

Abstract
We recently established an in vitro assay that monitors the fusion between latex-bead phagosomes and endocytic organelles in the presence of J774 macrophage cytosol (Jahraus et al., 1998). Here, we show that different reagents affecting the actin cytoskeleton can either inhibit or stimulate this fusion process. Because the membranes of purified phagosomes can assemble F-actin de novo from pure actin with ATP (Defacque et al., 2000a), we focused here on the ability of membranes to nucleate actin in the presence of J774 cytosolic extracts. For this, we used F-actin sedimentation, pyrene actin assays, and torsional rheometry, a biophysical approach that could provide kinetic information on actin polymerization and gel formation. We make two major conclusions. First, under our standard in vitro conditions (4 mg/ml cytosol and 1 mM ATP), the presence of membranes actively catalyzed the assembly of cytosolic F-actin, which assembled into highly viscoelastic gels. A model is discussed that links these results to how the actin may facilitate fusion. Second, cytosolic actin paradoxically polymerized more under ATP depletion than under high-ATP conditions, even in the absence of membranes; we discuss these data in the context of the well described, large increases in F-actin seen in many cells during ischemia.
INTRODUCTION
Actin is an exceedingly complex cellular molecule in terms of its dynamics and interactions with other proteins. This cytoskeletal protein is known to be essential for a wide array of cell functions such as motility, chemotaxis, phagocytosis, macropinocytosis, and cytokinesis, as well as for cell polarity and differentiation processes (White and Borisy, 1983; Condeelis, 1993; Cramer et al., 1994; Small et al., 1999). For these processes cells have evolved an ever-increasing list of >150 actin-binding proteins, which can modulate the behavior of actin (Pollard et al., 1994; Sheterline et al., 1995) and which are closely linked to dynamic networks of cellular signaling pathways (Burridge and Chrzanowska-Wodnicka, 1996; Hall, 1998; Machesky and Insall, 1999). Many of these actin-based functions are intimately associated with membranes, but the resulting phenomena are poorly understood (DeRosier and Tilney, 2000).
Actin is also clearly involved in intracellular membrane trafficking events, although it has proven extremely difficult to elucidate its precise role in these processes. Movement of membrane organelles such as the Golgi complex along actin filaments, has been seen in plants (Simon and Pon, 1996; Boevink et al., 1998), and many studies in animal cells have also provided evidence for actomyosin transport, both in vivo and in vitro (Kuznetsov et al., 1992; Simon and Pon, 1996; Goodson et al., 1997; Baker and Titus, 1998; Mermall et al., 1998; Rogers and Gelfand, 1998). More elusive, but noteworthy, is the possible role of actin in membrane organelle vesiculation, aggregation, docking, or fusion, especially during exo- and endocytosis (Burgoyne and Cheek, 1987; Riezman et al., 1996; Simon and Pon, 1996; Lamaze et al., 1997). One prominent idea that has been proposed is that F-actin provides a physical barrier that must be removed for exocytic vesicles to dock and fuse (Orci et al., 1972; Burgoyne and Cheek, 1987; Trifaro et al., 1992, 1993; Burgoyne et al., 1993; Muallem et al., 1995; Vitale et al., 1995; Aunis, 1998). However, not all studies on membrane organelle fusion would fit into this simple model because, in many systems actin is a positive effector of exocytosis (Orci et al., 1972; Muallem et al., 1995), as well as endocytosis (Lamaze et al., 1997). Moreover, the cytochalasins, which are actin-specific drugs, can stimulate or inhibit exocytosis, depending on the cell system (Orci et al., 1972; Burgoyne and Cheek, 1987; Aunis, 1998). That actin is somehow involved in membrane fusion is also consistent with a recent study by Bernstein et al. (1998), which showed that cycles of neuronal exocytosis correlated with cycles of actin polymerization and depolymerization. This theory is further supported by a recent high-resolution fluorescence microscopy study of exocytosis by Lang et al. (2000) in PC-12 cells. One of the main goals of the present, and related studies is to understand more about the links between actin and membrane fusion.
We have extensively used 1-μm latex beads as markers for phagosomes in J774 mouse macrophages, an approach that facilitates isolation of these organelles (Desjardins et al., 1994a,b). We found, both in vivo and in vitro, that phagosomes, after maturing intracellularly for up to 4–8 h (after uptake), fuse well with early endosomes, late endosomes, and lysosomes, whereas older phagosomes fuse poorly (Desjardins et al., 1997; Claus et al., 1998; Jahraus et al., 1998). These fusion events depend on the ability of phagosomes to bind to and move bidirectionally along microtubules, processes that we have also reconstructed using in vitro systems (Blocker et al., 1996, 1997, 1998). Phagosomes can also nucleate actin in vitro, a process that varies considerably with their maturation state in macrophages. Ezrin and/or moesin (Defacque et al., 2000a), as well as the phosphoinositide PI4,5 P2 (PIP2) (Defacque, H., Bos, E., Garvalov, B., Baret, C., Roy, C., Mangeat, P., Shin, H.W., Rybin, V., and Griffiths, G., submitted), were found to be essential for this membrane- and ATP-dependent actin assembly process.
In the present study we focus exclusively on 2-h phagosomes, which are active in all the processes we have studied. We first provide evidence that cytochalasin D (cyto D) and latrunculin A inhibit fusion between phagosomes and endosomes in vivo. Second, we demonstrate that this fusion process, reconstituted in vitro (Jahraus et al., 1998), could be either stimulated or inhibited by different reagents that affect the actin cytoskeleton. Third, we show by different approaches that, in the context of macrophage cytosolic extracts and high levels of ATP, the presence of membranes is crucial for efficient nucleation and polymerization of actin. Finally, data show that in the absence of membranes cytosolic actin paradoxically polymerizes more at low than at high ATP.
MATERIALS AND METHODS
Reagents and Cells
All chemicals were purchased from Sigma (St. Louis, MO), Boehringer (Mannheim, Germany), or Merck (Darmstadt, Germany) in analytical quality unless stated otherwise. For culture of J774A.1 mouse macrophages and preparation of cytosolic extracts, see Blocker et al. (1996).
In Vivo Fusion of Phagosomes with Endocytic Organelles
For this assay, carboxylated latex beads (Seradyne, Indianapolis, IN) were internalized by J774 macrophages for 1-h pulse and 1-h chase (2-h phagosomes) (Desjardins et al., 1994a; Jahraus et al., 1998). After washing, cells were fed with 5 mg/ml horseradish peroxidase (HRP) for 15 min and washed again on ice with phosphate-buffered saline and PBS/0.5% bovine serum albumin. In some experiments prewarmed media containing either cyto D (Sigma), latrunculin A (Molecular Probes, Leiden, The Netherlands), or no additions were then incubated with the cells at 37°C for a further 60 min. Cells were homogenized in 600 μl of homogenization buffer (HB) containing 250 mM sucrose, 3 mM imidazole, pH 7.4 with a protease inhibitor cocktail (PIC) (containing a final concentration of 1 μg/ml Pepstatin, 0.5 μg/ml TPCK, 0.5 μg/ml Leupeptin, and 4 μg/ml aprotinin), and 1 mM dithiothreitol. The nuclei were removed by centrifugation (Jahraus et al., 1998). This post nuclear supernatant (PNS, 500 μl) was collected, and one aliquot was saved for the HRP assay to determine the total HRP amount that served as a reference for the phagosome values. The rest was submitted to phagosome isolation by using the standard sucrose gradient (Jahraus et al., 1998). The phagosome band was collected, and the bead number was counted. The protein content of the PNS was determined using the Bio-Rad Protein assay (Bio-Rad, Munich, Germany). Finally, the HRP content of this fraction and the PNS were determined by an enzyme assay (ImmunoPure TMB substrate kit; Pierce, Rockford, IL), adapted to the same number of beads (phagosome fraction) or the same amount of protein (PNS) for the different drug concentrations, respectively. Values are expressed as relative amount of HRP in phagosomes and related to the total concentration of HRP in the PNS.
In Vitro Fusion of Phagosomes with Endocytic Organelles
The basic biochemical in vitro fusion assay is described in detail in Jahraus et al. (1998). In brief, 2-h phagosomes containing 1-μm latex beads coated with avidin (internalized for 1 h and chased for 1 h) and PNS membranes in which the endocytic organelles had been filled with biotinylated HRP (bHRP) (40-min internalization) were mixed on ice. Additionally, macrophage cytosol at a final concentration of 4 mg/ml was added. The reaction was carried out either with an ATP-regenerating system (final concentrations: 1.8 mM ATP, 14.7 mM creatine phosphate, and 73.3 μg/ml creatine phosphokinase) (high ATP state) or with an ATP-depleting system (final concentrations: 41.2 U/ml hexokinase [Boehringer Mannheim] and 27.5 mM d-glucose) (low ATP state). The mixture was adjusted to contain 0.05 mg/ml biotinylated insulin, 60 mM KOAc, 1.5 mM MgOAc, 1 mM DTT, and 12.5 mM HEPES, pH 7.4, and the volumes were balanced with HB. After 80-min incubation at 37°C fusion was assessed by estimating the amount of bHRP that binds to a fixed number of beads (Jahraus et al., 1998). Drugs, or other actin-modifying reagents were added to the complete in vitro fusion sample immediately before the start of the 37°C incubation. For preparing phalloidin-stabilized actin, 5.2 mg/ml purified skeletal muscle actin was mixed in 1:1 molar ratios with phalloidin for 10 min at room temperature to stabilize the actin filaments. The F-actin was centrifuged at 100000 × g for 1 h and, after resuspension, added at the indicated concentrations to the fusion assay samples. Recombinant rho GDP-dissociation inhibitor (rhoGDI) (kindly provided by O. Ullrich and M. Zerial, European Molecular Biology Laboratory), and the Ca2+-insensitive G1–3 domain of gelsolin (kindly provided by M. Way, European Molecular Biology Laboratory [Way et al., 1989]) were dialyzed against HB containing 1 mM DTT and PICs overnight before use. Thymosin-β4 (Tβ4) (a gift from W. Voelter, University of Tuebingen) that had been chemically synthesized (Echner and Voelter, 1988) was dissolved in distilled water. 2,3-Butanedione-2-monoxime (BDM) (Sigma) was dissolved freshly as a 0.5 M stock in water and added to 10 mM final concentration in the fusion assay.
