

Abstract
INTRODUCTION
Erythropoietin promotes myoblast proliferation and inhibits fibrosis, thus it could impede the pathogenesis of muscle degenerative diseases. However, its stimulation of erythropoiesis limits its use as a therapeutic. An erythropoietin analog, carbamylated erythropoietin (C-EPO), retains these protective actions yet does not interact with the erythropoietin receptor.
METHODS
To determine whether treatment with C-EPO alleviates the signs of muscular dystrophy in an animal model of Duchenne muscular dystrophy, we treated mdx mice with intraperitoneal injections of 50 μg/kg and 100 μg/kg C-EPO for 4 and 12 weeks, and we monitored weight, serum creatine kinase levels, and changes in muscle histology.
RESULTS
We observed moderate histological improvement at 4 weeks which did not translate into a significantly decreased level of serum creatine kinase.
DISCUSSION
At the dosages tested, C-EPO is not an effective therapeutic for the treatment of a mouse model of Duchenne muscular dystrophy.
Keywords: C-EPO, mdx, muscular, dystrophy, fibrosis
Introduction
Muscular dystrophies are a group of diseases characterized by muscle weakness and wasting. One of the most common forms of muscular dystrophies is Duchenne muscular dystrophy (DMD), a disease caused by mutations in the dystrophin gene.1 This disease is often modeled using mdx mice,2 which carry a point mutation in the dystrophin gene that results in premature termination of the protein product.3–6 By three weeks of age, mdx mice exhibit DMD-like histological changes in limb muscles, including variation in myofiber size, an increase in inflammatory cells, and regenerating myofibers which can be distinguished by the presence of centrally located nuclei.7–10 The histological changes in the diaphragm muscle of mdx mice at three weeks of age are similar to those of the limb muscles, but there is progressive muscle degeneration after this age that is accompanied by greater connective tissue infiltration. Functionally, the diaphragm muscle in mdx mice becomes the most severely affected,9,10 and it parallels the degeneration observed in the human disease.11 In both mdx mice and humans with DMD, reduced muscle membrane integrity leads to leakage of muscle proteins, usually measured as creatine kinase (CK), into the bloodstream. Measurement of serum CK levels is an accepted test for supporting a diagnosis of DMD.12–15
Thus far, no effective specific therapy is available for DMD. Many types of interventions are undergoing testing in both animal studies and human clinical trials, and these include approaches aimed at slowing the disease progression via correction of dystrophin expression in the myofibers,16–20 or by alteration of activity of pathways known to promote myofiber growth or inhibit myofiber loss and fibrosis.21,22
In the hematopoietic system, the cytokine erythropoietin (EPO) prevents apoptosis of late erythroid progenitors, and it also supports the proliferation of progenitor cells by signaling through the EPO receptor. EPO can act through other receptors, such as the GM-CSF and IL-3 receptors.23,24 Several studies have highlighted EPO’s protective biological effects in organs other than the hematopoietic system,25 and it is hypothesized that these effects occur through EPO binding to the common beta receptor, a subunit of GM-CSF, IL3 and IL5 receptors.26 In particular, EPO has shown an anti-apoptotic effect in both the heart and the central nervous system following ischemic- or trauma-induced injury.26,27
EPO has several effects that could aid in repair of skeletal muscle injury and prevention of fibrosis. In vitro, EPO promotes proliferation of C2C12 and primary mouse myoblasts, suggesting that it can stimulate the progenitor cell population during muscle repair.28 In vivo, EPO administration for treatment of chronic renal failure increased muscle fiber diameter.29 It promoted angiogenesis30 and increased capillary density in the muscle of a mouse injury model of sepsis.31
One side effect of therapeutic EPO administration is a potentially harmful rise in hemoglobin concentration.32,33 Overexpression of EPO in mice causes excessive erythrocytosis that leads to multiple organ degeneration, including skeletal muscle degeneration.34 To avoid the hematopoietic effects of EPO and target its other protective biological effects, EPO analogs have been developed, such as carbamylated EPO (C-EPO). These analogs do not bind to the EPO receptor and lack erythropoietic activity.35 Proof-of-principle experiments demonstrated that they retain anti-apoptotic, neuroprotective and regenerative activity in tissues other than the hematopoietic system by binding to the common β-receptor.26 C-EPO has been effective in preventing tissue degeneration in a number of disease models, including protecting against motor neuron death in a mouse model of amyotrophic lateral sclerosis.36 C-EPO also reduces the inflammatory response in cortical37 and cerebral infarcts,35 as well as in experimental autoimmune encephalomyelitis,38 and it has been indicated to reduce fibrosis in renal damage caused by ureteral obstruction.39
In a proof-of-principle experiment for the treatment of DMD, we tested whether administration of C-EPO to mdx mice could ameliorate dystrophic signs in muscle. mdx mice were injected intraperitoneally 3 times/week with 50 μg/kg or 100 μg/kg C-EPO for 4 and 12 weeks. Following treatment, serum CK levels were assessed, and histological evaluations of myofiber size, percentage of regenerating myofibers and presence of fibrotic tissue were performed in the diaphragm, gastrocnemius, tibialis anterior and quadriceps muscles of treated and control mdx mice. Our studies demonstrated that, although moderate histological improvement was observed 4 weeks following C-EPO treatment in some muscles, lowered serum creatine kinase levels did not accompany these changes.
