

Abstract
We examined the cardiomyopathy-causing tropomyosin mutations E180G, D175N, and V95A to determine their effects on actomyosin regulation. V95A reduced the ATPase rate when filaments were saturated with regulatory proteins both in the presence and absence of calcium, indicating either a stabilization of the inactive state or an inability to fully populate the active state. Effects of E180G and D175N were more complex. These two mutations increased ATPase rates at sub-saturating concentrations of troponin and tropomyosin as compared to wild type tropomyosin. At higher concentrations of regulatory proteins, ATPase rates became similar to wild type. Normal activation was achieved with the tight-binding myosin analog N-ethylmaleimide-S1, at saturating regulatory protein concentrations. These results suggest that the E180G and D175N mutations reduce the affinity of tropomyosin for actin and also destabilize troponin binding to the actin thin filaments.
Keywords: Cardiomyopathy, tropomyosin mutations, cardiac regulation, V95A, E180G, D175N
INTRODUCTION
Familial hypertrophic cardiomyopathy is a serious disorder that results from mutations in any one of several cardiac contractile proteins (1–4). We are particularly interested in mutations of the calcium-sensing regulatory complex of troponin and tropomyosin that binds to actin. Mutations of these regulatory proteins can either increase or decrease thin filament-activated ATPase activity of myosin. In several previously studied cases, changes in ATPase activities were shown to result from changes in the equilibrium transitions among the active, intermediate and inactive states of regulated thin filaments (5–7). The three structural states of regulated actin are defined by the position of tropomyosin with respect to the groove along the actin helix. Unique positions of tropomyosin on actin have been observed in the absence of calcium, at saturating calcium concentrations, and when fully activated with rigor myosin binding (8, 9).
Protein kinase C phosphomimetic mutants of TnI also alter the equilibria among states (7), indicating that changes in actomyosin state distributions may be a common response to changes in regulatory protein structure. We therefore investigated regulatory changes associated with mutations in tropomyosin that lead to congenital cardiomyopathies, particularly whether troponin and tropomyosin have a common mechanism for causing hypertrophic cardiomyopathy by altering the equilibrium between actin states.
We studied three mutations on tropomyosin that cause hypertrophic cardiomyopathy: V95A, E180G and D175N. Cardiomyopathy caused by V95A is highly lethal, but produces minimal cardiac gross and microscopic pathology (10). Deletions of this portion of tropomyosin also reduce the ability of regulated thin filaments to be fully activated by calcium (11).
Patients with cardiomyopathy due to E180G mutations show a higher mortality than those with D175N (12). Transgenic mouse lines with the D175N mutation show mild myocardial disorganization and hypertrophy along with impairment of both contraction and relaxation. These morphological and functional changes are not apparent in mouse lines expressing D180G (12, 13). Both E180G and D175N mutations lead to partial unwinding of the tropomyosin coiled-coil, and alter tropomyosin binding (14, 15). The juxtaposition of similar structural effects and proximity with disparate clinical outcomes leads to the question of correlating the effects of these two mutations on underlying sarcomeric regulation to better understand the differences between them.
We examined these mutations using ATPase assays in the presence and absence of calcium, and with increasing concentrations of NEM-S1. The measured ATPase rate is dependent on the cycle time for an individual myosin S1, the fraction of myosin that is bound to actin, and the distribution of actin among the three states of regulated actin (6). Calcium has little effect on the binding of S1 to actin during ATP hydrolysis (16). Actin in the intermediate state has a higher kcat for actin-activated ATPase activity than actin in the inactive state, and actin in the active state has the highest value of kcat. The higher rate constant is likely due to a larger rate constant for product release (17, 18).
The V95A mutation reduced ATPase activities in the presence and absence of calcium. These reductions were maintained at saturating concentrations of troponin and tropomyosin, and at varying concentrations of NEM-S1. We concluded that V95A either stabilizes the inactive state of actin, or cannot fully reach the active state. E180G and D175N had different effects on ATPase rates. ATPase rates for both these mutations were increased in the absence and presence of calcium at sub-saturating concentrations of troponin and tropomyosin. The ATPase rates, however, were similar to wild type when the regulatory proteins were at saturating concentrations. The rates in the presence on NEM-S1 were also similar to wild type. These results indicate that these mutations reduce tropomyosin interactions with actin and troponin.
