

Abstract
Using a functional high-throughput screening (HTS) and subsequent solution-phase parallel synthesis approach, we have discovered a novel series of positive allosteric modulators for mGlu4, a G-protein coupled receptor. This series is comprised of a homopiperazine central core. The solution-phase parallel synthesis and SAR of analogs derived from this series will be presented. This series of positive allosteric modulators of mGlu4 provide critical research tools to further probe the mGlu4-mediated effects in Parkinson’s disease.
1. Introduction
The development of new strategies for the treatment of Parkinson’s disease (PD) continues to be a major focus of attention.1–4 PD is a neurodegenerative disease caused by the degeneration of dopaminergic neurons in the substantia nigra pars compacta of the basal ganglia which leads to a debilitating movement disorder. Metabotropic glutamate receptor 4 (mGlu4) is expressed at a key synapse in the indirect pathway of the basal ganglia circuitry.5, 6 Several studies have shown that the activation of mGlu4 has anti-parkinsonian and neuroprotective effects in rodent PD models.6, 7 These results have established this receptor as a viable target for the symptomatic and disease modifying treatment of PD. We have taken the approach of selectively activating mGlu4 using positive allosteric modulators (PAMs), compounds which increase the potency of the endogenous neurotransmitter glutamate at mGlu4.8, 9
Compounds in Vanderbilt’s small molecule library were screened in a triple-add assay format at 10 µM to identify agonist, antagonist, or allosteric modulator activity in CHO cells stably expressing human mGlu4 and the chimeric G protein Gqi5. Test compounds were added to cells 140 seconds prior to addition of an EC20 concentration of glutamate; 90 seconds later an EC80 concentration of glutamate was added. Response was measured in a fluorometric calcium assay, and data were analyzed by finding the maximum value for each fluorescence trace. This “triple add” approach to HTS is utilized in order to identify compounds with different modes of pharmacology (agonists prior to EC20 addition, antagonists and negative allosteric modulators after EC80 addition, and positive allosteric modulators after EC20 addition) in a single experiment.
Using this functional high-throughput screening protocol, we have previously identified a number of novel mGlu4 PAM scaffolds. In addition to known mGlu4 PAM, PHCCC, 1,7, 10 we have reported a number of distinct structural classes of mGlu4 PAMs, such as VU0155041, 2,11, 12 VU0080241, 3,13 VU0001171, 4,14 VU0092145, 5,14 VU0361737, 6,15 VU0364439, 7,16 and VU0366037, 8 (Figure 1); of which, two analogs (2 and 6) have been shown to display efficacy in anti-Parkinsonian animal models.11, 15 However, all of the reported compounds bear striking resemblance in that they all contain polyaromatic moieties, making these compounds planar, and they do not contain any basic nitrogens. We now report a novel scaffold, represented by VU0105737, 9, as possessing mGlu4 PAM activity (Figure 2). This new chemotype is comprised of a nonplanar homopiperazine central core which contains a basic nitrogen (which can presumably lead to more soluble compounds than previously identified). Due to this structural novelty and the encouraging calculated properties, this scaffold provided an excellent opportunity to explore the SAR around this initial HTS lead as part of our hit-to-lead program.
Figure 1.

Structures of PHCCC, 1, and other recently reported mGlu4 PAMs, 2–9.
Figure 2.

Areas of SAR Exploration of VU0105737, 9.
2. Results and Discussion
The SAR surrounding compound 9 centered around 4 structural motifs: 1, exploring the 2,4-dimethoxyphenylamide; 2, exploring the linker chain length; 3, exploring the central homopiperazine ring; and 4, exploring the 4-methoxyphenylsulfonamide (Figure 2). The homopiperazine, 9, was readily modified using commercially available acid chlorides, carboxylic acids, sulfonyl chlorides and other core starting materials. The parallel synthesis and biological evaluation of this structure class is detailed below. All parallel synthetic procedures were conducted utilizing either test-tube reaction blocks or stir-plate mounted vial racks.
