

Abstract
Between 2% to 5% of all colon cancers arise in the setting of well defined inherited syndromes, including Lynch syndrome, familial adenomatous polyposis, MUTYH-associated polyposis, and certain hamartomatous polyposis conditions. Each is associated with a high risk of colon cancer. In addition to the syndromes, up to one-third of colon cancers exhibit increased familial risk, likely related to inheritance. A number of less penetrant, but possibly more frequent susceptibility genes have been identified for this level of inheritance. Clarification of predisposing genes allows for accurate risk assessment and more precise screening approaches. This review examines the colon cancer syndromes, their genetics and management, and also the common familial colon cancers with current genetic advances and screening guidelines.
Introduction
Of common malignancies, colorectal cancer (CRC) has one of the largest proportions of familial cases. Kindred and twin studies estimated that approximately 30% of all CRC cases are an inherited form of the disease [1, 2]. Approximately 5% of cases are associated with highly penetrant inherited mutations and clinical presentations that have been well characterized. The etiologies of the remaining 20%–30% of inherited CRCs are not completely understood. They are likely to be caused by alterations in single genes that are less penetrant but more common than those associated with the well-characterized syndromes. Examples include common polymorphisms in genes that regulate metabolism or genes that are regulated by environmental or other genetic factors. Inherited CRCs are also likely to be caused by alterations in multiple susceptibility loci that have additive effects. A precise understanding of the genetics of inherited CRCs is important for identifying at-risk individuals, improving cancer surveillance and prevention strategies, and developing better diagnostic and therapeutic approaches.
We review the genetics, clinical features, diagnosis, and management of the well-described colon cancer syndromes. Identification of the genes involved with these syndromes has led to genetic testing for diagnosis and improved understanding of the molecular mechanisms of colon cancer. Studies of the less-penetrant, but more common, causes of familial and inherited colon cancers have led to recommendations for colon cancer screening. Furthermore, genetic study in this group is improving our understanding of colon cancer risk, pathogenesis, and prevention.
INHERITED COLORECTAL CANCER SYNDROMES WITH ADENOMATOUS POLYPS
The syndromes of CRC are defined on the basis of clinical, pathological, and more recently, genetic findings. Conditions that express adenomatous polyps include Lynch syndrome (also called hereditary nonpolyposis colorectal cancer [HNPCC]), familial adenomatous polyposis (FAP), attenuated FAP, and MUTYH-associated polyposis (MAP). Hamartomatous polyps are the primary lesions in Peutz-Jeghers syndrome (PJS) and juvenile polyposis syndrome (JPS). Finally, hyperplastic polyposis (HPP) is an unusual condition that has a substantial cancer risk and must be distinguished from the other conditions. All of these conditions are inherited, autosomal dominant disorders, except MAP, which is autosomal recessive, and HPP, which is rarely inherited. Both attenuated FAP and MAP are associated with varying numbers of adenomas, so their phenotypes can be confused not only with each other, but also with Lynch syndrome, sporadic polyps, and other polyposis syndromes. Although clinical similarities do exist, each syndrome has distinct cancer risks, characteristic clinical features, and separate genetic etiologies. Diagnosis and management recommendations are based on these divergent features.
LYNCH SYNDROME
Individuals with Lynch syndrome are predisposed to various types of cancers, especially colon and endometrial (see Table 1) [3]. The syndrome accounts for 2%–4% of all CRCs [4]. Although affected individuals can develop colonic adenomas with greater frequency than the general population, polyposis is rare. The lifetime CRC risk is estimated to be 50%–80% [5].
Table 1.
Characteristic Features of High Risk Colorectal Cancer Conditions
| Condition: Inheritance | Gene | Lifetime Cancer Risks | Non-Malignant Features | |
|---|---|---|---|---|
|
Non-polyposis | ||||
| Lynch syndrome: Autosomal dominant | hMLH1 | Colon | 50–80% | Physical or non-malignant features, besides keratoacanthomas and sebaceous adenomas/carcinomas, are rare. |
| hMLH2 | Endometrium | 40–60% | ||
| hMSH6 | Stomach | 11–19% | ||
| hPMS2 | Ovary | 9–12% | ||
| EpCAMa | Hepatobiliary tract | 2–7% | ||
| Upper urinary tract | 4–5% | |||
| Pancreatic | 3–4% | |||
| Small bowel | 1–4% | |||
| CNS (glioblastoma) | 1–3% | |||
|
Adenomatous polyposis | ||||
| Familial adenomatous polyposis: Autosomal dominant | APC | Colon | 100% | 100s to 1,000s of colorectal adenomas |
| Duodenum/periampullary | 4–12% | Gastric fundic gland and duodenal | ||
| Stomach | <1% | adenomatous polyposis | ||
| Pancreas | 2% | CHRPE, epidermoid cysts, osteomas | ||
| Thyroid | 1–2% | Dental abnormalities | ||
| Liver (hepatoblastoma) | 1–2% | Desmoid tumors | ||
| CNS (medulloblastoma) | <1% | |||
| Attenuated FAP: Autosomal dominant | APC | Colon | 70% | < 100 colonic adenomas (range 0 to 100s) |
| Duodenum/periampullary | 4–12% | Upper GI polyposis similar to FAP | ||
| Thyroid | 1–2% | Other non-malignant features are rare in AFAP | ||
| MUTYH-associated polyposis: Autosomal recessive | MUTYH | Colon | 80% | Colonic phenotype similar to AFAP |
| Duodenum | 4% | Duodenal polyposis | ||
|
Hamartomatous polyposis | ||||
| Peutz-Jeghers syndrome: Autosomal dominant | STK11 | Breast | 54% | Mucocutaneous pigmentation |
| Colon | 39% | GI hamartomatous (Peutz-Jeghers) polyps | ||
| Pancreas | 11–36% | |||
| Stomach | 29% | |||
| Ovaryb | 21% | |||
| Lung | 15% | |||
| Small bowel | 13% | |||
| Uterine/cervixc | 9% | |||
| Testicled | <1% | |||
| Juvenile polyposis syndrome: Autosomal dominant | SMAD4 | Colon | 39% | GI hamartomatous (juvenile) polyps |
| BMPR1A | Stomach, pancreas, and small bowel | 21% | Features of HHT Congenital defects |
|
|
Hyperplastic polyposis | ||||
| Hyperplastic polyposis: Inheritance unknown | ? | Colon | ? >50% | Hyperplastic polyps, sessile serrated polyps, traditional serrated adenomas and mixed adenomas |
CNS, central nervous system; FAP, familial adenomatous polyposis; CHRPE, congenital hypertrophy of the retinal pigment epithelium; GI, gastrointestinal; HHT, hereditary hemorrhagic telangiectasia
Risks associated with EpCAM mutations are not yet known;
Sex cord tumors with annular tubules;
Adenoma malignum;
Sertoli cell tumors
Colon cancers and polyps arise in Lynch syndrome at a younger age of onset and a more proximal location compared to sporadic neoplams. Histologically, cancers are often poorly differentiated, mucinous, and -have large numbers of tumor-infiltrating lymphocytes. They are also characterized by a high level of microsatellite instability (MSI-H), a feature of cancers that arise in the setting of defective DNA mismatch repair (MMR) genes. Of interest is that colon cancers with MSI-H overall have a better prognosis compared to those without MSI [6].
Extra-Colonic Features
Endometrial cancer is the most common extra-colonic malignancy associated with Lynch syndrome. The lifetime risk is 40–60%, which is comparable to or even greater than the estimated risk for CRC in women with Lynch syndrome. Lynch syndrome is responsible for approximately 2% of all endometrial cancers [7]. Other cancers associated with Lynch syndrome are listed in Table 1 and include gastric, ovarian, biliary, urinary tract, small bowel, brain and pancreatic [8, 9]. A number of these cancer risk estimates are based on studies predominately consisting of highly penetrant Lynch syndrome families. More recent studies suggest that the risk for various cancers is likely lower than previously reported.
Genetics
Lynch syndrome is the result of a germline mutation in a class of genes involved in DNA MMR, including hMSH2, hMLH1, hMSH6, and hPMS2. The MMR system is necessary for maintaining genomic stability by correcting single-base mismatches and insertion-deletion loops that form during DNA replication. Mutations in hMSH2 and hMLH1 account for the up to 90% of Lynch syndrome cases; mutations in hMSH6 account for approximately 10% and mutations in hPMS2 are detected on rare occasions [8, 10]. Differences in cancer risks have been reported among the MMR genes, including MSH6 where colon cancer risk may be slightly lower and endometrial cancer risk possibly higher compared to hMSH2 and hMLH1 carriers [11]. The most striking difference is for hPMS2 mutation carriers which have recently been shown to have a 15%–20% risk for CRC, 15% for endometrial cancer, and 25%–32% for any Lynch syndrome-associated cancer to age 70 years [12]. These risk estimates are substantially lower when compared to other MMR genes.
Recently, germline deletions in the EpCAM (epithelial cell adhesion molecule) gene, also known as TACSTD1, were found in a subset of families with Lynch syndrome [13, 14]. These families displayed a Lynch syndrome phenotype with early onset and/or multiple cancers, in addition to hMSH2 deficient colorectal tumors, although no MMR gene mutations were identified. Of interest was that promoter hypermethylation of hMSH2 in tumors was detected, which is an uncommon finding in CRCs. These Lynch syndrome families were subsequently found to have germline deletions in the 3′ region of EpCAM, which resulted in EpCAM-hMSH2 fusion transcripts [13]. One estimate suggests that these deletions account for 6.3% of Lynch syndrome cases [15].
Diagnosis
Several tools are available to assist clinical diagnosis of Lynch syndrome, including analyses of family histories, tumor testing, mutation prediction models, and genetic testing.
It is important to obtain a detailed personal and family. Specific factors indicate patients have a high-risk CRC condition (see Table 2) and should be referred for genetic counseling. The Amsterdam criteria I (see Table 3) were originally developed for research purposes to identify families likely to have Lynch syndrome. More than 50% of families with Lynch syndrome, however, fail to meet these criteria. To increase sensitivity, the Amsterdam criteria II and the Bethesda guidelines were developed (see Table 3).
