

Abstract
Leiomyomas are believed to derive from the transformation of myometrial smooth muscle cells/connective tissue fibroblasts. Although the identity of the molecule(s) that initiate such cellular transformation and orchestrate subsequent growth is still unknown, conventional evidence indicates that ovarian steroids are essential for leiomyoma growth. Ovarian steroid action in their target cell/tissue is mediated in part through local expression of various growth factors, cytokines, and chemokines. These autocrine/paracrine molecules with proinflammatory and profibrotic activities serve as major contributing factors in regulating cellular transformation, cell growth and apoptosis, angiogenesis, cellular hypertrophy, and excess tissue turnover, events central to leiomyoma growth. This review addresses the key regulatory functions of proinflammatory and profibrotic mediators and their molecular mechanisms, downstream signaling that regulates cellular events that result in transformation, and commitments of specific cells into forming a cellular environment with a possible role in development and subsequent growth of leiomyomas.
Keywords: Inflammation, cytokines, chemokines, proteases, fibrosis, leiomyoma
Leiomyomas are benign uterine tumors of unknown etiology believed to arise from myometrial cellular transformation. Despite the presence of multiple tumors in the same uterus, they occur independent of metastasis and display a limited capacity of malignant transformation (<1%). Histologically, leiomyomas are well-encapsulated tissues that primarily consist of smooth muscle cells and connective tissue fibroblasts, with a limited vascularization network. Cytogenetic analysis consistently identified several nonrandom chromosomal rearrangements in leiomyomas that are considered to account for their tumorigenesis; however, inconsistency in their occurrence implies an indirect participation in the development of leiomyomas.1–4 Currently, the identity of factor(s) that initiate the transformation of myometrial cells into leiomyomas and orchestrate their subsequent development remains unknown. However, the development of leiomyomas during the reproductive years and regression following menopause implicate a direct role for ovarian steroids as their key growth-promoting factors. Substantial evidence has also been generated in support of the expression and function of various locally expressed growth mediators, including oncogenes and tumor suppressor genes, in regulating leiomyoma growth and regression. Whether these mediators participate in initial cellular transformation or development of leiomyomas is unknown; their differential expression in leiomyomas and regulation of leiomyoma cells under in vitro conditions implicate their potential function in growth and regression.
Despite the presence of multiple tumors in the same uterus, a significant percentage of women with leiomyomas are symptom free. However, the presence of symptomatic leiomyomas, due to their rapid growth, uterine bleeding, pelvic pain, and pelvic masses, is the leading reason for hysterectomy.5–7 Clinical and epidemiological observations indicate that symptomatic leiomyomas account for more than a third of all hysterectomies performed.7,8 Whether performed by laparotomy, laparoscopy, or robot-assisted laparoscopy, hysterectomies are major abdominal surgical procedures that carry an increased risk of postoperative morbidity. Intraoperative/postoperative complications due to injuries to the bowel, bladder, blood vessels, or other sites are the leading cause of postsurgical scar formation initiated by various proinflammatory and profibrotic mediators at the site of injury following hysterectomy and/or myomectomy.
In recent years, considerable progress has been made toward several new nonsurgical alternatives to hysterectomy. These interventional procedures include minimally invasive uterine artery embolization (UAE), using particulate emboli to occlude the uterine arteries and to disrupt the blood supply to fibroids, as well as magnetic resonance (MR) imaging-guided focused ultrasound surgery to reduce leiomyoma volume. These procedures are effective in alleviating fibroid-related symptoms and are gaining popularity,9,10 although MR imaging-guided focused ultrasound is only effective in the management of large tumors. However, particulates used in UAE and excessive cellular damage following the conclusion of MR imaging-guided focused ultrasound leaves debris and cellular fragments within the uterine environment that can cause tissue reactivity and a local inflammatory response, respectively. Tissue reactivity to foreign materials and cellular damage are the main cause of local inflammation, and it remains to be determined whether such procedures, through the generation of local inflammatory mediators, result in other site-specific complications. Although it may be rare, aberrant implantation of leiomyoma fragments in the peritoneal environment or abdominal wall after laparoscopic-assisted surgeries can result in the growth of the tumors outside the uterus.11
MEDICAL MANAGEMENT OF LEIOMYOMAS
In addition to the surgical procedures just described, considerable progress has been made toward the medical management of leiomyoma growth through therapeutic interventions. These efforts have been centered on the development of therapeutics that interfere with ovarian steroid biosynthesis or steroid receptor-mediated actions, due to the fact that leiomyoma growth is sensitive to ovarian steroids. Among these agents are the long-acting analogues of gonadotropin-releasing hormone agonist (GnRHa) therapy, which act through the pituitary-ovarian axis and create a hypoestrogenic condition. GnRHa therapies have proven effective in causing leiomyoma regression, but with considerable side effects, thus prolonged therapy is not advised. Because discontinuation of GnRHa therapy results in the rapid return of the tumor to its original size, cotherapy with low doses of cyclic or continuous estrogen “add back” has been effective in reducing GnRHa side effects and allows prolonged therapy. However, cotreatment with medroxyprogesterone acetate reverses the beneficial effect of GnRHa therapy.7 In addition to GnRHa, preclinical and clinical trials with therapeutics that are selective estrogen receptor modulators (SERMs) and, more specifically, selective progesterone receptor modulators (SPRMs), have shown potential as additional therapeutic avenues for managing the growth of leiomyomas.12–16 Several of these trials are investigating the effects of the SERMs raloxifene and tamoxifen and the SPRM’s CDB2914, asoprisnil (J865), and CDB4124, as well as RU-486.12–21 Similar to GnRHa therapy, prolonged therapy with these agents is not advised beyond a few months due to associated negative endometrial and liver side effects.21
There is limited information on how current hormonal therapeutic interventions act at the molecular level and why their use results in the regression of some leiomyomas for some women, but not all, who receive therapy. It is clear that GnRHa therapy acts at the level of the pituitary-ovarian axis to regress leiomyoma growth; however, evidence also supports the direct action of GnRHa on various peripheral tissues, including the uterus, through direct interaction with GnRH receptors. Considerable evidence also exists to support that GnRHa acts directly on cell growth and apoptosis in myometrial and leiomyoma cells by influencing the expression and function of a large number of genes, including proinflammatory and profibrotic mediators.22–25 Several studies have also provided evidence for the direct regulatory action of raloxifene, tamoxifen, RU-486, ZK98299, CDB2914, and asoprisnil on the expression of many genes involved in inflammatory response, fibrosis, and apoptosis, which results in leiomyoma regression.14,26–35 However, unlike RU-486 and ZK98299, which affect both leiomyoma and myometrial cells, CDB2914 and asoprisnil have been reported only to affect leiomyoma cell growth and the expression of several growth factors, cytokines, and proteases, without affecting the same factors in myometrial cells.26,27,29,31,33–35 Although the in vitro evidence is convincing, clinical observations do not support such cell-specific actions for these agents because both leiomyomas and myometrium and their isolated primary cell cultures express progesterone receptors (PRs), and these compounds are developed based on their potential interactions with PRs. The most likely explanation may lie in our limited understanding of the molecular mechanisms underlying ovarian steroid receptor activation and mediated signaling in leiomyomas as compared with other steroid-sensitive cells and tissues. Studies with crystal structures of the progesterone receptor (PR) ligand binding domain complexed with asoprisnil and the corepressors nuclear receptor corepressor (NCOR) and silencing mediator of retinoic acid and thyroid hormone receptor (SMAET) indicated that asoprisnil differentially recruits coactivators and corepressors when compared with RU-486 or progesterone (P4) in a breast cancer cell line; this specific cofactor interaction profile is apparently insufficient to oppose estrogenic activity in the rat uterus.36 In ELT3 leiomyoma cells, asoprisnil demonstrated partial P4-like inhibition of cyclooxygenase expression and enzymatic activity. Considering that leiomyoma and myometrium express several nuclear receptor coactivators and corepressors,37 it is conceivable that common molecular mechanisms may explain the actions of ovarian steroids in all their target cells and tissues, although accumulated evidence supports their unique functions in cell- and tissue-specific manners. This is particularly important because all cell types contain the same genome but only use a selective portion of it to implement their specific function in a time- and cell-dependent context. As such, the biological relevance of ovarian steroid actions in leiomyoma may become highly relevant for gaining a better understanding of the modes of action of current therapeutics if we identify their specific target genes, specifically proinflammatory and profibrotic genes, and the molecular mechanisms of their actions that result in the growth and regression of leiomyomas.
CELLULAR ORIGIN, PHENOTYPIC TRANSFORMATION, AND MEDIATORS OF LEIOMYOMAS
In addition to having benign uterine tumorigenic characteristics, leiomyomas are fibrotic disorders commonly characterized by excess accumulation of extracellular matrix (ECM), which is similarly observed in a wide spectrum of organ-specific fibrotic disorders such as dermal, renal, pulmonary, hepatic, and myocardial fibrosis. Although the etiology of fibrosis is either unknown or poorly understood, it is clear that a complex and multifaceted interactions among various cell types, including epithelial, endothelial, smooth muscle, infiltrating immune/inflammatory related cells, and, most importantly, fibroblasts, are critical for the establishment and progression of the disease. In this context, a strong association between inflammation and an increased risk of fibrinogenesis and tumorigenesis has been well established.
Inflammation involves a well-coordinated response to cellular injuries caused by infection, chemicals, mechanical insult, radiation, and chemotherapeutic drugs, resulting in either local or systematic activation of an innate and adaptive immune response. The innate immune cells (e.g., macrophages, mast cells, dendritic cells, and natural killer cells) are involved in initiating the inflammatory response by releasing various cytokines, chemokines, matrix-remodeling proteases, eicosanoids, and reactive oxygen and nitrogen species at the site of injury. Inflammatory reactions and maintenance of precise cellular homeostasis are critical for normal defense mechanisms and tissue repair processes to proceed; any failure in regulatory mechanisms leading to chronic inflammation could result in establishment of a conductive microenvironment favoring the initiation and progression of fibrinogenesis and tumorigenesis (Figs. 1 and 2).
Figure 1.
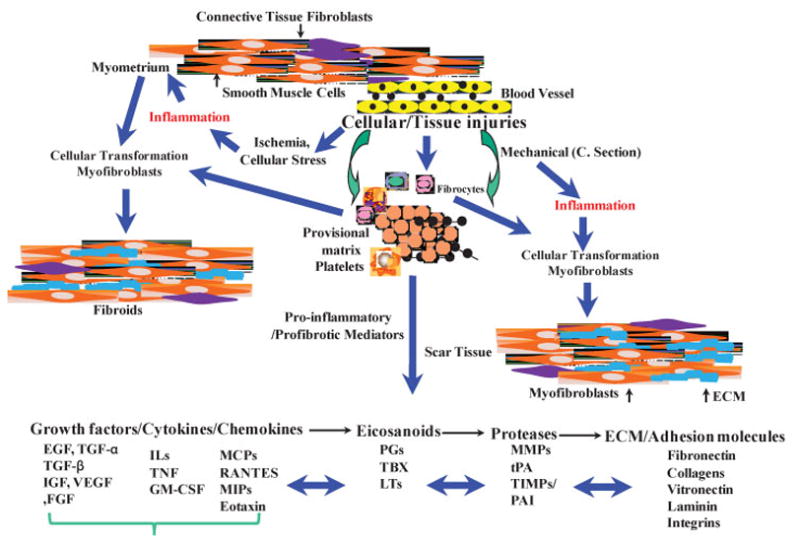
Schematic diagram illustrating an overview of cellular injury and inflammatory-mediated steps in development of leiomyoma as compared with uterine scar formation caused by mechanical injury (i.e., myomectomy and cesarean delivery). Cellular/tissue injuries and generation of an inflammatory response, either at a small scale (microenvironment) or an extended area (myomectomy/cesarean delivery), results in individual and combined regulatory interactions among several proinflammatory and profibrotic mediators. These mediators, including cytokines, chemokines, growth factors, eicosanoids, proteases, and extracellular matrix (ECM), activate and cause myoblasts and resident fibroblasts differentiation into myofibroblastic phenotype. In addition, possible participation of fibrocytes, which are derived from bone marrow and through circulation reside at the site of inflamed/injury, as well as transformation of vascular endothelial cell into mesenchymal cells, also exist to transform into myofibroblastic phenotype. Collectively, myofibroblasts are highly responsive to the action of various mediators, including cytokines and chemokines, and they produce and deposit various components of ECM that are essential for tissue repair process. Continuous inflammation, excess myofibroblastic transformation, and production of large quantity of ECM with concurrent reduced degradation represent a pathway that leads to development of either leiomyoma and/or scar tissue formation. EGF, endothelial growth factor; FGF, fibroblast growth factor; GM-CSF, granulocyte-macrophage colony-stimulating factor; IGF, insulinlike growth factor; IL, interleukin; LT, leukotriene; MCP, monocyte chemoattractant protein; MIP, macrophage inflammatory protein; MMP, metalloproteinase; PAI, plasminogen activator inhibitor; PG, prostaglandin; RANTES, regulated upon activation, normal T-cell expressed and secreted; TBX, thromboxane; TGF, transforming growth factor; TNF, tumor necrosis factor; TIMP, tissue inhibitor of metalloproteinase; tPA, tissue plasminogen activator; VEGF, vascular endothelial growth factor.
Figure 2.

Schematic diagram of pathways involving cellular injury and inflammatory-mediated regulation of fibrinolysis through the individual and combined actions of several cytokines and chemokines. Because fibrinolysis is an essential component of provisional matrix establishment and resolution, and angiogenesis and a key part of cellular/tissue repair involving the activation of transforming growth factor (TGF-β), any alteration in this process can often lead to defective fibrinogenic activity and tumorigenesis, including leiomyomas. IFN, interferon; IL, interleukin; MMP, metalloproteinase; PAI, plasminogen activator inhibitor; TIMP, tissue inhibitor of metalloproteinase; TNF, tumor necrosis factor.
Uterine tissue is susceptible to fibrinogenesis, as seen in rare occasions in women affected by Asherman’s syndrome, or in response to mechanical injury in women undergoing endometrial ablation and cesarean delivery.38–41 However, no evidence exists that uterine mechanical injuries result in development of leiomyomas, although histologically, they resemble fibrotic tissues that develop in response to such injuries at various sites throughout the body. Unlike leiomyomas, fibrinogenesis in other tissues occurs in an age-independent manner and from a common end point of defective wound repair. Fibrinogenesis requires a sustained but not chronic inflammatory condition that results in defective wound repair and tumorigenesis. It is intriguing that similar mechanical injury that causes fibrosis in adults results in near-normal tissue regeneration in fetal skin and is a gestational-dependent event.42 However, graft transplantation of adult skin flaps into fetal skin results in scar formation, whereas homologous transplantation of fetal skin flaps do not, despite well-developed neovascularization in both cases. The molecular mechanism that results in fetal skin regeneration is not well understood; however, the fibroblastic phenotype and genetic imprinting are considered potential mechanisms responsible for the differential outcome of fibrosis in fetal versus adult skin.42
CELL TYPES WITH POTENTIAL OF MYOFIBROBLASTIC TRANSFORMATION
Tissue fibrosis occurs throughout the body as part of a complex defensive mechanism against injuries to isolate surrounding cells from undergoing further cellular damage. Whether they develop as a result of systemic connective tissue diseases or in response to injuries, cellular events leading to fibrosis involve delicate inter-play among many distinct cell types and mediators. In recent years, our understanding of the biology of fibrosis has increased precipitately, primarily through various in vitro and in vivo models, including transgenic and knockout mice models. These studies implicated the existence of a common cellular component among all tissues undergoing fibrosis involving epithelial cell injury, activation and survival of fibroblasts, alternatively activated macrophages, and the infiltration of bone marrow–derived progenitor cells.43–51 Although in specific cases the activation of the epithelium is a key mediator of initial fibrinogenic signaling, in an overwhelming number of cases fibroblasts are the principal cell type involved in the establishment and progression of tissue fibrosis. In addition to resident fibroblasts, evidence implicates possible participation of circulating fibroblast precursors, or fibrocytes, in events leading to fibrinogenesis. Circulating fibrocytes have been detected during wound healing and are considered to ultimately participate in fibrotic disorders such as hypertrophic scars and keloids, scleroderma, renal fibrosis, airway remodeling in asthma, and experimental models of lung fibrosis.44–46,48–50,52,53
The myometrium consists of mainly myometrial smooth muscle cells (MSMCs), connective tissue fibroblasts, vascular cells, and a detectable number of bone marrow–derived progenitor cells.54,55 Although the roles of stem cells, fibrocytes, and vascular-derived cells in the development of leiomyomas are yet to be proven, in the setting of local inflammatory-mediated cellular injury, MSMC and resident connective tissue fibroblasts, because of their sheer numbers, are arguably the most likely cell populations to undergo cellular damage and phenotypic transformation and participate in the process of leiomyoma development. Cellular transformation into a myofibroblastic phenotype is key to the establishment and progression of fibrinogenesis (Fig. 1).43,46,56,57 In the case of tissues undergoing epithelial injury, epithelial to mesenchymal transition (EMT), is a determining factor in the outcome of fibrinogenesis.43,46,56,57 Considerable evidence exists with respect to the molecular mechanisms of EMT, endothelial-MT, and mesenchymal to epithelial (MET) transition; more specifically, during the developmental stage in which cells display a remarkable phenotypic plasticity. Under this context, loss of cell-cell adhesion, increased motility, cytoskeletal and morphological changes, and resistance to apoptosis are among changes that occur as a result of EMT during the development, events that also occur during wound healing and tissue fibrosis. In tissues such as lung, kidney, liver, and skin, all with a prominent epithelial cell layer, EMT, endo-MT, and MET are integrated parts of the tissue repair process and fibrosis, and appear to involve similar cellular changes and molecular pathways.43,46,56,57 In the absence of EMT, alternative cellular transition from other cell types into the fibroblastic phenotype, potentially including endo-MT and macrophage transformation into fibroblasts, can also participate in this process. In either case, fibroblasts require an enriched microenvironment to proliferate and undergo differentiation before developing into reparative or fibrotic mechanisms at the site of tissue damage.