Modification of the central homopiperazine core was performed in order to address the basicity of the homopiperazine nitrogens (piperazine, oxohomopiperazine, oxopiperazine) as well as to change the shape by introduction of the bridged [3.3.0] scaffold (Scheme 1. All experimental procedures are contained in the Supplemental Materials). The piperazine compound, 11, and [3.3.0]-compound, 13, were prepared from N-Boc starting materials, 10 and 12, via a three step protocol of sulfonylation, Boc removal and alkylation. The oxo-containing compounds started from piperazin-2-one (14a) or 1,4-diazepan-5-one (14b). Thus, sulfonamide formation with 4-methoxybenzenesulfonyl chloride (CH2Cl2, DIEA, 43%-quant.), followed by alkylation of the amide with methyl bromoacetate (Cs2CO3, CH3CN, 80°C, 45–50%) and saponification of the ester (LiOH, THF/MeOH/H2O, 35%-quant.) afforded the penultimate acid. Finally, HATU-mediated amide coupling with 2,4-dimethoxyaniline (DMF, DIEA) provided the final compounds, 15a and 15b.
Scheme 1.

Results from Table 1 show that little modification of the core portion of the molecule is tolerated. All modifications of the core structure led to compounds that were inactive and showed no potentiation of glutamate (as assessed by the %GluMax). We utilized the Chinese hamster ovary cells expressing human mGlu4 and the chimeric G protein Gqi5 to induce calcium mobilization as our pharmacological assay to determine mGlu4 potency. This assay does show fluctuation in the day-to-day maximal PAM response; due to this, all data have been normalized to the control compound, 1, as a comparison of relative efficacy (as noted in % PHCCC).10, 16
Table 1.
SAR Evaluation of Linker and Scaffold
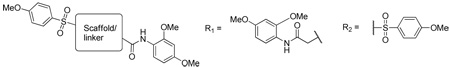 | ||||
|---|---|---|---|---|
| Cmpd | R | hEC50 (µM)a | % PHCCCa | Yieldc (%) |
| 11 | 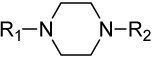 |
Inactiveb | 20.8 ± 1.1 | 45 |
| 13 | 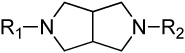 |
Inactiveb | 25.3 ± 2.3 | 49 |
| 15a | 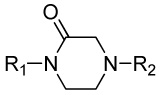 |
Inactiveb | 18.1 ± 1.3 | 19 |
| 15b | 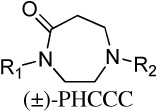 |
Inactiveb 3.1 ± 0.3 |
16.4 ± 0.8 | 60 |
EC50 and GluMax, are the average of at least three independent determinations performed in triplicate (Mean ± SEM shown in table). PHCCC is run as a control compound each day and it, and the maximal response generated in mGlu4 CHO cells in the presence of mGlu4 PAMs varies slightly in each experiment. Therefore, efficacy data were further normalized to the relative PHCCC response obtained in each day’s run.
Inactive compounds are defined as %GluMax did not surpass 2X the EC20 value for that day’s run.
All yields were obtained by reverse phase preparative HPLC and were optimized for purity (>95%) not yield.
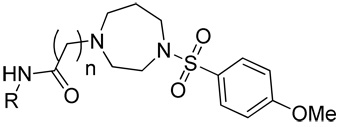
Keeping the right-side of the molecule constant, investigation of replacement of the amide aromatic ring with various heterocycles and alkyl groups, and exploration of the linker was next undertaken (Scheme 2, see Supplemental Material for full experimental procedures). To this end, commercially available N-Boc homopiperazine, 16, was reacted with 4-methoxybenzenesulfonyl chloride (CH2Cl2, DIEA) to give 17 in high yield (98%). Deprotection of the Boc group (4 M HCl, dioxane, quant.) furnished the key intermediate 18 which was converted to the urea, 20i, via reaction with 2,4-dimethoxyphenyl isocyanate (CH2Cl2, DIEA) in good yield (80%). Alternatively, 18 was N-alkylated with methyl bromoacetate (Cs2CO3, CH3CN, 80 °C, 76%) followed by saponification of the ester (LiOH, THF/H2O/MeOH, quant.) which gave the penultimate intermediate, 19, in good overall yield. The carboxylic acid was then coupled to a variety of anilines and amines utilizing HATU as the coupling reagent (HATU, DMF, DIEA, 45–85%) to provide the desired compounds (20a–j).