Table 2.
Indicators for Evaluation of High-Risk Colorectal Cancer Conditionsa
|
CRC, colorectal cancer; MSI-H, microsatellite instability high; LS, Lynch syndrome; GI, gastrointestinal
See also NCCN Guidelines for evaluation algorithms of inherited syndromes (www.nccn.org)
Tumor-infiltrating lymphocytes, Crohn’s-like lymphocytic reaction, mucinous or signet ring cell differentiation, or a medullary growth pattern
Endometrial, ovarian, gastric, small bowel, brain, hepatobiliary tract, upper uroepithelial, sebaceous gland, and pancreas cancer
serrated polyps include hyperplastic polyps, sessile serrated polyps (also known as sessile serrated adenomas), traditional serrated adenomas and mixed adenomas
Table 3.
Amsterdam Criteria and Revised Bethesda Guidelines
| Amsterdam Criteria I |
At least three relatives with CRC; all of the following must be met:
|
| Amsterdam Criteria II |
At least three relatives with colorectal, endometrial, small bowel, ureter, or renal pelvis cancer; all of the following must be met:
|
| Revised Bethesda Guidelines |
Requires at least one of the following:
|
CRC, Colorectal cancer; LS, Lynch syndrome; MSI-H, Microsatellite instability high
Endometrial, stomach, ovarian, pancreas, ureter, and renal pelvis, biliary tract, and brain tumors, sebaceous gland adenomas and keratoacanthomas, and carcinoma of the small bowel
Tumor infiltrating lymphocytes, Crohn’s-like lymphocytic reaction, mucinous/signet-ring differentiation, or medullary growth pattern
The Amsterdam criteria and revised Bethesda guidelines are used in clinical practice to identify individuals at risk for Lynch syndrome who require further evaluation. Commercial tests are available to analyze the distal portion of EpCAM and the 4 clinically relevant MMR genes. One approach to identify patients with Lynch syndrome is to perform genetic testing when individuals meet the personal and/or family history criteria mentioned above. Since germline mutations in hMLH1 and hMSH2 account for the majority of cases, genetic testing typically starts with analysis of these genes. Limitations of this strategy include high costs and reduced sensitivity compared with other diagnostic approaches.
A second approach for identifying Lynch syndrome, which has shown to be cost-effective, is to perform tumor testing when any of the Bethesda guidelines are identified. Various tumor testing strategies exist, most of which begin with MSI and/or immunohistochemistry (IHC) analysis of colorectal tumors. Approximately 90% of Lynch syndrome-associated CRCs will have MSI-H making this analysis is very sensitive. The specificity is much lower; however, as approximately 15% of sporadic CRCs are also MSI-H. Sporadic MSI-H CRCs are the result of somatic hypermethylation of the hMLH1 promoter region, as opposed to Lynch syndrome tumors which are the result of a germline gene mutation.
Tumor testing with IHC utilizes four antibodies specific for hMLH1, hMSH2, hMSH6 and hPMS2 proteins to evaluate tumors for MMR deficiency. The sensitivity of IHC is comparable to that of MSI analysis. However, IHC analysis can direct genetic testing to the appropriate MMR gene when loss of MMR protein expression is identified. Additional tumor testing, including BRAF mutation and hMLH1 promoter methylation analyses can be helpful in differentiating sporadic versus Lynch syndrome-associated CRCs. Tumor testing in endometrial cancers has proven to be equally as effective at identifying Lynch syndrome [7]. Other Lynch syndrome-associated tumors also frequently display MSI-H and loss of MMR protein expression, although their sensitivity and specificity in the clinical setting is not well established.
A number of models have recently been developed to help facilitate the diagnosis of Lynch syndrome, including but not limited to PREMM(1,2), MMRpro, and MMRpredict [16]. These models utilize personal and family history to estimate the probability that an individual carries a MMR gene mutation. Although, these models have been validated in CRC cases, they also performed well in one study using endometrial cancer cases [16].
A fourth approach for identifying Lynch syndrome has recently been incorporated at a number of centers. In this approach all colorectal and/or endometrial cancers are screened for MMR deficiency, regardless of age of onset or family history with MSI and/or IHC analysis. This approach has revealed that the revised Bethesda guidelines still miss approximately 28% of Lynch syndrome cases [4]. This reduced sensitivity for the revised Bethesda guidelines may in part be explained by the lower risk of CRC seen in hMSH6 and hPMS2 mutation carriers.
Management
It is important to identify individuals with Lynch syndrome because of the early onset and high penetrance of cancer and the demonstrated effectiveness of screening for this condition (Table 4). Surveillance decreases both the incidence of CRC and related deaths. After a median follow up of five years in 178 individuals with Lynch syndrome, those undergoing regular screening colonoscopy had a lower CRC incidence (11%) and CRC mortality (2%) compared to those who declined surveillance (27% and 12%, respectively) [17]. Colonoscopies at three year intervals have shown to decrease the risk of CRC by greater than 50% and prevent CRC deaths [18]. The reduction in CRC risk and death is expected to be even greater when screening intervals are reduced to every one-to- two years. Finally, long term aspirin use has been shown to decrease CRC incidence in Lynch syndrome and may be recommended in the near future [19].
Table 4.
Management Recommendations for High Risk Conditions
| Condition | Cancer | Recommendations |
|---|---|---|
| Lynch syndrome | Colon | • Colonoscopy, every 1–2 years, beginning at age 20–25 years |
| Endometrium/Ovary | • Consider prophylactic TAH/BSO after childbearing complete • Referral to specialist for consideration of gynecologic cancer screening |
|
| Upper urinary tract | • Consider annual urinalysis, beginning at age 30–35 years | |
| Upper GI tract | • Consider EGDa every 1–2 years, beginning at age 30–35 years | |
| Other | • Annual physical exam that includes a review for symptoms suggestive of related cancers and a skin examination to look for sebaceous carcinoma • There are no data to support routine screening for other related cancers, but approaches may be considered after counseling about the risks and benefits of available approaches |
|
| Familial adenomatous polyposis | Colon | • Colonoscopy every 1–2 yearsb, beginning at age 10–12 years • Screening can be delayed until the late teenage years in families with attenuated FAP • Prophylactic colectomy when polyps become unmanageable • If remaining rectum or ileal pouch, screen every 6 months to two yearsb |
| Upper GI tract | • EGDa every 1–3 yearsb starting at age 20–25 years | |
| Other | • Annual physical examination that includes an exam of the thyroid • There are no data to support routine screening for other related cancers, but approaches may be considered after counseling about the risks and benefits of available approaches |
|
| MUTYH-associated polyposis | Colon | • Colonoscopy every 2–3 yearsb beginning at age 25 years • Prophylactic colectomy when polyps become unmanageable |
| Duodenum | • EGDa every 1–3 yearsb starting at age 20–25 years | |
| Peutz-Jeghers syndrome | Colon | • Colonoscopy every 2–3 yearsb, beginning with symptoms or in late teens if no symptoms occur. |
| Breast | • Annual mammogram and breast MRI beginning by age 25 • Biannual clinical breast exam beginning by age 25 |
|
| Pancreas | • MRCP and/or endoscopic ultrasound of the pancreas every 1–2 years beginning by age 30 | |
| Stomach/Small bowel | • EGDa and abdominal CT with oral contrast every 2–3 yearsb, beginning at age 10 years | |
| Cervix/Uterus/Ovary | • Annual pelvic examination, Pap smear and transvaginal ultrasound beginning at age 18 years | |
| Testes | • Annual testicular exam beginning at age 10 and observation for feminizing changes | |
| Juvenile polyposis syndrome | Colon | • Colonoscopy every 2–3 yearsa, beginning with symptoms or in late teens if no symptoms occur. |
| Stomach | • EGDa every 1–3 yearsb | |
| Hyperplastic polyposis | Colon | • Colonoscopy every 1–2 yearsb in affected individuals • Prophylactic colectomy when polyps become unmanageable |
TAH/BSO, total abdominal hysterectomy and bilateral salpingo-oophorectomy; GI, gastrointestinal; FAP, familial adenomatous polyposis; EGD, esophagogastroduodenoscopy; MRCP, magnetic resonance cholangiopancreatography
Include side-viewing examination;
Frequency depends on polyp burden
Screening colonoscopy in affected individuals should be initiated by 20–25 years of age and repeated every 1–2 years. Subtotal colectomy with ileorectal anastomosis is advised with the appearance of colon cancer. Annual rectal surveillance is indicated thereafter. Segmental resection can be considered if annual colonoscopy can be assured thereafter.
Prophylactic hysterectomy and bilateral salpingo-oophorectomy reduce the incidence of endometrial and ovarian cancer in patients with Lynch syndrome and should be discussed as an option with women after completion of childbearing [20]. Screening for these malignancies is also used but is less effective [21, 22]. Annual screening starting at age 30 followed by prophylactic surgery at age 40 years was shown to have the highest net benefit measured in quality-adjusted life years [21]. However, the incremental cost-effectiveness ratio was approximately $195,000 per quality-adjusted life year compared with the next best strategy which was prophylactic surgery without screening at age 40 years. These findings highlight the need for improved screening measures during childbearing years. Although there are few evidence-based data regarding the efficacy of screening for other Lynch syndrome-associated tumors, including gastric, the National Comprehensive Cancer Network (NCCN) suggests several options to be considered in view of clinical need [23] (see Table 4).
FAMILIAL ADENOMATOUS POLYPOSIS: CLASSIC AND ATTENUATED
Familial adenomatous polyposis (FAP) is the second-most common inherited CRC syndrome with a prevalence of 1 in 10,000 individuals. Characteristic features of FAP include the development of hundreds to thousands of colonic adenomas, beginning in early adolescence, and inevitable CRC in untreated individuals. The average age of CRC diagnosis if untreated is 39 years; 7% develop CRC by age 21 and 95% by age 50. Attenuated FAP is a less-severe form of the disease, characterized by an average 69% lifetime risk of CRC, an average of approximately 30 colonic adenomatous polyps (range 0 to 100s), the tendency to develop proximal colonic neoplasms, and a later age of polyp and CRC development [24]. FAP, attenuated FAP, and Gardner syndrome (FAP with epidermoid cysts, osteomas, dental anomalies, and/or desmoid tumors) all result from germline mutations in APC (Table 1).