Cellular insult caused by various injuries often results in the infiltration of platelet-rich plasma into the tissue, forming a fibrinous clot at the site of injury. The extent of fibrinous clot formation, also referred as provisional matrix, depends on the degree of tissue damage.58 The provisional matrix is a rich source of many active molecules and an essential component of the normal tissue repair process (Fig. 1). The infiltration of inflammatory- and immune-related cells from peripheral blood into the provisional matrix and induction of a local inflammatory response are important features of soft tissue repair because removal of this matrix or depletion of macrophages at the site of injury results in defective tissue debridement, fibroblast proliferation, and wound repair.58 The recruitment of inflammatory-related cells, more specifically, macrophages, to the site of injury is further sustained by platelet-derived cytokines, as well as other chemoattractant factors. These factors include fibrinopeptides cleaved from fibrinogen by thrombin, degradation products of fibrin produced by plasmin, platelet factor 4, eicosanoids (leukotriene (LT)B4, LTC4, and prostaglandin E2 [PGE2]), and platelet activating factor released from endothelial cells or activated neutrophils.59,60 The recruitment of inflammatory cells to the site of injury is also facilitated by adhesive molecules such as fibrin, fibronectin, vitronectin, and other ECM that are present in the provisional matrix, as well as integrins that recognize these molecules.59–64 The infiltrating monocytes that become activated and differentiate into macrophages are a major source of growth factors, cytokines, chemokines, eicosanoids, and proteases. In addition to macrophages, neutrophils, T cells and mast cells, smooth muscle cells and connective tissue fibroblasts are the source of many of these molecules, or one may consider them as nonprofessional inflammatory related cells.
Fibroblasts are derived from the surrounding connective tissue and migrate into the provisional matrix, proliferate, and simultaneously undergo differentiation, acquiring a myofibroblastic phenotype defined by the expression of α-smooth muscle actin.65,66 Unlike connective tissue fibroblasts, fibrocytes express α-smooth muscle actin, which is characteristic of fibroblasts after transition into the myofibroblastic phenotype. Ultimately, fibroblasts, fibrocytes, and progenitor stem cells derived from circulation, as well as other cell types, are transformed and differentiate into the myofibroblastic phenotype. In the case of smooth muscle cells, their transformation into the myofibroblastic phenotype results in the expression of vimentin, a somewhat common characteristic of fibroblasts. Myofibroblasts, through the production of ECM, form a myofibroblast-enriched tissue with the ability to respond to various growth-promoting factors and to produce more ECM that can develop into fibrotic tissue such as leiomyoma. However, under normal tissue repair processes with limited inflammatory response, remodeling of the provisional matrix by various proteases, removal of fibroblasts through apoptosis, and re-epithelialization of the damaged cells results in near-normal tissue repair.67
The development of leiomyoma and its transformation into fibrotic benign tumors could also occur in response to local inflammatory reactions and the involvement of similar cellular components, despite the lack of evidence of the existence of chronic inflammation in the myometrium. There are reasons to believe that a micro-inflammatory environment with a constant supply of a variety of inflammatory mediators, by maintaining interplay between the extrinsic and intrinsic pathways, could promote myometrial cellular transformation into tumorigenesis and fibrinogenesis, as in leiomyomas. Such local inflammation could occur in response to cellular stress caused by either exogenous or endogenous agents, and by cells undergoing apoptosis. Sustained local inflammation can cause the generation of reactive oxygen and nitrogen species, which can function as chemical effectors in such a micro-environment. Inflammatory-and immune-related cell migration into the inflamed site, as well as local myometrial smooth muscle cells, fibroblasts within the connective tissue, and other vasculatures, all express various types of inflammatory mediators including a large number of cytokines, chemokines, eicosanoids, oxygen and nitrogen radicals, and proteases.22,68–73
The individual and combined actions of these mediators function to initiate, amplify, and control many molecular events that ultimately lead to resolution of the inflammatory response. At the same time, excess generation of these mediators and prolonged inflammation also serve as plausible mechanisms that initiate cellular transformation of the neighboring cells through DNA damage, leading to the activation of oncogenes and/or inactivation of tumor suppressor genes. In the case of myometrial cellular transformation into leiomyoma cells, the outcome is the generation of the myofibroblastic phenotype, which is the main cellular component of fibrinogenesis in all fibrotic tissue. Thus, conceptually, many cellular and molecular events that initiate progression of tissue fibrosis may occur during leiomyoma growth, but modalities that result in their development place leiomyomas into a different category, being unique tumors with fibrotic characteristic.
PROINFLAMMATORY MEDIATORS AND THEIR POTENTIAL ROLE IN LEIOMYOMA DEVELOPMENT AND GROWTH
Considerable evidence exists to support the role of proinflammatory and profibrotic mediators in normal cellular activities and in the pathophysiology of many diseases, including tumorigenesis and fibrinogenesis. In this context, localized cellular exposure to inflammatory mediators often results in cell surface activation of phospholipids that initiate the formation of many intrinsic substrates, including eicosanoids such as prostacyclin, thromboxane, leukotrienes, antithrombin III, protein C, plasminogen activators, and plasminogen activator inhibitors (Fig. 3).60–63,74–78 The expression and activities of these substances are regulated by the local release of cytokines, chemokines, and oxygen radicals produced by activated inflammatory and immune cells (Fig. 3). In addition, some cytokines and chemokines released and retained in the extracellular compartment before local cellular injury can become activated and serve as chemotactic factors, further potentiating the migration of inflammatory cells into the area.
Figure 3.

Schematic diagram of pathways involving cellular injury and inflammatory-mediated regulation of proteolytic activities involving matrix metalloproteinases (MMPs) and their regulation by several cytokines and chemokines. MMPs and their tissue inhibitors play a central role in provisional matrix degradation, cellular migration, angiogenesis, as well as tissue turnover, processes required for normal cellular/tissue regeneration and repair. Aberrant expression and activation of MMPs has been associated with various fibrinogenic and tumorigenic disorders, including leiomyomas. ECM, extracellular matrix; tPA, tissue plasminogen activator; uPA, urokinase plasminogen activator.
Many cytokines and chemokines regulate the production of eicosanoids and proteases, which are essential factors that mediate the inflammatory response.60–63,74–78 Eicosanoids act as intracellular mediators of ovarian steroids, cytokines, chemokines, and growth factor signaling in various cell types, and their elevated production has been associated with an increased incidence of tissue fibrosis; inhibition of their actions using nonsteroidal anti-inflammatory drugs reduces the effect. Leukotrienes are potent chemoattractant factors for many inflammatory-related cells and have been implicated in the pathogenesis of a variety of inflammatory processes.60,79–83 Myometrium and leiomyomas express all of the components of cyclooxygenase and lipo-oxygenase pathways, and they contain prostaglandin, thromboxane, and leukotriene receptors that mediate their actions in the tissues.69,84–87 In addition, it is generally accepted that the actions of ovarian steroids, specifically the mitogenic action of estrogen, are mediated in part through the expression of autocrine/paracrine growth factors, cytokines, and chemokines. Interaction of these mediators with their receptors activate various signaling pathways, resulting in consequent regulation of cell growth and differentiation, apoptosis, and expression of ECM and adhesion molecules, proteases, and proteases inhibitors that regulate the outcome of tissue turnover. In the absence of ovarian steroids, many of these events are directly influenced by individual and combined actions of growth factors, cytokines, chemokines, and other active molecules. These molecules collectively regulate the local myometrial micro-inflammatory condition as it proceeds through either a reparative stage or a degradative phase, ultimately resulting in the development of leiomyoma and fibrotic characteristics.
Cytokines act in a predominantly paracrine/autocrine manner and are produced and act at a local micro-environment.88,89 Some of the key characteristics of cytokines are their wide pleiotropy and their element of redundancy in biological function; all cytokines possess many overlapping functions that potentially could be mediated by other cytokines. Many cytokines are rarely produced individually; rather, they are expressed along with other cytokines. Thus the effects of one cytokine may be influenced by others released simultaneously by the same cell or by neighboring cells following activation, resulting in either synergistic or antagonistic effects. What distinguishes cytokines from growth factors is their ability to activate selective intracellular-signaling pathways that are not linked to mitogenesis. These distinctions are not always clear because some downstream cytokine targets are also linked to cell growth, such as MAPK/ERK. Cytokines act through specific cell surface receptors that are grouped into superfamilies based on the presence of homologous regions. These receptors are often present in low numbers; however, they become upregulated following cell activation (for review, see O’Garra and Robinson88 and Stetson et al89). Cytokines are produced in response to cellular stress caused by either exogenous or endogenous agents, and they function to control and minimize cellular damage. However, an uncontrolled and sustained generation of cytokines can lead to altered cell growth, differentiation, and apoptosis.
Chemokines represent a large superfamily of small peptides that are divided into several subgroups according to the spatial arrangement of the first two cysteine residues in the NH2-terminal region, including CXC or α, CC or β, C or γ, and CX3C or δ.90–92 Functionally, chemokines are divided into two groups: inducible inflammatory chemokines, which are produced at sites of inflammation and function to recruit various inflammatory- and immune-related cell types, and constitutive chemokines, which are produced in bone marrow and secondary lymphoid organs where they regulate leukocyte trafficking under noninflammatory physiological conditions. Regulation of these processes is complex and involves both differential secretion and presentation of chemokines in target tissues, as well as variably regulated expression of chemokine receptors during differentiation and activation of inflammatory cells. Chemokines also play regulatory functions in angiogenesis, hematopoiesis, infectious response, and fibrosis (Fig. 4).90,91,93,94 The biological actions of chemokines are mediated through a distinct family of G-protein-coupled receptors that have been identified in various cells types and include 10 CC chemokine receptors (CCR1-CCR10), 6 CXC chemokine receptors (CXCR1-CXCR6), and one CX3C receptor (for review, see Gerard and Rollins,90 Lau et al,91 and Zlotnik and Yoshie92). Although the origin of cytokines and chemokines is broadly known, the precise contribution of each cell type to their production in acute and chronic inflammation remains elusive.
Figure 4.

Schematic diagram of pathways involving cellular injury and inflammatory-mediated regulation through the action of individual or combined interaction of several cytokines and chemokines, including ELR- and ELR+chemokines. Because angiogenesis is a key step in neovacuolization and the reparative process of cellular/tissue injury, any alteration in this process can often lead to increased fibrinogenesis and tumorigenesis, including leiomyomas. IFN, interferon; IL, interleukin; TNF, tumor necrosis factor.
Considerable progress has been made in understanding the molecular etiology of leiomyomas during the past decade. Using various conventional as well as large-scale gene expression profiling and proteomic analysis has led to the identification of a large number of genes and proteins with altered expression, including many growth factors, cytokines, chemokines, eicosanoids, and proteases that exhibit patterns of expression, which, to an extent, may allow the differentiation of myometrium into leiomyomas.95–100 Despite the discrepancies among the results generated from various expression profiling strategies, each individual study allowed, to an extent, the sorting of genes in leiomyomas from matched myometrium based on the expression profile of a set of differentially expressed genes. Thus far, the gene array studies have indicated that the levels of expression, rather than the presence or absence of a selective number of genes, including many growth factors, cytokines, and chemokines, differentiate leiomyomas from myometrium, independent of tumor size.95–100 This phenomenon is not unique to leiomyomas and myometrium because studies with gene array analyses in many other cell types and tissues have revealed similar results, including the comparative analysis between leiomyomas and keloids.24,101–103
Because leiomyomas develop more frequently among African Americans as compared with other ethnicities, subjecting paired leiomyoma and myometrium from African Americans and whites to genomic and proteomic analyses yielded considerable similarity, relative to the molecular environments regulating fibroids, with differences seen in a gene-specific manner rather than in an ethnic-specific manner.98,104,105 Because many of the differentially expressed genes identified in different cohorts are known to regulate inflammatory response, angiogenesis, cell cycle, apoptosis, and ECM turnover, their products may account for the rapid growth of leiomyomas and their associated symptoms in African Americans as compared with other ethnic groups.
Despite these advances, we are still far from the identification of specific genes and the molecular mechanisms of action of their products that initiate the development and subsequent growth of leiomyoma. Rather, such efforts, along with rapid progress in various aspects of gene functional analysis in other fields, has led to the identification of genes whose products act as mediators of cell growth and differentiation, apoptosis, angiogenesis, inflammation, oxidative stress, ECM accumulation and tissue turnover, oncogenes and tumor suppression, and so on. These processes, either directly or indirectly, have been associated with the establishment and progression of many pathological disorders, including tumor development and fibrinogenesis, two well-established characteristics of leiomyomas.
IMPLICATION OF INFLAMMATORY-RELATED MEDIATORS
Although the direct contribution of constitutive inflammation to the development of leiomyoma remains speculative, considerable evidence exists in support of the involvement of proinflammatory and profibrotic mediators in the pathogenesis of leiomyomas. The results generated from conventional and gene expression profiling strategies have demonstrated the expression of a large number of growth factors, cytokines, and chemokines with proinflammatory and profibrotic properties in the myometrium and leiomyomas. Among the major cytokines, the expression of interleukin (IL)-1, IL-6, IL-11, IL-13, IL-15, interferon (IFN)γ, tumor necrosis factor (TNF)-α, granulocyte-macrophage colony-stimulating factor (GM-CSF), and erythropoietin have been documented in leiomyomas, with some evidence implicating their biological relevance to leiomyoma patho-physiology.72,106–108 These and other cytokines are recognized as key regulators of inflammation, angiogenesis, and tissue remodeling; events that are central to leiomyoma growth. Of particular interest, overexpression of IL-11, a member of the IL-6 family, and IL-13, a Th2 cytokine with homology to IL-4, serve as key regulators of subepithelial airway fibrosis, particularly through interaction with transforming growth factor (TGF)-β.89,109–117 IL-11 and IL-13, similar to TGF-β, are overexpressed in leiomyomas and are differentially regulated by ovarian steroids, GnRHa, RU-486, and TGF-β. GM-CSF has been shown to induce its own expression and the expression of TGF-β1 in myometrial and leiomyoma smooth muscle cells.106 TGF-β is a potent chemoattractant factor for macrophages and fibroblasts, whereas GM-CSF promotes macrophage uptake of apoptotic neutrophils.118–127 The interactions between TGF-β and GM-CSF may act as an important regulator in maintaining a balance between the inflammatory and immunosuppressive activities of GM-CSF and TGF-β, respectively. Furthermore, GM-CSF markedly increases the expression of TGF-β, and IL-1α, IFN-γ, IL-6, and TNF-α induce the expression of platelet-derived growth factor (PDGF), resulting in progression of tissue fibrosis.118–127
The expression profiles of many chemokines have also been characterized through large-scale gene expression profiling of leiomyomas and matched myometrium. Further evaluation of chemokine expression indicates local production of macrophage inflammatory protein (MIP)-lα, MIP-1β, regulated upon activation, normal T-cell expressed and secreted (RANTES), eotaxin, IL-8, CCR1, CCR3, CCR5, CXCR1, and CXCR2, in both leiomyoma and myometrium, with lower content of eotaxin, MIP-1α, MIP-1β, and CCR5 mRNA in leiomyomas, all with some degree of dependency on the menstrual cycle.128 Eotaxin mRNA expression in myometrial tissue from patients with single nodes was found to be much higher than in those with multiple nodes and higher than in those with proliferating leiomyomas. It has been observed that IL-8 expression in leiomyoma, as well as MIP-1α and CCR3 expression in myometrium, increases in patients with submucosal nodes, with a direct correlation between tumor size and the level of IL-8 and MIP-1β expression.128 The expression of IL-8 and monocyte chemoattractant protein 1 (MCP-1) mRNA levels in myometrial cells were also higher than observed in leiomyoma cells, with highest expression observed during the secretory phase of the menstrual cycle;70,129 expression decreased in GnRHa-treated tissues. Treatment with IL-1α and TNF-α caused a time- and dose-dependent increase in IL-8 production by myometrial cells; treatment with E2 and P4, alone or in combination, resulted in a decrease in MCP-1 production.70
The possibility of cytokines and chemokines participating in cellular events that result in myometrial cellular transformation into leiomyoma cells is conceivable. Plasma-derived fibrinogen released in response to cellular injury at the site of inflammation has been shown to elicit the release of several chemokines, including MIP-1α, MIP-1β, MIP-2, and MCP-1.130,131 MCP-1, in turn, stimulates local cellular LTB-4 production, and the LTB-4 receptor antagonist CP-105–696 inhibits the recruitment of neutrophils and macrophages, which is accompanied by a reduction in MCP-1 expression.60,90,132–141 These results suggest that induction of chemokines following the initiation of cellular and/or tissue injury mediates inflammatory response, occurring directly or indirectly through the production of LTB-4. Interestingly, MIP-2 expression was found to be limited to localized inflamed regions, independent of the expression of proinflammatory cytokines such as TNF-α or IL-1.142 IFN-γ treatment has been shown to selectively inhibit LPS-induced MCP-1, MIP-1α, MIP-1β, and MIP-2 expression and to induce IP-10 expression, which is an antiangiogenic chemokine. IL-3, IL-4, GM-CSF, IL-10, and IL-13 are expressed by myometrial and leiomyoma cells, and they are known to induce the expression of C10, a CC chemokine that regulates the chronic stage of host defense reaction. In addition, MCP-1 and MIP-1α expression is induced by IL-3 and GM-CSF, and it is inhibited by IL-4 and IFN-γ. In contrast, MCP-1 and MIP-1α inhibit IL-3- and GM-CSF-induced C-10 expression, which consequently induces rapid production of TNF-α and MCP-1, and later, an increase in IL-13.139,143–146 IL-13, in addition to its role as a potent anti-inflammatory molecule, serves as a key profibrotic cytokine through its induction of TGF-β. Another cytokine with anti-inflammatory properties is IL-10, which effectively inhibits the expression of MCP-1, MCP-5, MIP-1α, MIP-1β, MIP-2, IP-10, KC, and RANTES. These observations indicate the importance of complex interactions among cytokines and chemokines in inflammatory responses suggest their possible influence in myometrial inflammatory reactions that lead to development of and growth of leiomyomas. Furthermore, polymorphisms in several cytokine genes, including those encoding IL-1β, IL-6, and TNF-α, have been associated with an increased incidence of leiomyoma development.147–150 Further studies correlating polymorphisms in IL-1, IL-2, IL-4, IL-8, IL-12, and IL-18 genes with leiomyoma susceptibility indicated that proportions of the IL-12Rβ1 codon 378, but not other genes, was associated with the disease.148
The production of proteolytic enzymes in response to inflammation is also fundamental to angiogenesis, not only for the degradation of perivascular matrix, but also for the migration and proliferation of fibroblasts and endothelial cells (Figs. 3 and 4). During angiogenesis, the initial migration and proliferation of endothelial cells occurs in a fibronectin-rich ECM, whereas in vascular maturation, which takes place at later stages, it is rich in laminin.58 These processes also involve integrins, the essential components of ECM-cell and cell-cell interactions, which promote cell migration, gene expression, cell differentiation, and other cellular activities.63,75,151–156 At the initial stage of angiogenesis, the production of proteases such as matrix metalloproteinases (MMPs) and serine proteases (fibrinolytic system; plasminogen activators) by endothelial cells is necessary to degrade components of the ECM, including fibronectin and laminin.157–162 However, these proteases are produced in inactive forms and must become activated to initiate their local actions. The proteolytic activities of these enzymes are regulated by naturally occurring physiological inhibitors, tissue inhibitors of MMPs (TIMPs), and plasminogen activator inhibitors (PAIs). Cytokines, chemokines, and growth factors, such as IL-1, IL-8, TNF-α, GM-CSF, vascular endothelial growth factor (VEGF), fibroblast growth factors (FGFs), endothelial growth factor (EGF), TGF-α, TGF-β, PDGF, and IGF-I, all serve as angiogenic enhancing factors due to their ability to regulate the expression of MMPs, the fibrinolytic system and their inhibitors, and endothelial cell proliferation and migration. MMPs, tissue plasminogen activator, urokinase plasminogen activator, TIMPs, and PAIs are expressed by myometrial and leiomyoma cells and, similar to observed in other systems, their expression is regulated by various cytokines, chemokines, and growth factors.22,103,163–168
Hypoxia, a condition that promotes cellular stress and the inflammatory response, alters the expression of several cytokines, chemokines, and eicosanoids, as well as the expression of several proteases and angiogenic factors that enhance the development and progression of fibrosis.169–176 VEGF, FGF, EGF, and TGF-β, either alone or through synergistic interactions, stimulate plasminogen activator (PA) expression, which converts latent TGF-β into its active form.169–176 The activation of TGF-β by inhibiting PA expression provides a feedback loop that modulates FGF and VEGF expression, and, whereas FGF-2 and TGF-β have opposing effects on PA activity, FGF acts as a potent inducer of uPA expression with a relatively modest effect on PAI-I synthesis, and TGF-β downregulates uPA and upregulates PAI-I synthesis.169–176 Several cytokines and chemokines also regulate the expression of the fibrinolytic system, including several interleukins, M-CSF, GM-CSF, and MCP-1.63,64,66,74,75,90,93,111,132,153,158,169,177–182
Angiogenesis also depends on the balance between angiogenic factors and their inhibitors. Among the angiogenic suppressors are cytokines, such as TGF-β, TNF-α, and the interferons.77,93,115,169,171,173,175,176,183–187 Other factors with angiogenic inhibitory activities include collagen synthesis modifiers, protamine that inhibits the mitogenic action of FGF, PF-4, HA, thrombospondin, and angiostatin. The expression of these mediators in the myometrium and leiomyoma implies that both their direct and indirect actions can alter angiogenesis during leiomyoma development and growth. Several antiangiogenic factors known to control and terminate the multistage process of angiogenesis, including protease inhibitors, inhibitors of growth factors, cytokines, and chemokines, are being investigated for their potential in controlling angiogenesis and inflammation. These compounds may be useful as potential agents in the management of leiomyoma growth.