Scheme 2.

Synthetic procedure for the initial homopiperazine analogs, 20a–j.
The results in Table 2 show that the original hit with the 2,4-dimethoxyphenyl substituent was weakly active (9, >10 µM, 69.5% PHCCC) as was the 2-methoxyphenyl substituent (20a, >10 µM, 63.9% PHCCC); however, both compounds showed good efficacy (>100% PHCCC). Replacing the 2-methoxyphenyl with 4-methoxyphenyl led to an inactive compound (20b, 14.8% PHCCC). Other substitution patterns, such as 2,4-difluorophenyl (20c), 2-fluorophenyl (20d), 4-pyridyl (20e), cycloalkyl (20f, 20g) and cyclicdioxoaryl compound (20h) were all inactive. In addition, changing the length of the carbon linker also produced inactive compounds (20i, 20j). This rather ‘shallow’ SAR is a common occurrence with allosteric modulators.4,5
Table 2.
SAR Evaluation of Amide Aromatic Ring
 | |||||
|---|---|---|---|---|---|
| Cmpd | n | R | hEC50 (µM)a | % PHCCCa | Yieldc (%) |
| 9 | 1 | 2,4-dimethoxyphenyl | >10 | 69.5 ± 7.7 | 61 |
| 20a | 1 | 2-methoxyphenyl | >10 | 63.9 ± 3.6 | 64 |
| 20b | 1 | 4-methoxyphenyl | Inactiveb | 14.8 ± 0.8 | 20d |
| 20c | 1 | 2,4-difluorophenyl | Inactiveb | 17.7 ± 1.6 | 3d |
| 20d | 1 | 2-fluorophenyl | Inactiveb | 13.2 ± 0.4 | 2d |
| 20e | 1 | 4-pyridyl | Inactiveb | 13.9 ± 1.2 | 2d |
| 20f | 1 | Cyclohexyl | Inactiveb | 15.5 ± 1.3 | 12d |
| 20g | 1 | Isopropyl | Inactiveb | 15.0 ± 0.8 | 19d |
| 20h | 1 | 2,3-dihydrobenzo[b][1,4]dioxin-6-yl | Inactiveb | 17.6 ± 1.0 | 12d |
| 20i | 0 | 2,4-dimethoxyphenyl | Inactiveb | 13.5 ± 0.7 | 80 |
| 20j | 3 | 2,4-dimethoxyphenyl (±)-PHCCC |
Inactiveb 3.1 ± 0.3 |
14.8 ± 1.3 | 70 |
EC50 and GluMax, are the average of at least three independent determinations performed in triplicate (Mean ± SEM shown in table). PHCCC is run as a control compound each day, and the maximal response generated in mGlu4 CHO cells in the presence of mGlu4 PAMs varies slightly in each experiment. Therefore, efficacy data were further normalized to the relative PHCCC response obtained in each day’s run.
Inactive compounds are defined as %GluMax did not surpass 2X the EC20 value for that day’s run.
All yields were obtained by reverse phase preparative HPLC unless otherwise stated and were optimized for purity (>95%) not yield.
Yields obtained by mass directed HPLC17
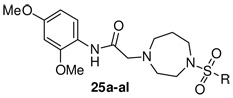
The last portion of the molecule for modification was the right-hand sulfonamide moiety (Scheme 3, Table 3, see Supplemental Material for full experimental details). Starting with 2,4-dimethoxyaniline, 21, amide formation with bromoacetyl bromide (CH2Cl2, DIEA, 81%) gave the desired α-bromo-amide, 22, which was N- alkylated with Boc-homopiperazine (Cs2CO3, CH3CN, 80 °C, 79%) to give 23 in 64% yield (2 steps). The Boc protecting group was removed (4 M HCl, dioxane, quant.) giving the key homopiperazine intermediate, 24, which was coupled with the appropriate sulfonyl chloride (CH2Cl2, DIEA, 45–85%) to furnish the desired sulfonamides 25a–al.
Scheme 3.

Table 3.