Extra-Colonic Features
Upper gastrointestinal (GI) tract polyposis occurs frequently with FAP or attenuated FAP. Gastric fundic gland polyps are found in approximately 50% of affected individuals and can be profuse [25]. Adenomatous polyps in the stomach also develop, but are much less common than fundic gland polyps and are usually in the antrum. Dysplastic changes in fundic gland polyps are common, but are of little concern unless the dysplasia is severe. The lifetime risk of gastric cancer is about 1% [26]. Adenomatous polyps of the duodenum are observed in more than 50% of individuals and are commonly found in the second and third portions. Duodenal cancer is the second most common malignancy in FAP or attenuated FAP, with a lifetime risk of approximately 4%–12% [27]. Additional cancers (see Table 1) also arise in FAP. Benign growths also occur including osteomas (most commonly found on the skull and mandible), congenital hypertrophy of the retinal pigment epithelium (CHRPE), epidermoid cysts, fibromas, dental abnormalities (un-erupted teeth or congenital absence of teeth), and desmoids (found in approximately 10% of FAP patients) [28]. These findings are less common in attenuated FAP. These benign lesions are usually of little concern, except for desmoid tumors, which can result in significant morbidity and even mortality, usually by compression of intra abdominal structures.
Diagnosis
A diagnosis of FAP is made when at least 100 colonic adenomas are identified, although individuals at younger ages with fewer polyps might also have FAP. The presence of extra-colonic lesions can also contribute to the initial diagnosis. The identification of APC mutations in a proband confirms the diagnosis, allowing precise identification of other relatives who are at risk.
Attenuated FAP is suspected when 10 or more, but fewer than 100 adenomas, are found in a person over 40 or 50 years of age [24, 29] (see Table 2). A precise diagnosis is often difficult in a single patient; polyp numbers vary with this disorder; attenuated FAP can mimic typical FAP, MAP, or even sporadic polyp development. Examinations of multiple family members can often determine the phenotype. Genetic testing is useful in that specific APC mutations are associated with attenuated FAP. Attenuated FAP and MAP, respectively, account for 10% to 20% of persons with 10 to 100 adenomatous polyps who do not have early classic FAP. It is common not to find a genetic etiology in this group.
Genetics
FAP and attenuated FAP are caused by germline mutations in APC, which encodes a tumor suppressor that is part of the WNT signaling pathway. New or de novo APC mutations are responsible for approximately 25% of FAP cases. In addition, approximately 20% of individuals with an apparent de novo APC mutation have somatic mosaicism (when two or more cell lines in the same individual differ genetically) [30].
The location of the mutation within APC has been associated with the severity of colonic polyposis, the degree of cancer risk, and the presence and/or frequency of non-malignant findings, including desmoids and CHRPE (genotype–phenotype correlations) [31]. Studies have failed to show a correlation between genotype and upper GI tumor development. Debate exists regarding the utility of basing management strategies on genotype-phenotype correlations.
Management
Individuals who are at risk for, or have a genetic diagnosis of, FAP should be examined by colonoscopy every 1–2 years, beginning at age 10–12 years (see Table 4). Once adenomatous polyps emerge, and annual follow-up colonoscopy is recommended until a decision is made to perform a colectomy.
Colectomy should be considered when more than 20 adenomas develop, when adenomas > 1 cm are found, or when advanced histology appears. Restorative procto-colectomy (also called total procto-colectomy) with ileal pouch anal anastamosis (IPAA) is recommended when large numbers of adenomas are found in the rectum. A 1–2 cm cuff of rectal mucosa is left just above the anus for air-liquid-solid discrimination. If adenomas are already present in that location, then mucosal stripping is done as part of the procedure. Some surgeons prefer to do mucosal stripping with all restorative procto-colectomy and IPAA procedures [32]. However, it is also common to preserve the rectum with ileo-rectal anastamosis if few or no rectal adenomas are present. Annual or more frequent endoscopic follow-up must be assured if any rectal tissue remains. If numerous adenomas appear, sulindac (and possibly celecoxib) can be used to effectively regress polyps, making surveillance easier. There are few data on the use of NSAIDs to delay surgery or as a primary treatment for FAP. Such use should be limited to investigative trials. Up to 33% of patients with a preserved rectum need completion proctectomy later, because of diffuse polyposis.
Patients with attenuated FAP should always be screened by colonoscopy because of the frequency of proximal colonic polyps. The screening should be done every 1–2 year, beginning in the late teenage years. Approximately 33% of patients with attenuated FAP can be managed over the long term with colonoscopy and polypectomy because of small polyp numbers [24]. About 66% will eventually require colectomy, although colectomy with ileo-rectal anastamosis is virtually always done to spare the rectum. Annual post-operative surveillance is required for polyp ablation, but subsequent proctectomy is rarely needed.
Upper GI endoscopy, including side-viewing endoscopy, should be performed every 1–3 years for patients with FAP or attenuated FAP, followed by endoscopic ultrasound for suspicious lesions at the ampulla [33]. This should begin around an age of 25 to 30 years [34]. Polyp biopsy and/or removal should be done, depending on size and appearance. The Spigelman staging criteria for duodenal adenomatosis can assist in determining the appropriate interval for follow-up. Over time, 20% or more of patients will require therapeutic endoscopic or surgery to treat duodenal adenomas and adenomas at the duodenal papilla. Gastric fundic gland polyps should be sampled, especially if they are large or erythematous. Gastrectomy is rarely needed, and usually only if severe dysplasia appears. Annual physical examination and possibly thyroid ultrasound are also routinely offered to individuals affected with attenuated FAP or FAP, given the increased risk for thyroid cancer [35] (See Tables 1 and 4).
MUTYH-ASSOCIATED POLYPOSIS
MAP is characterized by the presence of adenomatous polyposis of the colorectum and an increased risk of CRC. MAP is caused by biallelic mutations in MUTYH (also referred to as MYH) (Table 1). The colonic phenotype of MAP mimics attenuated FAP, including a propensity for proximal colonic neoplasms [36]. Colonic polyposis typically occurs by the time patients reach their 40s although polyps and cancer can occur at earlier ages. Biallelic MUTYH mutations have also been found in individuals with early onset CRC and few-to-no polyps [37].
Adenomatous polyps predominate in MAP, however unlike attenuated FAP, hyperplastic polyps are common. In a small series of 17 MAP patients, 8 (47%) displayed hyperplastic polyps and sessile serrated polyps [38]. Additionally, G:C to T:A transversions in KRAS were identified in 51 of 73 (70%) hyperplastic or sessile serrated polyps in individuals with MAP, compared with only 7 of 41 (17%) in their sporadic counterparts [38]. This evidence indicates an association between MAP and hyperplastic or sessile serrated polyps. Clearly, our understanding of the MAP phenotype and its pathway(s) to CRC is evolving.
Additional features
A European study showed that gastric and duodenal polyps occurred in approximately 11% and 17% of patients, respectively, with an estimated lifetime risk of duodenal cancer of 4% [39]. FAP-associated extra-colonic features such as osteomas, desmoids, CHRPE and thyroid cancer did not occur but an excess of ovarian, bladder, skin, sebaceous gland tumors and possibly breast cancer was observed [39].
Genetics and diagnosis
The MUTYH gene product is part of the base-excision repair pathway, which is involved in defending against oxidative DNA damage. Functionally, MUTYH helps prevent G:C to T:A transversions caused by oxidative stress to highly mutagenic DNA bases.
Clinical diagnostic criteria for MAP have not yet been fully established, but at present, the colonic phenotype may be considered similar to attenuated FAP. Genetic testing for MAP is warranted in individuals with greater than 10 colorectal adenomas but without an identifiable mutation in APC. Genetic testing strategies target the specific ethnic allelic frequencies of MUTYH variants. A genetic diagnosis of MAP confirms the diagnosis and allows genetic testing in family members. Siblings of individuals with biallelic MUTYH mutations have a 25% chance of having MAP. Therefore, unlike attenuated and classical FAP, parents and children of individuals with MAP are rarely affected but should be counseled about the risks.
Management
Patients with MAP often develop proximally located CRCs, so at-risk individuals should receive colonoscopic surveillance [36] (see Table 4). Surveillance is typically initiated when patients are in their mid-20s or mid-30s. Subtotal colectomy is advised for patients who develop colon cancer and should also be considered when colonoscopic management becomes problematic or when polyps become large or exhibit high-grade dysplasia. Screening for upper-GI tract tumors is similar to that for FAP because the risk for duodenal cancer is comparable.
HAMARTOMATOUS POLYPOSIS CONDITIONS
Peutz-Jeghers syndrome (PJS) and juvenile polyposis syndrome (JPS) are hamartomatous polyposis conditions that are both associated with an increased risk for colorectal and other malignancies [40] (Table 1). There are several other hamartomatous polyposis conditions that are very rare and confer little, if any, colon cancer risk. Cowden syndrome is one of these and it arises from mutations of PTEN [41].
The characteristic gastrointestinal lesions in PJS are small bowel, histologically distinctive hamartomatous polyps (96% of PJS patients) [42]. Gastric and colonic Peutz-Jeghers polyps are found in approximately 25% and 30% of cases respectively. GI symptoms first occur on average in the early teenage years and include small bowel obstruction, intussusceptions, and GI bleeding. The most consistent extra-colonic feature of PJS is the characteristic mucocutaneous pigmentation, typically presenting in childhood on the lips, buccal mucosa, and periorbital area.
A clinical diagnosis of PJS can be made when an individual has 2 or more of the following features: 2 or more Peutz-Jeghers polyps of the small intestine; typical mucocutaneous hyperpigmentation; and a family history of PJS.