F-Actin Sedimentation Assay
In vitro fusion of phagosomes with endosomes was performed as described above. Prior to the 37°C incubation, 20-μl aliquots from each sample were collected from the final mix as a “total actin” reference. After the fusion reaction, the samples were put on ice for 5 min. Triton X-100 and F-actin stabilization buffer, pH 7.4) (PHEM), were added to the mix at a final concentration of 1% Triton and 1× PHEM buffer. The whole reaction was carefully mixed by pipetting three times with a cut yellow tip and transferred directly into a TLA 100 centrifuge tube (Beckman Instruments, Palo Alto, CA). Centrifugation at 400,000 × g for 1 h at 4°C in a table top ultracentrifuge (Beckman Instruments) led to a clear supernatant. At these high centrifugal forces all F-actin in the system is expected to pellet, leaving G-actin in the supernatant. The two fractions were separated, and resuspended in equal volumes in the presence of 1× Laemmli buffer. Fifteen microliters of each fraction brought up to the same dilutions (total, pellet, and supernatant) were loaded on a 12.5% SDS-PAGE in parallel with a G-actin standard ranging from 0 to 100 ng. The gel was blotted onto nitrocellulose and probed with an anti-actin antibody (A2066; Sigma) and the respective secondary antibody linked to HRP. Signals were detected by chemiluminescence by using the ECL kit (Nycomed Amersham, Buckinghamshire, United Kingdom). In parallel, macrophage cytosol was loaded to assess the actin concentration.
Nucleotide Determination by High Performance Liquid Chromatography (HPLC)
Cytosol extracts were deproteinized and analyzed by HPLC as described in Peveri et al. (1992) before and after 80-min incubation at 37°C. All incubations prior to HPLC analysis, including the analysis of G-actin bound nucleotides (see below), were done in the same salt and buffer conditions as in the biochemical in vitro fusion assay. For HPLC analysis, the Waters 2690 separation module with the Waters 996 photodiode array detector was used. To integrate peaks we used Waters Millennium32 software. To calibrate the system nucleotides obtained from Sigma were used.
To isolate and measure the nucleotide state of G-actin from cytosol extracts we slightly modified the protocol established by Rosenblatt et al. (1995). In brief, cytosol with [32P]α-ATP (100 μCi in a total volume of 100 μl) was incubated for 80 min at 37°C with an ATP-regenerating or -depleting system in the presence or absence of PNS membranes. After the incubation F-actin was removed by centrifuging the solutions at 279,000 × g for 1 h. The supernatant was then spun through a Bio-Gel P-6 column (Bio-Rad Laboratories, Richmond, CA) pre-equilibrated in G-buffer without ATP to remove the majority of unbound nucleotides. The flow through (100 μl) was added to 50 μl of DNase 1 (Boehringer-Mannheim) coupled Affi-Gel 10 beads (Bio-Rad Laboratories), and incubated at 4°C for 1 h with frequent vortexing. Nonspecific proteins were removed with two 0.5-ml washes of wash buffer (0.4 M NH4Cl2, 10 mM Tris, pH 8.0, 0.2 mM CaCl2, 0.2 mM DTT) for 1 min and the washed beads were recovered by centrifugation for 1 min at 10,000 × g in an Eppendorf microfuge. The actin was denatured and eluted with 8 M urea, 10 mM Tris, pH 7.4, 0.2 mM CaCl2 at 95°C for 5 min before filtration through a 10,000 kDa cut-off spin filter unit. That actin was the major species eluted was confirmed by a Comassie-stained SDS-PAGE gel. The isolated nucleotides in the filtrate were then analyzed by HPLC.
Pyrene Actin Assay
Unless specified, for estimating polymerization of pure actin, increase of fluorescence upon polymerization of 1 μM actin (10% pyrenyl-labeled) was followed in G-buffer (10 mM Tris pH 8, 0.1 mM ATP, 0.1 mM DTT, 0.1 mM CaCl2) at 25°C for 15 min, immediately after addition of salts (50 mM KCl, 1 mM MgCl2) in a Aminco-Bowman Series 2 luminescence spectrometer (SLM-Aminco, Northampton, MA). Excitation and emission wavelengths were 365 and 407 nm, respectively. Actin purified from rabbit muscle was labeled with pyrenyl iodoacetamide (Molecular Probes, Eugene, OR) according to established protocols (Kouyama and Mihashi, 1981; Pardee et al., 1982). Pyrene G-actin was stored in liquid nitrogen in G-buffer containing 1 mM DTT and 10% glycerol. After rapidly thawing, the pyrene actin was centrifuged at 100,000 × g for 15 min at 4°C to remove any aggregates. A mixture of cytosol and pyrene actin was then incubated for 15 min at 4°C. Each sample containing 8.7 μl of cytosol (final concentration: 4 mg/ml) and pyrene actin (final concentration: 1 μM) was mixed with 10.7 μl of PNS at 4°C. In the presence of PNS we could not use <20% final concentration of pyrene actin to get a reliable signal. Salt and buffer conditions were always the same as in the biochemical in vitro fusion assay.
During these studies we found that with cytosolic extracts, and more prominently when membranes were present, the first few minutes of incubation with pyrene actin at 37°C always gave erratic fluorescence traces, presumably due to some light scattering events. To avoid this, the samples were incubated at 37°C for 5 min in the fluorimeter cuvette, before an ATP-regenerating or -depleting system was added (2.5 μl each) in a total volume of 45 μl. Increase of fluorescence at 37°C was then immediately monitored, as described above.
Rheological Measurements
Quantitative measurements of the macroscopic viscoelastic properties of the fusion assay were made by using a rotating disc rheometer, described in detail by Müller et al. (1991). The actin solution is contained in a cylindrical cuvette and is covered by a lipid monolayer to prevent denaturing of actin due to exposure to air. A silanized disc is placed on top of the solution. Torsional oscillations of the disc are excited by an oscillatory magnetic field acting on a small magnet fixed on the center of the top of disc. Time-dependent measurements were conducted at one fixed frequency (ω = 0.2/rad/s) at 37°C to monitor predominantly the polymerization of actin in cytosol and to study the effect of the presence of phagosomes, PNS membranes, and ATP on the polymerization. As a reference we also tested 4 μM pure G-actin in F-buffer. In all cases our conventional fusion assay was scaled up to a volume of 500 μl while keeping the relative concentrations of reagents constant except that only one-tenth of the usual phagosomes concentration was used to a final O.D.600 of 0.024. When we used phagosomes at the usual concentration used for the fusion assay we observed an aggregation and sedimentation, making rheological measurements impossible. After mixing, the fusion assay was kept on ice for 5 min, and then carefully pipetted into the prewarmed measuring chamber. Measurements were taken every 3 min during the 80-min incubation time.
Polymeric fluids (like F-actin solutions) are described by the
frequency-dependent complex viscoelastic modulus, G*(ω),
where G*(ω) = G′(ω) +
iG"(ω), which is composed of a “real” part,
G′(ω), and an “imaginary” part, iG"(ω) (i
denotes the imaginary unit, i = 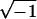 , and ω
is the angular frequency). The real part, the so-called storage
modulus, G′(ω), is a measure for the elastic component of
the network. The imaginary part, the so-called loss modulus
G"(ω), is related to the viscosity of the network by
G"(ω) = ωη(ω). The complex quantity
G*(ω) is characterized by its absolute value
‖G*‖ =
, and ω
is the angular frequency). The real part, the so-called storage
modulus, G′(ω), is a measure for the elastic component of
the network. The imaginary part, the so-called loss modulus
G"(ω), is related to the viscosity of the network by
G"(ω) = ωη(ω). The complex quantity
G*(ω) is characterized by its absolute value
‖G*‖ = 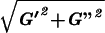 and by the phase shift ϕ = arctan (G"/G′).
‖G*‖ is determined by measuring the angular deflection
of the disc (as described above) as a function of the shear force and
is simply given by the ratio of the shear stress within the solution
and the angular deflection angle. ϕ is the phase shift angle between
the oscillatory force and the angular deflection of the disc.
G′(ω) and G"(ω) are finally obtained from the
two quantities ‖G*‖ and ϕ by using the relations
G′ = ‖G*‖cosϕ and G" =
‖G*‖sinϕ, respectively.
and by the phase shift ϕ = arctan (G"/G′).
‖G*‖ is determined by measuring the angular deflection
of the disc (as described above) as a function of the shear force and
is simply given by the ratio of the shear stress within the solution
and the angular deflection angle. ϕ is the phase shift angle between
the oscillatory force and the angular deflection of the disc.
G′(ω) and G"(ω) are finally obtained from the
two quantities ‖G*‖ and ϕ by using the relations
G′ = ‖G*‖cosϕ and G" =
‖G*‖sinϕ, respectively.
The viscoelastic moduli G′(ω) and G"(ω) are complex functions of frequency that depend on the concentration of polymerized actin and on the degree of cross-linking between filaments in a complex manner. For a full characterization of the viscoelastic behavior one has to measure ‖G*‖ and ϕ or G′(ω) and G"(ω) over several frequency decades. However, numerous studies of purely entangled and cross-linked actin networks showed that the degree of actin polymerization can be well characterized measuring the above parameters at a single frequency of ω = 0.2/rad/s (Sackmann, 1997).
The phase shift, ϕ, depends on the fluidity of the sample and is ϕ = π/2 for a pure fluid (such as G-actin solution) and ϕ = 0 for a solid (which responds instantaneously to a stepwise force). Therefore, the formation of an interconnected network of F-actin results in a decrease of the phase shift tgϕ = G"/G′ and the buildup of a finite elastic modulus G′(ω). Based on previous studies (Sackmann, 1997) measurements at a frequency of ω = 0.2/rad/s are chosen for such measurements.
It should be noted that we also measured full frequency curves of ‖G*‖ and ϕ in some cases to ensure that measurements at ω = 0.2/rad/s are well suited to study the buildup of viscoelastic networks in our cell extracts. However, because these measurements require 90 min for one lowest frequency applied they are not generally suited to study the time evolution of the generation of interconnected actin filaments.
