

Abstract
LR11, also known as SorLA, is a mosaic low-density lipoprotein receptor that exerts multiple influences on Alzheimer disease susceptibility. LR11 interacts with the amyloid-β precursor protein (APP) and regulates APP traffic and processing to amyloid-β peptide (Aβ). The functional domains of LR11 suggest that it can act as a cell surface receptor and as an intracellular sorting receptor for trans-Golgi network to endosome traffic. We show that LR11 over-expressed in HEK293 cells is radiolabeled following incubation of cells with [32Pi]orthophosphate. Liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) was used to discover putative LR11 interacting kinases. Rho-associated coiled-coil containing protein kinase (ROCK) 2 was identified as a binding partner and a candidate kinase acting on LR11. LR11 and ROCK2 co-immunoprecipitate from post-mortem human brain tissue and drug inhibition of ROCK activity reduces LR11 phosphorylation in vivo. Targeted knockdown of ROCK2 with siRNA decreased LR11 ectodomain shedding while simultaneously increasing intracellular LR11 protein level. Site-directed mutagenesis of serine 2206 in the LR11 cytoplasmic tail reduced LR11 shedding, decreased LR11 phosphorylation in vitro, and abrogated LR11 mediated Aβ reduction. These findings provide direct evidence that LR11 is phosphorylated in vivo and indicate that ROCK2 phosphorylation of LR11 may enhance LR11 mediated processing of APP and amyloid production.
Keywords: Alzheimer Disease, Lipoprotein Receptor, Protein Phosphorylation, Rho, Shedding, Rho Kinase
Introduction
LR11 is a multi-domain low-density lipoprotein receptor (LDLR)3 family member expressed prominently in brain (1). All members of the LDLR family (including LR11) bind apolipoprotein E (ApoE). Structural elements in LR11 also place it in the vacuolar protein sorting 10 protein (VPS10p) homology domain family of intracellular sorting receptors (2). We first identified this receptor as a transcript that was found to be down-regulated in microarray studies examining gene expression in patients with Alzheimer disease (AD), and we subsequently demonstrated preferential neuronal loss of LR11 protein in brain regions vulnerable to AD pathology (3, 4). More recent studies suggest early loss of neuronal LR11 in individuals with mild cognitive impairment (MCI), a condition which often represents prodromal AD (5). Additionally, genetic evidence directly links variants in the LR11 gene, SORL1, to AD risk (6).
Sequential enzymatic cleavages of amyloid-β precursor protein (APP) yield amyloid-β peptide (Aβ), the major constituent of senile plaques. Cell biological studies indicate that APP processing is determined by its intracellular traffic and exposure to secretase enzymes, and that Aβ is produced in the endosomal compartment as well as the early secretory pathway (7–10). A number of studies have reported insights into the mechanism by which LR11 may influence AD pathogenesis. LR11 overexpression results in a highly consistent, dose-dependent, reduction in APP processing to Aβ (4, 11). Conversely, loss of LR11 expression, as occurs in sporadic AD brain, increases Aβ levels in cultured cells and accelerates amyloid pathology in a transgenic mouse model of AD (6, 12, 13). LR11 interacts directly with APP and co-localization between these proteins has been shown in endosomal and Golgi compartments in a variety of cell types, including primary neurons (4, 11). Based on the established interactions between the lumenal domains of LR11 and APP, we hypothesize that LR11 acts as an endosomal chaperone to increase APP traffic in a non-amyloidogenic pathway.
To define the mechanisms by which LR11 modulates APP processing, it is important to analyze the functional motifs in the cytoplasmic tail of LR11 and study proteins that regulate the intracellular traffic of LR11. The domain structure of LR11 suggests that it can act as an endocytic cell surface receptor and as a sorting receptor for trans-Golgi network (TGN) to endosome traffic (2, 14, 15). The cytoplasmic tail possesses a Golgi-localizing, gamma-adaptin ear homology domain, ADP-ribosylation factor (GGA)-binding domain found on proteins, such as sortilin and mannose 6-phosphate receptors (MPRs), that shuttle between the TGN and endosomes. In addition, LR11 harbors a putative internalization sequence, analogous to the NPXY motif for coated pit-mediated endocytosis (14, 16–18). LR11's NPXY-like motif (FANSHY) does not appear to be an essential internalization signal, but an acidic cluster in the cytoplasmic tail is required for AP-2 complex-dependent endocytosis (19). Phosphorylation of VPS10p family members, including sortilin and LR11, as well as MPRs is suggested to regulate protein-protein interactions with GGA adaptors to facilitate receptor traffic from Golgi to endosomes; however, direct demonstration of LR11 phosphorylation is lacking (20). To verify that LR11 is indeed phosphorylated in vivo, LR11 was expressed in human embryonic kidney (HEK) 293 cells and incubated with [32Pi]orthophosphate. Using LC-MS/MS we identified Rho-associated coiled-coil containing protein kinase (ROCK) 2 as a putative LR11-interacting kinase. LR11 interaction with ROCK2 was validated in post-mortem human frontal cortex brain tissue. Further, we show drug inhibition of ROCK2 reduces LR11 phosphorylation in vivo, and targeted knockdown with siRNA reveals a ROCK2 associated mechanism of LR11 ectodomain shedding. Site-directed mutagenesis of potential ROCK2 phosphorylation sites in the LR11 cytoplasmic tail indicate that serine 2206 is necessary for LR11 shedding as well as LR11-mediated Aβ reduction.