SAR Evaluation of Sulfonamide Aromatic Ring
 | ||||
|---|---|---|---|---|
| Cmpd | R | hEC50 (µM) | % PHCCC | Yieldc (%) |
| 25a | phenyl | 3.5 ± 0.7 | 69.7 ± 5.1 | 80 |
| 25b | 2-fluorophenyl | 3.5 ± 0.5 | 36.6 ± 1.8 | 3d |
| 25c | 2-chlorophenyl | >10 | 25.8 ± 2.5 | 58 |
| 25d | 2-methoxyphenyl | Inactiveb | 21.8 ± 1.0 | 87 |
| 25e | 2-(trifluoromethyl)phenyl | Inactiveb | 20.1 ± 0.2 | 24d |
| 25f | 3-(trifluoromethyl)phenyl | Inactiveb | 18.2 ± 1.9 | 6d |
| 25g | 4-(trifluoromethyl)phenyl | 2.5 ± 0.5 | 26.6 ± 2.3 | 36 |
| 25h | 4-(trifluoromethoxy)phenyl | >10 | 48.2 ± 4.6 | 5d |
| 25i | 4-methylphenyl | 3.3 ± 0.2 | 48.7 ± 1.1 | 77 |
| 25j | 4-fluorophenyl | >10 | 27.6 ± 1.3 | 33d |
| 25k | 4-chlorophenyl | 4.1 ± 0.6 | 43.6 ± 4.4 | 41 |
| 25l | 4-tertbutylphenyl | >10 | 32.6 ± 3.1 | 32 |
| 25m | 4-acetylphenyl | >10 | 42.9 ± 2.7 | 65 |
| 25n | 2,4-dimethylphenyl | 1.3 ± 0.5 | 42.8 ± 4.1 | 58 |
| 25o | 2,5-dimethylphenyl | 2.7 ± 0.6 | 32.5 ± 1.2 | 67 |
| 25p | 2,5-dichlorophenyl | Inactiveb | 22.7 ± 0.7 | 67 |
| 25q | 2-chloro-6-methylphenyl | Inactiveb | 18.1 ± 0.9 | 75 |
| 25r | 2,6-dichlorophenyl | Inactiveb | 22.3 ± 1.7 | 23 |
| 25s | 3,4-dimethylphenyl | 3.2 ± 0.2 | 42.4 ± 5.0 | 67 |
| 25t | 3-chloro-4-methylphenyl | 3.4 ± 0.2 | 39.2 ± 1.2 | 43 |
| 25u | 3,4-dichlorophenyl | >10 | 24.3 ± 1.5 | 31 |
| 25v | 3,4-difluorophenyl | >10 | 27.0 ± 1.8 | 64 |
| 25w | 4-chloro-3-fluorophenyl | 4.5 ± 0.7 | 25.5 ± 1.2 | 53 |
| 25x | 2,4-dichloro-5-methylphenyl | 3.1 ± 0.9 | 43.6 ± 4.7 | 44 |
| 25y | 4-chloro-2-fluoro-5-methylphenyl | 2.7 ± 0.2 | 36.9 ± 2.7 | 36 |
| 25z | benzyl | >10 | 46.5 ± 4.8 | 12d |
| 25aa | 2,4-dichlorobenzyl | 2.8 ± 0.01 | 34.4 ± 3.5 | 44 |
| 25ab | isopropyl | Inctiveb | 17.5 ± 0.7 | 22d |
| 25ac | isobutyl | Inactiveb | 21.3 ± 0.9 | 27d |
| 25ad | 2-pyridyl | Inactiveb | 20.6 ± 0.3 | 36 |
| 25ae | 2-thiophene | 1.8 ± 0.3 | 52.9 ± 5.0 | 83 |
| 25af | 2-furyl | 3.3 ± 0.3 | 36.2 ± 2.2 | 98 |
| 25ag | 3-pyridyl | >10 | 25.6 ± 4.6 | 20 |
| 25ah | 3-furyl | >10 | 34.1 ± 3.6 | 70 |
| 25ai | 3-thiophene | >10 | 35.0 ± 3.3 | 79 |
| 25aj | 2-acetamidothiazol-5-yl | >10 | 46.4 ± 5.5 | 60 |
| 25ak | 2,4-dimethylthiazol-5-yl | >10 | 47.7 ± 2.1 | 73 |
| 25al | 2-methyl-5-(trifluoromethyl)thiazol-5-yl (±)-PHCCC |
2.5 ± 0.4 3.1 ± 0.3 |
28.1 ± 4.8 | 49 |
EC50 and GluMax, are the average of at least three independent determinations performed in triplicate (Mean ± SEM shown in table). PHCCC is run as a control compound each day, and the maximal response generated in mGlu4 CHO cells in the presence of mGlu4 PAMs varies slightly in each experiment. Therefore, data were further normalized to the relative PHCCC response obtained in each day’s run.