TRANSFORMING GROWTH FACTOR-β: THE PRINCIPAL MEDIATOR OF LEIOMYOMA FIBROSIS
TGF-β is universally recognized as a key profibrotic cytokine due to its multifunctional role in cell growth and differentiation, angiogenesis, apoptosis, inflammation, regulation of ECM, and in regulating the expression of adhesion molecules, proteases, and protease inhibitors. TGF-βs consist of three isoforms: TGF-β1, TGF-β2, and TGF-β3, produced by many cell types as latent proteins and intracellularly cleaved to form two mature homodimeric peptides. The mature TGF-β is noncovalently associated with latency-associated peptide, which is required for efficient secretion and prevents the binding of TGF-β to cell surface receptors, maintaining bioavailability in the ECM compartment. ECM proteins such as latent TGF-β-binding protein 1 (LTBP-1), LTBP-2, -3, and -4 and fibrillin-1 and –2 are involved in sequestration of latent TGF-βs in ECM and functionally regulate their activation.188–202 Activation of latent TGF-β is a prerequisite for binding to specific cell surface receptors, which consist of TGF-β type I, type II, and type III receptors. TGF-β type IIIR, also known as betaglycan, exists in a soluble form and associates with TGF-β in the ECM, serving as a regulatory mechanism in maintaining bioavailability of TGF-β at the tissue level.188,189,203–205 TGF-β mediates its actions through binding to and activation of TGF-β type IIR, resulting in activation of TGF-β type IR and recruitment and activation of several signaling pathways, specifically Smad signaling. Smads are composed of pathway-specific regulatory Smads (Smad1, 2, 3, 5, and 8), the common Smad (Smad4), and the inhibitory Smads (Smad6 and 7). TGF-β type IR activates Smad2/3, which associates with Smad4, causing translocation of the signaling molecules into the nucleus where they direct specific transcriptional responses to TGF-β actions. The inhibitory Smad7 interacts with TGF-β type IR and prevents phosphorylation of regulatory Smads, interrupting TGF-β-induced signaling. TGF-β also activates the MAPK pathway; as such, activation of Ras/MEK signaling acts as a positive regulator of cell growth, whereas activation of the p38 MAPK/JUK pathways elicits negative regulation of cell growth.
TGF-β both stimulates and inhibits cell growth, and its mitogenic activity appears to be indirect and due to the induction of growth factors such as EGF, PDGF, and PDGF α receptor.188,204,205 The role of TGF-β in angiogenesis, similar to its action on cell growth, displays a biphasic effect; at low doses, it synergistically enhances, whereas at high concentrations it decreases vascular invasion of cultured endothelial cells induced by VEGF and FGF. TGF-β inhibits the activities of other angiogenic factors in endothelial proliferation and migration under most cell culture conditions and can stimulate the production of ECM and proteinase inhibitors that are involved in angiogenic activity.190–202 Deletion of the TGF-β1 gene through homologous recombination suggests a fundamental role for TGF-β in regulating inflammatory response, and studies conducted with mice heterozygous for TGF-β1 (+/−) implicate TGF-β’s role in initiation and progression of fibrosis. As such, overexpression of TGF-β is widely accepted as a key factor in several fibrotic disorders, including pulmonary fibrosis, glomerulonephritis, cirrhosis of the liver, and dermal scarring. In various fibrotic disorders, the apoptotic process has been associated with progression of this abnormality.188,203–206
Apoptosis is a mechanism by which cells undergo programmed cell death and is recognized by its characteristic morphological and biochemical changes. In general, apoptosis is mediated through extrinsic (death receptor) and intrinsic (mitochondrial) pathways.207,208 The extrinsic pathway involves binding of a death ligand to its cognate cell surface receptor, where the ligand may be an integral membrane protein on a neighboring cell (e.g., Fas ligand) or a soluble extracellular protein (e.g., TNFα). The intrinsic pathway involves several proteins released from the mitochondrial intermembrane into the cytoplasm (i.e., cytochrome c). TGF-β receptor signaling directly or indirectly participates in the apoptotic process by regulating various components of extrinsic and intrinsic apoptotic pathways.206–208 For instance, the apoptotic process is accompanied by a decrease in the level of Bcl-X L protein and a low Bax/Bcl-X L ratio. TGF-β downregulates Bcl-2 and Bcl-X L while it increases the expression of p53 and Bax; this action of TGF-β occurs through interactions with TNF-α, a key regulator of Bax and p53 expression. TGF-β also activates caspases 3, 8, and 9. TGF-β exerts inhibitory actions on cell growth primarily by arresting cells at the G1 stage of the cell cycle(a stage where cells undergo apoptosis), as well as by downregulation of proliferative proteins, such as c-myc, coupled with upregulation of cell cycle inhibitory proteins p15INK4b, p21CIP1, or p27KIP.203,206 Enhanced Smad2 expression and Smad7 nuclear translocation has been implicated as a mechanism of TGF-β-induced apoptosis in vitro and in vivo.203 Because the gene promoters for TGF-β2 and -3 contain TATA boxes and a common proximal CRE-ATF site, they are subject to hormonal and developmental regulation; however, the TGF-β1 promoter lacks the classic TATA box and has multiple regulatory sites that can be immediately activated by early genes, such as c-jun, c-fos, and EGR-1, and by several oncogenes, such as Abl, Fos, Jun, Ras, and Src.188,203–206 Several different gene products participate in mediating the apoptotic process, inflammatory response, and fibrinogenesis, and they are regulated downstream from these immediate early genes and oncogenes.
The expression, regulation, and potential role of the TGF-β system in leiomyoma has been well documented.17,19,26,28,106,164,165,209–215 TGF-β isoforms, TGF-β receptors, TGF-β-binding proteins, and all of the components involved in their signaling pathways, including Smads, are expressed in leiomyomas and myometrium and their isolated smooth muscle cells. To an extent, the expression of TGF-β systems are elevated in leiomyomas, with a menstrual cycle–dependent occurrence, implicating their regulation by ovarian steroids. In addition to ovarian steroids, TGF-β self-regulates its own expression in leiomyoma and myometrial cells, and GM-CSF, a key cytokine known to regulate fibroblast transition into the myofibroblastic phenotype, regulates the expression of TGF-β as well. Leiomyoma cells were characterized by their ability to release more TGF-β when compared with myometrial cells, which retained more TGF-β, a mechanism that could account for the difference in the local bioavailability of TGF-β in leiomyoma.106 Under culture conditions where leiomyoma and myometrial smooth muscle cells were maintained in a quiescent state, TGF-β isoforms do not act as mitogens; however, they increased the rate of DNA synthesis displaying a higher response to TGF-β1 and TGF-β3 when compared with TGF-β2.106 Other studies have demonstrated a higher level of TGF-β3 mRNA in leiomyomas and a higher mitogenic response to TGF-β3, as compared with TGF-β1 in leiomyoma smooth muscle cells and to myometrial smooth muscle cells.17,19 Comparatively, TGF-β1 has been identified as a dominant isoform in other fibrotic tissues, and TGF-β3-associated fibrotic activity has been found to be due to, and mediated through, the induction of TGF-β1,216–219 although controversial observations in rat dermal wound healing implicate antifibrotic activity for TGF-β3. Because TGF-β isoforms are subject to posttranscriptional regulation, differences in the levels of protein expression may reflect their tissue-specific functions, which are mediated through the same receptor system.
Leiomyoma is characterized by overexpression of TGF-β type IIR when compared with the myometrium and pretreatment of leiomyoma smooth muscle cells (LSMC) with TGF-β type IIR antisense oligomers and/or neutralizing antibodies prevents TGF-β receptor-mediated actions.26 In addition, E2 and TGF-β interactively regulate LSMC growth, and inhibition of endogenous TGF-β or blocking TGF-β receptor action (TGF-β exogenous action) reduces E2 action, TGF-β self-regulation, and PAI-1 expression. These results suggest that cross talk between TGF-β and downstream ER signaling may regulate these and other cellular events in leiomyoma. In addition, the expression of MMPs and their tissue inhibitors, TIMPs, in leiomyoma and myometrium was differentially regulated by TGF-β in MSMCs. MMPs and TIMPs play a central role in inflammatory response, angiogenesis, apoptosis, and ECM turnover, and their regulation by TGF-β further points out the importance of TGF-β in the pathophysiology of leiomyoma. Microarray studies have provided additional evidence of the importance of TGF-β regulatory actions in the expression of many genes, some of which have not been known previously to be a target of TGF-β action.107 GnRHa therapy and treatment of isolated LSMCs and MSMCs with GnRHa has been shown to inhibit TGF-β, TGF-β receptor expression, as well as the expression of several other cytokines and chemokines. In addition to GnRHa, available evidence also indicates that treatment with antiestrogens, P4, SERMs, and SPRMs alters the growth and expression of several proinflammatory and profibrotic mediators in leiomyoma and myometrium, as well as in their isolated smooth muscle cells.26,220,221 Treatment with asoprisnil decreases the expression of proliferating cell nuclear antigen-positive, EGF, IGF-I, TGF-β, EGF receptor, IGF-IRa, TGF-β RII, and Bcl-2 expression, and it increases cleaved caspase-3 in leiomyoma cells but not in myometrial cells. These results suggest that asoprisnil selectively inhibits proliferation by downregulating the expression of growth factors and their receptors and induces apoptosis in leiomyoma cells without affecting proliferation and apoptosis in normal myometrial cells.29 It has also been reported that blockade of TGF-β signaling with the TGF-β type IR kinase inhibitor SB-525334 significantly decreases tumor incidence and multiplicity, and reduces the size of these tumors in the EKR rat model, although pharmacological inhibition of TGF-β signaling was observed to promote the development of epithelial tumors,222 a phenomenon that is also observed in other systems.
TGF-β regulates the expression of Smad3 and Smad7, and this self-regulatory mechanism controls TGF-β receptor-mediated action. We have reported that TGF-β and GnRHa activate Smad in LSMCs and MSMCs, and their cross talk alters the expression, induction, and activation of these and other pathways.215 Because GnRHa also targets the expression of TGF-β-, TGF-β receptors, and, more selectively, Smad7 expression, this suggests that downstream genes regulated by TGF-β receptors are also the targets of GnRHa action. Smad signaling results in the transcriptional activation of ECM, adhesion molecules, proteases and protease inhibitors, as well as cell cycle regulators; alteration in the cellular distribution of the expression of Smads has been associated with tumorigenesis.188,203–206 TGF-β receptors also use components of other pathways, including mitogen-activated protein kinase (MAPK), as part of their intracellular signaling, with functional interactions with PKC, Ca2+/CaM, and Smads. Functional interactions between MAPK and Smad signaling pathways mediate the biological actions of GnRH, TGF-β, and activin (a member of the TGF-β family), which, in turn, regulate the expression of gonadotropins, GnRH, and GnRH receptors.223,224 GnRHa and TGF-β1 treatment increases nuclear translocation of Smads and ERK1/2 in LSMCs and MSMCs, and the differences observed in the mRNA expression of immediate early response genes c-fos and c-jun in LSMCs and MSMCs suggests a postreceptor divergence of signaling in these cells. GnRHa and TGF-β1, through these signaling pathways, alter the expression of type I collagen, fibronectin, and PAI-1 mRNA in LSMCs and MSMCs in a cell- and gene-specific manner.28 Because the products of these genes regulate diverse biological activities, and specifically, ECM turnover, their differential expression may indicate a critical role for these genes in leiomyoma growth and regression.28 In recent microarray studies, several genes in this category were identified displaying differential expression in leiomyoma and myometrium, and they were targeted by GnRH therapy and TGF-β treatment in vitro.23,107,213,225–229
Results generated predominantly from in vitro studies have led to the hypothesis that TGF-β action in tissue fibrosis is indirect and is mediated through the induction of connective tissue growth factor (CTGF or CCN2). Similar to TGF-β, elevated expression of CCN2 has been documented in several fibrotic tissues and is considered a key regulator of connective tissue formation, angiogenesis, and tissue fibrosis.230,231 However, the expression of CCN2 in leiomyomas is inversely correlated with not only TGF-β1, but also with TGF-β3 expression.102 Such an inverse correlation was also observed between the expression of TGF-β1 and TGF-β3, and that of CCN3. However, under in vitro conditions, TGF-β1 increased the expression of CCN2 and CCN4 in LSMCs and MSMCs, whereas it inhibited the expression of CCN3 in the same cells. TGF-β3 has been shown to increase CCN2 expression in LSMCs,232 suggesting that both TGF-β isoforms may have similar functions in regulating the expression of CCNs in LSMCs and MSMCs. Gene expression profiling studies have also revealed a lower expression of CCN2 in hypertrophic scars, which express elevated levels of TGF-β1 as compared with that observed in normal skin.233 These observations in leiomyoma and hypertrophic scars suggest that CCN2 may not serve as a common downstream mediator of TGF-β action in all fibrotic disorders. To understand the biological significance of the variable expression profiles of CCNs and their regulation in leiomyoma and myometrium requires a more detailed investigation, considering the wide range of biological activities for these signaling molecules observed in other cell types.
OTHER PROFIBROTIC MEDIATORS
The EGF family members, including EGF, TGF-α, and heparin-binding (HB)-EGF, serve as mitogenic and differentiation factors for several cell types of ectodermal, mesodermal, and endodermal origins, and they play a key role in the wound healing process and in tissue fibrosis.234–237 Leiomyoma and myometrium express EGF, TGF-α, HB-EGF, and EGF receptor with relatively stable expression throughout the menstrual cycle, although it has been shown that higher levels of EGF are expressed during the luteal phase in patients with leiomyoma.108 The local production of EGF, TGF-α, and HB-EGF, as well as the presence of their receptors, imply an autocrine/paracrine role for these growth factors in regulating biological processes in the myometrium and leiomyomas. The EGF family of growth factors participates in many cellular activities associated with tissue fibrosis, including cell growth and ECM expression by fibroblasts and myofibroblasts, more specifically, through interaction with other growth factors such as PDGFs and IGFs, in both leiomyoma and myometrial smooth muscle.