RESULTS
Effects of Actin-Depolymerizing Agents on In Vivo Fusion
We first asked whether polymerized actin plays some role in the intracellular fusion of phagosomes with endocytic organelles in J774 cells. For this, we tested the effect of cyto D, which blocks (barbed-end) actin polymerization, or latrunculin A, which sequesters actin monomers, on the in vivo fusion of latex bead phagosomes with endocytic organelles by using a previously established assay (Desjardins et al., 1994b). Accordingly, J774-macrophages were allowed to sequentially internalize latex beads (1-h pulse, 1-h chase) followed by HRP as an endocytic marker for 15 min, a time point insufficient to allow significant fusion between phagosomes and endosomes, before adding the drugs. The fusion efficiency was then estimated by the amount of HRP that was associated with isolated phagosomes after in vivo incubation for 80 min. As shown in Figure 1, treatment of cells after phagocytic uptake with ≥0.5 μM of either latrunculin A or cyto D inhibited in vivo fusion by 50–60%. With results from S. Kuznetsov (personal communication) showing that cyto D inhibits movement of phagosomes from the cell periphery towards the perinuclear region in mouse macrophages, and Blocker et al. (1997, 1998) showing that phagosomes use microtubules for movement, both in vivo and in vitro, these data argue that both actin filaments and microtubules are used by phagosomes for intracellular transport in J774 cells, and that both these transport events facilitate membrane fusion.
Figure 1.

Actin-disrupting drugs inhibit in vivo phagosome-endosome fusion. Effect of increasing concentration of cytochalasin D and latrunculin A on in vivo fusion of phagosomes and endosomes in J774 macrophages, estimated by the transfer of HRP from endocytic organelles to preformed phagosomes. Cells were fed first for 1-h pulse and 1-h chase with latex beads followed by a 15-min pulse with 5 mg/ml HRP. Cytochalasin D or latrunculin A (range: 0–10 μM) were then added to the cells and incubated at 37°C for further 60 min. The HRP content of isolated phagosomes was measured and the data are expressed as the relative amount of HRP related to the number of phagosomes.
Effects of Actin Reagents on In Vitro Fusion
In the remaining part of this article we investigated the role of actin in in vitro fusion. For this, we first describe the effects of a variety of reagents affecting actin dynamics (and myosin functions) on the in vitro fusion between phagosomes and endocytic organelles. Our biochemical in vitro fusion assay measures the content mixing of bHRP internalized into endosomes and lysosomes with 2 h phagosomes containing avidin-coated latex beads (internalized for 1 h and chased for 1 h) (Jahraus et al., 1998) (Figure 2). The fusion was carried out in the presence of J774 cytosol, which at 4 mg/ml protein contains ∼4 μM G-actin, with no detectable F-actin seen by electron microscopy at the beginning of the incubation (our unpublished results). In all experiments we compared conditions by using an ATP-regenerating system with an ATP-depleting system. Fusion was assessed by the amount of bHRP that binds to a fixed number of avidin beads, after lysing the phagosomal membrane with Triton X-100.
Figure 2.

Schematic illustration of the phagosome-endosome in vitro fusion assay.
We found that four different reagents that target the actin cytoskeleton gave a significant inhibition of the fusion signal in the in vitro fusion assay. First, cyto D gave a 40% inhibition, with most of its effects being apparent at 3 μM (Figure 3A). Second, the addition of phalloidin-stabilized fragments of F-actin led to a concentration-dependent inhibition of fusion, with ∼85% inhibition seen at 10 μM phalloidin-actin (Figure 3B). This inhibition is presumably a consequence of actin filaments providing a physical barrier to fusion.
Figure 3.

Conditions that inhibit in vitro phagosome-endosome fusion. The effects are shown of cyto D (A), phalloidin-stabilized F-actin (B), rhoGDI (C), and 10 mM BDM (D) on the in vitro fusion of phagosomes with endocytic organelles in the presence of an ATP-regenerating system. These reagents were mixed into the standard fusion assay and the transfer of bHRP to phagosomes were expressed as nanograms of HRP bound to a fixed number of avidin beads. The low-ATP arrow shows the average values obtained under ATP-depleting conditions for three concentration points. In each case the means of one duplicate experiment is shown out of a total of three experiments done in duplicates. Although the absolute values obtained varied from one experiment to the next the patterns shown were consistently seen.
The family of rho GTPases is known to be intimately involved in the regulation of actin assembly, disassembly and dynamic reorganization events (Hall, 1998). They have also been strongly implicated in the regulation of exocytic fusion (Norman et al., 1994; Komuro et al., 1996; Brown et al., 1998; Gasman et al., 1999). A 60% inhibition was seen when 18 μM rhoGDI was added to the fusion mix (Fig. 3C). Under these in vitro conditions this protein should extract the membrane-bound rho family proteins (in their GDP state) from membranes and thereby inactivate them in the assay.
We asked whether myosins might play a role in the fusion assay by testing the effect of the low-specificity, myosin ATPase inhibitor BDM. This inhibited fusion with ∼45% at 10 mM (Figure 3D). BDM also inhibits the inward movement of phagosomes in mouse bone marrow macrophages (S. Kuznetson, personal communication). This drug had no effect on the low fusion signal we routinely find using an ATP-depleting system (Jahraus et al., 1998).
In contrast to these inhibitory effects, the addition of two other actin-binding proteins led to a significant and reproducible stimulation of fusion (Figure 4). The first was the N-terminal half of gelsolin (G1–3), a protein that can sever actin filaments and then bind to the newly exposed barbed ends in a calcium-independent fashion (Way et al., 1989). In our phagosome nucleation assay (that operates independently of cytosol) this protein greatly stimulates actin nucleation (Defacque et al., 2000b). When this recombinant protein was added to the fusion assay, we also saw a concentration-dependent increase in the extent of fusion, with ∼200% at 2.5 μM (a concentration that is roughly half the molar ratio of the total actin in the cytosol), and still higher levels reproducibly gave a smaller stimulation (Figure 4A). Both stimulatory and inhibitory effects of gelsolin G1–3 have been described in previous in vitro studies of exocytosis (Muallem et al., 1995).
Figure 4.

Two actin-binding proteins stimulate in vitro fusion. Increasing amounts of gelsolin (G1–3) (A) and Tβ4 (B) were titrated into the fusion assay. The mean and SD are shown for one experiment in duplicate (out of a total of four experiments) and the high- and low-ATP values are indicated.
The second reagent was the 5-kDa polypeptide Tβ4. This polypeptide, which is generally present in cells at concentrations significantly higher than G-actin itself, is considered to be the major cellular G-actin–binding protein (Cassimeris et al., 1992). As shown in Figure 4B, with increasing concentrations of Tβ4 added (in the presence of high ATP) the total extent of fusion seen was stimulated by >2-fold at 30 μM. These data resemble the stimulatory effects of Tβ4 seen in in vitro studies of coated-pit assembly (Lamaze et al., 1997) and exocytosis (Muallem et al., 1995).
Although these data are difficult to interpret, they collectively make a strong argument that actin assembly, structure or dynamics is strongly influencing both in vivo and in vitro membrane fusion. The same conclusion was made recently in an elegant study by Lang et al. (2000). Because Tilney and others have shown that membranes can nucleate actin assembly (Tilney and Cardell, 1970; Tilney, 1975), we decided to investigate in more detail the role of membranes in the polymerization of actin in this system. In a parallel study (Egeberg, M., Kjeken, R., Habermann, A., Jahraus, A., Defacque, H., Kuznetsov, S.A., and Griffiths, G., in preparation) we have made complementary microscopic analyses of this system and the data by both approaches are consistent with a testable model (see DISCUSSION).
Actin Polymerization and the Role of Membranes
Sedimentation Analysis of G- and F-Actin and Nucleotide Analyzes
At the end of the biochemical in vitro fusion assay (80 min) we had noticed that the contents of the tubes appeared to be more viscous than at the start, with the ones containing an ATP-regenerating system (high ATP) showing a more gel-like consistency and a visible aggregation of the opaque phagosomes. This observation led us to determine the amount of F-actin polymerization in this system by using a sedimentation protocol for filamentous actin. In these, and the subsequent series of experiments we also describe unexpected effects of low ATP levels, by using an ATP-depleting system that was initially used only as a control for the ATP-regenerating system. In all subsequent experiments we focused in detail on both the high- and low-ATP states (defined below, see “HPLC analyses”). It should be noted that the low-ATP state is not a “dead” state because a low level of fusion consistently occurs with ATP depletion, as determined by biochemical and by quantitative electronmicroscopy analyses (Jahraus et al., 1998).
We first evaluated the amount of actin that pelleted upon high-speed centrifugation from cytosol, both with and without membranes, and with high versus low ATP. This analysis must be considered as a crude estimate of how much F-actin is present in the various pellets. There is an extensive literature on a poorly characterized pool of actin that is intimately associated with membranes (Carraway and Carraway, 1989; Tranter et al., 1991). There is also a pool of actin that routinely copurifies with phagosomes (Desjardins et al., 1994a,b) that does not bind phalloidin (our unpublished results). In our sedimentation analyses we made no distinction between free F-actin and the G- or F-actin pools associated with membranes. Even in the experiments with pure cytosol alone the pellets may conceivably contain actin in various forms that are bound to large protein complexes (e.g., ARP 2/3–Wasp complexes). These data, therefore only give a qualitative estimate of total pelletable actin.
As shown in Figure 5, there was considerably more pelletable F-actin at low-ATP than at high-ATP levels, under all conditions tested at 37°C, and the addition of membranes did not significantly increase the pelletable actin over that seen with cytosol alone. However, in the high-ATP state the membranes clearly facilitated F-actin assembly, with no detectable signal seen when cytosol was incubated alone (F-actin, lanes 1 and 5). A distinct F-actin band was also evident when the complete fusion mix was incubated at 4°C (a temperature where no fusion occurs), but this was clearly less intense than that seen at 37°C. Only a faint signal for F-actin was evident when cytosol was incubated at 37°C with control avidin beads (without membranes), plus cytosol and an ATP-regenerating system.
Figure 5.