Materials and Methods
Animals
C57BL/10ScSn-Mdx/J mice (mdx) were purchased from the Jackson Laboratory (Bar Harbor, ME). The mice were 5–8 week old females that were homozygous for the mdx mutation (a nonsense mutation in exon 23 of the dystrophin gene).4 Doses of 0 μg/kg (control), 50 μg/kg or 100 μg/kg weight of carbamylated-erythropoietin (C-EPO) in 0.2% bovine serum albumin in phosphate-buffered saline were administered 3 times weekly via intraperitoneal (IP) injection, for 4 or 12 weeks. C-EPO was provided by Shire Human Genetic Therapies. Previous studies found beneficial effects at these dosages in the central and peripheral nervous systems and in the myocardium.35,38,40,41 The total volume of each IP injection did not exceed 100 μL. Animals were weighed once a week for the duration of the study. Prior to euthanasia, blood was collected from the tail vein to measure serum CK. Animals were euthanized via CO2 asphyxiation, and the diaphragm, gastrocnemius, quadriceps, and tibialis anterior muscles were collected from each animal and snap frozen in Optimal Cutting Temperature (OCT) for histological analysis. Animal groups consisted of 5–7 mice. All animals were handled in accordance with protocols approved by Animal Resources at Children’s Hospital, Boston.
Histological analyses of muscle tissue sections
For each muscle collected, 10 μm thick tissue sections were stained with hematoxylin and eosin. Measurements were made from 3 to 4 representative fields per muscle. Individual myofiber size was determined by measuring the shortest diameter of the fiber (n = 200 to 600 myofibers per muscle). Measurement of shortest diameter was chosen over measurement of cross-sectional area to reduce the bias in size due to obliquely cut fibers. To determine the fraction of regenerating muscle fibers, the number of myofibers with centrally located nuclei was counted and divided by the total number of myofibers in a field (n = 50 to 200 per field, n = 150 to 600 per muscle). The presence of fibrotic tissue was assessed by determining the area of extracellular spacing between muscle fibers using the NIH ImageJ software and dividing it by the total area of the muscle tissue within each section. The use of interstitial area as a measure for fibrotic area was validated by staining some sections for the intermediate filament protein, vimentin. The area of extracellular spacing between fibers calculated from H&E-stained tissue corresponded to the area expressing vimentin (data not shown).
Evaluation of CK levels in serum
One day prior to euthanasia, 200 μL of blood was collected via tail vein nick into BD Microtainer serum separator tubes from at least 5 mice per group. Blood was allowed to coagulate for 1 hour at room temperature and then centrifuged. Five to 25 μL of plasma serum were used in the Stanbio CK-NAC (UV-Rate) CK test to determine serum CK levels. For each blood sample, two measurements were taken and averaged.
Statistics
Data is presented as mean values; error bars denote standard error of measurement. For all tests, one-way ANOVA with alpha = 0.05 was calculated using the ANOVA: Single Factor analysis tool in Excel. If significance was found (p < 0.05), pair wise comparisons were made using Student’s t-test. The resulting t-test p-value is reported.