Individuals with PJS have an estimated 81% to 93% lifetime risk of cancer, including an almost 70% risk of GI cancer [43]. The lifetime risk of breast cancer approaches 50% and of pancreatic cancer is 11%–36%. Additional cancers are also common (see Table 1).
Unlike PJS, JPS does not typically have obvious physical findings that facilitate diagnosis. The key feature of JPS is multiple juvenile polyps, most prominently in the colon but also in the stomach, duodenum, and small bowel. The lifetime risk of CRC in individuals with JPS is estimated to be 39% [44]. Gastric cancer risk approaches 21% in those with multiple gastric polyps. Small bowel and pancreatic cancers may also develop.
A diagnosis of JPS is considered for anyone who has at least 3 juvenile polyps of the colon, multiple juvenile polyps throughout the GI tract, or any number of juvenile polyps and a family history of the condition.
Congenital defects occur in approximately 15% of JPS cases. A subset of patients with JPS also has hereditary hemorrhagic telangiectasias (HHT). Such often have mucocutaneous telangiectasias, GI arteriovenous malformations and pulmonary arterio-venous malformations.
Genetics
Germline mutations in STK11 (also called LKB1) are the only known cause of PJS, whereas JPS is caused by mutations in either SMAD4 or BMPR1A. Disease-associated mutations are identified in as many as 70% of patients with PJS [45], but in only 40% those with clinically defined JPS.
Certain features seen in JPS, such gastric polyposis and HHT, are more common with SMAD4 mutations than with BMPR1A and might guide genetic testing strategies. Individuals with a clinical diagnosis of JPS but without an identifiable mutation in SMAD4 or BMPR1A could have another hamartomatous polyposis condition or an unidentified mutation. Patients with Cowden syndrome, for example, can present with multiple juvenile colonic polyps and therefore be misdiagnosed as having JPS. Practice guidelines for the management of JPS and PJS have recently been reviewed by NCCN [23] and can be found at www.nccn.org (see Table 4). Patients should be referred to a specialized team, because of the rarity of these conditions, the complexities of screening diagnosis and management, and the limited data on the effectiveness of various screening modalities.
HYPERPLASTIC POLYPOSIS
Hyperplastic polyposis (HPP) is a rare condition characterized by multiple and/or large hyperplastic polyps of the colon. The etiology of HPP is unknown. The World Health Organization’s criteria for HPP include must 30 cumulative hyperplastic polyps of any size distributed throughout the colon (the number 20 is now often used [46]); 5 or more hyperplastic polyps proximal to the sigmoid colon with at least 2 being greater than 10 mm in diameter; or at least 1 hyperplastic colonic polyp in an individual with a first-degree relative with HPP. Sessile serrated polyps (also called sessile serrated adenomas) have also been added to the polyp histologic type [46]. Little is known about the etiology, natural history and incidence of HPP.
In the patients without cancer, HPP is generally asymptomatic and often identified during screening colonoscopy. The characteristic diminutive polyps infrequently bleed and sessile lesions may be difficult to detect via standard white-light colonoscopy.
Cancer Risk and Pathogenesis
HPP exhibits an increased risk of CRC [46], which occurs on average in the 50s or 60s, although very early age of onset has been reported. Metachronous and synchronous cancers are frequently observed [47] and lesions have a tendency to form in the proximal colon [48].
The literature on HPP consists mainly of case reports and small case series, so ascertainment bias must be considered when these studies are used to estimate CRC risk. Jass [49] reviewed all of the cases of HPP reported until 2006 and determined that 88 of 207 of these (43%) had CRC. However, the frequency of CRC was 69% in small case series and 37% in large case series, probably a consequence of ascertainment bias in the first group [49]. The heterogeneity of HPP could also affect calculations of CRC incidence among people with HPP. Considerable individual variations have been reported in the type, size, location, and number of polyps and the frequency of CRCs. One recent study showed that the number of hyperplastic polyps and serrated adenomas was significantly associated with CRC incidence [46].
It is now widely accepted that a serrated neoplasia pathway exists in addition to the traditional adenoma–carcinoma sequence of CRCs. [50]. Accumulated somatic changes within sessile serrated polyps, such as activating BRAF mutations and widespread methylation of CpG islands (referred to as the CpG-island methylator phenotype), with or without MSI, are important events in this pathway to carcinoma development [51]. Additional research is needed in order to determine if defects in the regulation of promoter methylation, lifestyle behaviors such as smoking, or other factors are involved in pathogenesis.
Familial cases of HPP have been reported, although these are rare [47]. The reported frequency of patients with HPP that have a family history of CRC (not a family history of hyperplastic polyposis) varies from 0 to 63% [48]. The evidence supporting the inheritance of HPP is weak, although both recessive and dominant transmission patterns have been proposed [52]. Curiously, individuals with biallelic MUTYH mutations have, on occasion, been shown to meet criteria for HPP [38].
Management
Although precise surveillance strategies have not been established, regular colonoscopy every 1–2 years appears appropriate in view of the cancer risk and propensity for proximal colonic lesions (see Table 4). Chromoendoscopy and narrow-band imaging could possibly improve hyperplastic polyp detection rates [53]. Tumors can arise from polyps that are smaller than 5 mm, so removal of only large polyps could be inadequate [38]. Evidence indicates that sessile serrated polyps should be treated as adenomas, in terms of cancer risk. When polyps larger than 5mm cannot be cleared, or if advanced dysplasia is found, subtotal colectomy should be considered [54]. It is difficult to know whether relatives of patients with HPP are at increased risk for colon cancer. Colonoscopic screening, starting at age 40 and repeated every 5 years, would be a reasonable recommendation for relatives until more information is known about familial risk.
COMMON FAMILIAL COLORECTAL CANCERS
Inherited forms of CRC account for as much as 30% of all CRC cases [2]. Several different categories of less-penetrant, but potentially more common, causes of susceptibility have become apparent, based on family history and population studies (Figure 1). These include high-risk familial, non-syndromic colon cancers and common familial-risk colon cancers which are defined by family history. Population studies have also identified genetic factors associated with increased risk for CRC that include low-penetrant susceptibility loci (identified in genome-wide association studies) and specific polymorphisms; no surveillance guidelines are presently given for these CRC risk factors. Interestingly, the low-penetrance susceptibility loci identified by genome-wide association studies have additive effects on CRC risk, whereas CRC risk associated with the common polymorphisms is affected by gene–gene and gene–environmental interactions. Gaining a better understanding of these factors will improve both the understanding of colon cancer genetic pathogenesis and the development of genetic-based colon cancer screening guidelines.
Figure 1.
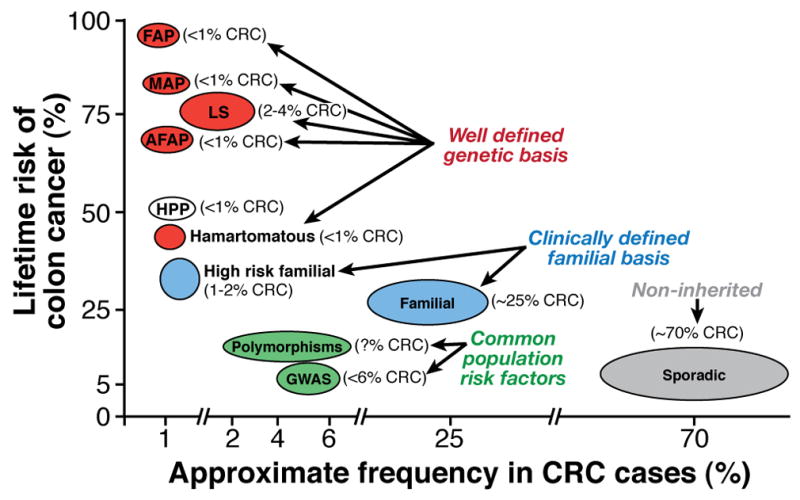
High-Risk Familial, Non-Syndromic Colon Cancer
Population-based studies have led to estimates that 0.8% to 2.3% of all CRC cases meet the Amsterdam I or II criteria for Lynch syndrome [55]. A subset of these “Amsterdam positive” cases, estimated at 40% to 70%, do not have MMR deficiency and therefore have been termed ‘familial colorectal cancer type X’ [55–57]. Study of these kindreds has revealed that CRC risk is lower than in Lynch syndrome and that CRC diagnosis averages 10 years later. Additionally, tumors do not exhibit MSI, and there is no increased incidence of extra-colonic malignancies. Surveillance recommendations are based on family history and are described below. The yet-to-be identified susceptibility gene or genes for this “type X” are likely to be uncommon but sufficiently penetrant to give rise to the observed autosomal dominant segregation patterns. Identification of the relevant genes will allow development of genetic testing and more precise surveillance strategies.
Common Familial Risk Colon Cancer
Individuals who have a first-degree relative with CRC diagnosed over age 50 years have a 2–3-fold increased risk for this malignancy. Population-based studies have demonstrated that approximately 20% of all CRC cases occur in a higher risk setting; CRC under age 50 or a first-degree relative pair with CRC [55]. Furthermore, having one first-degree relative with CRC under age 45 years, or having two first-degree relatives affected with CRC confers a 3–6-fold CRC risk compared to the general population [58]. Sibling pair and parent/child pair studies have identified chromosomal regions that could contain genes that confer this level of risk; these include 7q31, 9q22.33, 3q21-24, and 11q23, with some minor loci that have been identified in more than one study [59–62]. CRAC1 (also known as hereditary mixed polyposis syndrome, HMPS) has a similar risk level and has been associated with colon cancer by linkage analysis in Ashkenazi Jewish families [63] and in sibling pair studies [64]. These reports support the paradigm that common familial CRCs arise from a number of different, lower-penetrance susceptibility genes than those associated with the well-defined but rare inherited syndromes. These intermediate penetrant genes likely constitute only a small portion of the colon cancer population. Otherwise they would be detected by genome-wide association studies, where commonly occurring but very low penetrant susceptibility loci are detected. [65]; Indeed the genetic loci identified through family linkage and affected relative pair studies do not usually overlap with the susceptibility loci identified in genome-wide studies---see below.
