bhakdi S. Delivery of proteins into living cells by reversible membrane permeabilization with streptolysin-O. Proc Natl Acad Sci U S A. 2001;98:3185–3190. doi: 10.1073/pnas.051429498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walev I, Martin E, Jonas D, Mohamadzadeh M, Muller-Klieser W, Kunz L, Bhakdi S. Staphylococcal alpha-toxin kills human keratinocytes by permeabilizing the plasma membrane for monovalent ions. Infect Immun. 1993;61:4972–4979. doi: 10.1128/iai.61.12.4972-4979.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Seeley ES, Wickner W, Merz AJ. Vacuole fusion at a ring of vertex docking sites leaves membrane fragments within the organelle. Cell. 2002;108:357–369. doi: 10.1016/s0092-8674(02)00632-3. [DOI] [PubMed] [Google Scholar]
- Wei JZ, Hale K, Carta L, Platzer E, Wong C, Fang SC, Aroian RV. Bacillus thuringiensis crystal proteins that target nematodes. Proc Natl Acad Sci U S A. 2003;100:2760–2765. doi: 10.1073/pnas.0538072100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodford N, Livermore DM. Infections caused by Gram-positive bacteria: a review of the global challenge. J Infect. 2009;59(Suppl 1):S4–16. doi: 10.1016/S0163-4453(09)60003-7. [DOI] [PubMed] [Google Scholar]
- Wu JZ, Prausnitz JM. Osmotic pressures of aqueous bovine serum albumin solutions at high ionic strength. Fluid Phase Equilibria. 1999;155:139–154. [Google Scholar]
- Zitzer A, Wassenaar TM, Walev I, Bhakdi S. Potent membrane-permeabilizing and cytocidal action of Vibrio cholerae cytolysin on human intestinal cells. Infect Immun. 1997;65:1293–1298. doi: 10.1128/iai.65.4.1293-1298.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.