Estimation of G- and F-actin in the fusion assay by sedimentation analysis. G- and F-actin in the in vitro fusion assay was estimated after incubation under the four different conditions indicated. F-actin was sedimented at 400000 × g for 1 h, whereas the G-actin remained in the supernatant. The material was separated on a SDS gel and blotted with an anti-actin antibody.
These data demonstrate first, unexpectedly, that the preferred conditions for actin polymerization from our cytosol extracts, both with and without membranes, was in ATP-depleting, rather than ATP-regenerating conditions. Second, and more relevant for the main goal of this study, they show that in the presence of high ATP the membranes facilitated an enhanced polymerization of actin.
HPLC Analyses of Nucleotides in J774 Cytosol with and without PNS Membranes
Because the effects of high and low ATP were so different we decided to determine the concentration of nucleotides in our system under the different conditions by using HPLC. We compared the amounts of different nucleotides at the beginning of the incubation (before adding ATP-regenerating or -depleting system) with the amount of nucleotides after 80-min incubation at 37°C. We quantified the peaks for ATP, ADP, AMP, GTP, and GDP for equal volumes of material. The basal level of ATP in freshly thawed cytosol in the absence of added ATP was very low (4 μM), but it was significantly higher when PNS was added (33 μM). The levels of GTP and GDP were also low (1–5 μM), whereas ADP was 20–29 μM and AMP, 56–76 μM (Table 1). When cytosol was incubated with an ATP-regenerating system, the level of ATP reached ∼1 mM, and the addition of PNS consistently lowered this level by ∼200 μM. The addition of membranes with high ATP also led to a significant increase in the level of ADP over that found in cytosol alone. The total level of GTP increased consistently with high ATP, to 42 μM with cytosol alone and 90 μM when PNS membranes were added.
Table 1.
Nucleotide measurements by HPLC
| Time of incubation (min) | Sample | ATP | ADP | AMP | GTP | GDP |
|---|---|---|---|---|---|---|
| μM | ||||||
| 0a | Cytosol | 4.2 | 20.3 | 55.7 | 1.4 | 4.9 |
| Cytosol +PNS | 33.0 | 29.0 | 76.5 | 2.3 | 6.8 | |
| 80 | Cytosol + ATPb | 1154 | 65 | 9.0 | 41.8 | 3.4 |
| Cytosol − ATPc | 19.5 | 2.3 | 58.9 | 1.2 | 1.0 | |
| Cytosol, PNS+ ATP | 939.8 | 192.2 | 20.4 | 90.6 | 12.5 | |
| Cytosol, PNS − ATP | 4.4 | 1.6 | 62.7 | 0.5 | 1.4 | |
Estimations of nucleotide concentrations by HPLC in cytosol, with and without PNS membranes, in the presence of an ATP-regenerating system or an ATP-depleting system. We compare the system at time point 0 (freshly thawed cytosol) and after an 80-min incubation at 37°C. The data indicate one of at least two independent experiments on the same batch of cytosol, which showed similar values.
Measured before addition of ATP-regenerating/deleting system.
+ ATP denotes ATP-regenerating system.
− ATP denotes ATP-depleting system.
The presence of the ATP-depletion system reduced the total ATP to ∼20 μM with cytosol alone, and to 4 μM when PNS membranes were also present. Surprisingly, the cytosol under low ATP conditions had a ∼5-fold higher level of ATP than the starting cytoplasm without additions. At low ATP all other nucleotides were reduced to low micromolar levels with the exception of AMP, whose levels far exceeded those seen with high ATP, but not exceeding that detected in the starting cytosol extract.
Most importantly for the rest of this study is that high ATP means ∼1 mM ATP and 40–90 μM GTP (both close to physiological levels), whereas the low-ATP state means below 20 μM ATP and only ∼1 μM of GTP, in a cytosolic system that contains ∼4 μM total actin.
Nucleotide State of G-Actin
Rosenblatt et al. (1995) described an elegant method for quantifying the relative amounts of adenine nucleotides bound to G-actin from cytoplasmic extracts from Xenopus eggs. Applying a modified version of this method (see MATERIALS AND METHODS) we quantified the amount of ATP, ADP, or AMP bound to G-actin in cytosolic extracts at high and low levels of ATP by HPLC.
Under both high- and low-ATP conditions the bulk (∼80%) of the total G-actin carried ATP, whereas ∼10% bound ADP and 2–9% bound AMP (Table 2). These results agree well with the study by Rosenblatt et al. (1995), showing that the bulk of G-actin in Xenopus egg extracts is ATP-bound. Presumably, this is the form that preferentially incorporates into filaments at both high and low ATP. These data thus provided us with an unexpected result: even with ATP-depletion, the bulk of the G-actin is bound to ATP, and not ADP.
Table 2.
32P-α-labeled nucleotides bound to G-actina
| Sample | ATP
|
ADP
|
AMP
|
|||
|---|---|---|---|---|---|---|
| % | SD | % | SD | % | SD | |
| ATP-regenerating system | ||||||
| − PNS | 89.1 | 7.4 | 9.2 | 4.9 | 1.8 | 2.5 |
| +PNS | 88.2 | 5.8 | 6.1 | 0.6 | 5.7 | 5.3 |
| ATP-depleting system | ||||||
| − PNS | 76.6 | 12.4 | 16.5 | 8.3 | 7.0 | 4.0 |
| +PNS | 75.7 | 6.1 | 13.7 | 5.2 | 9.4 | 3.8 |
Quantification of 32P-α-labeled adenine nucleotides bound to G-actin, estimated by HPLC. Cytosol, with or without PNS membranes, was incubated for 80 min at 37°C in the presence of [32P]-α-ATP and either an ATP-regenerating system or an ATP-depleting system. The values shown reflect the bound nucleotides at the end of the incubation. The data indicate the mean and SD for at least two independent experiments for each value.
Following incubation with 32P-α-ATP.
Pyrene Actin Assay
To provide a more quantitative and kinetic description of actin polymerization in our system, we used pyrene labeled G-actin from muscle. This actin is known to have an increased fluorescence when it incorporates into F-actin. Therefore the kinetics of actin polymerization, as well as the relative amounts of total F-actin can be easily and sensitively estimated by spectrofluorimetry (Kouyama and Mihashi, 1981). We were unable to use intact phagosomes in this assay because the beads promote light scattering, which strongly interferes with the analysis. Therefore, in preliminary experiments phagosomal membranes were separated from the beads after sonication of the phagosomes (Wetzel and Korn, 1969). In the presence of high ATP, these purified membranes also significantly stimulated both the rate and extent of pyrene actin polymerization from pure actin (our unpublished results).
The overall summary of the data is given in Table 3, all from 15-min incubation with pyrene actin. By using pure actin by itself there was significantly more polymerization in the presence of high ATP than in the presence of low ATP, as expected from previous studies (Pollard, 1986; Carlier, 1990). In contrast, when polymerization of actin present in J774 cell cytosol was monitored (without membranes), actin polymerized more with low ATP than with high ATP. Overall, these data are in excellent agreement with the sedimentation analyses (Figure 5). At low ATP, the addition of PNS membranes led to an ∼20% increase in the total amount of polymerized actin, whereas with high ATP, the addition of these membranes stimulated the total extent of polymerization by 2-fold.
Table 3.
Quantification of pyrenea actin analysis
| ATPb | Relative rate of actin polymerization (fluorescence units × 10−4) | Total F-actin (fluorescence units) | |
|---|---|---|---|
| G-actin alone, F-actin buffer | + | 7.8 | 0.4 |
| G-actin alone, F-actin buffer | − | 3 | 0.15 |
| Cytosolc | − | 0.7 | 0.11 |
| Cytosolc | + | 2.4 | 0.36 |
| Fusion assayd | − | 1 | 0.25 |
| Fusion assay | + | 3 | 0.45 |
| Fusion assay + 1 μM cyto D | − | 0.2 | 0.01 |
| Fusion assay + 1 μM cyto D | + | 2 | 0.2 |
Fluorometric quantitation of the rate of pyrene actin polymerization and the total F-actin polymerized under the different conditions over a 15-min time period.
When used without cytosol 10% pyrene labeled muscle actin was used. For the experiments with cytosol, 1 μM of 100% labeled pyrene actin was mixed with 4 mg/ml cytosol, giving a final amount of 20% pyrene actin.
+ denotes ATP-regenerating system (1.0 mM ATP); − denotes ATP-depleting system.
4 mg/ml cytosol.
Fusion assay: PNS and cytosol.
Since the fusion assay, and the rheological studies (see below), are standardized to 80-min incubation time we also extended the time course of our pyrene actin measurements from 15 to 80 min in one set of experiments. As showed in Figure 6A, when cytosol alone was incubated with high ATP, the amount of polymerized actin only began to increase after ∼50 min. In contrast, in the cytosol with low ATP, after an initial drop in fluorescence, the rate of actin polymerization showed a steep increase between 5 and 20 min before it reached a plateau, followed by a second, slow elevation after 50 min. The latter had a similar slope to the late-phase polymerization seen with high ATP.
Figure 6.

Pyrene actin assembly in cytosol alone (A) or in cytosol plus PNS (B) at high and low ATP. Pyrene actin (10%) (1 μM) was added to 4 mg/ml cytosol. (A) Labeled pyrene actin was used, giving a final concentration of 2% labeling, whereas in B the pyrene actin added was 100% labeled, giving a final concentration of 20% labeling. Cytosol alone (A), or cytosol mixed with PNS membranes (B) were either incubated with an ATP-regenerating system (high ATP) or an ATP-depleting system (low ATP). After a 5-min preincubation at 37°C the increase in pyrene fluorescence (which indicates pyrene actin polymerization) was estimated by spectrofluoremetry. Relative fluorescence increase was expressed in arbitrary units.
As shown in Fig. 6B when PNS membranes were incubated with cytosol and low ATP the rate of actin polymerization was considerably enhanced, with a linear increase during the first ∼15 min before the rate of polymerization reached a plateau after ∼50 min of incubation. With PNS membranes, cytosol, and high ATP, the rate of actin polymerization also showed a linear increase during the first ∼15 min, before a consistent drop in polymerization was seen. This drop was followed by a second linear increase, starting at ∼25 min, which persisted throughout the rest of the 80-min incubation time.