Results
To test the effectiveness of C-EPO in reducing the signs of muscular dystrophy, 5–8 week old mdx female mice were injected IP with vehicle only, 50 μg/kg, or 100 μg/kg C-EPO. Throughout the study, the weight of the animals was monitored to determine whether treatment promoted muscle growth and concurrent gains in muscle weight. Administration of C-EPO did not significantly change the weight of the animals compared to vehicle-only controls (Fig. 1). Due to the sacrifice of some of the mice for the 4 week time point, the relative weights between treatment groups at 5 weeks changed. Outside of this imbalance, the weights of treated mice did not change significantly over time compared to control mice.
Figure 1.

Gross weight of female mdx mice treated with C-EPO. Black squares: 0 μg/kg. Gray squares: 50 μg/kg. White squares: 100 μg/kg C-EPO.
After 4 weeks, H&E-stained muscle sections were examined for improvements in muscle histology (Fig. 2, A–F). The percentage of regenerating fibers and percent area of fibrosis were measured in the diaphragm, gastrocnemius, quadriceps, and tibialis anterior muscles of each animal. The myofiber diameter was measured in the diaphragm only. The percentage of myofibers with centrally located nuclei was significantly higher in the diaphragm of both the 50 μg/kg and 100 μg/kg treatment groups, as compared to the control group (48%, 43%, and 34%, respectively; p < 0.05; Fig 2G). The average diameter size of the myofibers in the diaphragm was not changed in the 50 μg/kg group compared to control, while in the 100 μg/kg group it was decreased (p<0.05; Fig 2H). The distribution of myofiber diameters in the diaphragm demonstrates that the smaller average size is due to an overall shift towards all myofibers being smaller (Fig. 2I). In the gastrocnemius muscle, the percentage of centrally located nuclei was also slightly increased with administration of 50 μg/kg C-EPO (80% vs 65%, p = 0.039; Fig. 2G). However, no differences were seen in the tibialis anterior or quadriceps muscles. Evaluation of the percent area of interstitial connective tissue in the diaphragms showed a statistically significant increase in the group administered 100 μg/kg C-EPO (p = 0.007; Fig. 2J). However, no changes were found in any other muscles analyzed.
Figure 2.

Muscle histology of mdx mice treated with C-EPO for 4 weeks. A–C: H&E stained diaphragm, scale bar = 50 μm. D–F: H&E stained gastrocnemius, scale bar = 100 μm. A, D: 0 μg/kg treatment. B,E: 50 μg/kg treatment. C,F: 100 μg/kg treatment. G: Percentage of centrally located nuclei in the diaphragm (Dia), tibialis anterior (TA), quadriceps (Quad), and gastrocnemius (GA) muscles. H: Average myofiber diameter in the diaphragm. I: Distribution of fiber diameters in the diaphragm. J. Percentage of interstitial area in diaphragm, tibialis anterior, quadriceps, and gastrocnemius muscles. G–J: Black squares: 0 μg/kg. Gray squares: 50 μg/kg. White squares: 100 μg/kg C-EPO. ‘*’ denotes statistically significant difference versus control (p <0.05).
After 12 weeks of treatment, each group underwent similar histological analyses (Fig. 3). Unlike what was seen at 4 weeks, there was no significant difference in the percentages of regenerating myofibers in treated versus vehicle-treated mice (Fig. 3G). There was also no significant difference in the average myofiber diameter (Fig. 3H) or in the distribution of myofiber diameters (Fig. 3I). An apparent increase in area of the interstitial tissue was observed in the diaphragms of treated mice compared to untreated mice, although this difference was not statistically significant (Fig. 3J).
Figure 3.
