

Abstract
A convenient one-pot C-H alkenylation/ electrocyclization/ aromatization sequence has been developed for the synthesis of highly substituted pyridine derivatives from alkynes and α,β-unsaturated N-benzyl aldimines and ketimines that proceeds through dihydropyridine intermediates. A new class of ligands for C-H activation was developed, providing broader scope for the alkenylation step than could be achieved with previously reported ligands. Substantial information was obtained about the mechanism of the reaction. This included the isolation of a C-H activated complex and its structure determination by X-ray analysis; in addition, kinetic simulations using the Copasi software were employed to determine rate constants for this transformation, implicating facile C-H oxidative addition and slow reductive elimination steps.
Introduction
The synthesis of N-heterocycles is an important area of research due to their prevalence in natural products and drugs.1 Of the N-heterocycles, pyridines are the most extensively used in pharmaceutical research,2 and much effort has been devoted to their synthesis. Despite these efforts, the selective preparation of non-symmetrically substituted pyridines continues to be a significant challenge in synthesis.3 Traditional methods for pyridine synthesis involve the condensation of amines with carbonyl compounds, but high yields of single products can often only be obtained for symmetrically substituted systems.1 Cycloaddition reactions can also provide rapid access to substituted pyridines; however, there are significant constraints on the steric and electronic nature as well as the location of the substituents.3a,i–j Recently, a few elegant methods have appeared in the literature for the synthesis of selectively substituted pyridine derivatives. In particular, the triflic anhydride-mediated pyridine synthesis from N-vinyl amides and alkene or alkyne inputs provides a convenient route into the synthesis of highly substituted pyridines.4 Iminoannulation is also a mild method for the synthesis of pyridines from β-bromo-α,β-unsaturated imines and alkynes, though this route requires the pre-functionalization of the imine moiety.3b
Results and Discussion
Dihydropyridine Synthesis
We now report a new one-pot C-H alkenylation/ electrocyclization/ aromatization sequence that provides access to highly substituted pyridines from α,β-unsaturated imines and alkynes.5,6 In contrast to other methods for pyridine synthesis, rhodium-catalyzed C-H activation does not require the use of activated precursors and is tolerant of a variety of functional groups. As part of this work, a new class of ligands for C-H activation has been developed to achieve broad scope in the C-H alkenylation step.
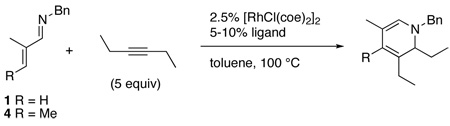
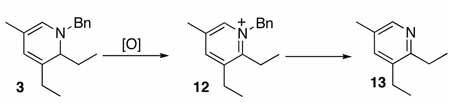
We began our investigation into this reaction by utilizing catalyst and reaction parameters optimized for the alkylation of α,β-unsaturated imines with alkenes.7 Specifically, using the electron-donating (dicyclohexylphosphinyl)ferrocene ligand in a 2:1 ligand to rhodium ratio provided the aza-triene product 2, which subsequently underwent in-situ electrocyclization to yield dihydropyridine (DHP) 3 (eq 1).8 While this catalyst system did prove successful with aldimine 1, only trace product was seen using aldimines bearing a β-substituent, e.g. 4 (Table 1, entry 2), even at elevated temperatures.
 |
(1) |
Table 1.
A Brief Survey of Catalyst Conditions.