MATERIALS AND METHODS
Protein Preparation
Actin and myosin were isolated from rabbit back muscle (19). Tissues were obtained with the approval of the Animal Care and Use Committee of East Carolina University. S1 was produced by digesting myosin with chymotrypsin (Worthington Biochemical) (20). WT human cardiac troponin and WT and mutant human cardiac (αα) tropomyosins were expressed in E. coli and purified as previously described (21). Ca2+-sensitivity of regulated cardiac thin filament sliding does not depend on the myosin isoform. These tropomyosins have two extra amino acids Gly-Ser at the N-terminal to substitute for N-terminal acetylating.
Protein concentrations were determined by absorbance measurements at 280 nm, corrected for scattering at 340 nm, using the following extinction coefficients (ε0.1%) for 280 nm: actin (1.15), myosin-S1 (0.75), tropomyosin (0.23), and troponin (0.37).
Measuring ATPase Rates
Rates of (γ32P) ATP hydrolysis were determined in the presence and absence of calcium at 25°C, pH 7.0 by measuring the release of 32Pi. Four time points were taken over a 10–15 minute period over which the production of 32Pi was linear with time so that the measured velocities were initial velocities. The buffer generally contained 1 mM ATP, 3 mM MgCl2, 10 mM MOPS, 34 mMNaCl, 1 mM EGTA or 0.5 mM CaCl2 and 1 mM dithiothreitol. Assays normally contained 0.1μM S1 and 10 μM actin. The proportions of actin: tropomyosin: troponin were 7:3:3 unless otherwise noted.
The conditions used to evaluate ATPase activity were chosen to explore the major states of regulated actin. In the absence of calcium the ATPase rates are at their minimum value and the inactive state is predominant. The fraction of intermediate state is thought to be 0.6–0.7 at saturating calcium with the inactive and active states being populated to varying degrees. NEM-S1 binding to regulated actin stabilizes the fully active state. In the presence of optimal concentrations of NEM-S1 the active state appears to be virtually fully populated.
ATPase rates measured in the presence of NEM-S1 were recorded at variable concentrations of NEM-S1. Virtually all of the NEM-S1 added bound to actin under these conditions (22). To prevent competitive inhibition of S1 binding, the actin concentration was increased by an equal amount to the NEM-S1 added to maintain the same level of free actin (5). ATPase rates were corrected for the activity of free S1 (0.06 s−1) and of NEM-S1 (0.002 s−1). The hydrolytic activity of NEM-S1 is not significantly increased upon binding to actin (< 1.4 fold change).
Statistics
Data in figures are shown as means with error bars showing standard deviation. Statistical significance (p< 0.05) was determined using the two sample significance t-test to compare means.
RESULTS
ATPase rates were first measured in the absence of calcium and increasing concentrations of tropomyosin to insure that actin was saturated with each of the tropomyosin mutants(Figure 1A). The troponin concentration was fixed at 2.1 μM, a 1.5:7 ratio of troponin to actin. At all concentrations of tropomyosin, the rates for D175N and E180G were significantly higher than wild type. The rates for V95A were slightly higher than wild type at all concentrations of tropomyosin.
Figure 1.

ATPase rates in the absence of calcium. [S1] = 0.1 μM, [Actin] = 10 μM, temperature = 25°C, pH = 7.(A) Troponin concentration was fixed at 2.1μM. Legend shows stoichiometry of troponin and tropomyosins per 7 actin monomers. E180G and D175N show increased ATPase rates compared to wild type at all tropomyosin concentrations. V95A shows a slightly increased rate compared to WT at all concentrations of tropomyosin. (B)Tropomyosin concentration was fixed at 4.3 μM with varying troponin concentrations. Legend shows ratios of tropomyosin and troponin per 7 actin. E180G and D175N show a higher ATPase rate as compared to wild type at low troponin concentration. V95A has a lower ATPase rate than wild type at all troponin concentrations.