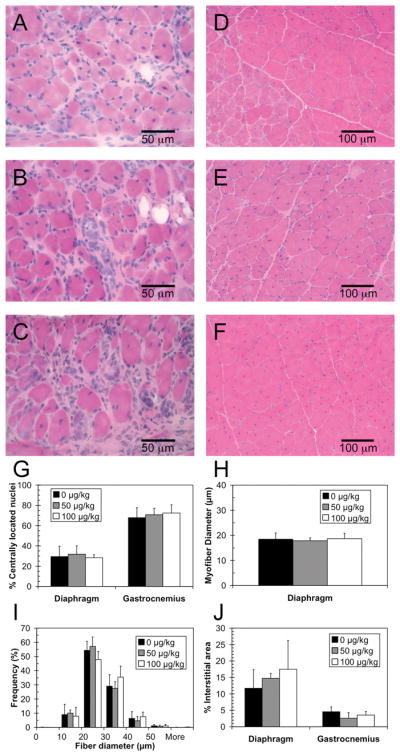
Muscle histology of mdx mice treated with C-EPO for 12 weeks. A–C: H&E stained diaphragm, scale bar = 50 μm. D–F: H&E stained gastrocnemius, scale bar = 100 μm. A, D: 0 μg/kg treatment. B,E: 50 μg/kg treatment. C, F: 100 μg/kg treatment. G: Percentage of centrally located nuclei in the diaphragm and gastrocnemius muscles. H: Average myofiber diameter in the diaphragm. I. Distribution of fiber diameters in the diaphragm. J. Percentage of interstitial area in diaphragm and gastrocnemius muscles. G–J: Black squares: 0 μg/kg. Gray squares: 50 μg/kg. White squares: 100 μg/kg C-EPO.
Previous studies using mdx mice have shown that a growth in the number of regenerating fibers can be accompanied by a gain in muscle function and a decrease in the levels of serum CK.42 To test whether C-EPO administration led to such changes, serum CK levels were assayed (Fig. 4). At neither 4 nor 12 weeks was there a significant decrease in the levels of serum CK in C-EPO-treated compared to untreated mdx animals.
Figure 4.

Serum creatine kinase activity in mdx mice treated with C-EPO for 4 and 12 weeks. Serum creatine kinase levels significantly increased in the 50 μg/kg-treated group at 4 weeks (p = 0.02) but returned to normal levels by 12 weeks. ‘*’ denotes a statistically significant difference versus control.
Discussion
Because of its known neuroprotective effects, as well as indications from several studies that erythropoietin had a stimulatory effect on processes that could promote muscle regeneration and repair, it was proposed that erythropoietin could be a useful therapeutic for muscular disorders.43 To harness the neuroprotective effects of EPO, nonhematopoietic derivatives were created, including C-EPO.35 C-EPO has shown effectiveness in preventing damage in numerous injury models, including experimental myocardial infarction,41 and brain and spinal cord injuries.44 In the myocardium after ischemia, C-EPO stimulates PI3-kinase and Akt,45 genes that are also involved in skeletal muscle growth.46,47
We hypothesized that administration of C-EPO would alleviate the pathology of disease in the mdx mouse model of Duchenne muscular dystrophy by promoting progenitor cell proliferation in vivo.28 The increased pool of cells would fuse into existing myofibers, thus increasing myofiber size, or it would form new (regenerating) myofibers. In addition, it was thought that C-EPO could deter the development of fibrosis in muscular dystrophy. Two dosages of C-EPO were selected based on studies by others that had demonstrated efficacy in central and peripheral nervous system injuries.35,38,40,41
We found that 50 μg/kg of C-EPO stimulated a significant increase in muscle regeneration following 4 weeks of administration, however this effect did not persist at 12 weeks. The observed short-term increase in regenerating myofibers in the diaphragm and gastrocnemius muscles was not accompanied by a significant increase in myofiber diameter. Surprisingly, the average myofiber diameter of the diaphragm in the 100 μg/kg group was decreased. This finding could indicate that, in the presence of C-EPO, activated muscle progenitors formed new, small myofibers rather than fusing into and increasing the size of existing muscle fibers. However, the distribution of fiber sizes indicates a slight shift towards all existing fibers being smaller rather than induction of new small regenerating fibers.
Overall, C-EPO administration did not significantly reduce the amount of interstitial (fibrotic) tissue in the muscles of treated mdx mice compared to controls. Unexpectedly, at the highest dosage the amount of fibrosis increased significantly in the diaphragm muscle 4 weeks following C-EPO treatment. This increase was not observed in any other muscle analyzed at this time point, nor in any of the muscles analyzed from animals that received 12 weeks of C-EPO treatment.