 | |||||
|---|---|---|---|---|---|
| Entry | Imine | Ligand | Mol% L |
Time (h) |
Yielda (%) |
| 1 | 1 | FcPCy2 | 10 | 2 | quant. |
| 2 | 4 | FcPCy2 | 10 | 24 | trace |
| 3 | 4 | 5 | 10 | 8 | 84 |
| 4 | 4 | 6a | 10 | 24 | 20 |
| 5 | 4 | 6b | 10 | 24 | 36 |
| 6 | 4 | 6c | 10 | 24 | 0 |
| 7 | 4 | 5 | 5 | 8 | 82 |
| 8 | 4 | 6b | 5 | 6 | 88 |
| 9 | 4 | 7a | 5 | 24 | 42 |
| 10 | 4 | 7b | 5 | 8 | 55 |
| 11 | 4 | 7c | 5 | 8 | 73 |
All yields were determined by NMR integration relative to 2,6-dimethoxytoluene as an internal standard
Ligand optimization was therefore performed. An extensive screen of commercially available phosphine ligands did not produce an ideal catalyst system, and we began to consider new ligand design. To ascertain the influence of steric effects, (diethylphosphinyl)ferrocene, (Figure 1, 5) was prepared and showed improved reactivity with β-substituted imine 4 providing the DHP product in 84% yield in 8 hours (Table 1, entry 3). In several examples of imine-directed alkylations and alkenylations, electron-donating ligands have exhibited improved reactivity.9 We were therefore intrigued by a novel class of ligands with a 4-(N,N-dimethylamino)phenyl substituent (DMAPh) that recently were reported for use in Suzuki-Miyaura couplings.10 A series of analogous aniline-substituted phosphine ligands with dialkyl substituents possessing various steric properties were thus prepared (Figure 1; 6a–c). Using a 2:1 ratio of ligand to rhodium, the diethyl-(DMAPh)phosphine ligand 6b (Table 1, entry 5) provided a more efficient catalyst than either the dicyclohexyl (6a) or dimethyl (6c) derivatives (Table 1, entries 4 and 6).
Figure 1.

Ligands for C-H activation.
We next anticipated that decreasing the ligand loading might improve the reaction efficiency by avoiding a required ligand dissociation step from the 6-coordinate Rh species prior to alkyne binding. Indeed, decreasing the ratio of ligand to rhodium from 2:1 to 1:1 (Table 1, entries 7 and 8) decreased the reaction time and considerably improved the yield for 6b (entry 8). To further investigate the effect of the electronic properties of the ligand, the corresponding diaryl alkyl phosphines 7a–c were prepared and screened (Table 1, entries 9 – 11). While these ligands did produce active catalyst systems, the yields were inferior to those obtained with ligand 6b, which was used in all subsequent experiments.
Under optimized conditions for catalysis, the scope of this transformation with other imines and alkynes was found to be quite good.11 Aldimine 1 and β-substituted aldimines 4, 8, and 9 each gave good DHP yields (Table 2, entries 1, 5 – 7, columns 5 and 6). Additionally, ketimines 10 and 11 reacted rapidly with 3-hexyne to give the desired DHP products (entries 9 and 15, columns 5 and 6). Alkyne scope was also good with non-symmetric alkynes giving DHP products in most cases as single regioisomers when one substituent on the alkyne is α-branched. In the simplest case, 4-methyl-2-pentyne reacted with 1 and 9 to give single regioisomers of DHP products (entries 2 and 8, columns 5 and 6), although this alkyne coupled with β-substituted imine 8 with poor regiocontrol (data not shown). The more sterically hindered TMS–substituted alkynes consistently exhibited good regioselectivity with the TMS substituent proximal to the DHP nitrogen for all imines evaluated (entries 3, 4, 10–12, columns 5 and 6). In some cases, the electronic character of the alkyne was found to override steric control to give major DHP products with the bulkier substituents distal to the nitrogen (entries 13 and 14, columns 5 and 6). When terminal alkynes were employed, alkyne dimerization was competitive with C-H alkenylation, leading to complex product mixtures.
Table 2.