Screening and Surveillance
As no specific genetic markers are presently available for common familial CRCs, screening and surveillance are based on family history. In view of the family risks given above, screening recommendations based on family history are as follows: 1) patients with a single first-degree relative over the age of 60 years with colon cancer should receive standard, average-risk colon cancer screening, but starting at age 40 years; 2) patients who have 1 relative with CRC under 60 years or 2 first-degree relatives with CRC should be screened every 5 years by colonoscopy, starting at age 40 years, or at an age 10 years younger than the earliest case in the family; and 3) patients with only second- or third-degree relatives with CRC should receive average-risk screening [66]. It is noted that these strategies are empirical, differ somewhat between health policy organizations and may need to be adapted based on the severity of the individual’s family history.
These screening guidelines will evolve as more genes are identified that predispose to CRC. Until then, family history is the best indicator of risk levels and appropriate screening strategies.
Low-Penetrance Loci
A recent model constructed from Scottish, genome-wide association study data estimated that as many as 170 common but separate genetic variations could confer susceptibility to CRC [67]. Other large genome-wide association and gene-specific association studies have associated only 10 specific loci with increased risk for CRC. These are estimated to account for only 6% of CRC cases [67]. These loci were identified based on single nucleotide polymorphism (SNP) markers. The odds ratio for each individual SNP is very low, ranging from 1.10 to 1.26, indicating a minimal increase in risk for each. However, these low-risk loci have an additive effect. Co-inheritance of SNPs on 8q24, 11q23 and 18q21, for example, results in an overall odds ratio or risk of 2.6 [68]. Several of the 10 CRC-associated loci, including 8q24, 9p24 and 18q21, have also been identified in multiple analyses [69].
One of the major challenges to genome-wide association studies, however, is that the variants identified at susceptibility loci are not ‘mutations’ that would be predicted to change function or expression of the gene product. They might affect gene expression through non-coding changes or lead to linkage disequilibrium with other genes that affect CRC risk [67, 70]. Another challenge is that genome-wide association studies do not often detect moderate- or high-penetrant alleles that are uncommon in the population [65]. These are best detected in family or sib-pair and parent-child pair studies.
Genetic Variants or Polymorphisms
Certain genetic variants or polymorphisms in a number of genes have been associated with increased colon cancer risk [71]. The genes with CRC-associated variants include APC-I1307K; HRAS1-VNTR; MTHFR; those encoding the N-acetyl transferases 1 and 2 (NAT1, NAT2); and those encoding the glutathione-S transferases Mu (GSTM1), Theta (GSTT1), and Pi (GSTP1) [72, 73]. The risks conferred by variants of these genes are modest, but their high frequency in the population could affect overall CRC incidence.
Polymorphisms that confer CRC risk are frequently reported; for example, some but not all diabetes-associated variants of transcription factor 7-like 2 (TCF7L2), which encodes a component of the WNT signaling pathway, are also associated with increased risk for colon cancer [74, 75]. Alleles of IGF-1 and IGFBP-3 were shown to modify risk in association with a number of lifestyle factors [76]. The type I TGF-β receptor gene variant TGFBR1*6A, is a reported risk factor for CRC and was proposed to account for as much as 3% of all CRC cases [77]. These findings are consistent with results from studies showing that Apc(Min/+);Tgfbr1(+/−) mice (haploinsufficient for Apc and Tgfbr1) develop 2-fold more intestinal tumors than the parent strain, Apc(Min/+) [78]. Furthermore, variants of SMAD7 (at 18q21) have been shown to affect CRC risk [79]; the rs12953717 variant appears to modulate SMAD7 expression, which could affect TGF-β signaling.
These and other genetic variants also modify the expression of known colon cancer susceptibility genes, so gene–gene interactions are important determinants of risk. In many cases for these variants, gene–environment interactions increase risk [71]. Polymorphisms or variants that influence colon cancer susceptibility are summarized in Table 5. Multiple polymorphisms or variants influence colon cancer susceptibility, but may differ in their affect depending on the genetic, environmental or clinical context. These overlapping and interacting affects are illustrated in Figure 2 and will also be described next in several subcategories.
Table 5.
Examples of genetic variants that modify colorectal cancer and/or polyp riska
| Gene | Symbol | Locus | Ref | Associations and effects on colon cancer and polyp risk |
|---|---|---|---|---|
| adenomatous polyposis coli | APC I1307K | 5q21 | 84, 87 | Moderate risk for adenomas |
| APC D1822V | Increases colon adenoma risk in smokers | |||
| APC T1493T | Modifies presentation of FAP | |||
| colorectal associated cancer 1 | CRAC1/HMPS | 15q13.3 | 63, 64 | Associated with some cases of hyperplastic polyposis and there are probable environmental modifier(s) |
| cyclin D1 | CCND1 P241P | 11q13.3 | 91 | Causes younger age on onset in Lynch syndrome; but decreased risk in sporadic cases with exposure to estrogen |
| cytochrome P450 family 24 subfamily A | CYP24A1 | 20q13.2 | 92 | Associated with increased site-specific CRC risk, with possible ethnicity influence. |
| cytochrome P450 family 24 subfamily B | CYP24B1 | 20q13.2 | 92 | Alters proximal CRC risk when associated with UV-spectrum sun exposure |
| glutathione S-transferase mu 1 | GSTM1 | 1p13.3 | 83 | Modifies presentation of Lynch syndrome, Ethnic and smoking factors modulate population risk in null allele carriers. |
| glutathione S-transferase theta 1 | GSTT1 | 22q11.23 | 83 | Modifies presentation of Lynch syndrome, Ethnic and smoking factors modulate population risk in null allele carriers. |
| glutathione S-transferase Pi | Gstp1 (mouse) | 73 | Modifies adenoma number and age-of-onset in Min mice | |
| hemochromatosis protein | HFE | 6p22.2 | 82 | Modifies presentation of Lynch syndrome |
| insulin-like growth factor 1A | IGF-1 | 12q23.2 | 81 | Modifies presentation of Lynch syndrome, possibly interacting with diabetes |
| insulin-like growth factor binding protein-3 | IGFBP-3 | 7p12.3 | 76 | Modifies colon cancer risk interacting with diabetes |
| N-acetyltransferase 1 | NAT1 | 8p22 | 84 | Modifies presentation of FAP and affects sporadic cancer risk in association with certain certain environmental factors |
| N-acetyltransferase 2 | NAT2 | 8p22 | 84 | Modifies presentation of FAP and affects sporadic cancer risk in association with certain certain environmental factors |
| ornithine decarboxylase 1 | ODC | 2p16.3 | 97 | Modifies adenoma risk interacting with NSAID use |
| selenoprotein P | SEPP1 | 5p12 | 95, 96 | Modifies adenoma risk interacting with certain dietary factors and smoking |
| thioredoxin reductase 1 | TXNRD1 | 12q23.3 | 95 | Modifies adenoma risk interacting with certain dietary factors and smoking |
| transcription factor 7-like 2 isoform 2 | TCF7L2 | 10q25 | 74, 75 | Modifies CRC risk, possibly in association with diabetes |
| transforming growth factor, beta receptor I | TGFBR1 | 9q22.33 | 77, 78 | Modifies CRC risk in high risk families, and in Min mice. |
| tumor protein 53 | TP53 | 17p13.1 | 90 | Causes Li Fraumeni syndrome; certain alleles also modify sporadic site-specific colon cancer risk in a gender-dependent manner |
The data in this table and paper refer to recent research studies, which require further study to justify clinical application.
Figure 2.

The figure illustrates how many genetic variants play both overlapping and different roles in different clinical and genetic settings. There are also multiple gene-gene and gene environmental interactions in these categories.
Variants and Well-Defined Inherited Colon Cancer Syndromes
Independent genetic factors have been described that modify the severity and penetrance of the high-risk CRC genes, such as those associated with Lynch syndrome and FAP. In a recent study, 2 non-coding SNPs that affect risk for CRC also modified risk among individuals with mutations that cause Lynch syndrome, with some differences between sexes [80]. A variant of insulin-like growth factor-I (IGF1) appears to be responsible for a more severe phenotype of Lynch syndrome [81]. Other reports showed that increased severity of Lynch syndrome was associated with variants of HFE (which also causes hereditary hemochromatosis) and genes that encode glutathione-S-transferases in [82] [83]. Different variants of NAT1 and NAT2 were associated up to a 2-fold difference in polyp number among patients with FAP, whereas the APC-T1493T variant increased polyp formation to a lesser extent [84]. Therefore, many polymorphisms by themselves confer either a small increase in risk or no risk at all unless they occur in combination with gene mutations known to cause one of the high-risk syndromes.
Variants of Known CRC Susceptibility Genes
Several studies have examined the possible role of variants or polymorphisms within APC that increase risk of CRC but do not cause the severe syndrome of FAP. Ashkenazi Jewish carriers of the APC-I1307K variant have been estimated to have a 2-fold increase in CRC risk, compared with the general population, as well as an increased risk for adenomas [85]. Molecular studies have shown that this variant makes APC particularly susceptible to additional mutation, providing a mechanism for the increased cancer risk in carriers [86]. Although controversial, the APC-D1822V variant seems to protect against CRC among post-menopause females that receive hormone supplements [87].
Variants in mismatch repair genes have recently been reported to alter colon cancer risk but not cause Lynch syndrome, including variants of hMSH6 and hMLH1 that increase or decrease rectal and colon cancer risk in Czech populations [88, 89]. Monoallelic MUTYH mutations have also been shown to increase risk for CRC, although the significance is modest [37].
Interactions with Environment Factors
The differential effects of some gene variants on CRC risk depend on environmental and other genetic factors. The R72P variant of TP53 has been associated with increased risk for a number of tumors, including adenomas and colon cancer, but only when the analysis was stratified by gender, location of lesion, and hormonal effects [90]. Furthermore, the synonymous P241P variant of Cyclin D1 (CCND1 c.870A>G) has been reported to increase the protective effects of post-menopause estrogen use on the development of colorectal neoplasia [91]. Different alleles of CYP24A1 and CYP24B1 were reported to affect colon cancer risk in association with intake of vitamin D and calcium, exposure to ultraviolet rays, gender, and estrogen replacement therapy [92, 93]. These results are consistent with molecular studies that identified a vitamin D response element within the CYP24A1 promoter that reduces protein binding and gene expression [94].