These data confirm the two main conclusions of the sedimentation assay: 1) at high ATP cytosolic actin polymerizes poorly in vitro, unless membranes are present; and 2) irrespective of the presence of membranes, cytosolic actin polymerizes much more under low-ATP than under high-ATP conditions.
Actin polymerization induced by purified phagosomes is strongly inhibited by cyto D (Defacque et al., 2000a). When pyrene actin polymerization was examined in the presence of PNS membranes and cytosol, actin assembly was also significantly inhibited by 1 μM cyto D. In the presence of high ATP this drug lowered the relative rate of actin polymerization by 80%, and the total F-actin content decreased by 96% (Table 3). This is likely due to cyto D blocking the incorporation of monomers into actin filament barbed ends that are closely associated with the nucleating machinery on the membrane surface. Such a scenario was shown for the phagosomal membrane in the presence of pure actin (Defacque et al., 2000a). Under the same conditions, but using an ATP-depleting system, cyto D inhibited the relative rate of actin polymerization by only ∼33% and the total F-actin content by ∼56% (Table 3). In these experiments membranes appear to have only a small positive effect on the low, ATP-dependent polymerization. Due to the complexity of this system, and the difficulties one faces in explaining the diverse effects of cyto D on cells (such as inducing, yet to be explained, membrane patches of actin) we did not attempt to ask more detailed questions using this drug.
A possible interpretation of the paradoxical increase in actin nucleation and polymerization at low ATP is that high ATP has an inhibitory effect on actin nucleation/polymerization. (e.g., by phosphorylating a capping or actin regulatory protein). Consistent with this possibility, when cytosol mixed with pyrene actin was incubated at 37°C with a range of ATP levels, it was evident that, although low ATP (4–30 μM) gave a robust polymerization, higher concentrations (50–100 μM) inhibited, and 1 mM ATP (the concentration obtained with the ATP-regenerating system) completely blocked nucleation (Figure 7A). In contrast, when the cytosol was omitted from the reaction mixture, we saw a significant polymerization of pure actin in the presence of 1 mM ATP (Figure 7B). Thus, the inhibitory effect of high ATP is most likely attributable to its effects on cytosolic components, rather than on actin itself.
Figure 7.

Effects of ATP on polymerization of cytosolic actin. Cytosol (4 mg/ml) was mixed with 1 μM pyrene actin (10% labeled, giving a final concentration of 2% labeling) and increasing concentrations of ATP (A). (B) Pyrene actin (1 μM) (10% labeled) incubated with or without cytosol in the presence of 1 mM ATP. After a 5-min preincubation at 37°C the increase in pyrene fluorescence was estimated by spectrofluoremetry. Relative fluorescence increase was expressed in arbitrary units.
Analysis of the Viscoelasticity and the Onset of Polymerization by Rheology
A complementary kinetic description of the polymerization of actin, with and without membranes could be provided by rheology. We therefore investigated the macroscopic, viscoelastic properties of the fusion mixture by using a rotating disc rheometer (Müller et al., 1991). This setup has been extensively applied to characterize the rheological properties of entangled solutions of pure actin, and their modifications by various actin-binding proteins (Sackmann, 1997). Because we did not detect significant polymerization of microtubules (Egeberg et al., in preparation) or intermediate filaments (our unpublished EM data) under our in vitro conditions, it seems likely that the major part of the rheological effects we observe are due to the actin cytoskeleton that develops in this system. Indeed, the results we describe are quantitatively similar in many aspects to previous data from pure actin systems (Sackmann, 1997).
Here, we analyzed those viscoelastic parameters generally used to describe the polymerization of actin (and other polymers) in vitro in terms of the absolute value of the complex modulus, ‖G*‖, and the phase shift, ϕ. ‖G*‖ is a measure for the stiffness of the system. This parameter increases with the concentration and length of the assembling polymers due to their sterical interactions with each other (see MATERIALS AND METHODS for more background). From ϕ one gains information on the ratio of the viscosity to the elasticity of the system. The onset of polymerization is indicated by a sudden decrease of the phase shift from the value ϕ = π/2, characteristic for pure fluids. This system is a highly sensitive and sophisticated indicator of both the kinetics of polymerization of actin in the system, and the assembly of cross-linked networks and gel formation. In the four data sets we provide (in Figure 8 A–D) ‖G*‖ are shown at two different scales (i and ii), whereas the curves for ϕ are given in iii.
Figure 8.


Rheology: the development of the rheological parameters, the complex modulus, ‖G*‖, and the phase shift, ϕ, of the different components are shown, all at one fixed frequency. In all figures we show two parameters on three different plots: i) magnitude of complex modulus, ii) magnification of i to better resolve the onset of the increase of ‖G*‖, and iii) phase shift of the complex modulus. Note that the point at which ϕ decreases abruptly indicates the beginning of polymerization. At the end of the reaction an indication of gelation state is reflected in how close the plot approaches the theoretical infinite gel at the lower right corner. Open symbols in all figures denote samples containing an ATP-regenerating system; closed symbols indicate samples with an ADP-depleting system. (A) Parameters for pure actin and actin in cytosol (indicated; both conditions contained 4 μM actin). Note the different behavior of pure actin from actin in cytosol. Under these conditions with high-ATP pure actin starts to polymerize after a short appreciable lag (first arrow in A, ii), whereas a 15-min lag time is seen with low ATP (second arrow in A, ii). This difference is more evident in the deflection of the phase angle (A, iii; arrows). Actin from cytosol showed a lag of 35 min with low ATP and 75 min with high ATP (A, iii). Although the pure actin showed only a moderate increase in viscoelasticity with both high and low ATP, the actin in cytosol formed a stiff gel with low ATP but not with high ATP (A, i and ii). (B) Comparison of the rheology of actin in cytosol alone and in cytosol in the presence of phagosomes (at one-tenth the concentration used in the standard fusion assay). Note that under high-ATP conditions the presence of phagosomes greatly enhances viscoelasticity (B, i and ii) over cytosol alone and reduces the lag phase before polymerization (B, iii). However, with high ATP the phagosomes did not increase the ‖G*‖ above the values obtained by cytosol alone with low ATP (i and ii). With both high and low ATP the membranes slightly delayed the onset of polymerization relative to cytosol with low ATP (iii). (C) Comparison of cytosol alone, cytosol plus phagosomes, and cytosol with phagosomes plus PNS. Note the striking effects of the mixture with phagosomes and PNS to increase both viscoelasticity (i and ii) and reduce the onset of polymerization (iii) relative to the other conditions, both with high and low ATP. Note also that although the stiffest actin gel is formed when membranes and ATP are present, the initial onset of polymerization occurs much faster with membranes at low ATP. (D) Comparison of cytosol alone, cytosol plus phagosomes with cytosol plus two aliquots of PNS membranes, all with high ATP only. Both the increase in viscoelasticity (i and ii) and the reduction in the lag before actin polymerization (iii) are significantly enhanced as the concentration of membranes increases.
Pure Actin
We first investigated the rheological properties of muscle actin alone at the concentration found in the standard fusion assay (4 μM) (Figure 8A, i–iii). After a lag phase of <5 min (see first arrow in Figure 8A, iii) actin polymerized rapidly with high ATP and developed a significant viscoelasticity that continued to grow steadily over 80 min (Figure 8A, i and ii). As expected, with low ATP, actin needed longer to start polymerizing (∼15-min lag), as was most clearly evident from the ϕ versus time plot (see second arrow in Figure 8A, iii), and stiffened slightly less than did the ATP actin (Figure 8A, i and ii). This agrees well with the study by Janmey et al. (1990).
Actin in Cytosol
The behavior of endogenous actin (∼4 μM) in cytosol (4 mg/ml protein) was different from that of pure actin. First, the lag phases in cytosol were significantly longer but more strikingly, it was under low ATP conditions that actin in cytosol first started to polymerize (at ∼35 min), and thereafter formed a resilient gel, which was stiffer than the corresponding pure actin solution by more than a factor of 2. Under high-ATP conditions we could detect the start of polymerization only after ∼70 min (Figure 8A, iii), without developing a significant viscoelasticity (Figure 8A, i and ii).
Cytosol plus Phagosomes
Figure 8B shows the rheological properties of the system when one-tenth of the usual concentration of phagosomes used in the fusion assay (for technical reasons described in MATERIALS AND METHODS) was added to the cytosol with high or low ATP. At the scale shown in (Figure 8B, i), it is clear that under high-ATP conditions, the phagosomes greatly enhanced the stiffness of the cytosol with a dramatic increase in viscoelasticity after ∼50 min of incubation (Figure 8B, ii). The addition of avidin beads (without membranes) did not influence the pattern of actin polymerization from cytosol.
As seen in Figure 8B, iii, of the four conditions tested, it was the cytosol with low ATP that polymerized fastest, and under this condition phagosomes slightly delayed its onset. However, also phagosomes with cytosol at low ATP showed a faster increase in polymerization relative to the high-ATP state. At steady state the overall stiffness achieved by phagosomes with high ATP was identical to that found for cytosol alone at low ATP (Figure 8B, i and ii).
Cytosol, Phagosomes plus PNS
When PNS membranes were added to phagosomes plus cytosol they had a dramatic effect on the system, both with high ATP and, surprisingly, also with low ATP. Under both conditions, the PNS significantly reduced the lag time for the onset of polymerization and led to a more developed stiffness (Figure 8C, i and ii). It was even more surprising to notice that the condition of low ATP and membranes gave the fastest onset of polymerization (15 min) of all the conditions we tested (Figure 8C, iii). In contrast to what was observed with (a low concentration of) phagosomal membranes the PNS membranes greatly facilitated actin polymerization, not only with high but also with low ATP. Nevertheless, it was in the presence of high ATP that the total system developed the greatest overall stiffness (Figure 8C, i). Although that combination initially polymerized more slowly than with low ATP, after a significant lag, the system started to stiffen at ∼30 min, and then rapidly developed the highest viscoelasticity we detected.