The PDGF family includes PDGF-AA, PDGF-BB, and PDGF-AB, which are derived from disulfide-bonded homo- and heterodimers of two structurally similar A- and B-polypeptide chains (PDGF-A and PDGF-B), as well as two additional isoforms, PDGF-C and PDGF-D, which are derived from protease activation of other isoforms.238–240 Although PDGF is expressed by a variety of cell types, including inflammatory-related cells, smooth muscle cells, fibroblasts, and myofibroblasts, platelets and macrophages serve as the major source of PDGF.241,242 Macrophage-derived PDGFs act as chemotactic factors for myofibroblasts, and abundant evidence supports the idea that PDGFs play a role in the progression of fibrotic disorders by promoting myofibroblast proliferation, angiogenesis, and ECM production (such as collagen, fibronectin, and glycosaminoglycans).238–240,243 Only limited data are available regarding the expression of PDGFs and their receptors in leiomyomas. The expression of PDGFs and PDGF receptors in myometrium and leiomyoma displays relatively similar abundance throughout the menstrual cycle. Myometrium and primary cultures of MSMCs express PDGF-AB as well as PDGF β receptor, but they express a low level of PDGF α receptors.108 Leiomyoma has been reported to contain more PDGF receptor binding sites than observed in the myometrium, but with the receptors they exhibit lower affinity for PDGF. Direct evidence is currently lacking that confirms that the expression of PDGFs and their receptors is regulated by the ovarian steroids, although reduction in their expression in leiomyoma and myometrium from women who have received GnRHa therapy indirectly supports the possibility for steroid-related regulation of their expression.244
PDGFs regulate their own expression and through their interactions with other growth factors and cytokines, they can induce various cellular activities that promote tissue fibrosis. Adenoviral overexpression of PDGF-B only causes a mild fibroproliferative response, whereas combination with TGF-β1 results in significant mesenchymal cell proliferation and collagen deposition. In addition, PDGF and PDGF receptors serve as mediators of the mitogenic actions of TGF-β and IL-β in fibroblasts and smooth muscle. IFN-γ is reported to alter the expression of PDGF-B,238–240 and evidence exists that that it also regulates IL-13 action in the pathogenesis of lung fibrosis, stimulating the production of PDGF through a STAT-6-dependent mechanism.115 This suggests that the paracrine actions of cytokines serve as important mechanisms for the maximal growth of and chemotactic responses of myofibroblasts. FGF-2 also induces the expression of PDGF receptor in bronchial smooth muscle cells. Soluble PDGF binding proteins such as α2-macroglobulin, PDGF-associated protein, and the extracellular part of the PDGF-a receptor, as well as ECM components, such as secreted protein acidic and rich in cysteine (SPARC), regulate the activity and availability of PDGF isoforms. PDGF receptor tyrosine kinase inhibitors have been used for controlling PDGF-induced tissue fibrosis.243 Administration of the PDGFR selective tyrosine kinase inhibitor AG1296, a tyrphostin analog, has been shown to reduce pulmonary fibrosis in a rat model. Imatinib is a PDGF receptor tyrosine kinase inhibitor that has been shown to ameliorate chronic allograft nephropathy.245
The insulinlike growth factor (IGF) system is composed of IGF-I and -II, the IGF receptors (types I and II), and IGF binding proteins (IGFBPs), as well as a range of IGFBP proteases,1,246,247 and are expressed in temporal and tissue-specific manners. The family is regulated by numerous factors, including hormones, growth factors, and physiological parameters, including nutrition, injury, and disease. IGF-I and IGF-II exhibit considerable homology in amino acid sequence, and both carry significant homology to insulin. IGFBPs exhibit widespread serum, tissue, and extravascular fluid distribution, and through high affinity binding, they regulate the biological activities of IGF. Many components of the IGF family also bind to the ECM where, upon release, they can bind to IGF receptors present on surrounding cell, specifically, to connective tissue fibroblasts. IGF and other growth factors are released from the ECM through proteolytic actions of various proteases, such as neutrophil proteases, cathepsin G, and elastase. Although IGFs have growth-promoting activity in specific cell types, they serve as strong antiapoptotic and differentiation factors for many cell types, including myoblasts.1,246,247 Similar to the mitogenic activity of EGF, IGF-1 mitogenic activity requires interaction with other growth factors, such as EGF and PDGF. PDGF, TGF-α, HB-EGF, and IGF-1, which are expressed and released by many cell types, can potentially act in this manner and influence cell migration, proliferation, and angiogenesis, even at the earliest stages of leiomyoma growth. IGFs, IGFBPs, and IGF receptors are expressed in both leiomyoma and myometrium throughout the menstrual cycle. However, no difference in expression of IGFs, IGFRs, and IGFBPs has been observed in leiomyoma when compared with myometrium.108
The VEGF and FGF families of growth factors are known as key regulating factors for angiogenesis, an important step in the pathogenesis of tissue fibrosis. The VEGF family includes VEGF-A, VEGF-B, VEGF-C, VEGF-D, VEGF-E, and placenta growth factor (PlGF). Alternative exon splicing of VEGF genes results in the generation of several isoforms of each factor, each with multiple and diverse biological functions.176,183 Through alternative splicing of pre-mRNA of the VEGF-A gene, several protein isoforms are generated, which include VEGF121, VEGF145, VEGF165, VEGF183, VEGF189, and VEGF206. VEGF165 is the predominant isoform; VEGF145 and VEGF183 are less frequent isoforms. Most VEGF-A isoforms contain a heparin-binding domain, and VEGF121 is predominantly detected as a soluble form, whereas VEGF189 and VEGF206 are relatively insoluble and are located in association with cells or are sequestered in ECM, binding heparin with high affinity. VEGF-C and VEGF-D induce capillary endothelial cell migration and proliferation, and they act as vascular permeability factors and are abundantly expressed in the heart, lung, skeletal muscle, colon, and small intestine. VEGF’s mediate their actions by binding to the tyrosine kinase receptors Flt-1 (FMS-like tyrosine kinase) and KDR (kinase domain region) receptors, although they may employ other different signal transduction properties. VEGF121, VEGF165, and VEGF189 are expressed in leiomyoma and myometrium and have been characterized by in situ hybridization and found to be localized smooth muscle cells. It has also been observed that levels of VEGF mRNA are significantly higher during the secretory phase than in the proliferative phase of the menstrual cycle in normal myometrium, and that these levels do not change significantly when compared with leiomyoma. Leiomyoma tissue collected from women treated with a GnRH analog did not express significantly different levels of VEGF mRNA as compared with leiomyoma tissue collected from untreated women.108,248 Although ovarian steroids regulate VEGF expression in other steroid-sensitive cells, the lack of direct evidence, as well as the observation that VEGF expression remains unaffected in GnRH tissues, indicates that factors other than ovarian steroid may regulate VEGF expression in leiomyoma and myometrium.
The FGF family is classified into subgroups according to structure, biochemical properties, and expression.249 The biological activity of FGFs is mediated through FGF receptors, which are members of the cell surface receptor tyrosine kinase family. These receptors contain Ig-like domains, which regulate binding affinity and ligand specificity, and a stretch of acidic amino acids (acidic box domain) located between Ig-like domains I and II, followed by a heparin binding region and a cell adhesion homology domain. FGFs are characterized by the lack of an extracellular export sequence, which has become an impediment of their role in angiogenesis. The alternative model of how bFGF can be transported out of the cell is based on binding of bFGF to a heparin sulfate proteoglycan that is located at the cell surface; the heparan sulfate-bound bFGF could then be activated and regulated in a manner through which the action of heparanase bypasses the need for a transport signal.249,250 The expression bFGF, FGF receptor 1, and FGF receptor 2 has been characterized by immunohistochemistry and Western blotting in leiomyomas and in myometrium, with detectable changes observed between leiomyomas and myometrium in staining intensity following GnRHa treatment. No tissue differences were observed when stained for FGF receptor 2 and no significant differences were observed between the untreated and GnRHa-treated groups. It was concluded that the expression and localization of bFGF and, to some extent, its receptors, are influenced by sex steroid hormones in leiomyomas and in myometrium, and lack of differences in expression between leiomyomas and myometrium favors the theory that bFGF may not necessarily contribute to the differences in growth regulation in these tissues.251
Signal transduction cascades activated by growth factor, cytokine, and chemokine receptors are mediated predominantly through Ras/MAPK, signaling, including phosphoinositol kinase, STATs, PKA, PKC, Src family tyrosine kinase, phospholipase C, and antiapoptotic kinase Akt signaling pathways, as well as several other transcriptional regulators. Translocation of the activated signals into the nucleus results in phosphorylation of several transcription factors, including AP-1, Elk-1, and SAP, which, in turn, regulate the expression of target genes. MAPK activation also results in activation of intranuclear proteins, including cyclin D1, which is important for the progression of the cell cycle from G1 to S-phase (for review, see234–240,246,249). MAPK is also activated by integrins, which are reported to be expressed in leiomyoma;252 however the biological significance of this finding remains to be investigated. Focal adhesion kinase (FAK) serves as an integrin signaling molecule and participates in promoting angiogenesis and cell migration, and it is expressed in both leiomyoma and myometrium, exhibiting altered expression during GnRHa-induced tumor regression.253 Integrins have also been associated with processes leading to activation of latent TGF-β. TGF-β receptor has also been implicated in IGFBP-3 cell signaling through Smad2 and Smad3, resulting in IGFBP-3-induced gene transcription and an inhibitory growth response.254 In addition, IGF-II/M-6-PR play a role in TGF-β1 activation by binding to latent TGF-β1.189
REGULATORY FUNCTIONS OF MICRO-RNAS IN INFLAMMATION AND FIBROSIS
Throughout the past several years, micro-RNAs (miR-NAs) have emerged as key regulators of gene expression stability. miRNAs are small (22 nucleotide) RNA that bind to the 3′ untranslated region of target genes, ultimately leading to mRNA cleavage and degradation. miRNAs are transcribed as precursor form from genes that constitute ~3% of the human genome, followed by extensive processing by Drosha, DGCR8/Pasha, and Dicer; in the nucleus and cytoplasm, precursor miRNAs form into mature miRNAs (19 to 24 nucleotides). The mature miRNAs, through interaction with RNA-induced silencing complex (RISC), regulate the expression of their specific target mRNAs.255,256 Evidence generated based on computational algorithms and conformational experiments indicate the presence of thousands of miRNAs, carrying potential to target the expression of 30% of all human genes.
Considerable evidence based on expression profiling supports the role of miRNA in the initiation and progression of many human cancers.257 Expression profiles of a few hundred miRNAs in leiomyomas and matched myometrium, as well as their isolated smooth muscle cells, also support their potential regulatory functions in leiomyoma gene expression stability.258,259 Among the miRNAs whose expression was significantly altered in leiomyomas as compared with myometrium are the let-7 family, miR-17, miR-21, miR-23b, miR-29b, miR-34a, miR-26a, miR-18a miR-125b, miR-139, miR-155, miR-206, miR-181a, and miR-142–5p. Altered expression of some of these miRNAs was also observed to be specifically different in leiomyomas from African Americans when compared with whites and other ethnic groups.258,259 These miRNAs are predicted to target the expression of many genes, including ovarian steroid receptors, TGF-β, TGF-β receptors, and several proinflammatory and immune-related genes. The regulatory functions of miRNAs in inflammation and immune system regulation occur through the well-coordinated control of specific genes, and this regulation is critical for initiation and termination of inflammatory and immune-related processes. Different patterns of expression of specific miRNAs have been observed in hematopoietic cell lineages, and their association with proliferation and differentiation, along with their altered expression in cancer and immune and inflammatory disorders, are evidence that miRNAs play important roles in these processes. Several miRNAs, specifically let7, miR-17–5p, miR-20a, miR-106a, miR-125b, miR-146, and miR-155, have been identified to influence the expression of inflammatory and immune response mediators.256,260 The expression of miR-155, miR-125, miR-17–5p, miR-20a, and miR-106a has been associated with inhibition of the differentiation and maturation of monocytes by regulating the expression of RUNX1 and macrophage colony-stimulating factor receptor (M-CSFR). Inflammation that results in nuclear factor (NF)-κB-dependent activation of monocyte activity results in the induction of miR-146, which, in turn, inhibits TNF receptor–associated factor 6 (TRAF6) and IL-1 receptor–associated kinase 1 (IRAK1) genes, the two key molecules downstream to the Toll-like receptor and cytokine signaling. Activation of murine macrophages by IFN-β also resulted in upregulation of miR-155 through the induction of TNF-α and JNK pathways, whereas miR-155-deficient mice are immunodeficient.261 Furthermore, IL-6 has been reported to induce the expression of let-7a and contribute to survival through the expression and activation of STAT3 phosphorylation.260–264 Hypoxia, which promotes angiogenesis, has been shown to reduce miR-15b and miR-16 expression; miR-221 and miR-222 indirectly reduce the expression of endothelial nitric oxide synthase (eNOS), which is an angiogenic inducer and contributor to other endothelial cell functions.217 The miR-17–92 cluster contains miR-17–5p, miR-17–3p, miR-18a, miR-19a, miR-20a, miR-19b, miR-92–1, and miR-18 and miR-19 have been found to target the expression of CTGF and thrombospondin-1 (TSP-1), respectively.94 The biological significance of miRNA-mediated CTGF regulation is due to the ability of CTGF in mediating the profibrotic actions of TGF-β in tissue fibrosis (for review, see Luo and Chegini258).
Further elucidation of the expression, regulation, and function of miRNAs in leiomyoma as compared with myometrium may allow for providing markers for diagnosis, mechanisms of inflammation-associated regulation of miRNA expression, and miRNA-mediated gene targeting, for therapeutic intervention to regulate leiomyoma growth. Furthermore, establishing the relationship between inflammatory response and miRNA expression profiles in the uterus could assist us in identifying novel links between inflammation, tumorigenesis, and tissue fibrosis.
SUMMARY AND CONCLUSIONS
Symptomatic uterine leiomyomas cause chronic pelvic pain and abnormal uterine bleeding. Medical therapies, which include analgesics or hormones for both pain and abnormal bleeding, are initially used to relieve these symptoms. The most common hormonal therapies involve the use of GnRH agonists. Recent clinical and preclinical investigation has provided evidence supporting the use of SERMs and SPRMs as possible alternative therapies to suppress leiomyoma growth. These therapies are not only expensive but have associated side effects that can be either intolerable to the patient or are capable of causing further morbidity, such as hot flashes and bone loss. However, many patients end up requiring hysterectomies when conservative medical therapy becomes poorly tolerated or ineffective. Hysterectomies are one of the most common operations, and many are directly due to the presence of symptomatic leiomyomas. Hysterectomies are major operations that carry potential risks, such as bleeding, infection and adhesion formation, and possible damage to other vital anatomical structures that can further add to the pain and suffering of the patient while increasing the overall cost to society. It is paradoxical that so little is known about the biology of a tumor that has such a costly morbidity.
In recent years, substantial progress has been made toward the identification and understanding of the molecular environment surrounding leiomyomas. Based on these studies, alongside comparative assessments with other systems, it appears that leiomyoma growth and regression are determined by the rate of cell proliferation, differentiation, apoptosis, angiogenesis, protease production, and ECM deposition. Ovarian steroids and various growth factors, cytokines and chemokines, peptide hormones and stress factors, and so on, are key regulators of many of these processes. However, the biological functions of many of these molecules in leiomyoma remain to be determined. Classically, ovarian steroid action is mediated through interactions of ER and PR with different DNA response elements that regulate transcriptional activation of specific genes that regulate these processes. Growth factors, cytokines, chemokines, however, mediate their actions through rapid recruitment and activation of intracellular signaling proteins that translocate into nucleus and regulate similar/different sets of genes. Ovarian steroids can also activate signaling pathways activated by growth factors/cytokines/chemokines, referred to as “nongenomic action” and is now recognized as a novel pathway that could be activated by these mediators even in the absence of E2. Investigation is needed to test many of the factors/pathways that are used by ovarian steroids, including both genomic and nongenomic pathways, which lead to inflammatory and fibrotic responses and, subsequently, to leiomyoma growth. In addition to leiomyoma smooth muscle cells, activation of many of these pathways and their downstream targeted genes are involved in regulating a wide variety of processes in other leiomyoma cell types that regulate matrix accumulation, angiogenesis, and local inflammatory response.
Leiomyomas develop from transformation of myometrial smooth muscle cells and/or connective tissue fibroblasts. However, the identity of molecule(s) that initiate myometrial cellular transformation and subsequently regulate their growth remains unknown. In vitro studies show that smooth muscle cells are highly plastic and can respond to changes in environmental factors by changing their phenotype. These changes depend on the integration of a large number of molecules that can aggregate and determine the patterns of gene expression appropriate for transformation of myometrial cells into leiomyoma. Recent microarray studies have provided evidence that many genes are differentially expression the myometrium and in leiomyoma, and they can be functionally categorized into transcription factors, as well as regulators of signal transduction pathways, apoptosis, cell and tissue structure, and other metabolic activities. In addition, isolated cells can participate in the transformation of myometrial cells into leiomyoma. However, the expression of many of the putatively identified genes requires confirmation, and definitive studies are needed to elucidate their precise molecular mechanisms of action in initiating the fibrotic response. Definitive studies in this area depend on use of sophisticated conditional gene targeting systems, where understanding their molecular mechanism regulating these genes would assist us to design new therapies complementing GnRHa, SERM, and SPRM that are being used as therapeutic medical management of leiomyoma but with many limitations.
Acknowledgments
I would like to thank Drs. Xiaoping Luo, Li Ding, and Qun Pan for their contributions to various aspects of this research, and Drs. Melinda S. Prucha and Maria B. Padua for their editorial assistance and suggestions. This work is supported by the National Institute of Health grants HD37432 and HD58664.