Specific SNPs within SEPP1 (selenoprotein P) and TXNRD1 (thioredoxin reductase 1) have recently been reported to modify the risk for adenomas [95]. Environmental interactions have also been reported; dietary selenium might reduce the risk of advanced colorectal adenomas, especially among recent smokers [96]. Furthermore, SNPs within ODC (ornithine decarboxylase) whose product regulates polyamine expression, have been reported to affect colon cancer risk [97]. The interaction observed between ODC variants, non-steroidal anti-inflammatory drug (NSAID) therapy, and colorectal adenoma formation and regression reveals the multi-factorial nature of colon cancer risk. Opportunities for therapeutic interventions that capitalize on these interactions, such as with combinations of drugs, are under investigation [98].
As progress has been made in elucidating the contribution of individual alleles to CRC and polyp phenotypes, it has become increasingly clear that many genes and alleles contribute differently in different contexts. Gene-gene and gene-environmental interactions are common in CRC susceptibility and may also differ depending on the underlying clinical, environmental or genetic setting. Figure 2 uses a Venn diagram approach to highlight these converging connections and to capture the multiple and overlapping contributions individual genes make to CRC risk and polyp phenotypes.
Modeling the Additive Risk of Multiple Variants
The CRC risk conferred by some SNPs might be recessive (only affect risk in individuals that are homozygous for the SNP) or are in linkage disequilibrium with other SNPs. However searches have not identified these types of SNPs [99]. Although data for co-inheritance of common and the rare variants at each locus were analyzed, it is possible that functional recessive variants are too rare to be detected in genome-wide association studies. A directed approach to examine the effects of coding SNPs on CRC risk revealed the greatest effects for rare SNPs with expected deleterious effects, suggesting that natural selection has removed most moderately and highly penetrant SNPs from the population [100].
These results indicate that a combination of variants could mediate the approximate 30% of CRC cases that appear to be familial; each variant alone is not highly penetrant or inherited in a Mendelian manner—their individual effects are ‘subclinical’. These types of variants could include polymorphisms in genes described above or even in genes that are associated with cancer syndromes, such as those in APC. However, when specific variants are co-inherited with 1 or more of these variants, cancer risk increases. Such SNP–SNP interactions have been proposed and modeled for cases where each SNP alone contributes some level of risk [101]. For example, if 3 polymorphisms that confer sub-clinical levels of risk occur with frequencies of 0.9, 0.8, and 0.35, and these are only associated with a phenotypic effect when all 3 are co-inherited, then the frequency of all 3 occurring together in the population is (0.9 * 0.8 * 0.35) ≅ 0.25; 25% of the population would be carriers of all 3 alleles and have increased susceptibility to CRC.
Conclusion
Genetics analyses of CRC risk need to be included mainstream clinical practice. A detailed analysis of family history is a fundamental component of this process; it is not only important for identifying patients that are at high risk for colorectal cancer (Table 2) and should receive genetic counseling, but also essential for identifying individuals with moderate risk that should receive more aggressive screening. Genetic features associated with moderate risk of colon cancer susceptibility will expand risk estimates and screening strategies beyond information collected from family histories. Genetic testing is available for most hereditary CRC syndromes and can be used to confirm suspected diagnosis, to clarify risks of extra-colonic cancers in affected individuals, and to identify relatives who are also at risk. Although CRC risk is increased in all of the syndromes reviewed, most have significant risk of developing extra-colonic cancer; these must be taken into account in determining medical management strategies. Diagnostic genomic applications are also evolving and will be increasingly integrated into patient care. The astute clinician will therefore need to stay abreast of these developments.
Acknowledgments
Grant support:
This research was supported by NCI grants P01-CA073992 (RWB), R01-CA040641 (RWB) and by the Huntsman Cancer Foundation.
Footnotes
Author contributions:
KWJ: study concept and design; acquisition of data; analysis and interpretation of data; drafting of the manuscript; critical revision of the manuscript for important intellectual content.
TMT: study concept and design; acquisition of data; analysis and interpretation of data; drafting of the manuscript; critical revision of the manuscript for important intellectual content.
DNW: study concept and design; acquisition of data; analysis and interpretation of data; drafting of the manuscript; critical revision of the manuscript for important intellectual content.
RWB: study concept and design; acquisition of data; analysis and interpretation of data; drafting of the manuscript; critical revision of the manuscript for important intellectual content; study supervision.
References
- 1.Lichtenstein P, et al. Environmental and heritable factors in the causation of cancer. New England Journal of Medicine. 2000;343:78–85. doi: 10.1056/NEJM200007133430201. [DOI] [PubMed] [Google Scholar]
- 2.Grady WM. Genetic testing for high-risk colon cancer patients. Gastroenterology. 2003;124(6):1574–94. doi: 10.1016/s0016-5085(03)00376-7. [DOI] [PubMed] [Google Scholar]
- 3.Lynch HT, de la Chapelle A. Hereditary colorectal cancer. N Engl J Med. 2003;348(10):919–32. doi: 10.1056/NEJMra012242. [DOI] [PubMed] [Google Scholar]
- 4.Hampel H, et al. Feasibility of screening for Lynch syndrome among patients with colorectal cancer. J Clin Oncol. 2008;26(35):5783–8. doi: 10.1200/JCO.2008.17.5950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stoffel E, et al. Calculation of risk of colorectal and endometrial cancer among patients with Lynch syndrome. Gastroenterology. 2009;137(5):1621–7. doi: 10.1053/j.gastro.2009.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ribic CM, et al. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N Engl J Med. 2003;349(3):247–57. doi: 10.1056/NEJMoa022289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hampel H, et al. Screening for Lynch syndrome (hereditary nonpolyposis colorectal cancer) among endometrial cancer patients. Cancer Res. 2006;66(15):7810–7. doi: 10.1158/0008-5472.CAN-06-1114. [DOI] [PubMed] [Google Scholar]
- 8.Rustgi AK. The genetics of hereditary colon cancer. Genes Dev. 2007;21(20):2525–38. doi: 10.1101/gad.1593107. [DOI] [PubMed] [Google Scholar]
- 9.Kastrinos F, et al. Risk of pancreatic cancer in families with Lynch syndrome. JAMA. 2009;302(16):1790–5. doi: 10.1001/jama.2009.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peltomaki P, Vasen H. Mutations associated with HNPCC predisposition --Update of ICG-HNPCC/INSiGHT mutation database. Dis Markers. 2004;20(4–5):269–76. doi: 10.1155/2004/305058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Plaschke J, et al. Lower Incidence of Colorectal Cancer and Later Age of Disease Onset in 27 Families With Pathogenic MSH6 Germline Mutations Compared With Families With MLH1 or MSH2 Mutations: The German Hereditary Nonpolyposis Colorectal Cancer Consortium. J Clin Oncol. 2004;22(22):4486–4494. doi: 10.1200/JCO.2004.02.033. [DOI] [PubMed] [Google Scholar]
- 12.Senter L, et al. The clinical phenotype of Lynch syndrome due to germ-line PMS2 mutations. Gastroenterology. 2008;135(2):419–28. doi: 10.1053/j.gastro.2008.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kovacs ME, et al. Deletions removing the last exon of TACSTD1 constitute a distinct class of mutations predisposing to Lynch syndrome. Hum Mutat. 2009;30(2):197–203. doi: 10.1002/humu.20942. [DOI] [PubMed] [Google Scholar]
- 14.Ligtenberg MJ, et al. Heritable somatic methylation and inactivation of MSH2 in families with Lynch syndrome due to deletion of the 3′ exons of TACSTD1. Nat Genet. 2009;41(1):112–7. doi: 10.1038/ng.283. [DOI] [PubMed] [Google Scholar]
- 15.Niessen RC, et al. Germline hypermethylation of MLH1 and EPCAM deletions are a frequent cause of Lynch syndrome. Genes Chromosomes Cancer. 2009;48(8):737–44. doi: 10.1002/gcc.20678. [DOI] [PubMed] [Google Scholar]
- 16.Backes FJ, et al. Prospective evaluation of DNA mismatch repair protein expression in primary endometrial cancer. Gynecol Oncol. 2009;114(3):486–90. doi: 10.1016/j.ygyno.2009.05.026. [DOI] [PubMed] [Google Scholar]
- 17.Stupart DA, et al. Surveillance colonoscopy improves survival in a cohort of subjects with a single mismatch repair gene mutation. Colorectal Dis. 2009;11(2):126–30. doi: 10.1111/j.1463-1318.2008.01702.x. [DOI] [PubMed] [Google Scholar]
- 18.Järvinen HJ, et al. Controlled 15-year trial on screening for colorectal cancer in families with hereditary nonpolyposis colorectal cancer. Gastroenterology. 2000;118:829–834. doi: 10.1016/s0016-5085(00)70168-5. [DOI] [PubMed] [Google Scholar]