Serum CK levels at both time points did not decrease in treated versus untreated animals. This finding supports the conclusion that despite an increase in the number of regenerating myofibers in some of the muscles of the C-EPO-treated mdx mice, the overall muscle integrity did not significantly improve. Therefore, we conclude that, although C-EPO has been effective in preventing damage in other disease conditions, it did not effectively alleviate the pathogenesis of muscular dystrophy in mdx mice. It is unclear as to why C-EPO-treatment did not result in the intended effect. Other studies using either erythropoietin or C-EPO for prolonged therapy in mice have noted development of anemia.38 In the case of erythropoietin, it has been speculated that anemia might have arisen from development of neutralizing antibodies that recognize endogenous proteins.48 In our study, possible signs of anemia, characterized by whiteness in the paws and a lack of blood flow in the tail, were noted at 12 weeks following treatment (data not shown). These effects seemed worse with the 100 μg/kg treatment group. It is thus possible that the development of anemia might have contributed to the lack of efficacy at 12 weeks.
While similar treatment regimens have proven beneficial in decreasing fibrosis and improving repair in other injury/disease models, we noted only nominal effects 4 weeks following treatment of mdx mice with C-EPO. It remains possible that lower dosages of C-EPO may result in beneficial effects in mdx skeletal muscle while reducing the chance of developing anemia. Future investigations may consider lower dosages to achieve long-term improvement in muscle diseases.
Acknowledgments
The authors would like to thank Matthew Mitchell and Mckenzie Wessen for technical help with the IP injections and with tissue harvesting, and Gregory S. Robinson and Arthur O. Tzianabos for insightful discussions and manuscript review. This work was supported by Shire Human Genetic Therapies. MPW was supported by the NIH/NIGMS T32 GM007226 Cellular and Developmental Biology Training Grant. EG is supported by grants from NIH/NINDS: 2R01NS047727 and 5P50NS040828.
Abbreviations
- C-EPO
carbamylated erythropoietin
- CK
creatine kinase
- DMD
Duchenne muscular dystrophy
- EPO
erythropoietin
- H&E
hematoxylin and eosin
References
- 1.Monaco AP, Kunkel LM. Cloning of the Duchenne/Becker muscular dystrophy locus. Adv Hum Genet. 1988;17:61–98. doi: 10.1007/978-1-4613-0987-1_3. [DOI] [PubMed] [Google Scholar]
- 2.Bulfield G, Siller WG, Wight PA, Moore KJ. X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc Natl Acad Sci U S A. 1984;81(4):1189–1192. doi: 10.1073/pnas.81.4.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hoffman EP, Brown RH, Jr, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51(6):919–928. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- 4.Sicinski P, Geng Y, Ryder-Cook AS, Barnard EA, Darlison MG, Barnard PJ. The molecular basis of muscular dystrophy in the mdx mouse: a point mutation. Science. 1989;244(4912):1578–1580. doi: 10.1126/science.2662404. [DOI] [PubMed] [Google Scholar]
- 5.Ryder-Cook AS, Sicinski P, Thomas K, Davies KE, Worton RG, Barnard EA, et al. Localization of the mdx mutation within the mouse dystrophin gene. Embo J. 1988;7(10):3017–3021. doi: 10.1002/j.1460-2075.1988.tb03165.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cavanna JS, Coulton G, Morgan JE, Brockdorff N, Forrest SM, Davies KE, et al. Molecular and genetic mapping of the mouse mdx locus. Genomics. 1988;3(4):337–341. doi: 10.1016/0888-7543(88)90124-3. [DOI] [PubMed] [Google Scholar]