The concentration of troponin was varied to ensure that troponin and tropomyosin were both optimized for maximal inhibition (Figure 1B). At low concentrations of troponin, ATPase rates with either E180G or D175N mutations were higher than with wild type tropomyosin. At high troponin concentrations, both tropomyosin mutants had rates similar to wild type. Thus these mutations reduced the affinity of troponin for tropomyosin-actin but did not shift the distribution of states.
The V95A mutant exhibited a different response to increasing troponin concentrations. ATPase rates were lower than wild type at high troponin concentrations, indicating that the V95A mutant may have destabilized the active state to a greater extent than seen with wild type troponin.
ATPase rates were also measured in the presence of calcium with varying concentrations of troponin and a fixed saturating tropomyosin concentration(Figure 2). At high concentrations of troponin both the D175N and E180G mutants had rates that were similar to wild type, while the rates were higher than wild type at low troponin concentrations. These data mirror results in the absence of calcium, indicating a weakened interaction of these two tropomyosin mutants with troponin.
Figure 2.
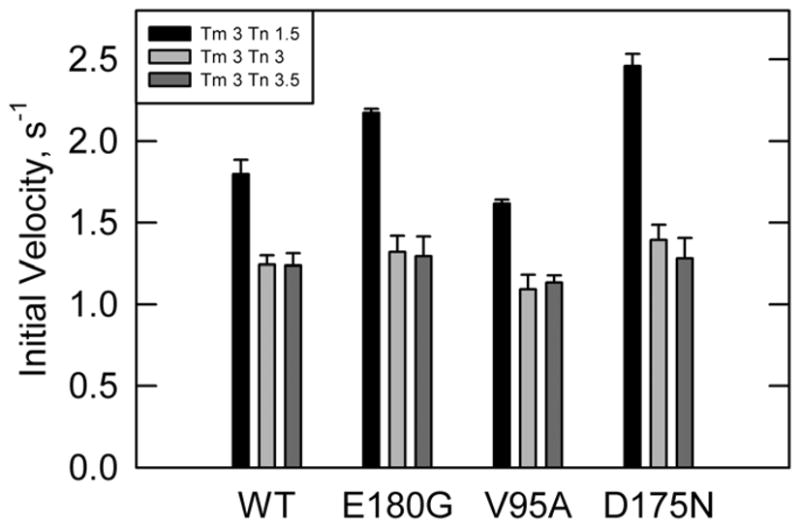
ATPase rates in the presence of calcium with a fixed tropomyosin concentration of 4.3 μM and varying concentrations of troponin in the presence of calcium. Legend shows stoichiometry of tropomyosin and troponin per 7 actins. E180G and D175N show increased rates at low troponin concentrations. V95A shows a reduced rate at all concentrations of troponin. [S1] = 0.1 μM, [Actin] = 10 μM, temperature = 25°C, pH = 7.
Actin filaments containing the V95A tropomyosin mutant had a lower S1 ATPase activity, relative to wildtype, in the presence of calcium at all troponin concentrations. Although the differences were small they were consistent with the behavior in the absence of calcium. That pattern indicated that either the inactive state was stabilized or that this mutation prevented regulated actin filaments from becoming fully activated. That distinction was made by examining the rate under conditions that maximized the population of the fully active state.
Figure 3 shows ATPase rates for wild type and all three tropomyosin mutations with increasing concentrations of NEM-S1 in the presence of calcium. NEM-S1 is a modified form of S1 that is capable of binding to and activating the thin filament in a similar fashion to unmodified rigor S1. However, NEM-S1 has a low ATPase activity which is not activated on binding to actin. As a result, increasing concentrations of NEM-S1 stabilize the fully active state of the actin-tropomyosin-troponin complex.
Figure 3.