- 19.Burn J, et al. Aspirin prevents cancer in Lynch syndrome. European Journal of Cancer Supplements. 2009;7(2):320–321. [Google Scholar]
- 20.Schmeler KM, et al. Prophylactic surgery to reduce the risk of gynecologic cancers in the Lynch syndrome. N Engl J Med. 2006;354(3):261–9. doi: 10.1056/NEJMoa052627. [DOI] [PubMed] [Google Scholar]
- 21.Kwon JS, et al. Cost-effectiveness analysis of prevention strategies for gynecologic cancers in Lynch syndrome. Cancer. 2008;113(2):326–35. doi: 10.1002/cncr.23554. [DOI] [PubMed] [Google Scholar]
- 22.Lynch HT, et al. Hereditary ovarian carcinoma: heterogeneity, molecular genetics, pathology, and management. Mol Oncol. 2009;3(2):97–137. doi: 10.1016/j.molonc.2009.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.NCCN. National Comprehensive Cancer Network (NCCN) Clinical Practice Guidelines in Oncology: Colorectal Cancer Screening. V.1.2010. 2009 doi: 10.6004/jnccn.2010.0003. http://www.nccn.org/ [DOI] [PubMed]
- 24.Burt RW, et al. Genetic testing and phenotype in a large kindred with attenuated familial adenomatous polyposis. Gastroenterology. 2004;127(2):444–51. doi: 10.1053/j.gastro.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 25.Burt RW. Gastric fundic gland polyps. Gastroenterology. 2003;125(5):1462–9. doi: 10.1016/j.gastro.2003.07.017. [DOI] [PubMed] [Google Scholar]
- 26.Bianchi LK, et al. Fundic gland polyp dysplasia is common in familial adenomatous polyposis. Clin Gastroenterol Hepatol. 2008;6(2):180–5. doi: 10.1016/j.cgh.2007.11.018. [DOI] [PubMed] [Google Scholar]
- 27.Bulow S, et al. Duodenal adenomatosis in familial adenomatous polyposis. Gut. 2004;53(3):381–6. doi: 10.1136/gut.2003.027771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Speake D, et al. Desmoid tumours in patients with familial adenomatous polyposis and desmoid region adenomatous polyposis coli mutations. Br J Surg. 2007;94(8):1009–13. doi: 10.1002/bjs.5633. [DOI] [PubMed] [Google Scholar]
- 29.Knudsen AL, Bisgaard ML, Bulow S. Attenuated familial adenomatous polyposis (AFAP). A review of the literature. Fam Cancer. 2003;2(1):43–55. doi: 10.1023/a:1023286520725. [DOI] [PubMed] [Google Scholar]
- 30.Hes FJ, et al. Somatic APC Mosaicism: An Underestimated Cause Of Polyposis Coli. Gut. 2008;57:71–76. doi: 10.1136/gut.2006.117796. [DOI] [PubMed] [Google Scholar]
- 31.Nieuwenhuis MH, Vasen HF. Correlations between mutation site in APC and phenotype of familial adenomatous polyposis (FAP): a review of the literature. Crit Rev Oncol Hematol. 2007;61(2):153–61. doi: 10.1016/j.critrevonc.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 32.Guillem JG, et al. ASCO/SSO review of current role of risk-reducing surgery in common hereditary cancer syndromes. J Clin Oncol. 2006;24(28):4642–60. doi: 10.1200/JCO.2005.04.5260. [DOI] [PubMed] [Google Scholar]
- 33.Rattner DW, et al. Defining the criteria for local resection of ampullary neoplasms. Arch Surg. 1996;131(4):366–371. doi: 10.1001/archsurg.1996.01430160024003. [DOI] [PubMed] [Google Scholar]
- 34.Gallagher MC, Phillips RK, Bulow S. Surveillance and management of upper gastrointestinal disease in Familial Adenomatous Polyposis. Fam Cancer. 2006;5(3):263–273. doi: 10.1007/s10689-005-5668-0. [DOI] [PubMed] [Google Scholar]
- 35.Herraiz M, et al. Prevalence of thyroid cancer in familial adenomatous polyposis syndrome and the role of screening ultrasound examinations. Clin Gastroenterol Hepatol. 2007;5(3):367–73. doi: 10.1016/j.cgh.2006.10.019. [DOI] [PubMed] [Google Scholar]
- 36.Lubbe SJ, et al. Clinical implications of the colorectal cancer risk associated with MUTYH mutation. J Clin Oncol. 2009;27(24):3975–80. doi: 10.1200/JCO.2008.21.6853. [DOI] [PubMed] [Google Scholar]
- 37.Cleary SP, et al. Germline MutY human homologue mutations and colorectal cancer: a multisite case-control study. Gastroenterology. 2009;136(4):1251–60. doi: 10.1053/j.gastro.2008.12.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boparai KS, et al. Hyperplastic polyps and sessile serrated adenomas as a phenotypic expression of MYH-associated polyposis. Gastroenterology. 2008;135(6):2014–8. doi: 10.1053/j.gastro.2008.09.020. [DOI] [PubMed] [Google Scholar]
- 39.Vogt S, et al. Expanded extracolonic tumor spectrum in MUTYH-associated polyposis. Gastroenterology. 2009 doi: 10.1053/j.gastro.2009.08.052. In Press, Accepted Manuscript. [DOI] [PubMed] [Google Scholar]
- 40.Schreibman IR, et al. The hamartomatous polyposis syndromes: a clinical and molecular review. Am J Gastroenterol. 2005;100(2):476–90. doi: 10.1111/j.1572-0241.2005.40237.x. [DOI] [PubMed] [Google Scholar]
- 41.Pilarski R. Cowden Syndrome: A Critical Review of the Clinical Literature. J Genet Couns. 2008 doi: 10.1007/s10897-008-9187-7. [DOI] [PubMed] [Google Scholar]
- 42.McGarrity TJ, Amos C. Peutz-Jeghers syndrome: clinicopathology and molecular alterations. Cell Mol Life Sci. 2006;63(18):2135–44. doi: 10.1007/s00018-006-6080-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gammon A, et al. Hamartomatous polyposis syndromes. Best Pract Res Clin Gastroenterol. 2009;23(2):219–31. doi: 10.1016/j.bpg.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brosens LA, et al. Risk of colorectal cancer in juvenile polyposis. Gut. 2007;56(7):965–7. doi: 10.1136/gut.2006.116913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Volikos E, et al. LKB1 exonic and whole gene deletions are a common cause of Peutz-Jeghers syndrome. J Med Genet. 2006;43(5):e18. doi: 10.1136/jmg.2005.039875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Boparai KS, et al. Increased colorectal cancer risk during follow-up in patients with hyperplastic polyposis syndrome: a multicentre cohort study. Gut. 2009 doi: 10.1136/gut.2009.185884. [DOI] [PubMed] [Google Scholar]
- 47.Yeoman A, et al. Hyperplastic polyposis in the New Zealand population: a condition associated with increased colorectal cancer risk and European ancestry. N Z Med J. 2007;120(1266):U2827. [PubMed] [Google Scholar]
- 48.Chow E, et al. Hyperplastic polyposis syndrome: phenotypic presentations and the role of MBD4 and MYH. Gastroenterology. 2006;131(1):30–9. doi: 10.1053/j.gastro.2006.03.046. [DOI] [PubMed] [Google Scholar]
- 49.Jass JR. Gastrointestinal polyposes: clinical, pathological and molecular features. Gastroenterol Clin North Am. 2007;36(4):927–46. viii. doi: 10.1016/j.gtc.2007.08.009. [DOI] [PubMed] [Google Scholar]
- 50.Snover DC, et al. Serrated polyps of the large intestine: a morphologic and molecular review of an evolving concept. Am J Clin Pathol. 2005;124(3):380–91. doi: 10.1309/V2EP-TPLJ-RB3F-GHJL. [DOI] [PubMed] [Google Scholar]
- 51.Minoo P, et al. Extensive DNA methylation in normal colorectal mucosa in hyperplastic polyposis. Gut. 2006 doi: 10.1136/gut.2005.082859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Young J, Jass JR. The case for a genetic predisposition to serrated neoplasia in the colorectum: hypothesis and review of the literature. Cancer Epidemiol Biomarkers Prev. 2006;15(10):1778–84. doi: 10.1158/1055-9965.EPI-06-0164. [DOI] [PubMed] [Google Scholar]
- 53.Adler A, et al. Narrow-band versus white-light high definition television endoscopic imaging for screening colonoscopy: a prospective randomized trial. Gastroenterology. 2009;136(2):410–6. e1. doi: 10.1053/j.gastro.2008.10.022. quiz 715. [DOI] [PubMed] [Google Scholar]
- 54.Doran D, et al. Prophylactic colectomy for hyperplastic polyposis. Ir J Med Sci. 2009 doi: 10.1007/s11845-009-0422-5. [DOI] [PubMed] [Google Scholar]
- 55.Kerber R, et al. Frequency of familial colon cancer and hereditary nonpolyposis colorectal cancer in a large population database. Familial Cancer. 2005;4(3):239–244. doi: 10.1007/s10689-005-0657-x. [DOI] [PubMed] [Google Scholar]
- 56.Lindor NM, et al. Lower cancer incidence in Amsterdam-I criteria families without mismatch repair deficiency: familial colorectal cancer type X. Jama. 2005;293(16):1979–85. doi: 10.1001/jama.293.16.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mueller-Koch Y, et al. Hereditary non-polyposis colorectal cancer: clinical and molecular evidence for a new entity of hereditary colorectal cancer. Gut. 2005;54(12):1733–1740. doi: 10.1136/gut.2004.060905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Johns LE, Houlston RS. A systematic review and meta-analysis of familial colorectal cancer risk. Am J Gastroenterol. 2001;96(10):2992–3003. doi: 10.1111/j.1572-0241.2001.04677.x. [DOI] [PubMed] [Google Scholar]
- 59.Neklason DW, et al. Common familial colorectal cancer linked to chromosome 7q31: a genome-wide analysis. Cancer Res. 2008;68(21):8993–7. doi: 10.1158/0008-5472.CAN-08-1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wiesner GL, et al. A subset of familial colorectal neoplasia kindreds linked to chromosome 9q22.2-31.2. Proc Natl Acad Sci U S A. 2003;100(22):12961–12965. doi: 10.1073/pnas.2132286100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kemp Z, et al. Evidence for a colorectal cancer susceptibility locus on chromosome 3q21-q24 from a high-density SNP genome-wide linkage scan. Hum Mol Genet. 2006;15(19):2903–10. doi: 10.1093/hmg/ddl231. [DOI] [PubMed] [Google Scholar]