- 7.Spencer MJ, Montecino-Rodriguez E, Dorshkind K, Tidball JG. Helper (CD4(+)) and cytotoxic (CD8(+)) T cells promote the pathology of dystrophin-deficient muscle. Clin Immunol. 2001;98(2):235–243. doi: 10.1006/clim.2000.4966. [DOI] [PubMed] [Google Scholar]
- 8.Spencer MJ, Tidball JG. Do immune cells promote the pathology of dystrophin-deficient myopathies? Neuromuscul Disord. 2001;11(6–7):556–564. doi: 10.1016/s0960-8966(01)00198-5. [DOI] [PubMed] [Google Scholar]
- 9.Karpati G, Carpenter S, Prescott S. Small-caliber skeletal muscle fibers do not suffer necrosis in mdx mouse dystrophy. Muscle Nerve. 1988;11(8):795–803. doi: 10.1002/mus.880110802. [DOI] [PubMed] [Google Scholar]
- 10.Tanabe Y, Esaki K, Nomura T. Skeletal muscle pathology in X chromosome-linked muscular dystrophy (mdx) mouse. Acta Neuropathol. 1986;69(1–2):91–95. doi: 10.1007/BF00687043. [DOI] [PubMed] [Google Scholar]
- 11.Stedman HH, Sweeney HL, Shrager JB, Maguire HC, Panettieri RA, Petrof B, et al. The mdx mouse diaphragm reproduces the degenerative changes of Duchenne muscular dystrophy. Nature. 1991;352(6335):536–539. doi: 10.1038/352536a0. [DOI] [PubMed] [Google Scholar]
- 12.Nichol CJ. Serum Creatine Phosphokinase Measurements in Muscular Dystrophy Studies. Clin Chim Acta. 1965;11:404–407. doi: 10.1016/0009-8981(65)90186-5. [DOI] [PubMed] [Google Scholar]
- 13.Pearce JM, Pennington RJ, Walton JN. Serum Enzyme Studies in Muscle Disease. Ii. Serum Creatine Kinase Activity in Muscular Dystrophy and in Other Myopathic and Neuropathic Disorders. J Neurol Neurosurg Psychiatry. 1964;27:96–99. doi: 10.1136/jnnp.27.2.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fowler WM., Jr Diagnostic and Prognostic Significance of Serum Enzymes. I. Muscular Dystrophy. Arch Phys Med Rehabil. 1964;45:117–124. [PubMed] [Google Scholar]
- 15.Swaiman KF, Sandler B. The Use of Serum Creatine Phosphokinase and Other Serum Enzymes in the Diagnosis of Progressive Muscular Dystrophy. J Pediatr. 1963;63:1116–1119. doi: 10.1016/s0022-3476(63)80193-6. [DOI] [PubMed] [Google Scholar]
- 16.Nelson SF, Crosbie RH, Miceli MC, Spencer MJ. Emerging genetic therapies to treat Duchenne muscular dystrophy. Curr Opin Neurol. 2009;22(5):532–538. doi: 10.1097/WCO.0b013e32832fd487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Muir LA, Chamberlain JS. Emerging strategies for cell and gene therapy of the muscular dystrophies. Expert Rev Mol Med. 2009;11:e18. doi: 10.1017/S1462399409001100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peault B, Rudnicki M, Torrente Y, Cossu G, Tremblay JP, Partridge T, et al. Stem and progenitor cells in skeletal muscle development, maintenance, and therapy. Mol Ther. 2007;15(5):867–877. doi: 10.1038/mt.sj.6300145. [DOI] [PubMed] [Google Scholar]
- 19.Heemskerk H, de Winter CL, van Ommen GJ, van Deutekom JC, Aartsma-Rus A. Development of antisense-mediated exon skipping as a treatment for duchenne muscular dystrophy. Ann N Y Acad Sci. 2009;1175:71–79. doi: 10.1111/j.1749-6632.2009.04973.x. [DOI] [PubMed] [Google Scholar]
- 20.Wood MJ, Gait MJ, Yin H. RNA-targeted splice-correction therapy for neuromuscular disease. Brain. 2010 doi: 10.1093/brain/awq002. in press. [DOI] [PubMed] [Google Scholar]
- 21.Patel K, Macharia R, Amthor H. Molecular mechanisms involving IGF-1 and myostatin to induce muscle hypertrophy as a therapeutic strategy for Duchenne muscular dystrophy. Acta Myol. 2005;24(3):230–241. [PubMed] [Google Scholar]