One pot synthesis of pyridines from α,β-unsaturated imines and alkynes
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Imine | Alkyne | Time (h) |
DHP | % Yielda (isolated) |
[O] Temp. (°C) |
[O] Time (h) |
Pyridine | Overall Yieldb |
| 1 | 2 |  |
98 (97) | 0 | 8 |  |
80% | ||
| 2 | 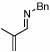 |
2 | 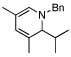 |
94 (99) | 0 | 8 | 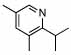 |
69% | |
| 3 | 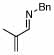 |
10 | 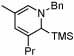 |
50 | 23 | 12 | 54% | ||
| 4 | 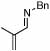 |
4 | 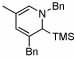 |
81 | 23 | 12 | 66% | ||
| 5 | 2 | 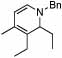 |
98 | 0 | 8 | 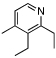 |
54% | ||
| 6 | 6 | 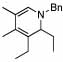 |
88 | 23 | 8 | 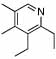 |
61% | ||
| 7 | 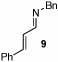 |
4 | 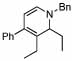 |
96 | 23 | 14 | 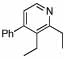 |
86% | |
| 8 | 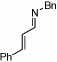 |
4 | 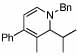 |
98 | 23 | 14 | 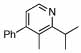 |
69% | |
| 9 | 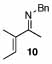 |
2 | 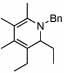 |
99 (78) | 75 | 16 | 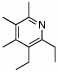 |
75% | |
| 10 | 2 | 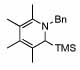 |
99 | 0 | 16 | 73% | |||
| 11 | 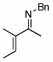 |
6 | 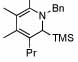 |
81 | 75 | 16 | 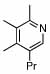 |
59% | |
| 12 | 8 | 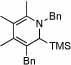 |
86 | 75 | 16 | 50% | |||
| 13 | 24 | 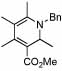 |
76 (70) | 120 | 16 | 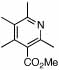 |
53%c | ||
| 14 | 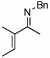 |
2 | 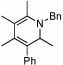 |
74 | 75 | 12 | 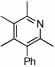 |
64%d | |
| 15 | 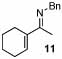 |
2 | 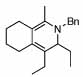 |
99 | 120 | 16 | 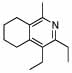 |
32% | |
Determined by NMR integration relative to 2,6-dimethoxytoluene as an internal standard.
Isolated overall yields based on starting α,β-unsaturated imine.
For this specific substrate, purified DHP was used for pyridine synthesis and the yield provided is the overall yield for the three steps.
The minor regioisomer with the Ph-substituent in the 2-position was isolated in 26% yield.
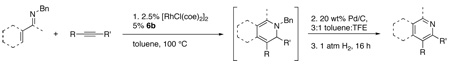
Oxidation to Pyridines
With a method to prepare highly substituted DHPs in hand, we next sought to develop an efficient method for their conversion to the corresponding highly substituted pyridines. We envisioned that this could be accomplished by oxidation of the DHPs to N-benzylpyridinium intermediates followed by debenzylation.12
Using purified DHP 3, a variety of oxidants were probed for this transformation. DDQ13 while effectively oxidizing the DHP, formed only small amounts of the pyridinium salt along with extensive decomposition (Table 3, entry 3). Manganese oxidants such as MnO214 and Mn(OAc)3/H5IO615, were also ineffective, with MnO2 providing only trace pyridinium with much DHP 3 remaining and Mn(OAc)3 primarily decomposing the reagents (entries 1 and 2). Tritylchloride was a very effective oxidant, producing the pyridinium salt cleanly and efficiently. We envisioned that debenzylation might occur in-situ by nucleophilic displacement, but this transformation was not realized (entry 4). Ideal conditions were ultimately found using 10% Pd/C and air as the oxidant in acetic acid.16 Under these conditions, the pyridinium was formed cleanly. Previous reports indicated that the addition of phenol could facilitate the debenzylation in-situ.17 While we did not find this additive to be effective in removing the benzyl group (data not shown), simply subjecting the reaction to an atmosphere of hydrogen following the oxidation did provide the deprotected pyridine via hydrogenolysis in a one-pot procedure with no additives, simplifying the overall transformation (entry 5).
Table 3.
Oxidant Screen for the Conversion of DHP 3 to Pyridine 13.
 | ||||
|---|---|---|---|---|
| Entry | Oxidant | Solvent | T (°C) | Result |
| 1 | MnO2 | CH2Cl2 | 23 | trace 12 |
| 2 | Mn(OAc)3/H3IO5 | AcOH | 23 | trace 12 |
| 3 | DDQ | EtOAc | 23 | trace 12 |
| 4 | Ph3CCl | CH2Cl2 | 23 | 12 |
| 5 | Pd/C, aira | AcOH | 50 | 13, 70% |
Followed by 1 atm H2 for 1 h at 23 °C.
While the DHPs can be isolated and purified in some cases, significant decomposition during chromatography is often seen, and in many cases, for example with the silyl substituted DHPs, the compounds are too unstable to isolate. We were therefore hopeful that conversion to the pyridines could be carried out using crude DHP reaction mixtures, alleviating the problem of isolation. This idea was validated when the crude reaction mixture from the formation of DHP 3 provided pyridine 13 in 66% yield.