Inactive compounds are defined as %GluMax did not surpass 2X the EC20 value for that day’s run.
All yields were obtained by reverse phase preparative HPLC unless otherwise stated and were optimized for purity (>95%) not yield.
Yields obtained by mass directed HPLC17
The compounds evaluated in Table 3 cover a range of phenyl, benzyl, alkyl and heteroaryl groups. The phenyl group showed good activity and efficacy (25a, 3.5 µM, 69.7% PHCCC). However, substitution at the 2- position only tolerated F (25b, 3.5 µM, 36.6% PHCCC) and Cl (25c, >10 µM, 25.8% PHCCC) with the latter being only a weak potentiator. The 4-position was the most tolerated with 4-CF3-phenyl (25g, 2.1 µM, 26.6% PHCCC) and 4-Me-phenyl (25i, 3.3 µM, 48.7% PHCCC) being the most potent. Disubstituted phenyl groups in general showed good activity especially with dimethylated phenyl groups where the 2,4-dimethylphenyl was the most potent compound of this series (25n, 1.3 µM, 42.8% PHCCC) along with 2,5-dimethyl phenyl (25o, 2.7 µM, 32.5% PHCCC). 2,4,5-Trisubstituted phenyl groups also showed potencies as good or better than the original HTS hit (25x, 3.1 µM, 43.6% PHCCC and 25y, 2.7 µM, 36.9% PHCCC).
The limited SAR around the benzyl group showed contrasting activity compared to the phenyl group. For example, the benzyl group was a weak potentiator (25z, >10 µM, 46.5% PHCCC). Furthermore the 2,4-dichlorobenzyl group showed good potency (25aa, 2.8 µM, 34.4% PHCCC) which in the case of the dihalogenated phenyl groups were all inactive. Compounds 25ab and 25ac showed that alkyl groups were not tolerated. Substitution with heteroaryl groups led to 3 compounds with acceptable potency. The most potent was the 2-thienyl (25ae, 1.8 µM, 52.9% PHCCC) followed by the 2-methyl-4-trifluoromethylthiazol-5-yl (25al, 2.5 µM, 28.1% PHCCC) and the 2-furyl (25af, 3.3 µM, 36.2% PHCCC).
Compounds 9, 20a, 25a, 25i, 25ae were selected based on potency and efficacy for further in vitro pharmacokinetic (PK) studies which included CYP450 inhibition, metabolic stability (ClINT)18 and rat plasma protein binding (rPPB) (Table 4, see Supplemental Material for assay details). Although these compounds possessed favorable free fraction when tested for plasma protein binding in rats (>3% free), this class of compounds was very unstable in liver microsomes (ClINT) and were equipotent with inhibiting CYP activity (2C9 and 3A4 < 10 µM).
Table 4.
In Vitro Pharmacokinetic Data for Selected Compounds.
| Cmpd | CYP | rPPB, %fua |
Human CLINT (CLHEP)b |
|||
|---|---|---|---|---|---|---|
| 1A2 | 2C9 | 2D6 | 3A4 | |||
| 9 | >30 µM | 1.8 µM | >20 µM | 4.2 µM | 3.9 | 908.58 (20.53) |
| 20a | >30 µM | 2.0 µM | >20 µM | 4.6 µM | 3.5 | 893.34 (20.52) |
| 25a | >20 µM | 2.9 µM | >20 µM | 7.0 µM | 11.5 | 559.86 (20.24) |
| 25i | 16.29 µM | 2.3 µM | 13.9 µM | 3.0 µM | 3.1 | 731.45 (20.41) |
| 25ae | 8.46 µM | 2.6 µM | 10.1 µM | 7.4 µM | 27.3 | 517.92 (20.18) |
rat protein binding, % free unbound.