- 62.Djureinovic T, et al. A genome wide linkage analysis in Swedish families with hereditary non-familial adenomatous polyposis/non-hereditary non-polyposis colorectal cancer. Gut. 2006;55(3):362–6. doi: 10.1136/gut.2005.075333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jaeger E, et al. Common genetic variants at the CRAC1 (HMPS) locus on chromosome 15q13.3 influence colorectal cancer risk. Nat Genet. 2008;40(1):26–8. doi: 10.1038/ng.2007.41. [DOI] [PubMed] [Google Scholar]
- 64.Daley D, et al. Identification of susceptibility genes for cancer in a genome-wide scan: results from the colon neoplasia sibling study. Am J Hum Genet. 2008;82(3):723–36. doi: 10.1016/j.ajhg.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dermitzakis ET, Clark AG. Genetics. Life after GWA studies. Science. 2009;326(5950):239–40. doi: 10.1126/science.1182009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Winawer S, et al. Colorectal cancer screening and surveillance: Clinical guidelines and rationale-Update based on new evidence. Gastroenterology. 2003;124(2):544–60. doi: 10.1053/gast.2003.50044. [DOI] [PubMed] [Google Scholar]
- 67.Tenesa A, Dunlop MG. New insights into the aetiology of colorectal cancer from genome-wide association studies. Nat Rev Genet. 2009 doi: 10.1038/nrg2574. [DOI] [PubMed] [Google Scholar]
- 68.Tenesa A, et al. Genome-wide association scan identifies a colorectal cancer susceptibility locus on 11q23 and replicates risk loci at 8q24 and 18q21. Nat Genet. 2008 doi: 10.1038/ng.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Curtin K, et al. Meta association of colorectal cancer confirms risk alleles at 8q24 and 18q21. Cancer Epidemiol Biomarkers Prev. 2009;18(2):616–21. doi: 10.1158/1055-9965.EPI-08-0690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hardy J, Singleton A. Genomewide association studies and human disease. N Engl J Med. 2009;360(17):1759–68. doi: 10.1056/NEJMra0808700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Houlston RS, I, Tomlinson P. Polymorphisms and colorectal tumor risk. Gastroenterology. 2001;121(2):282–301. doi: 10.1053/gast.2001.26265. [DOI] [PubMed] [Google Scholar]
- 72.Huang K, et al. GSTM1 and GSTT1 polymorphisms, cigarette smoking, and risk of colon cancer: a population-based case-control study in North Carolina (United States) Cancer Causes Control. 2006;17(4):385–94. doi: 10.1007/s10552-005-0424-1. [DOI] [PubMed] [Google Scholar]
- 73.Ritchie KJ, et al. Markedly enhanced colon tumorigenesis in ApcMin mice lacking glutathione S-transferase Pi. Proc Natl Acad Sci U S A. 2009 doi: 10.1073/pnas.0911351106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Folsom AR, et al. Variation in TCF7L2 and increased risk of colon cancer: the Atherosclerosis Risk in Communities (ARIC) Study. Diabetes Care. 2008;31(5):905–9. doi: 10.2337/dc07-2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hazra A, et al. Association of the TCF7L2 polymorphism with colorectal cancer and adenoma risk. Cancer Causes Control. 2008;19(9):975–80. doi: 10.1007/s10552-008-9164-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Morimoto LM, et al. Insulin-like growth factor polymorphisms and colorectal cancer risk. Cancer Epidemiol Biomarkers Prev. 2005;14(5):1204–11. doi: 10.1158/1055-9965.EPI-04-0695. [DOI] [PubMed] [Google Scholar]
- 77.Xu Y, Pasche B. TGF-beta signaling alterations and susceptibility to colorectal cancer. Hum Mol Genet. 2007;16(1):R14–20. doi: 10.1093/hmg/ddl486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zeng Q, et al. Tgfbr1 haploinsufficiency is a potent modifier of colorectal cancer development. Cancer Res. 2009;69(2):678–86. doi: 10.1158/0008-5472.CAN-08-3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Broderick P, et al. A genome-wide association study shows that common alleles of SMAD7 influence colorectal cancer risk. Nat Genet. 2007;39(11):1315–1317. doi: 10.1038/ng.2007.18. [DOI] [PubMed] [Google Scholar]
- 80.Wijnen JT, et al. Chromosome 8q23.3 and 11q23.1 variants modify colorectal cancer risk in Lynch syndrome. Gastroenterology. 2009;136(1):131–7. doi: 10.1053/j.gastro.2008.09.033. [DOI] [PubMed] [Google Scholar]
- 81.Reeves SG, et al. IGF1 is a modifier of disease risk in hereditary non-polyposis colorectal cancer. Int J Cancer. 2008;123(6):1339–43. doi: 10.1002/ijc.23668. [DOI] [PubMed] [Google Scholar]
- 82.Shi Z, et al. Haemochromatosis HFE gene polymorphisms as potential modifiers of hereditary nonpolyposis colorectal cancer risk and onset age. Int J Cancer. 2009;125(1):78–83. doi: 10.1002/ijc.24304. [DOI] [PubMed] [Google Scholar]
- 83.Felix R, et al. GSTM1 and GSTT1 polymorphisms as modifiers of age at diagnosis of hereditary nonpolyposis colorectal cancer (HNPCC) in a homogeneous cohort of individuals carrying a single predisposing mutation. Mutat Res. 2006;602(1–2):175–81. doi: 10.1016/j.mrfmmm.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 84.Crabtree MD, et al. Analysis of candidate modifier loci for the severity of colonic familial adenomatous polyposis, with evidence for the importance of the N-acetyl transferases. Gut. 2004;53(2):271–6. doi: 10.1136/gut.2003.015586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rennert G, et al. Colorectal polyps in carriers of the APC I1307K polymorphism. Dis Colon Rectum. 2005;48(12):2317–21. doi: 10.1007/s10350-005-0167-9. [DOI] [PubMed] [Google Scholar]
- 86.Zauber NP, et al. The characterization of somatic APC mutations in colonic adenomas and carcinomas in Ashkenazi Jews with the APC I1307K variant using linkage disequilibrium. J Pathol. 2003;199(2):146–51. doi: 10.1002/path.1226. [DOI] [PubMed] [Google Scholar]
- 87.Tranah GJ, et al. APC Asp1822Val and Gly2502Ser polymorphisms and risk of colorectal cancer and adenoma. Cancer Epidemiol Biomarkers Prev. 2005;14(4):863–70. doi: 10.1158/1055-9965.EPI-04-0687. [DOI] [PubMed] [Google Scholar]
- 88.Tulupova E, et al. Do polymorphisms and haplotypes of mismatch repair genes modulate risk of sporadic colorectal cancer? Mutat Res. 2008;648(1–2):40–5. doi: 10.1016/j.mrfmmm.2008.09.005. [DOI] [PubMed] [Google Scholar]
- 89.Allan JM, et al. MLH1 -93G>A promoter polymorphism and risk of mismatch repair deficient colorectal cancer. Int J Cancer. 2008;123(10):2456–9. doi: 10.1002/ijc.23770. [DOI] [PubMed] [Google Scholar]
- 90.Koushik A, et al. p53 Arg72Pro polymorphism and risk of colorectal adenoma and cancer. Int J Cancer. 2006;119(8):1863–8. doi: 10.1002/ijc.22057. [DOI] [PubMed] [Google Scholar]
- 91.Schernhammer ES, et al. Cyclin D1 A870G polymorphism and the risk of colorectal cancer and adenoma. Br J Cancer. 2006;94(6):928–34. doi: 10.1038/sj.bjc.6603007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Dong LM, et al. Vitamin D related genes, CYP24A1 and CYP27B1, and colon cancer risk. Cancer Epidemiol Biomarkers Prev. 2009;18(9):2540–8. doi: 10.1158/1055-9965.EPI-09-0228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ding EL, et al. Interaction of estrogen therapy with calcium and vitamin D supplementation on colorectal cancer risk: reanalysis of Women’s Health Initiative randomized trial. Int J Cancer. 2008;122(8):1690–4. doi: 10.1002/ijc.23311. [DOI] [PubMed] [Google Scholar]
- 94.Roff A, Wilson RT. A novel SNP in a vitamin D response element of the CYP24A1 promoter reduces protein binding, transactivation, and gene expression. J Steroid Biochem Mol Biol. 2008;112(1–3):47–54. doi: 10.1016/j.jsbmb.2008.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Peters U, et al. Variation in the selenoenzyme genes and risk of advanced distal colorectal adenoma. Cancer Epidemiol Biomarkers Prev. 2008;17(5):1144–54. doi: 10.1158/1055-9965.EPI-07-2947. [DOI] [PubMed] [Google Scholar]
- 96.Peters U, et al. High serum selenium and reduced risk of advanced colorectal adenoma in a colorectal cancer early detection program. Cancer Epidemiol Biomarkers Prev. 2006;15(2):315–20. doi: 10.1158/1055-9965.EPI-05-0471. [DOI] [PubMed] [Google Scholar]
- 97.Barry EL, et al. Ornithine decarboxylase polymorphism modification of response to aspirin treatment for colorectal adenoma prevention. J Natl Cancer Inst. 2006;98(20):1494–500. doi: 10.1093/jnci/djj398. [DOI] [PubMed] [Google Scholar]
- 98.Gerner EW, et al. A comprehensive strategy to combat colon cancer targeting the adenomatous polyposis coli tumor suppressor gene. Ann N Y Acad Sci. 2005;1059:97–105. doi: 10.1196/annals.1339.033. [DOI] [PubMed] [Google Scholar]
- 99.Spain SL, et al. Colorectal cancer risk is not associated with increased levels of homozygosity in a population from the United Kingdom. Cancer Res. 2009;69(18):7422–9. doi: 10.1158/0008-5472.CAN-09-0659. [DOI] [PubMed] [Google Scholar]
- 100.Webb E, et al. A genome-wide scan of 10 000 gene-centric variants and colorectal cancer risk. Eur J Hum Genet. 2009;17(11):1507–14. doi: 10.1038/ejhg.2009.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Goodman JE, et al. Exploring SNP-SNP interactions and colon cancer risk using polymorphism interaction analysis. Int J Cancer. 2006;118(7):1790–7. doi: 10.1002/ijc.21523. [DOI] [PMC free article] [PubMed] [Google Scholar]