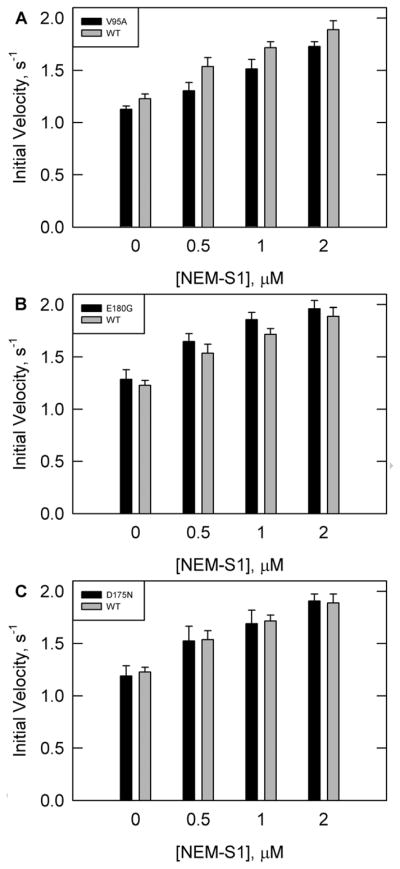
ATPase rates in the presence of calcium with increasing concentrations of NEM-S1 and saturating concentrations of troponin and tropomyosin. Conditions are similar to Figure 2 except for varying NEM-S1 and a corresponding increase in actin as detailed in Materials and Methods. Tropomyosin was either WT (solid grey bars in all panels) or mutants(solid black bars) (A) V95A, (B) E180G, or (C) D175N.
In the presence of calcium the rates with V95A were about 11% lower than wild type at all concentrations of NEM-S1 examined (Figure 3A). Therefore, it was possible that the V95A mutation prevented full activation.
The ATPase rates with the E180G mutant were 3% higher than wild type at the lowest and highest NEM-S1 concentrations examined, which is within the statistical error of measurement (Figure 3B). The rates with D175N were similar to wild type at all concentrations of NEM-S1 in the presence of calcium when the concentrations of troponin and tropomyosin were saturating(Figure 3C). There is no evidence of a significant change in ATPase activation in the fully active state for either mutant. The only effect of the D175N and E180G mutations was on the binding to the other components of the regulatory apparatus.
NEM-S1 activated ATPase rates in the low calcium condition are shown in Figure 4. Activation required higher concentrations of NEM-S1 than in the presence of calcium. At 4 μM NEM-S1 the maximum rate observed for wild type actin filaments was approximately 70% of the maximum value observed at saturating calcium. The rates for V95A were lower than wild type values at all concentrations of NEM-S1 showing that stabilization of the active state required higher NEM-S1 concentrations than in the wild type case or that the active state cannot be fully reached with the V95A mutant (Figure 4A). The rates of D175N and E180G were similar to wild type at all concentrations of NEM-S1 (Figure 4B-C). These data confirm the results of Figure 3, again indicating that there was no defect in reaching the fully active state.
Figure 4.

ATPase rates in the absence of calcium with increasing concentrations of NEM-S1 and saturating concentrations of troponin and tropomyosin. Conditions are similar to Figure 1 except for varying NEM-S1 and a corresponding increase in actin as detailed in Materials and Methods. Tropomyosin was either WT (solid grey bars in all panels) or mutants (solid black bars) (A) V95A, (B) E180G, or (C) D175N.
DISCUSSION
Small changes in the primary structure of tropomyosin and troponin affect regulation of cardiac muscle contraction. Post-translational modification may be an adaptive response whereas mutations may lead to various familial cardiac disorders. Our previous work with troponin showed that alterations in troponin changed the distribution of states of actin-tropomyosin-troponin (regulated actin) without having much effect on the stability of the regulated actin filaments, the attachment of myosin S1-ATP to actin or on transitions from one actin-S1 chemical state to another (5–7). The states of regulated actin are in rapid equilibrium with each other: Inactive = Intermediate = Active (23–26). We found that the Δ14 TnT mutation stabilized the active state of regulated actin (5), protein kinase C phosphomimetic mutations of TnI stabilized the inactive state (7) and R146G and R146W mutations of TnI stabilized the intermediate state (6).
Our approach was to look at regulatory function as a measure of changes in distribution of regulated actin states. That is, different forms of regulated actin have different abilities to stimulate S1 ATPase activity. The approximate relative activities are 0.04, 0.15 and 1 for the inactive, intermediate and active states, respectively. Estimation of changes in distributions requires that the activity of the fully active state be known. Measurement of the activity of the fully active state was done by using NEM-S1 to stabilize the fully active state. Differences in ATPase activation compared to wild type values in the absence of NEM-S1, but not at saturating NEM-S1, suggest changes in the equilibrium distribution among the inactive, intermediate and active states of regulated actin. Decreases in the maximally activated rate could occur if a mutation prevented full stabilization of the activate state. Because this type of analysis had not been previously applied to tropomyosin mutants we reexamined three tropomyosin mutants that had been studied previously.