Intrinsic clearance, human (CLINT). Predicted hepatic clearance (CLHEP).
Conclusions
In summary, through a functional HTS campaign, we identified a novel chemotype as an mGlu4 PAM and SAR was explored around 4-structural motifs. These compounds represent a series of homopiperazine sulfonamides. Limited SAR around this scaffold suggests a possible alternate allosteric binding site compared to previously disclosed mGlu4 PAMs where more robust SAR was identified. Unfortunately, these compounds possess less than ideal PK properties (poor metabolic stability, CYP 2C9 and CYP 2D6 inhibition) preventing their further study. However, as this class of compounds represent a novel chemotype in the mGlu4 PAM area, we anticipate these compounds will inform the community for future scaffold design.
Supplementary Material
Acknowledgment
The authors thank Emily L. Days, Tasha Nalywajko, Cheryl A. Austin and Michael Baxter Williams for their critical contributions to the HTS portion of the project. In addition, we would like to thank Katrina Brewer, Ryan Morrison and Matt Mulder for technical assistance with the PK assays, and Chris Denicola, Nathan Kett, and Sichen Chang for the purification of compounds utilizing the mass-directed HPLC system. This work was supported by the National Institute of Mental Health, National Institute of Neurological Disorders and Stroke, NIH/MLPCN (5U54MH084659-02), the Michael J. Fox Foundation, the Vanderbilt Department of Pharmacology and the Vanderbilt Institute of Chemical Biology.
Footnotes
Supporting Information Available. Details of experimental procedures and spectroscopic data for synthesized compounds and biological procedures. This material is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
- 1.Obeso JA, Rodriguez-Oroz MC, Goetz CG, Marin C, Kordower JH, Rodriguez M, Hirsch EC, Farrer M, Schapira AHV, Halliday G. Missing pieces in the Parkinson's disease puzzle. Nat. Med. 2010;16:653–661. doi: 10.1038/nm.2165. [DOI] [PubMed] [Google Scholar]
- 2.Schapira AHV, Bezard E, Brotchie J, Calon F, Collingridge GL, Ferger B, Hengerer B, Hirsch E, Jenner P, Le Novére N, Obeso JA, Schwarzchild MA, Spampinato U, Davidai G. Novel pharmacological targets for the treatment of Parkinson's disease. Nature Rev. Drug Discov. 2006;5:845–854. doi: 10.1038/nrd2087. [DOI] [PubMed] [Google Scholar]
- 3.Jankovic J. Parkinson's disease therapy: treatment of early and late disease. Chin. Med. J. 2001;114:227–234. [PubMed] [Google Scholar]
- 4.Conn PJ, Christopoulos A, Lindsley CW. Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nature Rev. Drug. Dis. 2009;8:41–54. doi: 10.1038/nrd2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schoepp DD. Unveiling the functions of presynaptic metabotropic glutamate receptors in the central nervous system. J. Pharm. Expt. Ther. 2001;299:12–20. [PubMed] [Google Scholar]
- 6.Marino MJ, Williams DL, Jr, O'Brien JA, Valenti O, McDonald TP, Clements MK, Wang R, DiLella AG, Kinney GG, Conn PJ. Allosteric modulation of group III metabotropic glutamate receptor 4: a potential approach to Parkinson's disease treatment. Proc. Nat. Acad. Sci. 2003;100:13668–13673. doi: 10.1073/pnas.1835724100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maj M, Bruno V, Dragic Z, Yamamoto R, Battaglia G, Inderbitzin W, Stoehr N, Stein T, Gasparini F, Vranesic I, Kuhn R, Nicoletti F, Flor PJ. (−)-PHCCC, a positive allosteric modulator of mGluR4: characterization, mechanism of action, and neuroprotection. Neuropharmacology. 2003;45:895–906. doi: 10.1016/s0028-3908(03)00271-5. [DOI] [PubMed] [Google Scholar]