Figure 8C, iii, shows that the onset of incipient gelation under high-ATP conditions developed fastest with the highest concentration of membranes (total PNS membranes plus phagosomes). This idea is extended by the data shown in Figure 8D, i–iii, demonstrating a strong correlation between the onset of rise in ‖G*‖ and the fall in ϕ with an increasing concentration of membranes in the system. The data in Figure 8, C and D, also suggest that membranes greatly enhance a process that appears to go on similarly, albeit more slowly in the cytosol by itself (leading to our conclusion that the membranes catalyze actin assembly). Whereas the cytosol alone showed the drop in the phase change at 70 min (indicative of the onset of polymerization), (one-tenth) phagosomes reduced this lag to 50 min; it was lowered to 30 min with one unit of PNS and to 20 min with two (PNS-PNS) (Figure 8D, iii). This is also reflected in the curves for ‖G*‖ in Figure 8D, i and ii.
Collectively, the rheological data make two important points that agree with, and extend the data from sedimentation analysis and pyrene actin. First, membranes catalyze the assembly of actin at ATP levels close to physiological conditions (1 mM). Second, they confirm that actin in cytosol spontaneously polymerizes much faster at low levels of ATP (and other nucleotides) than it does at high levels of ATP (and other nucleotides). The rheology data additionally provide evidence that PNS membranes seem also to have a capacity to enhance actin polymerization under low-ATP conditions. The contribution of the different membrane compartments in the PNS mixture to the processes we describe must await a more detailed investigation.
DISCUSSION
Actin and Fusion
The use of a spectrum of reagents affecting actin (and myosin) in an in vitro phagosome-endocytic organelle fusion assay, as well as the physical appearance of the system collectively convinced us that the actin cytoskeleton must play an important role in this fusion process. Moreover, two actin reagents, cyto D and latrunculin A, also inhibited the fusion in vivo. A detailed explanation of these phenomena is a difficult task. This can best be exemplified by the use of the cytochalasins, which could inhibit exocytic fusion in many studies but stimulate the process in many others (see INTRODUCTION). The effects of these drugs in cells, or cell extracts cannot easily be extrapolated from their well-characterized effects on pure actin. A reasonable explanation that has been offered to partly rationalize some of these findings is that, in some, and perhaps all, cells filamentous actin may locally form a structural barrier that can prevent exocytic fusion. In that case, a removal of some of this actin by drugs that depolymerize it (or by natural processes) may allow fusion to proceed (Burgoyne and Cheek, 1987; Aunis, 1998). This is our tentative explanation for why the addition of an excess of phalloidin-stabilized actin filaments could block fusion in our system.
More difficult to rationalize has been the positive effector function of the actin cytoskeleton, which has been extensively described in the process of exocytosis (Orci et al., 1972; Norman et al., 1994; Muallem et al., 1995; Lang et al., 2000). The results of this study, in conjunction with a parallel study have led us to a testable model (our unpublished results) for one possible mechanism by which membrane-catalyzed actin assembly could facilitate the aggregation of membrane organelles prior to fusion. The model being proposed is that the newly assembled, membrane nucleated actin can bind to (other) phagosomes, or endocytic organelles via a myosin. The subsequent transport of these organelles towards the barbed ends of actin, localized adjacent to the nucleating membrane, allows aggregation and docking of membrane organelles. Myosin V is important for this process (our unpublished results; Al-Haddad, A., Shonn, M.A., Redlich, B., Blocker, A., Burkhardt, J.K., Yu, H., Hammer III, J.A., Weiss, D., Steffen, W., Griffiths, G., and Kuznetsov, S.A., unpublished data). Some elements of our model have been independently proposed by Lang et al. (2000).
Parallel experiments in our group have led us to a rationale for our findings that one of the actin-binding proteins, gelsolin (G1–3), can facilitate membrane fusion. In our actin nucleation assay, by using phagosomes (without cytosol) we have recently found that if phagosomes are pretreated with gelsolin (G1–3) and these organelles are then re-isolated on a gradient, their subsequent ability to nucleate actin is enhanced up to 3-fold (Defacque et al., 2000b). We postulate that this enhancement of actin nucleation by phagosomal-bound gelsolin allows more organelles to aggregate and then fuse on the newly assembled F-actin, according to the above-described model.
Membranes Catalyze Actin Nucleation at High and Low ATP
Our data obtained by using sedimentation and gel analyses, pyrene actin, as well as rheology make a strong case that membranes can catalyze the nucleation and polymerization of F-actin. This point has been repeatedly made by Tilney (1975), and by others on the whole-cell level. Elegant in vitro experiments by Mooseker and Tilney (1975) also showed that isolated (and not sealed) microvilli could nucleate actin. In all cases the actin barbed end is adjacent to the membrane. Other groups have also provided evidence that isolated membranes facilitate actin polymerization (Hashimoto and Tatsumi, 1988; Shariff and Luna, 1990; Shariff and Luna, 1992; Katanaev and Wymann, 1998). The nucleation we describe here in vitro has all the hallmarks of a classical catalysis: without membranes and at physiological ATP levels, actin in cytosol needed ∼70 min to start polymerizing but in the presence of membranes the process was initiated 40 min faster. Moreover, from the rheological measurements, the actin that polymerized in the presence of membranes was much more entangled than that which assembled without membranes. As in enzyme catalysis, in the actin assembly process the membranes are not consumed.
The phagosome-actin nucleation process is undoubtedly complex and a crucial role for ezrin/moesin (Defacque et al., 2000a), PIP2 (Defacque, H., Bos, E., Garvalov, B., Barret, C., Roy, C., Mangeat, P., Shin, H.W., Rybin, V., and Griffiths, G., unpublished data), as well as gelsolin (see above) has been shown using the in vitro phagosome-actin nucleation assay that does not require cytosol. We have also detected ARP2/3 on phagosomes, but until now we have only negative evidence for a role of this complex and its associated proteins, N-Wasp and Cdc42, in the in vitro actin nucleation process. In agreement with this view, we also see no effect of tox B (which cleaves all rho proteins), nor of guanosine-5′-O-(3-thio)triphosphate (our unpublished results). Moreover, there is no GTP requirement in the process. It should be noted that in many recent models (Machesky and Insall, 1999; Blanchoin et al., 2000), ARP2/3 nucleates branching of actin filaments, with exposed barbed ends, from preexisting actin filaments whose origin we argue, is the membrane surface. We believe that the initial de novo nucleation/insertion process on a membrane surface (Gaertner and Wegner, 1991), that we observe in the phagosome system reconstitutes the growth of filaments upon which ARP2/3 could subsequently effect actin branching. We have as yet no evidence for such a mechanism in our system. However, such a link may be expected because N-Wasp and ARP2/3 have been shown to play a role in actin dependent motility of endosomes and lysosomes (Taunton et al., 2000).
Actin in Cytosol Behaves Differently to Pure Actin
We show here that actin, a protein that has been extensively characterized as a pure component behaves differently when it is in a cytosolic extract, and that membranes further modify the system. Our data from different approaches show that with high ATP (1 mM) there was little significant polymerization of cytosolic actin over the time course of our experiments (in the absence of membranes). In contrast, the rates and extents of polymerization were greatly increased when ATP was depleted to ∼20 μM (cytosol alone) or to 4 μM (cytosol plus PNS), coinciding with a drastic reduction in ADP, GTP, and GDP, but not AMP. This behavior, which we refer to as the “ATP paradox,” is the opposite to that found with pure actin that polymerizes more at higher ATP levels. Our data strongly suggest that physiological levels of ATP (1 mM) inhibit actin nucleation from cytosolic extracts. The addition of 100 μM GTP to 1 mM ATP rescued the polymerization of pyrene actin back to the levels seen at low ATP (our unpublished results). The detailed mechanisms of the high ATP inhibition and the derepression at low ATP require further analysis.
Actin and Ischemia
A survey of the literature revealed to us an extensive set of articles describing a general elevation of cellular F-actin under conditions of ischemia, a disease condition that has been especially studied in kidney epithelial cells (Hinshaw et al., 1988; Golenhofen et al., 1995; Kellerman et al., 1996; Sutton and Molitoris, 1998, and references therein). Many of these studies have focused on epithelial cells. Whereas the bulk of F-actin in these cells is predominantly in the apical microvilli and cortex, under physiological ATP, the increased amount of F-actin that polymerizes at low ATP is significantly depleted from the apical pole and mostly accumulates in the perinuclear region. Upon returning to normal ATP levels the normal, mostly apical, distribution of phalloidin-labeling reappeared in these studies. We suggest that this low ATP effect is predominantly due to a derepression of membrane actin nucleation. Rho and cofilin are two actin-regulating proteins that have recently been identified as being involved in the regulation of actin dynamics during ischemia and the recovery process (Raman and Atkinson, 1999; Schwartz et al., 1999).
By taking advantage of a method developed by Rosenblatt et al. (1995) to estimate the nucleotide state of G-actin, we were able to measure the nucleotides bound to G-actin in cytosol at the end of an incubation at 37°C. This analysis showed clearly that under both high- and low-ATP conditions the bulk of G-actin was associated with ATP rather than ADP (and possibly AMP). Thus, even after ATP depletion there was sufficient ATP available in the system (4–20 μM) to bind to the bulk of the G-actin (for which the total in the starting cytosol is only 4 μM). In our system, a major role for ADP-actin in polymerization under low-ATP conditions, as proposed by Carlier (1993), is unlikely. Moreover, the levels of ADP dropped to very low micromolar levels when ATP was depleted.
Our data provide two additional observations that may be general features of actin following ATP depletion in cells. First, membrane nucleation of actin is probably differently regulated at high versus low ATP, and second, as discussed, a condition of low ATP seems to derepress an inhibition of nucleation that is effective at high ATP. The mechanism of the high ATP inhibition of cytosolic actin nucleation is currently under investigation.
ACKNOWLEDGMENTS
We thank Sergei Kuznetsov and Marie-France Carlier for their many suggestions throughout this study that has been generously supported by network grants from the Human Frontier Science organization. The Tβ4 was a generous gift from H. Eichner and W. Voelter, the gelsolin G1–3 was kindly provided by Michael Way and the rhoGDI by Oliver Ullrich and Marino Zerial. We thank Ariel Blocker, Marie-France Carlier, Tony Hyman, and Alan Weeds for critically reading an earlier version of the manuscript, and Sergei Kusnetzov for critically reading many versions of the article. We also thank Enrique M. De La Cruz for helpful comments. Finally, a special thanks to Tom Pollard for intuition, persistence, and many helpful suggestions that led to a significant improvement of this manuscript.