References
- 1.Holly J. Physiology of the IGF system. Novartis Found Symp. 2004;262:19–26. 26–35, 265–268. [PubMed] [Google Scholar]
- 2.Levy B, Mukherjee T, Hirschhorn K. Molecular cytogenetic analysis of uterine leiomyoma and leiomyosarcoma by comparative genomic hybridization. Cancer Genet Cytogenet. 2000;121(1):1–8. doi: 10.1016/s0165-4608(00)00225-9. [DOI] [PubMed] [Google Scholar]
- 3.Ligon AH, Morton CC. Leiomyomata: heritability and cytogenetic studies. Hum Reprod Update. 2001;7(1):8–14. doi: 10.1093/humupd/7.1.8. [DOI] [PubMed] [Google Scholar]
- 4.Lobel MK, Somasundaram P, Morton CC. The genetic heterogeneity of uterine leiomyomata. Obstet Gynecol Clin North Am. 2006;33(1):13–39. doi: 10.1016/j.ogc.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 5.Parker WH. Uterine myomas: management. Fertil Steril. 2007;88(2):255–271. doi: 10.1016/j.fertnstert.2007.06.044. [DOI] [PubMed] [Google Scholar]
- 6.Parker WH. Etiology, symptomatology, and diagnosis of uterine myomas. Fertil Steril. 2007;87(4):725–736. doi: 10.1016/j.fertnstert.2007.01.093. [DOI] [PubMed] [Google Scholar]
- 7.Wallach EE, Vlahos NF. Uterine myomas: an overview of development, clinical features, and management. Obstet Gynecol. 2004;104(2):393–406. doi: 10.1097/01.AOG.0000136079.62513.39. [DOI] [PubMed] [Google Scholar]
- 8.Stewart EA. Uterine fibroids. Lancet. 2001;357(9252):293–298. doi: 10.1016/S0140-6736(00)03622-9. [DOI] [PubMed] [Google Scholar]
- 9.Bradley LD. Uterine fibroid embolization: a viable alternative to hysterectomy. Am J Obstet Gynecol. 2009;201(2):127–135. doi: 10.1016/j.ajog.2009.01.031. [DOI] [PubMed] [Google Scholar]
- 10.Lénárd ZM, McDannold NJ, Fennessy FM, et al. Uterine leiomyomas: MR imaging-guided focused ultrasound surgery—imaging predictors of success. Radiology. 2008;249(1):187–194. doi: 10.1148/radiol.2491071600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wada-Hiraike O, Yamamoto N, Osuga Y, Yano T, Kozuma S, Taketani Y. Aberrant implantation and growth of uterine leiomyoma in the abdominal wall after laparoscopically assisted myomectomy. Fertil Steril. 2009;92(5):1747, e13–e15. doi: 10.1016/j.fertnstert.2009.07.968. [DOI] [PubMed] [Google Scholar]
- 12.DeManno D, Elger W, Garg R, et al. Asoprisnil (J867): a selective progesterone receptor modulator for gynecological therapy. Steroids. 2003;68(10–13):1019–1032. doi: 10.1016/j.steroids.2003.09.008. [DOI] [PubMed] [Google Scholar]
- 13.Eisinger SH, Meldrum S, Fiscella K, le Roux HD, Guzick DS. Low-dose mifepristone for uterine leiomyomata. Obstet Gynecol. 2003;101(2):243–250. doi: 10.1016/s0029-7844(02)02511-5. [DOI] [PubMed] [Google Scholar]
- 14.Jirecek S, Lee A, Pavo I, Crans G, Eppel W, Wenzl R. Raloxifene prevents the growth of uterine leiomyomas in premenopausal women. Fertil Steril. 2004;81(1):132–136. doi: 10.1016/j.fertnstert.2003.06.009. [DOI] [PubMed] [Google Scholar]
- 15.Palomba S, Russo T, Orio F, Jr, et al. Effectiveness of combined GnRH analogue plus raloxifene administration in the treatment of uterine leiomyomas: a prospective, randomized, single-blind, placebo-controlled clinical trial. Hum Reprod. 2002;17(12):3213–3219. doi: 10.1093/humrep/17.12.3213. [DOI] [PubMed] [Google Scholar]
- 16.Palomba S, Orio F, Jr, Morelli M, et al. Raloxifene administration in premenopausal women with uterine leiomyomas: a pilot study. J Clin Endocrinol Metab. 2002;87(8):3603–3608. doi: 10.1210/jcem.87.8.8747. [DOI] [PubMed] [Google Scholar]
- 17.Arici A, Sozen I. Transforming growth factor-beta3 is expressed at high levels in leiomyoma where it stimulates fibronectin expression and cell proliferation. Fertil Steril. 2000;73(5):1006–1011. doi: 10.1016/s0015-0282(00)00418-0. [DOI] [PubMed] [Google Scholar]
- 18.Chwalisz K, Larsen L, Mattia-Goldberg C, Edmonds A, Elger W, Winkel CA. A randomized, controlled trial of asoprisnil, a novel selective progesterone receptor modulator, in women with uterine leiomyomata. Fertil Steril. 2007;87(6):1399–1412. doi: 10.1016/j.fertnstert.2006.11.094. [DOI] [PubMed] [Google Scholar]
- 19.Lee BS, Nowak RA. Human leiomyoma smooth muscle cells show increased expression of transforming growth factor-beta 3 (TGF beta 3) and altered responses to the antiproliferative effects of TGF beta. J Clin Endocrinol Metab. 2001;86(2):913–920. doi: 10.1210/jcem.86.2.7237. [DOI] [PubMed] [Google Scholar]
- 20.Levens ED, Potlog-Nahari C, Armstrong AY, et al. CDB-2914 for uterine leiomyomata treatment: a randomized controlled trial. Obstet Gynecol. 2008;111(5):1129–1136. doi: 10.1097/AOG.0b013e3181705d0e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spitz IM. Clinical utility of progesterone receptor modulators and their effect on the endometrium. Curr Opin Obstet Gynecol. 2009;21(4):318–324. doi: 10.1097/GCO.0b013e32832e07e8. [DOI] [PubMed] [Google Scholar]
- 22.Chegini N. Implication of growth factor and cytokine networks in leiomyomas. In: Hill J, editor. Cytokines in Human Reproduction. New York, NY: Wiley & Sons; 2000. pp. 133–159. [Google Scholar]
- 23.Chegini N, Verala J, Luo X, Xu J, Williams RS. Gene expression profile of leiomyoma and myometrium and the effect of gonadotropin releasing hormone analogue therapy. J Soc Gynecol Investig. 2003;10(3):161–171. doi: 10.1016/s1071-5576(03)00004-2. [DOI] [PubMed] [Google Scholar]
- 24.Luo X, Ding L, Xu J, Williams RS, Chegini N. Leiomyoma and myometrial gene expression profiles and their responses to gonadotropin-releasing hormone analog therapy. Endocrinology. 2005;146(3):1074–1096. doi: 10.1210/en.2004-1384. [DOI] [PubMed] [Google Scholar]
- 25.Walker CL, Stewart EA. Uterine fibroids: the elephant in the room. Science. 2005;308(5728):1589–1592. doi: 10.1126/science.1112063. [DOI] [PubMed] [Google Scholar]
- 26.Chegini N, Ma C, Tang XM, Williams RS. Effects of GnRH analogues, ‘add-back’ steroid therapy, antiestrogen and antiprogestins on leiomyoma and myometrial smooth muscle cell growth and transforming growth factor-beta expression. Mol Hum Reprod. 2002;8(12):1071–1078. doi: 10.1093/molehr/8.12.1071. [DOI] [PubMed] [Google Scholar]
- 27.Chen W, Ohara N, Wang J, et al. A novel selective progesterone receptor modulator asoprisnil (J867) inhibits proliferation and induces apoptosis in cultured human uterine leiomyoma cells in the absence of comparable effects on myometrial cells. J Clin Endocrinol Metab. 2006;91(4):1296–1304. doi: 10.1210/jc.2005-2379. [DOI] [PubMed] [Google Scholar]
- 28.Ding L, Xu J, Luo X, Chegini N. Gonadotropin releasing hormone and transforming growth factor beta activate mitogen-activated protein kinase/extracellularly regulated kinase and differentially regulate fibronectin, type I collagen, and plasminogen activator inhibitor-1 expression in leiomyoma and myometrial smooth muscle cells. J Clin Endocrinol Metab. 2004;89(11):5549–5557. doi: 10.1210/jc.2004-0161. [DOI] [PubMed] [Google Scholar]
- 29.Ohara N, Morikawa A, Chen W, et al. Comparative effects of SPRM asoprisnil (J867) on proliferation, apoptosis, and the expression of growth factors in cultured uterine leiomyoma cells and normal myometrial cells. Reprod Sci. 2007;14(8, suppl):20–27. doi: 10.1177/1933719107311464. [DOI] [PubMed] [Google Scholar]
- 30.Ohara N. Action of progesterone receptor modulators on uterine leiomyomas. Clin Exp Obstet Gynecol. 2008;35(3):165–166. [PubMed] [Google Scholar]
- 31.Stewart EA, Austin DJ, Jain P, Penglase MD, Nowak RA. RU486 suppresses prolactin production in explant cultures of leiomyoma and myometrium. Fertil Steril. 1996;65(6):1119–1124. [PubMed] [Google Scholar]
- 32.Williams AR, Critchley HO, Osei J, et al. The effects of the selective progesterone receptor modulator asoprisnil on the morphology of uterine tissues after 3 months treatment in patients with symptomatic uterine leiomyomata. Hum Reprod. 2007;22(6):1696–1704. doi: 10.1093/humrep/dem026. [DOI] [PubMed] [Google Scholar]
- 33.Xu Q, Takekida S, Ohara N, et al. Progesterone receptor modulator CDB-2914 down-regulates proliferative cell nuclear antigen and Bcl-2 protein expression and up-regulates caspase-3 and poly(adenosine 5′-diphosphate-ribose) polymerase expression in cultured human uterine leiomyoma cells. J Clin Endocrinol Metab. 2005;90(2):953–961. doi: 10.1210/jc.2004-1569. [DOI] [PubMed] [Google Scholar]
- 34.Xu Q, Ohara N, Chen W, et al. Progesterone receptor modulator CDB-2914 down-regulates vascular endothelial growth factor, adrenomedullin and their receptors and modulates progesterone receptor content in cultured human uterine leiomyoma cells. Hum Reprod. 2006;21(9):2408–2416. doi: 10.1093/humrep/del159. [DOI] [PubMed] [Google Scholar]
- 35.Xu Q, Ohara N, Liu J, et al. Progesterone receptor modulator CDB-2914 induces extracellular matrix metalloproteinase inducer in cultured human uterine leiomyoma cells. Mol Hum Reprod. 2008;14(3):181–191. doi: 10.1093/molehr/gan004. [DOI] [PubMed] [Google Scholar]
- 36.Madauss KP, Grygielko ET, Deng SJ, et al. A structural and in vitro characterization of asoprisnil: a selective progesterone receptor modulator. Mol Endocrinol. 2007;21(5):1066–1081. doi: 10.1210/me.2006-0524. [DOI] [PubMed] [Google Scholar]
- 37.Hussein-Fikret S, Fuller PJ, Gargett CE. Expression of steroid receptor coactivators in cultured cells from paired myometrial and fibroid tissues. J Soc Gynecol Investig. 2005;12(6):445–451. doi: 10.1016/j.jsgi.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 38.Berman JM. Intrauterine adhesions. Semin Reprod Med. 2008;26(4):349–355. doi: 10.1055/s-0028-1082393. [DOI] [PubMed] [Google Scholar]
- 39.Davies C, Gibson M, Holt EM, Torrie EP. Amenorrhoea secondary to endometrial ablation and Asherman’s syndrome following uterine artery embolization. Clin Radiol. 2002;57(4):317–318. doi: 10.1053/crad.2001.0846. [DOI] [PubMed] [Google Scholar]
- 40.Hare AA, Olah KS. Pregnancy following endometrial ablation: a review article. J Obstet Gynaecol. 2005;25(2):108–114. doi: 10.1080/01443610500040745. [DOI] [PubMed] [Google Scholar]
- 41.Magos A. Hysteroscopic treatment of Asherman’s syndrome. Reprod Biomed Online. 2002;4(suppl 3):46–51. doi: 10.1016/s1472-6483(12)60116-3. [DOI] [PubMed] [Google Scholar]
- 42.Buchanan EP, Longaker MT, Lorenz HP. Fetal skin wound healing. Adv Clin Chem. 2009;48:137–161. doi: 10.1016/s0065-2423(09)48006-5. [DOI] [PubMed] [Google Scholar]
- 43.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119(6):1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kisseleva T, Brenner DA. Hepatic stellate cells and the reversal of fibrosis. J Gastroenterol Hepatol. 2006;21(suppl 3):S84–S87. doi: 10.1111/j.1440-1746.2006.04584.x. [DOI] [PubMed] [Google Scholar]
- 45.Kisseleva T, Uchinami H, Feirt N, et al. Bone marrow-derived fibrocytes participate in pathogenesis of liver fibrosis. J Hepatol. 2006;45(3):429–438. doi: 10.1016/j.jhep.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 46.Kisseleva T, Brenner DA. Mechanisms of fibrogenesis. Exp Biol Med (Maywood) 2008;233(2):109–122. doi: 10.3181/0707-MR-190. [DOI] [PubMed] [Google Scholar]
- 47.Lee EY, Xia Y, Kim WS, et al. Hypoxia-enhanced wound-healing function of adipose-derived stem cells: increase in stem cell proliferation and up-regulation of VEGF and bFGF. Wound Repair Regen. 2009;17(4):540–547. doi: 10.1111/j.1524-475X.2009.00499.x. [DOI] [PubMed] [Google Scholar]
- 48.Martínez-Climent JA, Andreu EJ, Prosper F. Somatic stem cells and the origin of cancer. Clin Transl Oncol. 2006;8(9):647–663. doi: 10.1007/s12094-006-0035-7. [DOI] [PubMed] [Google Scholar]
- 49.Mehrad B, Burdick MD, Zisman DA, Keane MP, Belperio JA, Strieter RM. Circulating peripheral blood fibrocytes in human fibrotic interstitial lung disease. Biochem Biophys Res Commun. 2007;353(1):104–108. doi: 10.1016/j.bbrc.2006.11.149. [DOI] [PubMed] [Google Scholar]
- 50.Stappenbeck TS, Miyoshi H. The role of stromal stem cells in tissue regeneration and wound repair. Science. 2009;324(5935):1666–1669. doi: 10.1126/science.1172687. [DOI] [PubMed] [Google Scholar]
- 51.Mattoli S, Bellini A, Schmidt M. The role of a human hematopoietic mesenchymal progenitor in wound healing and fibrotic diseases and implications for therapy. Curr Stem Cell Res Ther. 2009 December 1; doi: 10.2174/157488809789649232. (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- 52.Chen Y, Xiang LX, Shao JZ, Pan RL, Wang YX, Dong XJ, et al. Recruitment of endogenous bone marrow mesenchymal stem cells towards injured liver. J Cell Mol Med. 2009 September 24; doi: 10.1111/j.1582-4934.2009.00912.x. (Epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Keeley EC, Mehrad B, Strieter RM. The role of circulating mesenchymal progenitor cells (fibrocytes) in the pathogenesis of fibrotic disorders. Thromb Haemost. 2009;101(4):613–618. [PMC free article] [PubMed] [Google Scholar]
- 54.Chang HL, Senaratne TN, Zhang L, et al. Uterine leiomyomas exhibit fewer stem/progenitor cell characteristics when compared with corresponding normal myometrium. Reprod Sci. 2009 October 5; doi: 10.1177/1933719109348924. (Epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ono M, Maruyama T, Masuda H, et al. Side population in human uterine myometrium displays phenotypic and functional characteristics of myometrial stem cells. Proc Natl Acad Sci U S A. 2007;104(47):18700–18705. doi: 10.1073/pnas.0704472104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Acloque H, Thiery JP, Nieto MA. The physiology and pathology of the EMT. Meeting on the epithelial-mesenchymal transition. EMBO Rep. 2008;9(4):322–326. doi: 10.1038/embor.2008.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Acloque H, Adams MS, Fishwick K, Bronner-Fraser M, Nieto MA. Epithelial-mesenchymal transitions: the importance of changing cell state in development and disease. J Clin Invest. 2009;119(6):1438–1449. doi: 10.1172/JCI38019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Singer AJ, Clark RA. Cutaneous wound healing. N Engl J Med. 1999;341(10):738–746. doi: 10.1056/NEJM199909023411006. [DOI] [PubMed] [Google Scholar]
- 59.Fitzpatrick FA, Soberman R. Regulated formation of eicosanoids. J Clin Invest. 2001;107(11):1347–1351. doi: 10.1172/JCI13241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fitzpatrick FA. Cyclooxygenase enzymes: regulation and function. Curr Pharm Des. 2004;10(6):577–588. doi: 10.2174/1381612043453144. [DOI] [PubMed] [Google Scholar]
- 61.Bennett JS. Platelet-fibrinogen interactions. Ann N Y Acad Sci. 2001;936:340–354. doi: 10.1111/j.1749-6632.2001.tb03521.x. [DOI] [PubMed] [Google Scholar]
- 62.Bennett JS, Berger BW, Billings PC. The structure and function of platelet integrins. J Thromb Haemost. 2009;7(suppl 1):200–205. doi: 10.1111/j.1538-7836.2009.03378.x. [DOI] [PubMed] [Google Scholar]
- 63.Clemetson KJ, Clemetson JM. Platelet receptor signalling. Hematol J. 2004;5(suppl 3):S159–S163. doi: 10.1038/sj.thj.6200444. [DOI] [PubMed] [Google Scholar]
- 64.Clemetson KJ, Clemetson JM. Collagen receptors as potential targets for novel anti-platelet agents. Curr Pharm Des. 2007;13(26):2673–2683. doi: 10.2174/138161207781662894. [DOI] [PubMed] [Google Scholar]
- 65.Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat ML, Gabbiani G. The myofibroblast: one function, multiple origins. Am J Pathol. 2007;170(6):1807–1816. doi: 10.2353/ajpath.2007.070112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hinz B. Formation and function of the myofibroblast during tissue repair. J Invest Dermatol. 2007;127(3):526–537. doi: 10.1038/sj.jid.5700613. [DOI] [PubMed] [Google Scholar]
- 67.Clark RA. Fibrin and wound healing. Ann N Y Acad Sci. 2001;936:355–367. doi: 10.1111/j.1749-6632.2001.tb03522.x. [DOI] [PubMed] [Google Scholar]
- 68.Hwu YM, Li SH, Lee RK, Tsai YH, Yeh TS, Lin SY. Increased expression of platelet-derived growth factor C messenger ribonucleic acid in uterine leiomyomata. Fertil Steril. 2008;89(2):468–471. doi: 10.1016/j.fertnstert.2007.02.031. [DOI] [PubMed] [Google Scholar]