- 22.Khurana TS, Davies KE. Pharmacological strategies for muscular dystrophy. Nat Rev Drug Discov. 2003;2(5):379–390. doi: 10.1038/nrd1085. [DOI] [PubMed] [Google Scholar]
- 23.Hanazono Y, Sasaki K, Nitta H, Yazaki Y, Hirai H. Erythropoietin induces tyrosine phosphorylation of the beta chain of the GM-CSF receptor. Biochem Biophys Res Commun. 1995;208(3):1060–1066. doi: 10.1006/bbrc.1995.1442. [DOI] [PubMed] [Google Scholar]
- 24.Jubinsky PT, Krijanovski OI, Nathan DG, Tavernier J, Sieff CA. The beta chain of the interleukin-3 receptor functionally associates with the erythropoietin receptor. Blood. 1997;90(5):1867–1873. [PubMed] [Google Scholar]
- 25.Ghezzi P, Brines M. Erythropoietin as an antiapoptotic, tissue-protective cytokine. Cell Death Differ. 2004;11 (Suppl 1):S37–44. doi: 10.1038/sj.cdd.4401450. [DOI] [PubMed] [Google Scholar]
- 26.Brines M, Grasso G, Fiordaliso F, Sfacteria A, Ghezzi P, Fratelli M, et al. Erythropoietin mediates tissue protection through an erythropoietin and common beta-subunit heteroreceptor. Proc Natl Acad Sci U S A. 2004;101(41):14907–14912. doi: 10.1073/pnas.0406491101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Joyeux-Faure M, Godin-Ribuot D, Ribuot C. Erythropoietin and myocardial protection: what’s new? Fundamental & Clinical Pharmacology. 2005;19(4):439–446. doi: 10.1111/j.1472-8206.2005.00347.x. [DOI] [PubMed] [Google Scholar]
- 28.Ogilvie M, Yu X, Nicolas-Metral V, Pulido SM, Liu C, Ruegg UT, et al. Erythropoietin stimulates proliferation and interferes with differentiation of myoblasts. J Biol Chem. 2000;275(50):39754–39761. doi: 10.1074/jbc.M004999200. [DOI] [PubMed] [Google Scholar]
- 29.Davenport A, King RF, Ironside JW, Will EJ, Davison AM. The effect of treatment with recombinant human erythropoietin on the histological appearance and glycogen content of skeletal muscle in patients with chronic renal failure treated by regular hospital haemodialysis. Nephron. 1993;64(1):89–94. doi: 10.1159/000187284. [DOI] [PubMed] [Google Scholar]
- 30.Vaziri ND. Cardiovascular effects of erythropoietin and anemia correction. Curr Opin Nephrol Hypertens. 2001;10(5):633–637. doi: 10.1097/00041552-200109000-00013. [DOI] [PubMed] [Google Scholar]
- 31.Kao RL, Xenocostas A, Rui TR, Yu P, Huang W, Rose J, et al. Erythropoietin improves skeletal muscle microcirculation and tissue bioenergetics in a mouse sepsis model. Crit Care. 2007;11(3):R58. doi: 10.1186/cc5920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fishbane S, Besarab A. Mechanism of increased mortality risk with erythropoietin treatment to higher hemoglobin targets. Clin J Am Soc Nephrol. 2007;2(6):1274–1282. doi: 10.2215/CJN.02380607. [DOI] [PubMed] [Google Scholar]
- 33.Rosner MH, Bolton WK. The mortality risk associated with higher hemoglobin: is the therapy to blame? Kidney Int. 2008;74(6):695–697. doi: 10.1038/ki.2008.263. [DOI] [PubMed] [Google Scholar]
- 34.Heinicke K, Baum O, Ogunshola OO, Vogel J, Stallmach T, Wolfer DP, et al. Excessive erythrocytosis in adult mice overexpressing erythropoietin leads to hepatic, renal, neuronal, and muscular degeneration. Am J Physiol Regul Integr Comp Physiol. 2006;291(4):R947–956. doi: 10.1152/ajpregu.00152.2006. [DOI] [PubMed] [Google Scholar]
- 35.Leist M, Ghezzi P, Grasso G, Bianchi R, Villa P, Fratelli M, et al. Derivatives of erythropoietin that are tissue protective but not erythropoietic. Science. 2004;305(5681):239–242. doi: 10.1126/science.1098313. [DOI] [PubMed] [Google Scholar]