Abbreviations used:
- BDM
2,3-butanedione-2-monoxime
- bHRP
biotinylated horseradish peroxidase
- BSA
bovine serum albumin
- cyto D
cytochalasin D
- HB
homogenization buffer
- HRP
horseradish peroxidase
- ‘high’ ATP = ∼1 μM ATP
‘low’ ATP = 5–20 μM ATP
- PHEM buffer
60 mM PIPES-KOH, 25 mM HEPES-KOH, 10 mM EGTA, 2 mM MgCl2, pH 7.4
- PICs
protease inhibitor cocktail
- PNS
postnuclear supernatant (= total cell membranes, excluding nuclei)
- rhoGDI
rho GDP-dissociation inhibitor
- Tβ4
thymosin β4
REFERENCES
- Aunis D. Exocytosis in chromaffin cells of the adrenal medulla. Int Rev Cytol. 1998;181:213–320. doi: 10.1016/s0074-7696(08)60419-2. [DOI] [PubMed] [Google Scholar]
- Baker JP, Titus MA. Myosins: matching functions with motors. Curr Opin Cell Biol. 1998;10:80–86. doi: 10.1016/s0955-0674(98)80089-6. [DOI] [PubMed] [Google Scholar]
- Bernstein BW, DeWit M, Bamburg JR. Actin disassembles reversibly during electrically induced recycling of synaptic vesicles in cultured neurons. Brain Res Mol Brain Res. 1998;53:236–251. doi: 10.1016/s0169-328x(97)00319-7. [DOI] [PubMed] [Google Scholar]
- Blanchoin L, Amann KJ, Higgs HN, Marchand JB, Kaiser DA, Pollard TD. Direct observation of dendritic actin filament networks nucleated by Arp2/3 complex and WASP/Scar proteins. Nature. 2000;404:1007–1011. doi: 10.1038/35010008. [DOI] [PubMed] [Google Scholar]
- Blocker A, Griffiths G, Olivo JC, Hyman AA, Severin FF. A role for microtubule dynamics in phagosome movement. J Cell Sci. 1998;111:303–312. doi: 10.1242/jcs.111.3.303. [DOI] [PubMed] [Google Scholar]
- Blocker A, Severin FF, Burkhardt JK, Bingham JB, Yu H, Olivo JC, Schroer TA, Hyman AA, Griffiths G. Molecular requirements for bi-directional movement of phagosomes along microtubules. J Cell Biol. 1997;137:113–129. doi: 10.1083/jcb.137.1.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blocker A, Severin FF, Habermann A, Hyman AA, Griffiths G, Burkhardt JK. Microtubule-associated protein-dependent binding of phagosomes to microtubules. J Biol Chem. 1996;271:3803–3811. doi: 10.1074/jbc.271.7.3803. [DOI] [PubMed] [Google Scholar]
- Boevink P, Oparka K, Santa Cruz S, Martin B, Betteridge A, Hawes C. Stacks on tracks: the plant Golgi apparatus traffics on an actin/ER network. Plant J. 1998;15:441–447. doi: 10.1046/j.1365-313x.1998.00208.x. [DOI] [PubMed] [Google Scholar]
- Brown AM, O'Sullivan AJ, Gomperts BD. Induction of exocytosis from permeabilized mast cells by the guanosine triphosphatases Rac and Cdc42. Mol Biol Cell. 1998;9:1053–1063. doi: 10.1091/mbc.9.5.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgoyne RD, Cheek TR. Reorganization of peripheral actin filaments as a prelude to exocytosis. Biosci Rep. 1987;7:281–288. doi: 10.1007/BF01121449. [DOI] [PubMed] [Google Scholar]
- Burgoyne RD, Morgan A, Robinson I, Pender N, Cheek TR. Exocytosis in adrenal chromaffin cells. J Anat. 1993;183:309–314. [PMC free article] [PubMed] [Google Scholar]
- Burridge K, Chrzanowska-Wodnicka M. Focal adhesions, contractility, and signaling. Annu Rev Cell Dev Biol. 1996;12:463–518. doi: 10.1146/annurev.cellbio.12.1.463. [DOI] [PubMed] [Google Scholar]
- Carlier MF. Actin polymerization and ATP hydrolysis. Adv Biophys. 1990;26:51–73. doi: 10.1016/0065-227x(90)90007-g. [DOI] [PubMed] [Google Scholar]
- Carlier MF. Dynamic actin. Curr Biol. 1993;3:30–32. doi: 10.1016/0960-9822(93)90192-q. [DOI] [PubMed] [Google Scholar]
- Carraway KL, Carraway CA. Membrane-cytoskeleton interactions in animal cells. Biochim Biophys Acta. 1989;988:147–171. doi: 10.1016/0304-4157(89)90017-8. [DOI] [PubMed] [Google Scholar]
- Cassimeris L, Safer D, Nachmias VT, Zigmond SH. Thymosin beta 4 sequesters the majority of G-actin in resting human polymorphonuclear leukocytes. J Cell Biol. 1992;119:1261–1270. doi: 10.1083/jcb.119.5.1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claus V, Jahraus A, Tjelle T, Berg T, Kirschke H, Faulstich H, Griffiths G. Lysosomal enzyme trafficking between phagosomes, endosomes, and lysosomes in J774 macrophages. Enrichment of cathepsin H in early endosomes. J Biol Chem. 1998;273:9842–9851. doi: 10.1074/jbc.273.16.9842. [DOI] [PubMed] [Google Scholar]
- Condeelis J. Life at the leading edge: the formation of cell protrusions. Annu Rev Cell Biol. 1993;9:411–444. doi: 10.1146/annurev.cb.09.110193.002211. [DOI] [PubMed] [Google Scholar]
- Cramer LP, Mitchison TJ, Theriot JA. Actin-dependent motile forces and cell motility. Curr Opin Cell Biol. 1994;6:82–86. doi: 10.1016/0955-0674(94)90120-1. [DOI] [PubMed] [Google Scholar]
- Defacque H, Egeberg M, Antzberger A, Ansorge W, Way M, Griffiths G. Actin assembly induced by polylysine beads or purified phagosomes: Quantitation by a new flow cytometry assay. Cytometry. 2000b;41:46–54. [PubMed] [Google Scholar]
- Defacque H, Egeberg M, Habermann A, Diakonova M, Roy C, Mangeat P, Voelter W, Marriott G, Pfannstiel J, Faulstich H, Griffiths G. Involvement of ezrin/moesin in de novo actin assembly on phagosomal membranes. EMBO J. 2000a;19:199–212. doi: 10.1093/emboj/19.2.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRosier DJ, Tilney LG. F-Actin bundles are derivatives of microvilli. What does this tell us about how bundles might form? [In Process Citation] J Cell Biol. 2000;148:1–6. [PMC free article] [PubMed] [Google Scholar]
- Desjardins M, Celis JE, van Meer G, Dieplinger H, Jahraus A, Griffiths G, Huber LA. Molecular characterization of phagosomes. J Biol Chem. 1994a;269:32194–32200. [PubMed] [Google Scholar]
- Desjardins M, Huber LA, Parton RG, Griffiths G. Biogenesis of phagolysosomes proceeds through a sequential series of interactions with the endocytic apparatus. J Cell Biol. 1994b;124:677–688. doi: 10.1083/jcb.124.5.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desjardins M, Nzala NN, Corsini R, Rondeau C. Maturation of phagosomes is accompanied by changes in their fusion properties and size-selective acquisition of solute materials from endosomes. J Cell Sci. 1997;110:2303–2314. doi: 10.1242/jcs.110.18.2303. [DOI] [PubMed] [Google Scholar]
- Echner H, Voelter W. Liebig's Annalen der Chemie. 1988. Eine neue Synthese von Thymosin a1; pp. 1095–1097. [Google Scholar]
- Gaertner A, Wegner A. Mechanism of the insertion of actin monomers between the barbed ends of actin filaments and barbed end-bound insertin. J Muscle Res Cell Motil. 1991;12:27–36. doi: 10.1007/BF01781171. [DOI] [PubMed] [Google Scholar]
- Gasman S, Chasserot-Golaz S, Popoff MR, Aunis D, Bader M. Involvement of P GTPases in calcium-regulated exocytosis from adrenal chromaffin cells. J Cell Sci. 1999;112:4763–4771. doi: 10.1242/jcs.112.24.4763. [DOI] [PubMed] [Google Scholar]
- Golenhofen N, Doctor RB, Bacallaø R, Mandel LJ. Actin and villin compartmentation during ATP depletion and recovery in renal cultured cells. Kidney Int. 1995;48:1837–1845. doi: 10.1038/ki.1995.482. [DOI] [PubMed] [Google Scholar]
- Goodson HV, Valetti C, Kreis TE. Motors and membrane traffic. Curr Opin Cell Biol. 1997;9:18–28. doi: 10.1016/s0955-0674(97)80147-0. [DOI] [PubMed] [Google Scholar]
- Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509–514. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- Hashimoto K, Tatsumi N. Regulation of actin polymerization by membrane fraction of platelets. Biochem Int. 1988;16:267–278. [PubMed] [Google Scholar]
- Hinshaw DB, Armstrong BC, Burger JM, Beals TF, Hyslop PA. ATP and microfilaments in cellular oxidant injury. Am J Pathol. 1988;132:479–488. [PMC free article] [PubMed] [Google Scholar]
- Jahraus A, Tjelle TE, Berg T, Habermann A, Storrie B, Ullrich O, Griffiths G. In vitro fusion of phagosomes with different endocytic organelles from J774 macrophages. J Biol Chem. 1998;273:30379–30390. doi: 10.1074/jbc.273.46.30379. [DOI] [PubMed] [Google Scholar]
- Janmey PA, Hvidt S, Oster GF, Lamb J, Stossel TP, Hartwig JH. Effect of ATP on actin filament stiffness. Nature. 1990;347:95–99. doi: 10.1038/347095a0. [DOI] [PubMed] [Google Scholar]
- Katanaev VL, Wymann MP. GTPgammaS-induced actin polymerisation in vitro: ATP- and phosphoinositide-independent signaling via Rho-family proteins and a plasma membrane-associated guanine nucleotide exchange factor. J Cell Sci. 1998;111:1583–1594. doi: 10.1242/jcs.111.11.1583. [DOI] [PubMed] [Google Scholar]
- Kellerman PS, Norenberg SL, Jones GM. Early recovery of the actin cytoskeleton during renal ischemic injury in vivo. Am J Kidney Dis. 1996;27:709–714. doi: 10.1016/s0272-6386(96)90107-9. [DOI] [PubMed] [Google Scholar]