- 69.Miura S, Khan KN, Kitajima M, et al. Differential infiltration of macrophages and prostaglandin production by different uterine leiomyomas. Hum Reprod. 2006;21(10):2545–2554. doi: 10.1093/humrep/del205. [DOI] [PubMed] [Google Scholar]
- 70.Sozen I, Olive DL, Arici A. Expression and hormonal regulation of monocyte chemotactic protein-1 in myometrium and leiomyomata. Fertil Steril. 1998;69(6):1095–1102. doi: 10.1016/s0015-0282(98)00072-7. [DOI] [PubMed] [Google Scholar]
- 71.Sozen I, Arici A. Cellular biology of myomas: interaction of sex steroids with cytokines and growth factors. Obstet Gynecol Clin North Am. 2006;33(1):41–58. doi: 10.1016/j.ogc.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 72.Suzuki M, Takamizawa S, Nomaguchi K, et al. Erythropoietin synthesis by tumour tissues in a patient with uterine myoma and erythrocytosis. Br J Haematol. 2001;113(1):49–51. doi: 10.1046/j.1365-2141.2001.02682.x. [DOI] [PubMed] [Google Scholar]
- 73.Mesquita FS, Dyer SN, Heinrich DA, Bulun SE, Marsh EE, Nowak RA. Reactive oxygen species mediate mitogenic growth factor signaling pathways in human leiomyoma smooth muscle cells. Biol Reprod. 2010;82(2):341–351. doi: 10.1095/biolreprod.108.075887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Collen D, Lijnen HR. The tissue-type plasminogen activator story. Arterioscler Thromb Vasc Biol. 2009;29(8):1151–1155. doi: 10.1161/ATVBAHA.108.179655. [DOI] [PubMed] [Google Scholar]
- 75.Holly SP, Larson MK, Parise LV. Multiple roles of integrins in cell motility. Exp Cell Res. 2000;261(1):69–74. doi: 10.1006/excr.2000.5040. [DOI] [PubMed] [Google Scholar]
- 76.Lijnen HR. Elements of the fibrinolytic system. Ann N Y Acad Sci. 2001;936:226–236. doi: 10.1111/j.1749-6632.2001.tb03511.x. [DOI] [PubMed] [Google Scholar]
- 77.Lijnen HR. Extracellular proteolysis in the development and progression of atherosclerosis. Biochem Soc Trans. 2002;30(2):163–167. doi: 10.1042/. [DOI] [PubMed] [Google Scholar]
- 78.Lijnen HR. Matrix metalloproteinases and cellular fibrinolytic activity. Biochemistry (Mosc) 2002;67(1):92–98. doi: 10.1023/a:1013908332232. [DOI] [PubMed] [Google Scholar]
- 79.Andreou A, Feussner I. Lipoxygenases—structure and reaction mechanism. Phytochemistry. 2009;70(13–14):1504–1510. doi: 10.1016/j.phytochem.2009.05.008. [DOI] [PubMed] [Google Scholar]
- 80.Chawengsub Y, Gauthier KM, Campbell WB. Role of arachidonic acid lipoxygenase metabolites in the regulation of vascular tone. Am J Physiol Heart Circ Physiol. 2009;297(2):H495–H507. doi: 10.1152/ajpheart.00349.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mochizuki N, Kwon YG. 15-lipoxygenase-1 in the vasculature: expanding roles in angiogenesis. Circ Res. 2008;102(2):143–145. doi: 10.1161/CIRCRESAHA.107.170191. [DOI] [PubMed] [Google Scholar]
- 82.O’Donnell VB, Maskrey B, Taylor GW. Eicosanoids: generation and detection in mammalian cells. Methods Mol Biol. 2009;462:5–23. [PubMed] [Google Scholar]
- 83.Wymann MP, Schneiter R. Lipid signalling in disease. Nat Rev Mol Cell Biol. 2008;9(2):162–176. doi: 10.1038/nrm2335. [DOI] [PubMed] [Google Scholar]
- 84.Aitokallio-Tallberg A. Prostacyclin and thromboxane synthesis by endometrial cancer and leiomyomas. Prostaglandins. 1990;39(3):259–266. doi: 10.1016/0090-6980(90)90045-w. [DOI] [PubMed] [Google Scholar]
- 85.Pinto S, Coppo M, Bruni V, Rosati D, Cirri R, Abbate R. Changes in thromboxane A2 generation and plasma lipid pattern in pseudomenopause induced by gonadotropin releasing hormone (GnRH) analogue buserelin. Prostaglandins Leukot Essent Fatty Acids. 1991;43(3):203–207. doi: 10.1016/0952-3278(91)90170-a. [DOI] [PubMed] [Google Scholar]
- 86.Rees MC, Turnbull AC. Leiomyomas release prostaglandins. Prostaglandins Leukot Med. 1985;18(1):65–68. doi: 10.1016/0262-1746(85)90051-4. [DOI] [PubMed] [Google Scholar]
- 87.Yamaguchi M, Mori N. Prostaglandin production by human myometrium, uterine cervix and leiomyoma. Prostaglandins Leukot Med. 1987;29(1):107–112. doi: 10.1016/0262-1746(87)90102-8. [DOI] [PubMed] [Google Scholar]
- 88.O’Garra A, Robinson D. Development and function of T helper 1 cells. Adv Immunol. 2004;83:133–162. doi: 10.1016/S0065-2776(04)83004-9. [DOI] [PubMed] [Google Scholar]
- 89.Stetson DB, Voehringer D, Grogan JL, et al. Th2 cells: orchestrating barrier immunity. Adv Immunol. 2004;83:163–189. doi: 10.1016/S0065-2776(04)83005-0. [DOI] [PubMed] [Google Scholar]
- 90.Gerard C, Rollins BJ. Chemokines and disease. Nat Immunol. 2001;2(2):108–115. doi: 10.1038/84209. [DOI] [PubMed] [Google Scholar]
- 91.Lau EK, Allen S, Hsu AR, Handel TM. Chemokine-receptor interactions: GPCRs, glycosaminoglycans and viral chemokine binding proteins. Adv Protein Chem. 2004;68:351–391. doi: 10.1016/S0065-3233(04)68010-7. [DOI] [PubMed] [Google Scholar]
- 92.Zlotnik A, Yoshie O. Chemokines: a new classification system and their role in immunity. Immunity. 2000;12(2):121–127. doi: 10.1016/s1074-7613(00)80165-x. [DOI] [PubMed] [Google Scholar]
- 93.Keeley EC, Mehrad B, Strieter RM. Chemokines as mediators of neovascularization. Arterioscler Thromb Vasc Biol. 2008;28(11):1928–1936. doi: 10.1161/ATVBAHA.108.162925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mehrad B, Keane MP, Strieter RM. Chemokines as mediators of angiogenesis. Thromb Haemost. 2007;97(5):755–762. [PMC free article] [PubMed] [Google Scholar]
- 95.Dimitrova IK, Richer JK, Rudolph MC, et al. Gene expression profiling of multiple leiomyomata uteri and matched normal tissue from a single patient. Fertil Steril. 2009;91(6):2650–2663. doi: 10.1016/j.fertnstert.2008.03.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wei T, Geiser AG, Qian HR, et al. DNA microarray data integration by ortholog gene analysis reveals potential molecular mechanisms of estrogen-dependent growth of human uterine fibroids. BMC Womens Health. 2007;7:5. doi: 10.1186/1472-6874-7-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wei JJ, Chiriboga L, Arslan AA, Melamed J, Yee H, Mittal K. Ethnic differences in expression of the dysregulated proteins in uterine leiomyomata. Hum Reprod. 2006;21(1):57–67. doi: 10.1093/humrep/dei309. [DOI] [PubMed] [Google Scholar]
- 98.Wei JJ, Chiriboga L, Mittal K. Expression profile of the tumorigenic factors associated with tumor size and sex steroid hormone status in uterine leiomyomata. Fertil Steril. 2005;84(2):474–484. doi: 10.1016/j.fertnstert.2005.01.142. [DOI] [PubMed] [Google Scholar]
- 99.Ahn WS, Kim KW, Bae SM, et al. Targeted cellular process profiling approach for uterine leiomyoma using cDNA microarray, proteomics and gene ontology analysis. Int J Exp Pathol. 2003;84(6):267–279. doi: 10.1111/j.0959-9673.2003.00362.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wang H, Mahadevappa M, Yamamoto K, et al. Distinctive proliferative phase differences in gene expression in human myometrium and leiomyomata. Fertil Steril. 2003;80(2):266–276. doi: 10.1016/s0015-0282(03)00730-1. [DOI] [PubMed] [Google Scholar]
- 101.Luo X, Ding L, Xu J, Chegini N. Gene expression profiling of leiomyoma and myometrial smooth muscle cells in response to transforming growth factor-beta. Endocrinology. 2005;146(3):1097–1118. doi: 10.1210/en.2004-1377. [DOI] [PubMed] [Google Scholar]
- 102.Luo X, Ding L, Chegini N. CCNs, fibulin-1C and S100A4 expression in leiomyoma and myometrium: inverse association with TGF-beta and regulation by TGF-beta in leiomyoma and myometrial smooth muscle cells. Mol Hum Reprod. 2006;12(4):245–256. doi: 10.1093/molehr/gal015. [DOI] [PubMed] [Google Scholar]
- 103.Luo X, Pan Q, Liu L, Chegini N. Genomic and proteomic profiling II: comparative assessment of gene expression profiles in leiomyomas, keloids, and surgically-induced scars. Reprod Biol Endocrinol. 2007;5:35. doi: 10.1186/1477-7827-5-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Pan Q, Luo X, Chegini N. Genomic and proteomic profiling I: leiomyomas in African Americans and Caucasians. Reprod Biol Endocrinol. 2007;5:34. doi: 10.1186/1477-7827-5-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wei JJ, Chiriboga L, Arslan AA, Melamed J, Yee H, Mittal K. Ethnic differences in expression of the dysregulated proteins in uterine leiomyomata. Hum Reprod. 2006;21(1):57–67. doi: 10.1093/humrep/dei309. [DOI] [PubMed] [Google Scholar]
- 106.Chegini N, Tang XM, Ma C. Regulation of transforming growth factor-beta1 expression by granulocyte macrophage-colony-stimulating factor in leiomyoma and myometrial smooth muscle cells. J Clin Endocrinol Metab. 1999;84(11):4138–4143. doi: 10.1210/jcem.84.11.6147. [DOI] [PubMed] [Google Scholar]
- 107.Luo X, Ding L, Xu J, Williams RS, Chegini N. Leiomyoma and myometrial gene expression profiles and their responses to gonadotropin-releasing hormone analog therapy. Endocrinology. 2005;146(3):1074–1096. doi: 10.1210/en.2004-1384. [DOI] [PubMed] [Google Scholar]
- 108.Maruo T, Ohara N, Wang J, Matsuo H. Sex steroidal regulation of uterine leiomyoma growth and apoptosis. Hum Reprod Update. 2004;10(3):207–220. doi: 10.1093/humupd/dmh019. [DOI] [PubMed] [Google Scholar]
- 109.Chen Q, Rabach L, Noble P, et al. IL-11 receptor alpha in the pathogenesis of IL-13-induced inflammation and remodeling. J Immunol. 2005;174(4):2305–2313. doi: 10.4049/jimmunol.174.4.2305. [DOI] [PubMed] [Google Scholar]
- 110.Fichtner-Feigl S, Young CA, Kitani A, Geissler EK, Schlitt HJ, Strober W. IL-13 signaling via IL-13R alpha2 induces major downstream fibrogenic factors mediating fibrosis in chronic TNBS colitis. Gastroenterology. 2008;135(6):2003–2013. doi: 10.1053/j.gastro.2008.08.055. [DOI] [PubMed] [Google Scholar]
- 111.Malavia NK, Mih JD, Raub CB, Dinh BT, George SC. IL-13 induces a bronchial epithelial phenotype that is profibrotic. Respir Res. 2008;9:27. doi: 10.1186/1465-9921-9-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Nishimura Y, Nitto T, Inoue T, Node K. IL-13 attenuates vascular tube formation via JAK2-STAT6 pathway. Circ J. 2008;72(3):469–475. doi: 10.1253/circj.72.469. [DOI] [PubMed] [Google Scholar]
- 113.Reiman RM, Thompson RW, Feng CG, et al. Interleukin-5 (IL-5) augments the progression of liver fibrosis by regulating IL-13 activity. Infect Immun. 2006;74(3):1471–1479. doi: 10.1128/IAI.74.3.1471-1479.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Sugimoto R, Enjoji M, Nakamuta M, et al. Effect of IL-4 and IL-13 on collagen production in cultured LI90 human hepatic stellate cells. Liver Int. 2005;25(2):420–428. doi: 10.1111/j.1478-3231.2005.01087.x. [DOI] [PubMed] [Google Scholar]
- 115.Wynn TA. Fibrotic disease and the T(H)1/T(H)2 paradigm. Nat Rev Immunol. 2004;4(8):583–594. doi: 10.1038/nri1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Yamagata S, Tomita K, Sato R, Niwa A, Higashino H, Tohda Y. Interleukin-18-deficient mice exhibit diminished chronic inflammation and airway remodelling in ovalbumin-induced asthma model. Clin Exp Immunol. 2008;154(3):295–304. doi: 10.1111/j.1365-2249.2008.03772.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zheng T, Oh MH, Oh SY, Schroeder JT, Glick AB, Zhu Z. Transgenic expression of interleukin-13 in the skin induces a pruritic dermatitis and skin remodeling. J Invest Dermatol. 2009;129(3):742–751. doi: 10.1038/jid.2008.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Galkowska H, Wojewodzka U, Olszewski WL. Chemokines, cytokines, and growth factors in keratinocytes and dermal endothelial cells in the margin of chronic diabetic foot ulcers. Wound Repair Regen. 2006;14(5):558–565. doi: 10.1111/j.1743-6109.2006.00155.x. [DOI] [PubMed] [Google Scholar]
- 119.Shephard P, Hinz B, Smola-Hess S, Meister JJ, Krieg T, Smola H. Dissecting the roles of endothelin, TGF-beta and GM-CSF on myofibroblast differentiation by keratinocytes. Thromb Haemost. 2004;92(2):262–274. doi: 10.1160/TH03-11-0669. [DOI] [PubMed] [Google Scholar]
- 120.Kikuchi T, Shively JD, Foley JS, Drazen JM, Tschumperlin DJ. Differentiation-dependent responsiveness of bronchial epithelial cells to IL-4/13 stimulation. Am J Physiol Lung Cell Mol Physiol. 2004;287(1):L119–L126. doi: 10.1152/ajplung.00365.2003. [DOI] [PubMed] [Google Scholar]
- 121.Robson MC, Dubay DA, Wang X, Franz MG. Effect of cytokine growth factors on the prevention of acute wound failure. Wound Repair Regen. 2004;12(1):38–43. doi: 10.1111/j.1067-1927.2004.012109.x. [DOI] [PubMed] [Google Scholar]
- 122.Mollah ZU, Aiba S, Nakagawa S, et al. Interleukin-3 in cooperation with transforming growth factor beta induces granulocyte macrophage colony stimulating factor independent differentiation of human CD34+hematopoietic progenitor cells into dendritic cells with features of Langerhans cells. J Invest Dermatol. 2003;121(6):1397–1401. doi: 10.1111/j.1523-1747.2003.12641.x. [DOI] [PubMed] [Google Scholar]
- 123.Jost MM, Ninci E, Meder B, et al. Divergent effects of GM-CSF and TGFbeta1 on bone marrow-derived macrophage arginase-1 activity, MCP-1 expression, and matrix metalloproteinase-12: a potential role during arteriogenesis. FASEB J. 2003;17(15):2281–2283. doi: 10.1096/fj.03-0071fje. [DOI] [PubMed] [Google Scholar]
- 124.Kelly M, Kolb M, Bonniaud P, Gauldie J. Re-evaluation of fibrogenic cytokines in lung fibrosis. Curr Pharm Des. 2003;9(1):39–49. doi: 10.2174/1381612033392341. [DOI] [PubMed] [Google Scholar]
- 125.Chen G, Grotendorst G, Eichholtz T, Khalil N. GM-CSF increases airway smooth muscle cell connective tissue expression by inducing TGF-beta receptors. Am J Physiol Lung Cell Mol Physiol. 2003;284(3):L548–L556. doi: 10.1152/ajplung.00091.2002. [DOI] [PubMed] [Google Scholar]
- 126.Suzuki M, Harashima A, Okochi A, et al. Transforming growth factor-beta(1) augments granulocyte-macrophage colony-stimulating factor-induced proliferation of umbilical cord blood CD34(+) cells with an associated tyrosine phosphorylation of STAT5. Exp Hematol. 2002;30(10):1132–1138. doi: 10.1016/s0301-472x(02)00902-5. [DOI] [PubMed] [Google Scholar]
- 127.Hanada T, Yoshimura A. Regulation of cytokine signaling and inflammation. Cytokine Growth Factor Rev. 2002;13(4–5):413–421. doi: 10.1016/s1359-6101(02)00026-6. [DOI] [PubMed] [Google Scholar]
- 128.Syssoev KA, Kulagina NV, Chukhlovin AB, Morozova EB, Totolian AA. Expression of mRNA for chemokines and chemokine receptors in tissues of the myometrium and uterine leiomyoma. Bull Exp Biol Med. 2008;145(1):84–89. doi: 10.1007/s10517-008-0038-1. [DOI] [PubMed] [Google Scholar]
- 129.Senturk LM, Sozen I, Gutierrez L, Arici A. Interleukin 8 production and interleukin 8 receptor expression in human myometrium and leiomyoma. Am J Obstet Gynecol. 2001;184(4):559–566. doi: 10.1067/mob.2001.111160. [DOI] [PubMed] [Google Scholar]
- 130.Allen GB, Cloutier ME, Larrabee YC, Tetenev K, Smiley ST, Bates JH. Neither fibrin nor plasminogen activator inhibitor-1 deficiency protects lung function in a mouse model of acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2009;296(3):L277–L285. doi: 10.1152/ajplung.90475.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Smiley ST, King JA, Hancock WW. Fibrinogen stimulates macrophage chemokine secretion through toll-like receptor 4. J Immunol. 2001;167(5):2887–2894. doi: 10.4049/jimmunol.167.5.2887. [DOI] [PubMed] [Google Scholar]
- 132.Doherty T, Broide D. Cytokines and growth factors in airway remodeling in asthma. Curr Opin Immunol. 2007;19(6):676–680. doi: 10.1016/j.coi.2007.07.017. [DOI] [PubMed] [Google Scholar]
- 133.Fujiwara K, Matsukawa A, Ohkawara S, Takagi K, Yoshinaga M. Functional distinction between CXC chemokines, interleukin-8 (IL-8), and growth related oncogene (GRO)alpha in neutrophil infiltration. Lab Invest. 2002;82(1):15–23. doi: 10.1038/labinvest.3780391. [DOI] [PubMed] [Google Scholar]