Previous work showed that the E180G and D175N mutants of tropomyosin had destabilized coiled-coils and reduced the affinity of tropomyosin to actin filaments (14). We also observed destabilization of regulated actin filaments. At concentrations of troponin and tropomyosin that give optimal regulation for wild type actin filaments these mutants produced actin filaments with greater stimulation of ATPase activity than wild type in both the absence and presence of calcium. The requirement for higher concentrations of troponin can be rationalized by the location of these mutations within the primary binding site for troponin (tropomyosin residues 160–220). At sufficiently high concentrations of troponin and tropomyosin the rates in EGTA and calcium were similar to wild type values. This result confirms the reported destabilizing effect of these mutations on regulated actin filament formation.
We observed no significant differences in activation with NEM-S1 between actin filaments formed with wild type tropomyosin and the E180G and D175N mutants in either the presence or absence of calcium. These mutants did not prevent actin filaments from moving fully to the active state. The NEM-S1 results also ruled out large changes in processes such as S1-ATP binding to actin or rates of product release. The previously reported destabilization of the coiled coil structure appears to have no effect other than that of the stability of regulated actin filaments.
Actin filaments containing E180G and D175N mutants of tropomyosin functioned normally as long as the actin filaments were fully saturated with regulatory proteins. The observation that these mutations lead to cardiac failure suggest that the physiological concentrations of troponin and tropomyosin in vivo are insufficient to allow binding of the full complement of regulatory proteins to cardiac actin filaments. The production of tropomyosin is reduced in the case of the D175N mutation making it more likely that the actin filaments are not properly regulated. This would lead to partial activation during diastole and cardiomyopathy. These results suggest that increasing troponin and tropomyosin expression could normalize contractile function.
The V95A tropomyosin mutant exhibited a slightly reduced affinity for troponin but continued to exhibit abnormal regulation at saturating protein concentrations. Actin filaments containing V95A tropomyosin were more active than wild type when the troponin concentration was 2.1 μM. However at 4.3 μM troponin the ATPase rates were slightly depressed with the V95A mutation in both the absence and presence of calcium (Figs. 1 and 2). The difference in activity between actin filaments containing wild type and V95A tropomyosin was small (10–20%). Previous studies of V95A have shown a reduced rate in the presence of saturating calcium concentrations for V95A, along with changes in calcium sensitivity and myosin cycling (11).
It is surprising that such a small difference could lead to the high rate of mortality linked to that mutation. We explored the possibility that the V95A mutant had a defective activation by tight binding S1. In the absence of calcium the activity of actin filaments containing V95A tropomyosin was about 0.8 of the wild type value over the range of 0 to 4 μM NEM-S1. At saturating calcium the relative activity of V95A tropomyosin containing actin filaments was about 0.89 from 0 to 2 μM NEM-S1. Thus actin filaments containing the V95A mutation have a slightly depressed activation of myosin S1-ATPase activity. This depression could be due to a shift in the equilibria among the states of regulated actin to favor the inactive state. It is also possible that actin filaments containing the V95A mutation cannot totally reach the active state.
Although the troponin mutants that we have studied exhibited changes in the distribution of states this same primary defect may not explain changes in activity of the three tropomyosin mutants studied here. This could have important consequences in treatment of cardiac disorders. Treatments directed against mutations in troponin that normalize the distribution of states of regulated actin may not be effective for tropomyosin mutations.
Acknowledgments
We thank Dr. Fang Wang for providing the tropomyosin clone and mutants, and Dr. Nicolas M. Brunet, Dr. Aya K. Takeda, and Nancy M. Meyer for assistance with regulatory protein preparations.