- Komuro R, Sasaki T, Takaishi K, Orita S, Takai Y. Involvement of Rho and Rac small G proteins and Rho GDI in Ca2+− dependent exocytosis from PC12 cells. Genes Cells. 1996;1:943–951. doi: 10.1046/j.1365-2443.1996.760276.x. [DOI] [PubMed] [Google Scholar]
- Kouyama T, Mihashi K. Fluorimetry study of N-(1-pyrenyl)iodoacetamide-labeled F-actin. Local structural change of actin protomer both on polymerization and on binding of heavy meromyosin. Eur J Biochem. 1981;114:33–38. [PubMed] [Google Scholar]
- Kuznetsov SA, Langford GM, Weiss DG. Actin-dependent organelle movement in squid axoplasm. Nature. 1992;356:722–725. doi: 10.1038/356722a0. [DOI] [PubMed] [Google Scholar]
- Lamaze C, Fujimoto LM, Yin HL, Schmid SL. The actin cytoskeleton is required for receptor-mediated endocytosis in mammalian cells. J Biol Chem. 1997;272:20332–20335. doi: 10.1074/jbc.272.33.20332. [DOI] [PubMed] [Google Scholar]
- Lang T, Wacker I, Wunderlich I, Rohrbach A, Giese G, Soldati T, Almers W. Role of actin cortex in the subplasmalemmal transport of secretory granules in PC-12 cells. Biophys J. 2000;78:2863–2877. doi: 10.1016/S0006-3495(00)76828-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machesky LM, Insall RH. Signaling to actin dynamics. J Cell Biol. 1999;146:267–272. doi: 10.1083/jcb.146.2.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mermall V, Post PL, Mooseker MS. Unconventional myosins in cell movement, membrane traffic, and signal transduction. Science. 1998;279:527–533. doi: 10.1126/science.279.5350.527. [DOI] [PubMed] [Google Scholar]
- Mooseker MS, Tilney LG. Organization of an actin filament-membrane complex. Filament polarity and membrane attachment in the microvilli of intestinal epithelial cells. J Cell Biol. 1975;67:725–743. doi: 10.1083/jcb.67.3.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muallem S, Kwiatkowska K, Xu X, Yin HL. Actin filament disassembly is a sufficient final trigger for exocytosis in nonexcitable cells. J Cell Biol. 1995;128:589–598. doi: 10.1083/jcb.128.4.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller O, Gaub HE, Bärmann M, Sackmann E. Viscoelastin moduli of sterically and chemically cross-linked actin networks in the dilute to semidilute regime: measurements by an oscillating disk rheometer. Macromolecules. 1991;24:3111–3120. [Google Scholar]
- Norman JC, Price LS, Ridley AJ, Hall A, Koffer A. Actin filament organization in activated mast cells is regulated by heterotrimeric and small GTP-binding proteins. J Cell Biol. 1994;126:1005–1015. doi: 10.1083/jcb.126.4.1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orci L, Gabbay KH, Malaisse WJ. Pancreatic beta-cell web: its possible role in insulin secretion. Science. 1972;175:1128–1130. doi: 10.1126/science.175.4026.1128. [DOI] [PubMed] [Google Scholar]
- Pardee JD, Simpson PA, Stryer L, Spudich JA. Actin filaments undergo limited subunit exchange in physiological salt conditions. J Cell Biol. 1982;94:316–324. doi: 10.1083/jcb.94.2.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peveri P, Heyworth PG, Curnutte JT. Absolute requirement for GTP in activation of human neutrophil NADPH oxidase in a cell-free system: role of ATP in regenerating GTP. Proc Natl Acad Sci USA. 1992;89:2494–2498. doi: 10.1073/pnas.89.6.2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard TD. Rate constants for the reactions of ATP- and ADP-actin with the ends of actin filaments. J Cell Biol. 1986;103:2747–2754. doi: 10.1083/jcb.103.6.2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard TD, Almo S, Quirk S, Vinson V, Lattman EE. Structure of actin binding proteins: insights about function at atomic resolution. Annu Rev Cell Biol. 1994;10:207–249. doi: 10.1146/annurev.cb.10.110194.001231. [DOI] [PubMed] [Google Scholar]
- Raman N, Atkinson SJ. Rho controls actin cytoskeletal assembly in renal epithelial cells during ATP depletion and recovery. Am J Physiol. 1999;276:C1312–C1324. doi: 10.1152/ajpcell.1999.276.6.C1312. [DOI] [PubMed] [Google Scholar]
- Riezman H, Munn A, Geli MI, Hicke L. Actin-, myosin- and ubiquitin-dependent endocytosis. Experientia. 1996;52:1033–1041. doi: 10.1007/BF01952099. [DOI] [PubMed] [Google Scholar]
- Rogers SL, Gelfand VI. Myosin cooperates with microtubule motors during organelle transport in melanophores [comment] Curr Biol. 1998;8:161–164. doi: 10.1016/s0960-9822(98)70063-6. [DOI] [PubMed] [Google Scholar]
- Rosenblatt J, Peluso P, Mitchison TJ. The bulk of unpolymerized actin in Xenopus egg extracts is ATP-bound. Mol Biol Cell. 1995;6:227–236. doi: 10.1091/mbc.6.2.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sackmann E. Viscoelasticity, rheology and molecular conformational dynamics of entangled and cross-linked actin networks. In: Isenberg G, editor. Modern Optics, Electronics, and High Precision Techniques in Cell Biology. Heidelberg: Springer Verlag; 1997. pp. 213–259. [Google Scholar]
- Schwartz N, Hosford M, Sandoval RM, Wagner MC, Atkinson SJ, Bamburg J, Molitoris BA. Ischemia activates actin depolymerizing factor: role in proximal tubule microvillar actin alterations. Am J Physiol. 1999;276:F544–F551. doi: 10.1152/ajprenal.1999.276.4.F544. [DOI] [PubMed] [Google Scholar]
- Shariff A, Luna EJ. Dictyostelium discoideum plasma membranes contain an actin-nucleating activity that requires ponticulin, an integral membrane glycoprotein. J Cell Biol. 1990;110:681–692. doi: 10.1083/jcb.110.3.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shariff A, Luna EJ. Diacylglycerol-stimulated formation of actin nucleation sites at plasma membranes [see comments] Science. 1992;256:245–247. doi: 10.1126/science.1373523. [DOI] [PubMed] [Google Scholar]
- Sheterline P, Clayton J, Sparrow J. Actin Protein Profile. 1995;2:1–103. [PubMed] [Google Scholar]
- Simon VR, Pon LA. Actin-based organelle movement. Experientia. 1996;52:1117–1122. doi: 10.1007/BF01952110. [DOI] [PubMed] [Google Scholar]
- Small JV, Rottner K, Kaverina I. Functional design in the actin cytoskeleton. Curr Opin Cell Biol. 1999;11:54–60. doi: 10.1016/s0955-0674(99)80007-6. [DOI] [PubMed] [Google Scholar]
- Sutton TA, Molitoris BA. Mechanisms of cellular injury in ischemic acute renal failure. Semin Nephrol. 1998;18:490–497. [PubMed] [Google Scholar]
- Taunton J, Rowning BA, Coughlin ML, Wu M, Moon RT, Mitchison TJ, Larabell CA. Actin-dependent propulsion of endosomes and lysosomes by recruitment of N-WASP. J Cell Biol. 2000;148:519–530. doi: 10.1083/jcb.148.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilney LG. The role of actin in nonmuscle cell motility. Soc Gen Physiol Ser. 1975;30:339–388. [PubMed] [Google Scholar]
- Tilney LG, Cardell Jr. RR. Factors controlling the reassembly of the microvillous border of the small intestine of the salamander. J Cell Biol. 1970;47:408–422. doi: 10.1083/jcb.47.2.408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tranter MP, Sugrue SP, Schwartz MA. Binding of actin to liver cell membranes: the state of membrane-bound actin. J Cell Biol. 1991;112:891–901. doi: 10.1083/jcb.112.5.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trifaro JM, Vitale ML. Cytoskeleton dynamics during neurotransmitter release. Trends Neurosci. 1993;16:466–472. doi: 10.1016/0166-2236(93)90079-2. [DOI] [PubMed] [Google Scholar]
- Trifaro JM, Vitale ML, Rodriguez Del Castillo A. Cytoskeleton and molecular mechanisms in neurotransmitter release by neurosecretory cells. Eur J Pharmacol. 1992;225:83–104. doi: 10.1016/0922-4106(92)90088-d. [DOI] [PubMed] [Google Scholar]
- Vitale ML, Seward EP, Trifaro JM. Chromaffin cell cortical actin network dynamics control the size of the release-ready vesicle pool and the initial rate of exocytosis. Neuron. 1995;14:353–363. doi: 10.1016/0896-6273(95)90291-0. [DOI] [PubMed] [Google Scholar]
- Way M, Gooch J, Pope B, Weeds AG. Expression of human plasma gelsolin in Escherichia coli and dissection of actin binding sites by segmental deletion mutagenesis. J Cell Biol. 1989;109:593–605. doi: 10.1083/jcb.109.2.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetzel MG, Korn ED. Phagocytosis of latex beads by Acahamoeba castellanii (Neff). 3. Isolation of the phagocytic vesicles and their membranes. J Cell Biol. 1969;43:90–104. doi: 10.1083/jcb.43.1.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JG, Borisy GG. On the mechanisms of cytokinesis in animal cells. J Theor Biol. 1983;101:289–316. doi: 10.1016/0022-5193(83)90342-9. [DOI] [PubMed] [Google Scholar]