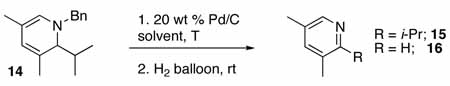
Applying this procedure to other DHPs, e.g. 14, we unfortunately found that this procedure was not general. Instead of isolating the expected pyridine 15, the major pyridine product 16 had lost the iso-propyl substituent and was formed in very low yield (Table 4, entry 1). This oxidative dealkylation is a known decomposition pathway in the oxidation of 1,4- and 1,2-DHPs.18 The oxidants used with DHP 3 were reexamined with DHP 14, but did not show any improvement. A screen of temperatures and solvents was then undertaken.
Table 4.
Optimization of the oxidation of DHP 12 to pyridine 13
 | |||||
|---|---|---|---|---|---|
| Entry | Sovent (M) | Temp (°C) |
Time (h) |
15:16 | Total Yield |
| 1 | AcOH (0.2) | 50 | 5 | 16 only | trace |
| 2 | MeOH (0.2) | 50 | 12 | 10:1 | <20% |
| 3 | TFEa (0.2) | 23 | 3 | 1:1 | 54% |
| 4 | TFEa (0.2) | 0 | 5 | 4:1 | 51% |
| 5 | TFE:Me-Phb 2:1 (0.1) | 0 | 5 | 4:1 | >50% |
| 6 | TFE:Me-Phb 1:1 (0.1) | 0 | 5 | 9:1 | >50% |
| 7 | TFE:Me-Ph 1:3 (0.1) | 0 | 8 | 15 only | 60% |
14 was not soluble in the solvent at this temperature.
TFE was not fully miscible in the co-solvent at this temperature.
Using the less acidic solvent methanol at 50 °C did oxidize 14 with little dealkylation; however, the reaction was substantially slowed in this solvent and did not go to completion, resulting in low conversion (Table 4, entry 2). Next, 2,2,2-trifluoroethanol (TFE) was used as a solvent and the temperature was lowered to 23 °C. Under these conditions, 15 and 16 were obtained in a 1:1 ratio with an overall yield of 54% (entry 3). Dropping the temperature to 0 °C decreased the dealkylation pathway even further, giving 15 and 16 in a 4:1 ratio without effecting the overall yield (entry 4). It was noted that 14 was not soluble in TFE at room temperature or below. To improve the solubility of the DHP, cosolvents were next investigated and the concentration was decreased. In a 2:1 solution of TFE:toluene at 0 °C, the overall selectivity increased only slightly (entry 5); however, by increasing the amount of cosolvent to a 1:1 mixture, a significant increase in selectivity was seen and pyridines 15 and 16 were obtained in a 9:1 ratio (entry 6). It was additionally noted that while TFE and toluene were miscible at room temperature, at 0 °C the solvents were no longer miscible and a fine suspension formed with vigorous stirring. Further increases in the relative amount of the cosolvent attenuated this problem, and using a 1:3 ratio of TFE:toluene provided 15 exclusively in 60% yield (entry 7). The use of toluene is particularly advantageous because it enables a one-pot synthesis of pyridines from α,β-unsaturated imines and alkynes via a C-H activation /electrocyclization /aromatization sequence with no intermediary isolation or solvent removal.
Applying this method to the crude reaction mixture from the formation of 14, we were pleased to find that 15 was formed in good yield over the three steps (please refer to Table 2, entry 2, columns 9 and 10). This one-pot method is quite general, delivering good to high overall yields of pyridine products for the three-step sequence (Table 2, columns 9 and 10). Increasing substitution on the DHP did require prolonged reaction times and increased temperature (e.g. entry 9), but all substrates were cleanly oxidized to the pyridine products. Interestingly, TMS-substituted DHP’s did not provide silylated pyridines, but instead, in-situ desilylation occurred to generate pyridine products without substitution at the ortho-position (entries 3, 4, 10 – 12, columns 9 and 10). In this manner, the silyl substituents can serve as blocking groups to enable bulky substituents to be located distal to the nitrogen. The presence of carboxy substituents is also well tolerated, leaving a site for further transformations (entry 13, columns 9 and 10).