Funding:
NIH F32HL090206 (MCM)
NIH HL63974 (PBC)
NIH R01AR044504 (JMC)
Abbreviations
- NEM-S1
N-ethylmaleimide-S1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Maron BJ, McKenna WJ, Danielson GK, Kappenberger LJ, Kuhn HJ, Seidman CE, Shah PM, Spencer WH, 3rd, Spirito P, Ten Cate FJ, Wigle ED Task Force on Clinical Expert Consensus Documents. American College of Cardiology, Committee for Practice Guidelines. European Society of Cardiology. American college of Cardiology/European society of cardiology clinical expert consensus document on hypertrophic cardiomyopathy. A report of the American college of cardiology foundation task force on clinical expert consensus documents and the European society of cardiology committee for practice guidelines. J Am Coll Cardiol. 2003;42:1687–1713. doi: 10.1016/s0735-1097(03)00941-0. [DOI] [PubMed] [Google Scholar]
- 2.Maron BJ. Hypertrophic cardiomyopathy: A systematic review. JAMA. 2002;287:1308–1320. doi: 10.1001/jama.287.10.1308. [DOI] [PubMed] [Google Scholar]
- 3.Ahmad F, Seidman JG, Seidman CE. The genetic basis for cardiac remodeling. Annu Rev Genomics Hum Genet. 2005;6:185–216. doi: 10.1146/annurev.genom.6.080604.162132. [DOI] [PubMed] [Google Scholar]
- 4.Seidman JG, Seidman C. The genetic basis for cardiomyopathy: From mutation identification to mechanistic paradigms. Cell. 2001;104:557–567. doi: 10.1016/s0092-8674(01)00242-2. [DOI] [PubMed] [Google Scholar]
- 5.Gafurov B, Fredricksen S, Cai A, Brenner B, Chase PB, Chalovich JM. The delta 14 mutation of human cardiac troponin T enhances ATPase activity and alters the cooperative binding of S1-ADP to regulated actin. Biochemistry. 2004;43:15276–15285. doi: 10.1021/bi048646h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mathur MC, Kobayashi T, Chalovich JM. Some cardiomyopathy-causing troponin I mutations stabilize a functional intermediate actin state. Biophys J. 2009;96:2237–2244. doi: 10.1016/j.bpj.2008.12.3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mathur MC, Kobayashi T, Chalovich JM. Negative charges at protein kinase C sites of troponin I stabilize the inactive state of actin. Biophys J. 2008;94:542–549. doi: 10.1529/biophysj.107.113944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Poole KJ, Lorenz M, Evans G, Rosenbaum G, Pirani A, Craig R, Tobacman LS, Lehman W, Holmes KC. A comparison of muscle thin filament models obtained from electron microscopy reconstructions and low-angle X-ray fibre diagrams from non-overlap muscle. J Struct Biol. 2006:273–284. doi: 10.1016/j.jsb.2006.02.020. [DOI] [PubMed] [Google Scholar]
- 9.Pirani A, Vinogradova MV, Curmi PM, King WA, Fletterick RJ, Craig R, Tobacman LS, Xu C, Hatch V, Lehman W. An atomic model of the thin filament in the relaxed and Ca2+-activated states. J Mol Biol. 2006;357:707–717. doi: 10.1016/j.jmb.2005.12.050. [DOI] [PubMed] [Google Scholar]
- 10.Karibe A, Tobacman LS, Strand J, Butters C, Back N, Bachinski LL, Arai AE, Ortiz A, Roberts R, Homsher E, Fananapazir L. Hypertrophic cardiomyopathy caused by a novel alpha-tropomyosin mutation (V95A) is associated with mild cardiac phenotype, abnormal calcium binding to troponin, abnormal myosin cycling, and poor prognosis. Circulation. 2001;103:65–71. doi: 10.1161/01.cir.103.1.65. [DOI] [PubMed] [Google Scholar]
- 11.Landis CA, Bobkova A, Homsher E, Tobacman LS. The active state of the thin filament is destabilized by an internal deletion in tropomyosin. J Biol Chem. 1997;272:14051–14056. doi: 10.1074/jbc.272.22.14051. [DOI] [PubMed] [Google Scholar]