- 134.Galligan CL, Matsuyama W, Matsukawa A, et al. Up-regulated expression and activation of the orphan chemokine receptor, CCRL2, in rheumatoid arthritis. Arthritis Rheum. 2004;50(6):1806–1814. doi: 10.1002/art.20275. [DOI] [PubMed] [Google Scholar]
- 135.Hicks A, Monkarsh SP, Hoffman AF, Goodnow R., Jr Leukotriene B4 receptor antagonists as therapeutics for inflammatory disease: preclinical and clinical developments. Expert Opin Investig Drugs. 2007;16(12):1909–1920. doi: 10.1517/13543784.16.12.1909. [DOI] [PubMed] [Google Scholar]
- 136.Hogaboam CM, Bone-Larson CL, Steinhauser ML, et al. Exaggerated hepatic injury due to acetaminophen challenge in mice lacking C-C chemokine receptor 2. Am J Pathol. 2000;156(4):1245–1252. doi: 10.1016/S0002-9440(10)64995-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hoshino A, Kawamura YI, Yasuhara M, et al. Inhibition of CCL1-CCR8 interaction prevents aggregation of macrophages and development of peritoneal adhesions. J Immunol. 2007;178(8):5296–5304. doi: 10.4049/jimmunol.178.8.5296. [DOI] [PubMed] [Google Scholar]
- 138.Jakubzick C, Wen H, Matsukawa A, Keller M, Kunkel SL, Hogaboam CM. Role of CCR4 ligands, CCL17 and CCL22, during Schistosoma mansoni egg-induced pulmonary granuloma formation in mice. Am J Pathol. 2004;165(4):1211–1221. doi: 10.1016/S0002-9440(10)63381-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.LaFleur AM, Lukacs NW, Kunkel SL, Matsukawa A. Role of CC chemokine CCL6/C10 as a monocyte chemo-attractant in a murine acute peritonitis. Mediators Inflamm. 2004;13(5–6):349–355. doi: 10.1155/S0962935104000511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Matsukawa A, Hogaboam CM, Lukacs NW, Lincoln PM, Strieter RM, Kunkel SL. Endogenous MCP-1 influences systemic cytokine balance in a murine model of acute septic peritonitis. Exp Mol Pathol. 2000;68(2):77–84. doi: 10.1006/exmp.1999.2296. [DOI] [PubMed] [Google Scholar]
- 141.Watanabe H, Numata K, Ito T, Takagi K, Matsukawa A. Innate immune response in Th1- and Th2-dominant mouse strains. Shock. 2004;22(5):460–466. doi: 10.1097/01.shk.0000142249.08135.e9. [DOI] [PubMed] [Google Scholar]
- 142.Call DR, Nemzek JA, Ebong SJ, et al. Differential local and systemic regulation of the murine chemokines KC and MIP2. Shock. 2001;15(4):278–284. doi: 10.1097/00024382-200115040-00005. [DOI] [PubMed] [Google Scholar]
- 143.Coelho AL, Schaller MA, Benjamim CF, Orlofsky AZ, Hogaboam CM, Kunkel SL. The chemokine CCL6 promotes innate immunity via immune cell activation and recruitment. J Immunol. 2007;179(8):5474–5482. doi: 10.4049/jimmunol.179.8.5474. [DOI] [PubMed] [Google Scholar]
- 144.Matsukawa A, Hogaboam CM, Lukacs NW, et al. Expression and contribution of endogenous IL-13 in an experimental model of sepsis. J Immunol. 2000;164(5):2738–2744. doi: 10.4049/jimmunol.164.5.2738. [DOI] [PubMed] [Google Scholar]
- 145.Orlofsky A, Wu Y, Prystowsky MB. Divergent regulation of the murine CC chemokine C10 by Th(1) and Th(2) cytokines. Cytokine. 2000;12(3):220–228. doi: 10.1006/cyto.1999.0535. [DOI] [PubMed] [Google Scholar]
- 146.Steinhauser ML, Hogaboam CM, Matsukawa A, Lukacs NW, Strieter RM, Kunkel SL. Chemokine C10 promotes disease resolution and survival in an experimental model of bacterial sepsis. Infect Immun. 2000;68(11):6108–6114. doi: 10.1128/iai.68.11.6108-6114.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Hsieh YY, Chang CC, Tsai FJ, Lin CC, Yeh LS, Tsai CH. Tumor necrosis factor-alpha-308 promoter and p53 codon 72 gene polymorphisms in women with leiomyomas. Fertil Steril. 2004;82(suppl 3):1177–1181. doi: 10.1016/j.fertnstert.2004.03.035. [DOI] [PubMed] [Google Scholar]
- 148.Hsieh YY, Chang CC, Tsai CH, Lin CC, Tsai FJ. Interleukin (IL)-12 receptor beta1 codon 378 G homozygote and allele, but not IL-1 (beta-511 promoter, 3953 exon 5, receptor antagonist), IL-2 114, IL-4-590 intron 3, IL-8 3′-UTR 2767, and IL-18 105, are associated with higher susceptibility to leiomyoma. Fertil Steril. 2007;87(4):886–895. doi: 10.1016/j.fertnstert.2006.07.1541. [DOI] [PubMed] [Google Scholar]
- 149.Litovkin KV, Domenyuk VP, Bubnov VV, Zaporozhan VN. Interleukin-6 -174G/C polymorphism in breast cancer and uterine leiomyoma patients: a population-based case control study. Exp Oncol. 2007;29(4):295–298. [PubMed] [Google Scholar]
- 150.Pietrowski D, Thewes R, Sator M, Denschlag D, Keck C, Tempfer C. Uterine leiomyoma is associated with a polymorphism in the interleukin 1-beta gene. Am J Reprod Immunol. 2009;62(2):112–117. doi: 10.1111/j.1600-0897.2009.00718.x. [DOI] [PubMed] [Google Scholar]
- 151.Carpenter KL, van der Veen C, Taylor SE, et al. Macrophages, lipid oxidation, ceroid accumulation and alpha-tocopherol depletion in human atherosclerotic lesions. Gerontology. 1995;41(suppl 2):53–67. doi: 10.1159/000213725. [DOI] [PubMed] [Google Scholar]
- 152.Cinel I, Opal SM. Molecular biology of inflammation and sepsis: a primer. Crit Care Med. 2009;37(1):291–304. doi: 10.1097/CCM.0b013e31819267fb. [DOI] [PubMed] [Google Scholar]
- 153.Furuichi K, Gao JL, Horuk R, Wada T, Kaneko S, Murphy PM. Chemokine receptor CCR1 regulates inflammatory cell infiltration after renal ischemia-reperfusion injury. J Immunol. 2008;181(12):8670–8676. doi: 10.4049/jimmunol.181.12.8670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Furuichi K, Kaneko S, Wada T. Chemokine/chemokine receptor-mediated inflammation regulates pathologic changes from acute kidney injury to chronic kidney disease. Clin Exp Nephrol. 2009;13(1):9–14. doi: 10.1007/s10157-008-0119-5. [DOI] [PubMed] [Google Scholar]
- 155.Hardwick SJ, Hegyi L, Clare K, et al. Apoptosis in human monocyte-macrophages exposed to oxidized low density lipoprotein. J Pathol. 1996;179(3):294–302. doi: 10.1002/(SICI)1096-9896(199607)179:3<294::AID-PATH590>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 156.Hodge JC, Morton CC. Genetic heterogeneity among uterine leiomyomata: insights into malignant progression. Hum Mol Genet. 2007;16(Spec No 1):R7–13. doi: 10.1093/hmg/ddm043. [DOI] [PubMed] [Google Scholar]
- 157.Coraux C, Roux J, Jolly T, Birembaut P. Epithelial cell-extracellular matrix interactions and stem cells in airway epithelial regeneration. Proc Am Thorac Soc. 2008;5(6):689–694. doi: 10.1513/pats.200801-010AW. [DOI] [PubMed] [Google Scholar]
- 158.Gill SE, Parks WC. Metalloproteinases and their inhibitors: regulators of wound healing. Int J Biochem Cell Biol. 2008;40(6–7):1334–1347. doi: 10.1016/j.biocel.2007.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Moali C, Hulmes DJ. Extracellular and cell surface proteases in wound healing: new players are still emerging. Eur J Dermatol. 2009;19(6):552–564. doi: 10.1684/ejd.2009.0770. [DOI] [PubMed] [Google Scholar]
- 160.Page-McCaw A, Ewald AJ, Werb Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat Rev Mol Cell Biol. 2007;8(3):221–233. doi: 10.1038/nrm2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Ra HJ, Parks WC. Control of matrix metalloproteinase catalytic activity. Matrix Biol. 2007;26(8):587–596. doi: 10.1016/j.matbio.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Toriseva M, Kähäri VM. Proteinases in cutaneous wound healing. Cell Mol Life Sci. 2009;66(2):203–224. doi: 10.1007/s00018-008-8388-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Behera MA, Feng L, Yonish B, Catherino W, Jung SH, Leppert P. Thrombospondin-1 and thrombospondin-2 mRNA and TSP-1 and TSP-2 protein expression in uterine fibroids and correlation to the genes COL1A1 and COL3A1 and to the collagen cross-link hydroxyproline. Reprod Sci. 2007;14(8, suppl):63–76. doi: 10.1177/1933719107309591. [DOI] [PubMed] [Google Scholar]
- 164.Dou Q, Tarnuzzer RW, Williams RS, Schultz GS, Chegini N. Differential expression of matrix metalloproteinases and their tissue inhibitors in leiomyomata: a mechanism for gonadotrophin releasing hormone agonist-induced tumour regression. Mol Hum Reprod. 1997;3(11):1005–1014. doi: 10.1093/molehr/3.11.1005. [DOI] [PubMed] [Google Scholar]
- 165.Ma C, Chegini N. Regulation of matrix metalloproteinases (MMPs) and their tissue inhibitors in human myometrial smooth muscle cells by TGF-beta1. Mol Hum Reprod. 1999;5(10):950–954. doi: 10.1093/molehr/5.10.950. [DOI] [PubMed] [Google Scholar]
- 166.Palmer SS, Haynes-Johnson D, Diehl T, Nowak RA. Increased expression of stromelysin 3 mRNA in leiomyomas (uterine fibroids) compared with myometrium. J Soc Gynecol Investig. 1998;5(4):203–209. doi: 10.1016/s1071-5576(98)00009-4. [DOI] [PubMed] [Google Scholar]
- 167.Takemura N, Yoshida S, Kennedy S, Deguchi M, Ohara N, Maruo T. Matrix metalloproteinase-1 and -9 promoter polymorphisms are not associated with an increased risk of uterine leiomyomas in a Japanese population. J Soc Gynecol Investig. 2006;13(3):232–236. doi: 10.1016/j.jsgi.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 168.Wolańska M, Sobolewski K, Bańkowski E, Jaworski S. Matrix metalloproteinases of human leiomyoma in various stages of tumor growth. Gynecol Obstet Invest. 2004;58(1):14–18. doi: 10.1159/000077177. [DOI] [PubMed] [Google Scholar]
- 169.Byrne AM, Bouchier-Hayes DJ, Harmey JH. Angiogenic and cell survival functions of vascular endothelial growth factor (VEGF) J Cell Mol Med. 2005;9(4):777–794. doi: 10.1111/j.1582-4934.2005.tb00379.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Hickey MM, Simon MC. Regulation of angiogenesis by hypoxia and hypoxia-inducible factors. Curr Top Dev Biol. 2006;76:217–257. doi: 10.1016/S0070-2153(06)76007-0. [DOI] [PubMed] [Google Scholar]
- 171.Knowles J, Loizidou M, Taylor I. Endothelin-1 and angiogenesis in cancer. Curr Vasc Pharmacol. 2005;3(4):309–314. doi: 10.2174/157016105774329462. [DOI] [PubMed] [Google Scholar]
- 172.Kopp HG, Ramos CA, Rafii S. Contribution of endothelial progenitors and proangiogenic hematopoietic cells to vascularization of tumor and ischemic tissue. Curr Opin Hematol. 2006;13(3):175–181. doi: 10.1097/01.moh.0000219664.26528.da. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Murdoch C, Muthana M, Lewis CE. Hypoxia regulates macrophage functions in inflammation. J Immunol. 2005;175(10):6257–6263. doi: 10.4049/jimmunol.175.10.6257. [DOI] [PubMed] [Google Scholar]
- 174.Rodriguez PG, Felix FN, Woodley DT, Shim EK. The role of oxygen in wound healing: a review of the literature. Dermatol Surg. 2008;34(9):1159–1169. doi: 10.1111/j.1524-4725.2008.34254.x. [DOI] [PubMed] [Google Scholar]
- 175.Sen CK. Wound healing essentials: let there be oxygen. Wound Repair Regen. 2009;17(1):1–18. doi: 10.1111/j.1524-475X.2008.00436.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Shibuya M. Structure and function of VEGF/VEGF-receptor system involved in angiogenesis. Cell Struct Funct. 2001;26(1):25–35. doi: 10.1247/csf.26.25. [DOI] [PubMed] [Google Scholar]
- 177.Arnout J, Hoylaerts MF, Lijnen HR. Haemostasis. Handb Exp Pharmacol. 2006;176(Pt 2):1–41. doi: 10.1007/3-540-36028-x_1. [DOI] [PubMed] [Google Scholar]
- 178.Fichtner-Feigl S, Strober W, Geissler EK, Schlitt HJ. Cytokines mediating the induction of chronic colitis and colitis-associated fibrosis. Mucosal Immunol. 2008;1(suppl 1):S24–S27. doi: 10.1038/mi.2008.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Fujimoto H, Gabazza EC, Taguchi O, et al. Thrombin-activatable fibrinolysis inhibitor deficiency attenuates bleo-mycin-induced lung fibrosis. Am J Pathol. 2006;168(4):1086–1096. doi: 10.2353/ajpath.2006.050610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Galkowska H, Wojewodzka U, Olszewski WL. Chemokines, cytokines, and growth factors in keratinocytes and dermal endothelial cells in the margin of chronic diabetic foot ulcers. Wound Repair Regen. 2006;14(5):558–565. doi: 10.1111/j.1743-6109.2006.00155.x. [DOI] [PubMed] [Google Scholar]
- 181.Johnson LL, Berggren KN, Szaba FM, Chen W, Smiley ST. Fibrin-mediated protection against infection-stimulated immunopathology. J Exp Med. 2003;197(6):801–806. doi: 10.1084/jem.20021493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Wolberg AS. Thrombin generation and fibrin clot structure. Blood Rev. 2007;21(3):131–142. doi: 10.1016/j.blre.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 183.Cross MJ, Claesson-Welsh L. FGF and VEGF function in angiogenesis: signalling pathways, biological responses and therapeutic inhibition. Trends Pharmacol Sci. 2001;22(4):201–207. doi: 10.1016/s0165-6147(00)01676-x. [DOI] [PubMed] [Google Scholar]
- 184.Lijnen HR. Metalloproteinases in development and progression of vascular disease. Pathophysiol Haemost Thromb. 2003;33(5–6):275–281. doi: 10.1159/000083814. [DOI] [PubMed] [Google Scholar]
- 185.Margosio B, Rusnati M, Bonezzi K, et al. Fibroblast growth factor-2 binding to the thrombospondin-1 type III repeats, a novel antiangiogenic domain. Int J Biochem Cell Biol. 2008;40(4):700–709. doi: 10.1016/j.biocel.2007.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Owen CA. Leukocyte cell surface proteinases: regulation of expression, functions, and mechanisms of surface localization. Int J Biochem Cell Biol. 2008;40(6–7):1246–1272. doi: 10.1016/j.biocel.2008.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Petreaca ML, Yao M, Ware C, Martins-Green MM. Vascular endothelial growth factor promotes macrophage apoptosis through stimulation of tumor necrosis factor superfamily member 14 (TNFSF14/LIGHT) Wound Repair Regen. 2008;16(5):602–614. doi: 10.1111/j.1524-475X.2008.00411.x. [DOI] [PubMed] [Google Scholar]
- 188.de Caestecker Mde CM. The transforming growth factor-beta superfamily of receptors. Cytokine Growth Factor Rev. 2004;15(1):1–11. doi: 10.1016/j.cytogfr.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 189.Hyytiäinen M, Penttinen C, Keski-Oja J. Latent TGF-beta binding proteins: extracellular matrix association and roles in TGF-beta activation. Crit Rev Clin Lab Sci. 2004;41(3):233–264. doi: 10.1080/10408360490460933. [DOI] [PubMed] [Google Scholar]
- 190.Barcellos-Hoff MH, Akhurst RJ. Transforming growth factor-beta in breast cancer: too much, too late. Breast Cancer Res. 2009;11(1):202. doi: 10.1186/bcr2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Gauldie J, Bonniaud P, Sime P, Ask K, Kolb M. TGF-beta, Smad3 and the process of progressive fibrosis. Biochem Soc Trans. 2007;35(Pt 4):661–664. doi: 10.1042/BST0350661. [DOI] [PubMed] [Google Scholar]
- 192.Gharaee-Kermani M, Hu B, Phan SH, Gyetko MR. Recent advances in molecular targets and treatment of idiopathic pulmonary fibrosis: focus on TGFbeta signaling and the myofibroblast. Curr Med Chem. 2009;16(11):1400–1417. doi: 10.2174/092986709787846497. [DOI] [PubMed] [Google Scholar]
- 193.Goodwin A, Jenkins G. Role of integrin-mediated TGFbeta activation in the pathogenesis of pulmonary fibrosis. Biochem Soc Trans. 2009;37(Pt 4):849–854. doi: 10.1042/BST0370849. [DOI] [PubMed] [Google Scholar]
- 194.Goumans MJ, van Zonneveld AJ, ten Dijke P. Transforming growth factor beta-induced endothelial-to-mesenchymal transition: a switch to cardiac fibrosis? Trends Cardiovasc Med. 2008;18(8):293–298. doi: 10.1016/j.tcm.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 195.Leivonen SK, Kähäri VM. Transforming growth factor-beta signaling in cancer invasion and metastasis. Int J Cancer. 2007;121(10):2119–2124. doi: 10.1002/ijc.23113. [DOI] [PubMed] [Google Scholar]
- 196.Matsuzaki K. Modulation of TGF-beta signaling during progression of chronic liver diseases. Front Biosci. 2009;14:2923–2934. doi: 10.2741/3423. [DOI] [PubMed] [Google Scholar]
- 197.Moustakas A, Heldin CH. The regulation of TGFbeta signal transduction. Development. 2009;136(22):3699–3714. doi: 10.1242/dev.030338. [DOI] [PubMed] [Google Scholar]
- 198.Pardali E, ten Dijke P. Transforming growth factor-beta signaling and tumor angiogenesis. Front Biosci. 2009;14:4848–4861. doi: 10.2741/3573. [DOI] [PubMed] [Google Scholar]