- 36.Mennini T, De Paola M, Bigini P, Mastrotto C, Fumagalli E, Barbera S, et al. Nonhematopoietic erythropoietin derivatives prevent motoneuron degeneration in vitro and in vivo. Mol Med. 2006;12(7–8):153–160. doi: 10.2119/2006-00045.Mennini. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Villa P, van Beek J, Larsen AK, Gerwien J, Christensen S, Cerami A, et al. Reduced functional deficits, neuroinflammation, and secondary tissue damage after treatment of stroke by nonerythropoietic erythropoietin derivatives. J Cereb Blood Flow Metab. 2007;27(3):552–563. doi: 10.1038/sj.jcbfm.9600370. [DOI] [PubMed] [Google Scholar]
- 38.Savino C, Pedotti R, Baggi F, Ubiali F, Gallo B, Nava S, et al. Delayed administration of erythropoietin and its non-erythropoietic derivatives ameliorates chronic murine autoimmune encephalomyelitis. J Neuroimmunol. 2006;172(1–2):27–37. doi: 10.1016/j.jneuroim.2005.10.016. [DOI] [PubMed] [Google Scholar]
- 39.Srisawat N, Manotham K, Eiam-Ong S, Katavetin P, Praditpornsilpa K, Eiam-Ong S. Erythropoietin and its non-erythropoietic derivative: do they ameliorate renal tubulointerstitial injury in ureteral obstruction? Int J Urol. 2008;15(11):1011–1017. doi: 10.1111/j.1442-2042.2008.02149.x. [DOI] [PubMed] [Google Scholar]
- 40.Bianchi R, Brines M, Lauria G, Savino C, Gilardini A, Nicolini G, et al. Protective effect of erythropoietin and its carbamylated derivative in experimental Cisplatin peripheral neurotoxicity. Clin Cancer Res. 2006;12(8):2607–2612. doi: 10.1158/1078-0432.CCR-05-2177. [DOI] [PubMed] [Google Scholar]
- 41.Fiordaliso F, Chimenti S, Staszewsky L, Bai A, Carlo E, Cuccovillo I, et al. A nonerythropoietic derivative of erythropoietin protects the myocardium from ischemia-reperfusion injury. Proc Natl Acad Sci U S A. 2005;102(6):2046–2051. doi: 10.1073/pnas.0409329102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bogdanovich S, Krag TO, Barton ER, Morris LD, Whittemore LA, Ahima RS, et al. Functional improvement of dystrophic muscle by myostatin blockade. Nature. 2002;420(6914):418–421. doi: 10.1038/nature01154. [DOI] [PubMed] [Google Scholar]
- 43.Scoppetta C, Grassi F. Erythropoietin: a new tool for muscle disorders? Medical Hypotheses. 2004;63(1):73–75. doi: 10.1016/j.mehy.2003.12.044. [DOI] [PubMed] [Google Scholar]
- 44.Lapchak PA. Carbamylated erythropoietin to treat neuronal injury: new development strategies. Expert Opin Investig Drugs. 2008;17(8):1175–1186. doi: 10.1517/13543784.17.8.1175. [DOI] [PubMed] [Google Scholar]
- 45.Xu X, Cao Z, Cao B, Li J, Guo L, Que L, et al. Carbamylated erythropoietin protects the myocardium from acute ischemia/reperfusion injury through a PI3K/Akt-dependent mechanism. Surgery. 2009;146(3):506–514. doi: 10.1016/j.surg.2009.03.022. [DOI] [PubMed] [Google Scholar]
- 46.Glass DJ. Molecular mechanisms modulating muscle mass. Trends Mol Med. 2003;9(8):344–350. doi: 10.1016/s1471-4914(03)00138-2. [DOI] [PubMed] [Google Scholar]
- 47.Lai KM, Gonzalez M, Poueymirou WT, Kline WO, Na E, Zlotchenko E, et al. Conditional activation of akt in adult skeletal muscle induces rapid hypertrophy. Mol Cell Biol. 2004;24(21):9295–9304. doi: 10.1128/MCB.24.21.9295-9304.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Casadevall N, Nataf J, Viron B, Kolta A, Kiladjian JJ, Martin-Dupont P, et al. Pure red-cell aplasia and antierythropoietin antibodies in patients treated with recombinant erythropoietin. N Engl J Med. 2002;346(7):469–475. doi: 10.1056/NEJMoa011931. [DOI] [PubMed] [Google Scholar]