- 12.Michele DE, Gomez CA, Hong KE, Westfall MV, Metzger JM. Cardiac Dysfunction in Hypertrophic Cardiomyopathy Mutant Tropomyosin Mice is Transgene-Dependent, Hypertrophy-Independent, and Improved by β-Blockade. Circulation Research. 2002;91:255–262. doi: 10.1161/01.res.0000027530.58419.82. [DOI] [PubMed] [Google Scholar]
- 13.Muthuchamy M, Pieples K, Rethinasamy P, Hoit B, Grupp IL, Boivin GP, Wolska B, Evans C, Solaro RJ, Wieczorek DF. Mouse model of a familial hypertrophic cardiomyopathy mutation in alpha-tropomyosin manifests cardiac dysfunction. Circ Res. 1999;85:47–56. doi: 10.1161/01.res.85.1.47. [DOI] [PubMed] [Google Scholar]
- 14.Kremneva E, Boussouf S, Nikolaeva O, Maytum R, Geeves MA, Levitsky DI. Effects of two familial hypertrophic cardiomyopathy mutations in alpha-tropomyosin, Asp175Asn and Glu180Gly, on the thermal unfolding of actin-bound tropomyosin. Biophys J. 2004;87:3922–3933. doi: 10.1529/biophysj.104.048793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Golitsina N, An Y, Greenfield NJ, Thierfelder L, Iizuka K, Seidman JG, Seidman CE, Lehrer SS, Hitchcock-DeGregori SE. Effects of two familial hypertrophic cardiomyopathy-causing mutations on alpha-tropomyosin structure and function. Biochemistry. 1997;36:4637–4642. doi: 10.1021/bi962970y. [DOI] [PubMed] [Google Scholar]
- 16.Chalovich JM. Actin mediated regulation of muscle contraction. Pharmacol Ther. 1992;55:95–148. doi: 10.1016/0163-7258(92)90013-p. [DOI] [PubMed] [Google Scholar]
- 17.Rosenfeld SS, Taylor EW. The mechanism of regulation of actomyosin subfragment 1 ATPase. J Biol Chem. 1987;262:9984–9993. [PubMed] [Google Scholar]
- 18.Heeley DH, Belknap B, White HD. Mechanism of regulation of phosphate dissociation from actomyosin-ADP-pi by thin filament proteins. Proc Natl Acad Sci U S A. 2002;99:16731–16736. doi: 10.1073/pnas.252236399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kielley WW, Harrington WF. A model for the myosin molecule. Biochim Biophys Acta. 1960;41:401–421. doi: 10.1016/0006-3002(60)90037-8. [DOI] [PubMed] [Google Scholar]
- 20.Weeds AG, Taylor RS. Separation of subfragment-1 isoenzymes from rabbit skeletal muscle myosin. Nature. 1975;257:54–56. doi: 10.1038/257054a0. [DOI] [PubMed] [Google Scholar]
- 21.Schoffstall B, Brunet NM, Williams S, Miller VF, Barnes AT, Wang F, Compton LA, McFadden, Taylor DW, Seavy M, Dhanarajan R, Chase PB. Ca2+ sensitivity of regulated cardiac thin filament sliding does not depend on myosin isoform. J Physiol. 2006;577:935–944. doi: 10.1113/jphysiol.2006.120105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schnekenbuhl S. PhD thesis. Universitat Ulm; Ulm, Germany: 1994. Calcium-independent activation of a membrane free muscle preparation. Is muscle contraction regulated by two different mechanisms? [Google Scholar]
- 23.Chalovich JM. Regulation of striated muscle contraction: A discussion. J Muscle Res Cell Motil. 2002;23:353–361. doi: 10.1023/a:1022066922922. [DOI] [PubMed] [Google Scholar]
- 24.McKillop DF, Geeves MA. Regulation of the interaction between actin and myosin subfragment 1: Evidence for three states of the thin filament. Biophys J. 1993;65:693–701. doi: 10.1016/S0006-3495(93)81110-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pirani A, Xu C, Hatch V, Craig R, Tobacman LS, Lehman W. Single particle analysis of relaxed and activated muscle thin filaments. J Mol Biol. 2005;346:761–772. doi: 10.1016/j.jmb.2004.12.013. [DOI] [PubMed] [Google Scholar]
- 26.Butters CA, Willadsen KA, Tobacman LS. Cooperative interactions between adjacent troponin-tropomyosin complexes may be transmitted through the actin filament. J Biol Chem. 1993;268:15565–15570. [PubMed] [Google Scholar]