- 8.Hopkins CR, Lindsley CW, Niswender CM. mGluR4 Positive Allosteric Modulation as Potential Treatment of Parkinson's Disease. Future Med. Chem. 2009;1:501–513. doi: 10.4155/fmc.09.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lindsley CW, Niswender CM, Engers DW, Hopkins CR. Recent Progress in the Development of mGluR4 Positive Allosteric Modulators for the Treatment of Parkinson's Disease. Curr. Top. Med. Chem. 2009;9:949–963. [PubMed] [Google Scholar]
- 10.Williams R, Zhou Y, Niswender CM, Luo Q, Conn PJ, Lindsley CW, Hopkins CR. Re-exploration of the PHCCC scaffold: discovery of improved positive allosteric modulators of mGluR4. ACS Chem. Neurosci. 2010;1:411–419. doi: 10.1021/cn9000318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Niswender CM, Johnson KA, Weaver CD, Jones CK, Xiang Z, Luo Q, Rodriguez AL, Marlo JE, de Paulis T, Thompson AD, Days EL, Nalywajko T, Austin CA, Williams MB, Ayala JE, Williams R, Lindsley CW, Conn PJ. Discovery, characterization, and antiparkinsonian effect of novel positive allosteric modulators of metabotropic glutamate receptor 4. Mol. Pharmacol. 2008;74:1345–1358. doi: 10.1124/mol.108.049551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Williams R, Johnson KA, Gentry PR, Niswender CM, Weaver CD, Conn PJ, Lindsley CW, Hopkins CR. Synthesis and SAR of a novel positive allosteric modulator (PAM) of the metabotropic glutamate receptor 4 (mGluR4) Bioorg. Med. Chem. Lett. 2009;19:4967–4970. doi: 10.1016/j.bmcl.2009.07.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Niswender CM, Lebois EP, Luo Q, Kim K, Muchalski H, Yin H, Conn PJ, Lindsley CW. Positive allosteric modulators of the metabotropic glutamate receptor subtype 4 (mGluR4): Part I. Discovery of pyrazolo[3,4-d]pyrimidines as novel mGluR4 positive allosteric modulators. Bioorg. Med. Chem. Lett. 2008;18:5626–5630. doi: 10.1016/j.bmcl.2008.08.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Williams R, Niswender CM, Luo Q, Le U, Conn PJ, Lindsley CW. Positive allosteric modulators of the metabotropic glutamate receptor subtype 4 (mGluR4). Part II: Challenges in hit-to-lead. Bioorg. Med. Chem. Lett. 2009;19:962–966. doi: 10.1016/j.bmcl.2008.11.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Engers DW, Niswender CM, Weaver CD, Jadhav S, Menon UN, Zamorano R, Conn PJ, Lindsley CW, Hopkins CR. Synthesis and evaluation of a series of heterobiarylamides that are centrally penetrant metabotropic glutamate receptor 4 (mGluR4) positive allosteric modulators (PAMs) J. Med. Chem. 2009;52:4115–4118. doi: 10.1021/jm9005065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Engers DW, Gentry PR, Williams R, Bolinger JD, Weaver CD, Menon UN, Conn PJ, Lindsley CW, Niswender CM, Hopkins CR. Synthesis and SAR of novel, 4-(phenylsulfamoyl)phenylacetamide mGlu4 positive allosteric modulators (PAMs) identified by functional high-throughput screening (HTS) Bioorg. Med. Chem. Lett. 2010;20:5175–5178. doi: 10.1016/j.bmcl.2010.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leister W, Strauss K, Wisnoski D, Zhao Z, Lindsley C. Development of a custom high-throughput preparative liquid chromatography/mass spectrometer platform for the preparative purification and analytical analysis of compound libraries. J. Comb. Chem. 2003;5:322–329. doi: 10.1021/cc0201041. [DOI] [PubMed] [Google Scholar]
- 18.Chang G, Steyn SJ, Umland JP, Scott DO. Strategic Use of Plasma and Microsome Binding To Exploit in Vitro Clearance in Early Drug Discovery. ACS Med. Chem. Lett. 2010;1:50–63. doi: 10.1021/ml900012h. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.