Isolation of a Potential Intermediate and Discussion of Mechanism
In considering whether the reaction was proceeding via a C-H activation mechanism, we were hopeful that a C-H activated species could be detected directly. In a stochiometric reaction of 1 (1 equiv), [RhCl(coe)2]2 (0.5 equiv), and FcPCy2 (2 equiv) at room temperature, a new complex 17 formed immediately upon combination of the reagents (eq 4). Complex 17 was identified based on the presence in its 1H NMR spectrum of an apparent quartet shifted strongly upfield to −14.15 ppm, which corresponds to the doublet of triplets expected for a [Rh]-H species with 2 equivalent coordinated phosphine ligands. In the 31P NMR spectrum, a doublet at 128.42 ppm with a coupling constant of 113 Hz (1J; Rh-H) is seen. These data are consistent with other Rh-H complexes with similar ligand environments.19
 |
(4) |
Orange, block-like crystals of 17 were obtained by vapor diffusion of pentane into a toluene solution at room temperature. X-ray diffraction studies provided the structure shown in Figure 2. The geometry of 17 is distorted octahedral, with a P-Rh-P angle of 157.69(4)°, which is common for hydride complexes bearing bulky phosphine ligands.19 The bond angle for the metallacyclic N-Rh-C bond is 78.4(2)°, and this distortion from 90° causes the remaining bond angles in the square plane to be slightly more obtuse than would be true for an ideal octahedral complex. The Rh-H bond length is 1.36(4) Å.
Figure 2.

X-ray crystal structure (ORTEP diagram) of 17 with thermal ellipsoids drawn at the 50% probability level. Hydrogen atoms distant from the metal center have been omitted for clarity.
Isolated 17 is only sparingly soluble in most solvents, but redissolving it in both toluene-d8 and THF-d8 resulted in a 2:1 equilibrium mixture of 17 and free imine 1. The 31P NMR spectrum accordingly shows a mixture of complex 17, RhCl(FcPCy2)2, and free FcPCy2.20 Because of the lability of 17 in solution, its involvement in the catalytic cycle cannot be readily ascertained. However, when used catalytically (5 mol%) in the reaction between 1 and 3-hexyne, DHP 3 is obtained quantitatively. Additionally, in a stoichiometric reaction between 17 and 3-hexyne, DHP 3 is formed rapidly and in quantitative yield.
While attempts were made to identify the analogous catalytic intermediate using the more active aniline-substituted phosphine ligand 6b, no peaks indicative of a [Rh]-H complex were seen by 1H or 31P NMR spectroscopy. However, it is reasonable that a facile and readily reversible C-H activation occurs with this ligand, and that the equilibrium lies in favor of the non-C-H activated substrate. Because ligand 6b was more active under catalytic conditions, it was used in further kinetic experiments.
A plausible mechanistic pathway is shown in Scheme 1.21 Coordination of the imine to the catalyst through the imine nitrogen followed by oxidative addition of the C-H bond gives intermediate 18. Association of the alkyne followed by migratory insertion of the Rh-H bond provides intermediate 19.22 Subsequent reductive elimination provides the azatriene intermediate 2. Electrocyclization of 2 produces the dihydropyridine product, 3. Reversible isomerization of 2 produces 20, which cannot cyclize. Under typical reaction conditions, 2 and 20 are not generally observable by NMR. Reducing the reaction temperature to room temperature slows the rate of electrocyclization such that the growth and decay of 2 can be traced. Furthermore, the isomerization pathway to 20 is not reversible at this temperature; thus a buildup of 20 is seen throughout the course of the reaction. Under these conditions, four of the species shown in Scheme 1 (1, 2, 3, and 20) could be identified and their concentrations monitored by NMR spectroscopy. A plot of the concentrations of these species over time during a typical reaction course is given in Figure 3, plot A. The disappearance of 1 obeys pseudo first-order kinetics, producing a linear plot when ln([1]) is plotted versus time. Plot A in Figure 3 shows the appearance of intermediate 2 preceding the formation of DHP 3. Intermediate 2 builds up to a maximum concentration of 0.02 M before decaying.
Scheme 1.

Proposed Mechanistic Pathway
Figure 3.