- 199.Pohlers D, Brenmoehl J, Löffler I, et al. TGF-beta and fibrosis in different organs - molecular pathway imprints. Biochim Biophys Acta. 2009;1792(8):746–756. doi: 10.1016/j.bbadis.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 200.Schnaper HW, Jandeska S, Runyan CE, et al. TGF-beta signal transduction in chronic kidney disease. Front Biosci. 2009;14:2448–2465. doi: 10.2741/3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Varga J, Pasche B. Transforming growth factor beta as a therapeutic target in systemic sclerosis. Nat Rev Rheumatol. 2009;5(4):200–206. doi: 10.1038/nrrheum.2009.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Wu MY, Hill CS. TGF-beta superfamily signaling in embryonic development and homeostasis. Dev Cell. 2009;16(3):329–343. doi: 10.1016/j.devcel.2009.02.012. [DOI] [PubMed] [Google Scholar]
- 203.Byfield SD, Roberts AB. Lateral signaling enhances TGF-beta response complexity. Trends Cell Biol. 2004;14(3):107–111. doi: 10.1016/j.tcb.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 204.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425(6958):577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 205.ten Dijke P, Hill CS. New insights into TGF-beta-Smad signalling. Trends Biochem Sci. 2004;29(5):265–273. doi: 10.1016/j.tibs.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 206.Siegel PM, Massagué J. Cytostatic and apoptotic actions of TGF-beta in homeostasis and cancer. Nat Rev Cancer. 2003;3(11):807–821. doi: 10.1038/nrc1208. [DOI] [PubMed] [Google Scholar]
- 207.Cummings J, Ward TH, Ranson M, Dive C. Apoptosis pathway-targeted drugs—from the bench to the clinic. Biochim Biophys Acta. 2004;1705(1):53–66. doi: 10.1016/j.bbcan.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 208.Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407(6805):770–776. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- 209.Arici A, Sozen I. Expression, menstrual cycle-dependent activation, and bimodal mitogenic effect of transforming growth factor-beta1 in human myometrium and leiomyoma. Am J Obstet Gynecol. 2003;188(1):76–83. doi: 10.1067/mob.2003.118. [DOI] [PubMed] [Google Scholar]
- 210.Chegini N, Luo X, Ding L, Ripley D. The expression of Smads and transforming growth factor beta receptors in leiomyoma and myometrium and the effect of gonadotropin releasing hormone analogue therapy. Mol Cell Endocrinol. 2003;209(1–2):9–16. doi: 10.1016/j.mce.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 211.Dou Q, Zhao Y, Tarnuzzer RW, et al. Suppression of transforming growth factor-beta (TGF beta) and TGF beta receptor messenger ribonucleic acid and protein expression in leiomyomata in women receiving gonadotropin-releasing hormone agonist therapy. J Clin Endocrinol Metab. 1996;81(9):3222–3230. doi: 10.1210/jcem.81.9.8784073. [DOI] [PubMed] [Google Scholar]
- 212.Dou Q, Williams RS, Chegini N. Inhibition of transforming growth factor-beta 1 alters the growth, anchor-dependent cell aggregation and integrin mRNA expression in human promonocytes: implications for endometriosis and peritoneal adhesion formation. Mol Hum Reprod. 1997;3(5):383–391. doi: 10.1093/molehr/3.5.383. [DOI] [PubMed] [Google Scholar]
- 213.Luo X, Ding L, Xu J, Chegini N. Gene expression profiling of leiomyoma and myometrial smooth muscle cells in response to transforming growth factor-beta. Endocrinology. 2005;146(3):1097–1118. doi: 10.1210/en.2004-1377. [DOI] [PubMed] [Google Scholar]
- 214.Tang XM, Dou Q, Zhao Y, McLean F, Davis J, Chegini N. The expression of transforming growth factor-beta s and TGF-beta receptor mRNA and protein and the effect of TGF-beta s on human myometrial smooth muscle cells in vitro. Mol Hum Reprod. 1997;3(3):233–240. doi: 10.1093/molehr/3.3.233. [DOI] [PubMed] [Google Scholar]
- 215.Xu J, Luo X, Chegini N. Differential expression, regulation, and induction of Smads, transforming growth factor-beta signal transduction pathway in leiomyoma, and myometrial smooth muscle cells and alteration by gonadotropin-releasing hormone analog. J Clin Endocrinol Metab. 2003;88(3):1350–1361. doi: 10.1210/jc.2002-021325. [DOI] [PubMed] [Google Scholar]
- 216.Blobe GC, Schiemann WP, Lodish HF. Role of transforming growth factor beta in human disease. N Engl J Med. 2000;342(18):1350–1358. doi: 10.1056/NEJM200005043421807. [DOI] [PubMed] [Google Scholar]
- 217.Hua Z, Lv Q, Ye W, et al. MiRNA-directed regulation of VEGF and other angiogenic factors under hypoxia. PLoS One. 2006;1:e116. doi: 10.1371/journal.pone.0000116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Ihn H. Autocrine TGF-beta signaling in the pathogenesis of systemic sclerosis. J Dermatol Sci. 2008;49(2):103–113. doi: 10.1016/j.jdermsci.2007.05.014. [DOI] [PubMed] [Google Scholar]
- 219.Yu L, Border WA, Huang Y, Noble NA. TGF-beta isoforms in renal fibrogenesis. Kidney Int. 2003;64(3):844–856. doi: 10.1046/j.1523-1755.2003.00162.x. [DOI] [PubMed] [Google Scholar]
- 220.Barbarisi A, Petillo O, Di Lieto A, et al. 17-beta estradiol elicits an autocrine leiomyoma cell proliferation: evidence for a stimulation of protein kinase-dependent pathway. J Cell Physiol. 2001;186(3):414–424. doi: 10.1002/1097-4652(2000)9999:999<000::AID-JCP1040>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 221.Rossi MJ, Chegini N, Masterson BJ. Presence of epidermal growth factor, platelet-derived growth factor, and their receptors in human myometrial tissue and smooth muscle cells: their action in smooth muscle cells in vitro. Endocrinology. 1992;130(3):1716–1727. doi: 10.1210/endo.130.3.1311246. [DOI] [PubMed] [Google Scholar]
- 222.Laping NJ, Everitt JI, Frazier KS, et al. Tumor-specific efficacy of transforming growth factor-beta RI inhibition in Eker rats. Clin Cancer Res. 2007;13(10):3087–3099. doi: 10.1158/1078-0432.CCR-06-1811. [DOI] [PubMed] [Google Scholar]
- 223.Norwitz ER, Xu S, Xu J, et al. Direct binding of AP-1 (Fos/Jun) proteins to a SMAD binding element facilitates both gonadotropin-releasing hormone (GnRH)- and activin-mediated transcriptional activation of the mouse GnRH receptor gene. J Biol Chem. 2002;277(40):37469–37478. doi: 10.1074/jbc.M206571200. [DOI] [PubMed] [Google Scholar]
- 224.Norwitz ER, Xu S, Jeong KH, et al. Activin A augments GnRH-mediated transcriptional activation of the mouse GnRH receptor gene. Endocrinology. 2002;143(3):985–997. doi: 10.1210/endo.143.3.8663. [DOI] [PubMed] [Google Scholar]
- 225.Ahn WS, Kim KW, Bae SM, et al. Targeted cellular process profiling approach for uterine leiomyoma using cDNA microarray, proteomics and gene ontology analysis. Int J Exp Pathol. 2003;84(6):267–279. doi: 10.1111/j.0959-9673.2003.00362.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Quade BJ, Wang TY, Sornberger K, Dal Cin P, Mutter GL, Morton CC. Molecular pathogenesis of uterine smooth muscle tumors from transcriptional profiling. Genes Chromosomes Cancer. 2004;40(2):97–108. doi: 10.1002/gcc.20018. [DOI] [PubMed] [Google Scholar]
- 227.Tsibris JC, Segars J, Coppola D, et al. Insights from gene arrays on the development and growth regulation of uterine leiomyomata. Fertil Steril. 2002;78(1):114–121. doi: 10.1016/s0015-0282(02)03191-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Wang H, Mahadevappa M, Yamamoto K, et al. Distinctive proliferative phase differences in gene expression in human myometrium and leiomyomata. Fertil Steril. 2003;80(2):266–276. doi: 10.1016/s0015-0282(03)00730-1. [DOI] [PubMed] [Google Scholar]
- 229.Weston G, Trajstman AC, Gargett CE, Manuelpillai U, Vollenhoven BJ, Rogers PA. Fibroids display an anti-angiogenic gene expression profile when compared with adjacent myometrium. Mol Hum Reprod. 2003;9(9):541–549. doi: 10.1093/molehr/gag066. [DOI] [PubMed] [Google Scholar]
- 230.Leask A, Abraham DJ. All in the CCN family: essential matricellular signaling modulators emerge from the bunker. J Cell Sci. 2006;119(Pt 23):4803–4810. doi: 10.1242/jcs.03270. [DOI] [PubMed] [Google Scholar]
- 231.Leask A. Targeting the TGFbeta, endothelin-1 and CCN2 axis to combat fibrosis in scleroderma. Cell Signal. 2008;20(8):1409–1414. doi: 10.1016/j.cellsig.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 232.Joseph DS, Malik M, Nurudeen S, Catherino WH. Myometrial cells undergo fibrotic transformation under the influence of transforming growth factor beta-3. Fertil Steril. 2009 March 26; doi: 10.1016/j.fertnstert.2009.01.081. (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- 233.Tsou R, Cole JK, Nathens AB, et al. Analysis of hypertrophic and normal scar gene expression with cDNA microarrays. J Burn Care Rehabil. 2000;21(6):541–550. doi: 10.1097/00004630-200021060-00012. [DOI] [PubMed] [Google Scholar]
- 234.Leahy DJ. Structure and function of the epidermal growth factor (EGF/ErbB) family of receptors. Adv Protein Chem. 2004;68:1–27. doi: 10.1016/S0065-3233(04)68001-6. [DOI] [PubMed] [Google Scholar]
- 235.Leu TH, Maa MC. Functional implication of the interaction between EGF receptor and c-Src. Front Biosci. 2003;8:s28–s38. doi: 10.2741/980. [DOI] [PubMed] [Google Scholar]
- 236.Iwamoto R, Mekada E. Heparin-binding EGF-like growth factor: a juxtacrine growth factor. Cytokine Growth Factor Rev. 2000;11(4):335–344. doi: 10.1016/s1359-6101(00)00013-7. [DOI] [PubMed] [Google Scholar]
- 237.Carpenter G. The EGF receptor: a nexus for trafficking and signaling. Bioessays. 2000;22(8):697–707. doi: 10.1002/1521-1878(200008)22:8<697::AID-BIES3>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 238.Bonner JC. Regulation of PDGF and its receptors in fibrotic diseases. Cytokine Growth Factor Rev. 2004;15(4):255–273. doi: 10.1016/j.cytogfr.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 239.Yu J, Ustach C, Kim HR. Platelet-derived growth factor signaling and human cancer. J Biochem Mol Biol. 2003;36(1):49–59. doi: 10.5483/bmbrep.2003.36.1.049. [DOI] [PubMed] [Google Scholar]
- 240.Li X, Eriksson U. Novel PDGF family members: PDGF-C and PDGF-D. Cytokine Growth Factor Rev. 2003;14(2):91–98. doi: 10.1016/s1359-6101(02)00090-4. [DOI] [PubMed] [Google Scholar]
- 241.Vegeto E, Ghisletti S, Meda C, Etteri S, Belcredito S, Maggi A. Regulation of the lipopolysaccharide signal transduction pathway by 17beta-estradiol in macrophage cells. J Steroid Biochem Mol Biol. 2004;91(1–2):59–66. doi: 10.1016/j.jsbmb.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 242.Moro L, Reineri S, Piranda D, et al. Nongenomic effects of 17beta-estradiol in human platelets: potentiation of thrombin-induced aggregation through estrogen receptor beta and Src kinase. Blood. 2005;105(1):115–121. doi: 10.1182/blood-2003-11-3840. [DOI] [PubMed] [Google Scholar]
- 243.Brekken RA, Sage EH. SPARC, a matricellular protein: at the crossroads of cell-matrix communication. Matrix Biol. 2001;19(8):816–827. doi: 10.1016/s0945-053x(00)00133-5. [DOI] [PubMed] [Google Scholar]
- 244.Di Lieto A, De Rosa G, De Falco M, et al. Relationship between platelet-derived growth factor expression in leiomyomas and uterine volume changes after gonadotropin-releasing hormone agonist treatment. Hum Pathol. 2002;33(2):220–224. doi: 10.1053/hupa.2002.31298. [DOI] [PubMed] [Google Scholar]
- 245.Heldin CH. Development and possible clinical use of antagonists for PDGF and TGF-beta. Ups J Med Sci. 2004;109(3):165–178. doi: 10.3109/2000-1967-083. [DOI] [PubMed] [Google Scholar]
- 246.LeRoith D, Roberts CT., Jr The insulin-like growth factor system and cancer. Cancer Lett. 2003;195(2):127–137. doi: 10.1016/s0304-3835(03)00159-9. [DOI] [PubMed] [Google Scholar]
- 247.Firth SM, Baxter RC. Cellular actions of the insulin-like growth factor binding proteins. Endocr Rev. 2002;23(6):824–854. doi: 10.1210/er.2001-0033. [DOI] [PubMed] [Google Scholar]
- 248.Gentry CC, Okolo SO, Fong LF, Crow JC, Maclean AB, Perrett CW. Quantification of vascular endothelial growth factor-A in leiomyomas and adjacent myometrium. Clin Sci (Lond) 2001;101(6):691–695. [PubMed] [Google Scholar]
- 249.Böttcher RT, Niehrs C. Fibroblast growth factor signaling during early vertebrate development. Endocr Rev. 2005;26(1):63–77. doi: 10.1210/er.2003-0040. [DOI] [PubMed] [Google Scholar]
- 250.Lin X. Functions of heparan sulfate proteoglycans in cell signaling during development. Development. 2004;131(24):6009–6021. doi: 10.1242/dev.01522. [DOI] [PubMed] [Google Scholar]
- 251.Wu X, Blanck A, Olovsson M, Möller B, Lindblom B. Expression of basic fibroblast growth factor (bFGF), FGF receptor 1 and FGF receptor 2 in uterine leiomyomas and myometrium during the menstrual cycle, after menopause and GnRHa treatment. Acta Obstet Gynecol Scand. 2001;80(6):497–504. [PubMed] [Google Scholar]
- 252.Taylor CV, Letarte M, Lye SJ. The expression of integrins and cadherins in normal human uterus and uterine leiomyomas. Am J Obstet Gynecol. 1996;175(2):411–419. doi: 10.1016/s0002-9378(96)70155-2. [DOI] [PubMed] [Google Scholar]
- 253.Chegini N, Kornberg L. Gonadotropin releasing hormone analogue therapy alters signal transduction pathways involving mitogen-activated protein and focal adhesion kinases in leiomyoma. J Soc Gynecol Investig. 2003;10(1):21–26. [PubMed] [Google Scholar]
- 254.Fanayan S, Firth SM, Baxter RC. Signaling through the Smad pathway by insulin-like growth factor-binding protein-3 in breast cancer cells. Relationship to transforming growth factor-beta 1 signaling. J Biol Chem. 2002;277(9):7255–7261. doi: 10.1074/jbc.M108038200. [DOI] [PubMed] [Google Scholar]
- 255.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 256.Bi Y, Liu G, Yang R. MicroRNAs: novel regulators during the immune response. J Cell Physiol. 2009;218(3):467–472. doi: 10.1002/jcp.21639. [DOI] [PubMed] [Google Scholar]
- 257.Calin GA, Pekarsky Y, Croce CM. The role of microRNA and other non-coding RNA in the pathogenesis of chronic lymphocytic leukemia. Best Pract Res Clin Haematol. 2007;20(3):425–437. doi: 10.1016/j.beha.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 258.Luo X, Chegini N. The expression and potential regulatory function of microRNAs in the pathogenesis of leiomyoma. Semin Reprod Med. 2008;26(6):500–514. doi: 10.1055/s-0028-1096130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Wang T, Zhang X, Obijuru L, et al. A micro-RNA signature associated with race, tumor size, and target gene activity in human uterine leiomyomas. Genes Chromosomes Cancer. 2007;46(4):336–347. doi: 10.1002/gcc.20415. [DOI] [PubMed] [Google Scholar]
- 260.Tili E, Croce CM, Michaille JJ. miR-155: on the crosstalk between inflammation and cancer. Int Rev Immunol. 2009;28(5):264–284. doi: 10.1080/08830180903093796. [DOI] [PubMed] [Google Scholar]
- 261.Tili E, Michaille JJ, Cimino A, et al. Modulation of miR-155 and miR-125b levels following lipopolysaccharide/TNF-alpha stimulation and their possible roles in regulating the response to endotoxin shock. J Immunol. 2007;179(8):5082–5089. doi: 10.4049/jimmunol.179.8.5082. [DOI] [PubMed] [Google Scholar]
- 262.Tili E, Michaille JJ, Costinean S, Croce CM. MicroRNAs, the immune system and rheumatic disease. Nat Clin Pract Rheumatol. 2008;4(10):534–541. doi: 10.1038/ncprheum0885. [DOI] [PubMed] [Google Scholar]
- 263.Chegini N, Zhao Y, Williams RS, Flanders KC. Human uterine tissue throughout the menstrual cycle expresses transforming growth factor-beta 1 (TGF beta 1), TGF beta 2, TGF beta 3, and TGF beta type II receptor messenger ribonucleic acid and protein and contains [125I]TGF beta 1-binding sites. Endocrinology. 1994;135(1):439–449. doi: 10.1210/endo.135.1.8013382. [DOI] [PubMed] [Google Scholar]
- 264.Chegini N, Ma C, Tang XM, Williams RS. Effects of GnRH analogues, ‘add-back’ steroid therapy, antiestrogen and antiprogestins on leiomyoma and myometrial smooth muscle cell growth and transforming growth factor-beta expression. Mol Hum Reprod. 2002;8(12):1071–1078. doi: 10.1093/molehr/8.12.1071. [DOI] [PubMed] [Google Scholar]