Experimental and simulated timecourses for the reaction of 1 with 5 equivalents of 3-hexyne using 2.5 mol% [RhCl(coe)2]2 and 5 mol% 6b at 25 ± 0.5 °C. A: experimental timecourse for Scheme 1. For plots B – D, the experimental and simulated data for Scheme 2 are overlaid, with experimental data designated using dashed lines and the corresponding simulated data using solid lines. Starting concentrations of 1: B; 0.10 M, C; 0.025 M, D; 0.50 M. The error in the values for the experimental data were <10%. See the Supporting Information for further details. Simulated data were obtained using the appropriate starting concentrations of reactants A, B and D, and using the following rate constants: k1 = 250 M−1h−1; k2 = 50 M−1h−1; k3 = 8 h−1; k4 = 0.6 h−1; k5 = 0.03 h−1.
To extract information regarding the rate constants of this reaction, the system was modeled computationally using the kinetic modeling program, Copasi.23 Because the proposed catalytic intermediates could not be detected by either 1H or 31P NMR spectroscopy, a simplified system was used in simulations. The modeled cycle is shown in Scheme 2.24 In addition to removing all of the ligand exchange steps from the mechanistic cycle, the reactions were all made irreversible for the simulation. Without the ability to detect and quantify catalytic intermediates, the simulated data obtained for the reverse reactions were highly underdetermined.
Scheme 2.

Simplified system for modeling in Copasi and the values obtained for k1 – k5.
Based on the experimental data obtained for the reaction in Scheme 2 at 25 ± 0.5 °C at three different reaction concentrations, 0.025 M, 0.10 M, and 0.50 M,25 the Copasi software was used to find rate constants k1 – k5 (Scheme 2). Iterative optimizations of k1 – k3 and k4 – k5 were performed at each concentration until simulated plots for each matched the observed plots (Figure 3, plots B – D). Errors were estimated by incrementally modifying each optimized rate constant independently until a reasonable fit was no longer obtained. As is often the case with numerical integrations, the rate constants generated by the simulation exhibited widely varying sensitivity to changes. For example, the simulated value of k2 could be varied over many orders of magnitude with deviations from the experimental data occurring only when k2 was set below 50 mmol·ml−1·h−1. This implies that the migratory insertion step is fast relative to the oxidative addition and reductive elimination steps. To corroborate this finding, the concentration of the alkyne was varied from 0.20 M to 1.0 M in a 0.10 M (relative to 1) reaction both experimentally and in the simulation. Experimentally, the rates of intermediate and product formation were found to be independent of alkyne concentration, and the plots obtained were nearly identical to the original plot at 0.50 M in alkyne. Likewise, in the modeled experiment no change in the observed rates was seen when the concentration of alkyne was varied.
Based on the obtained rate constants, the reductive elimination step is rate limiting with a rate constant on the order of 8 h−1. This is consistent with deuterium-labeling studies performed on other C-H alkylation systems that showed that C-H oxidative addition was rapid with reductive elimination being the rate limiting step.21 The rate constant for the electrocyclization is on the order of 0.6 h−1.26
Conclusion
A one-pot procedure has been developed for the synthesis of highly substituted pyridines from α,β-unsaturated imines and alkynes via a new C-H alkenylation/ electrocyclization/ aromatization sequence. Additionally, a new class of ligands was identified that greatly expands the scope of the C-H alkenylation reaction. The isolation and X-ray analysis of a C-H activated complex sheds some light on the mechanism of this transformation. Kinetic simulations were performed to find possible rate constants for the transformation and implicate facile oxidative addition with rate limiting reductive elimination.
Supplementary Material
Acknowledgement
This work was supported by NIH Grant GM069559 to J.A.E., an NSF predoctoral fellowship to D.A.C., and the Director and Office of Energy Research, Office of Basic Energy Sciences, Chemical Sciences Division, U.S. Department of Energy, under Contract DE-AC03-76SF00098 to R.G.B. Thanks to Dr. Allen G. Oliver at the UCB X-ray diffraction facility (CHEXRAY) for solving the structure of 17 and to Michael D. Pluth and Stuart E. Smith for helpful discussions.
Footnotes
Supporting Information Available: Complete experimental details and spectral data for all new compounds, and the X-ray crystallographic information in cif format for complex 17 are described. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Joule JA, Mills K. Heterocyclic Chemistry. 4th ed. Oxford, UK: Blackwell; 2000. [Google Scholar]
- 2.Carey JS, Laffan D, Thomson C, Williams MT. Org. Biomol. Chem. 2006;4:2337–2347. doi: 10.1039/b602413k. [DOI] [PubMed] [Google Scholar]
- 3.For recent reviews on pyridine syntheses see the following and references therein: Varela J, Saa C. Chem. Rev. 2003;103:3787–3801. doi: 10.1021/cr030677f. Zeni G, Larock RL. Chem. Rev. 2006;106:4644–4680. doi: 10.1021/cr0683966. For recent examples of transition-metal facilitated pyridine syntheses see: Trost BM, Gutierrez AC. Org. Lett. 2007;9:1473–1476. doi: 10.1021/ol070163t. Movassaghi M, Hill MD. J. Am. Chem. Soc. 2006;128:4592–4593. doi: 10.1021/ja060626a. McCormick MM, Duong HA, Zuo G, Louie J. J. Am. Chem. Soc. 2005;127:5030–5031. doi: 10.1021/ja0508931. Yamamoto Y, Kinpara K, Ogawa R, Nishiyama H, Itoh K. Chem. Eur. J. 2006;12:5618–5631. doi: 10.1002/chem.200600176. Tanaka R, Yuza A, Watai Y, Suzuki D, Takayama Y, Sato F, Urabe H. J. Am. Chem. Soc. 2005;127:7774–7780. doi: 10.1021/ja050261e. Takahashi T, Tsai F-Y, Li Y, Wang H, Kondo Y, Yamanaka M, Nakajima K, Kotora M. J. Am. Chem. Soc. 2002;124:5059–5067. doi: 10.1021/ja017507+. For pyridine syntheses from Diels Alder inputs see: Boger DL. Chem. Rev. 1986;86:781–793. Fletchter MD, Hurst TE, Miles TJ, Moody CJ. Tetrahedron. 2006;62:5454–5463..
- 4.Movassaghi M, Hill MD, Ahmad OK. J. Am. Chem. Soc. 2007;129:10096–10097. doi: 10.1021/ja073912a. [DOI] [PubMed] [Google Scholar]
- 5.For an example of pyridine synthesis from oxime and alkyne inputs via C-H activation see: Parthasarathy K, Jeganmohan M, Cheng C-H. Org. Lett. 2007 doi: 10.1021/ol7028367. ASAP.
- 6.For previous studies on chelation-assisted alkenylation proceeding via C-H activation, see the following: Lim S-G, Lee JH, Moon CW, Hong J-B, Jun C-H. Org. Lett. 2003;5:2759–2761. doi: 10.1021/ol035083d. Lim Y-G, Lee K-H, Koo BT, Kang J-B. Tetrahedron Lett. 2001;42:7609–7612. Kakiuchi F, Uetsuhara T, Tanaka Y, Chatani N, Murai S. J. Mol. Cat. A. 2002;183–183:511–514..
- 7.Colby DA, Bergman RG, Ellman JA. J. Am. Chem. Soc. 2006;128:5604–5605. doi: 10.1021/ja0584931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Similar reactivity was reported by Odom in the β-alkylation of the N-phenyl imine of 1-acetylcyclohexene, which was prepared in situ by Ti - mediated hydroamination of cyclohexenylacetylene. See: Cao C, Li Y, Shi Y, Odom AL. Chem. Commun. 2004:2002–2003. doi: 10.1039/b405620e..
- 9.(a) Thalji RL, Ahrendt KA, Bergman RG, Ellman JA. J. Org. Chem. 2005;70:6775–6781. doi: 10.1021/jo050757e. [DOI] [PubMed] [Google Scholar]; (b) O’Malley SJ, Tan KL, Watzke A, Bergman RG, Ellman JA. J. Am. Chem. Soc. 2005;127:13496–13497. doi: 10.1021/ja052680h. [DOI] [PubMed] [Google Scholar]
- 10.Guram AS, King AO, Allen JG, Wang X, Schenkel LB, Chan J, Bunel EE, Faul MM, Larsen RD, Martinelli MJ, Reider PJ. Org. Lett. 2006;8:1787–1789. doi: 10.1021/ol060268g. [DOI] [PubMed] [Google Scholar]
- 11.Many of the DHP products are not stable to chromatographic purification and therefore only NMR yields are reported.
- 12.The Diels-Alder reaction of aza-dienes with activated alkynes provides 1,4-DHP products, which can be converted to the corresponding pyridines. However, the scope of this transformation is limited to highly activated alkynes. (See references 3i, j.)
- 13.Wallace DJ, Gibb AD, Cottrell IF, Kennedy DJ, Brands KMJ, Dolling UH. Synthesis. 2001;12:1784–1789. [Google Scholar]
- 14.Eynde JJV, Delfosse F, Mayence A, van Haverbeke Y. Tetrahedron. 1995;23:6511–6516. [Google Scholar]
- 15.Lemire A, Grenon M, Pourashraf M, Charette AB. Org. Lett. 2004;6:3517–3520. doi: 10.1021/ol048624n. [DOI] [PubMed] [Google Scholar]
- 16.Nakamichi N, Kawashita Y, Hayashi M. Org. Lett. 2002;4:3955–3957. doi: 10.1021/ol0268135. [DOI] [PubMed] [Google Scholar]
- 17.Bennasar M-L, Zulaica E, Roca T, Alonso Y, Monerris M. Tetrahedron Lett. 2003;44:4711–4714. [Google Scholar]
- 18. Zhu X-Q, Zhao B-J, Cheng J-P. J. Org. Chem. 2000;65:8158–8163. doi: 10.1021/jo000484h. (b) For a review on the chemistry of dihydropyridines see: Lavilla R. J. Chem. Soc., Perkin Trans. 1. 2002:1141–1156..
- 19.(a) Albinati A, Arz C, Pregosin PS. J. Organomet. Chem. 1987;335:379–394. [Google Scholar]; (b) Hara T, Yamagata T, Mashima K, Kataoka Y. Organometallics. 2007;26:110–118. [Google Scholar]
- 20.See supporting information for further spectroscopic discussion and evidence for the identity and purity of isolated 17.
- 21.For mechanistic studies on other chelation-assisted C-H alkylations see: Jun C-H, Moon CW, Lee D-Y. Chem. Eur. J. 2002;8 2423-2328. Kakiuchi F, Ohtaki H, Sonoda M, Chatani N, Murai S. Chem. Lett. 2001:918–919. Lenges CP, Brookhart M. J. Am. Chem. Soc. 1999;121:6616–6623..
- 22.Alternatively, carbometalation of the alkyne could occur generating a seven-membered aza-metallacycle. In addition to the instability of the seven-membered intermediate, carbometallation should place the more sterically hindered substituent distal to the nitrogen, which is opposite to the regioselectivity observed in our case (Table 2 entries 2–4, 8, 10–12).
- 23.Copasi, version 4.2 was employed for these simulations. See Hoops S, Sahle S, Gauges R, Lee C, Pahle J, Simus N, Singhal M, Xu L, Mendes P, Kummer U. Bioinformatics. 2006;22:3067–3074. doi: 10.1093/bioinformatics/btl485.. For further information or to download Copasi, see http://www.copasi.org/tiki-index.php.
- 24.For recent examples of transition-metal mediated systems modeled using an early version of the Copasi software, Gepasi, see: Watson MP, Overman LE, Bergman RG. J. Am. Chem. Soc. 2007;129:5031–5044. doi: 10.1021/ja0676962. Ghosh R, Zhang X, Achord P, Emge TJ, Krogh-Jespersen K, Goldman AS. J. Am. Chem. Soc. 2007;129:853–866. doi: 10.1021/ja0647194. Wiedemann SH, Lewis JC, Ellman JA, Bergman RG. J. Am. Chem. Soc. 2006;128:2452–2462. doi: 10.1021/ja0576684..
- 25.Reaction concentrations are given with respect to imine 1. In all cases, 2.5 mol% [RhCl(coe)2]2, 5 mol% (DMAPh)PEt2, and 5 equivalents of alkyne were used.
- 26.For another example where rate constants were determined for 6π electrocyclizations of azatrienes see: Maynard DF, Okamura WH. J. Org. Chem. 1995;60:1763–1771. For examples of DHP formation via azatriene intermediates see the following and references therein: Sydorenko N, Hsung RP, Vera EL. Org. Lett. 2006;8:2611–2614. doi: 10.1021/ol060932t. Tanaka K, Mori H, Yamamoto M, Katsumura S. J. Org. Chem. 2001;66:3099–3110. doi: 10.1021/jo005779+..
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.