EXPERIMENTAL PROCEDURES
Antibodies
V5 epitope: monoclonal AbD Serotec (MCA1360); LR11: monoclonal BD Transduction Labs (611860), polyclonal anti-sera to C terminus generated against the peptide CEDAPMITGFSDDVPMVIA by Covance Research Products, Inc. (Denver, PA), preimmune sera collected before the first immunization; ROCK2 monoclonal Abcam (ab56661); APP monoclonal 6E10 Covance (Signet, SIG-39320–200); Calnexin polyclonal Assay Designs, Ann Arbor, MI (SPA-860).
Generation of LR11 Mutants
N-terminal V5-tagged LR11: human LR11 cDNA in pcDNA3.1 was a gift from Dr. Chica Schaller (Zentrum für Molekulare Neurobiologie, Universität Hamburg, Hamburg, Germany). LR11 cDNA was amplified using AccuPrime Pfx DNA polymerase Supermix (Invitrogen), a sense primer with an XbaI site (5′-ATA TTC TAG AAG CGC TGC CCT GCA GCC CGA-3′), and an antisense primer introducing an XhoI site (5′-AAT ACT CGA GTC AGG CTA TCA CCA TGG GGA-3′). The resulting PCR products were cut using the appropriate restriction enzymes and ligated into modified pcDNA3.1 (Invitrogen) plasmid that contained the LR11 signal peptide followed by the LR11 propeptide and V5 tag. To generate LR11 mutants, N-terminal V5-tagged LR11 was used as a template and QuikChange XL Site-directed Mutagenesis kit (Stratagene) was employed. For LR11 S2167A, the sense primer was 5′-GAA GCA CCG GAG GCT GCA GGC CAG CTT CAC CGC CTT CGC CAA CAG-3′ and the antisense was 5′-CTG TTG GCG AAG GCG GTG AAG CTG GCC TGC AGC CTC CGG TGC TTC-3′. For LR11 S2206A, the sense primer was 5′-TAT GAT AAC TGG ATT TGC AGA TGA CGT CCC CAT GGT GAT AGC CTG-3′ and the antisense was 5′-CAG GCT ATC ACC ATG GGG ACG TCA TCT GCA AAT CCA GTT ATC ATA-3′. All constructs were verified by restriction digest and sequencing.
Cell Culture and Transfection
Human embryonic kidney 293 (HEK293) cells were maintained in DMEM with 10% fetal bovine serum (FBS) (Invitrogen) and 1% penicillin/streptomycin (Cellgro/Mediatech Inc., Herndon, VA).). Equivalent amounts of cells were plated and transfected with the indicated constructs using Lipofectamine 2000 (Invitrogen). ROCK2 ON-TARGETplus SMARTpool (Dharmacon) and ON-TARGET Non-targeting Pool siRNA (Dharmacon) were transfected using DharmaFECT1 siRNA Transfection Reagent (Dharmacon) according to the manufacturer's instructions.
Media Conditioning, Cell Lysis, and Immunoblotting
Media was conditioned for 24 h, and protein precipitation was performed by adding 4:1 ratio of methanol to chloroform to conditioned media (CM). CM was vortexed and spun at 15,000 × g for 2 min. Aqueous layer was removed, 1:1 methanol added, and spun as before. Protein pellet was air dried and solubilized in 2× Laemmli sample buffer and equivalent amounts of sample were loaded for immunoblot analysis. Cells were lysed as previously described (4) in PBS plus protease inhibitor mixture (PIC) (Roche Diagnostics, Mannheim, Germany), Halt phosphatase inhibitor mixture (Pierce), and lysis buffer containing 0.5% Nonidet P-40, 0.5% deoxycholate, 150 mm sodium chloride, and 50 mm Tris, pH 7.4 (PBS+PIC+Halt+lysis buffer). Post-mortem human frontal cortex brain tissue was provided by the Emory University Brain Bank. Tissue was homogenized (Dounce homogenizer) in the PIC+Halt+lysis buffer described above and subjected to a 1000 × g spin to remove nuclei and debris. Cleared lysate was used for co-immunoprecipitations described below.
Immunoblotting was performed as previously described (4). To load equivalent amounts of soluble lysate per sample (Fig. 5), protein concentration was determined by bicinchoninic acid (BCA) method (Pierce). Images were captured using an Odyssey Image Station (LiCor, Lincoln, NE), and band intensities were quantified using Scion Image. Statistical analysis was performed using Student's t test for independent samples.
FIGURE 5.

ROCK2 knockdown decreases LR11 ectodomain shedding. A, HEK293 cells were transfected with ROCK2 or Scramble (non-targeting) siRNA smart pools and harvested after 96 h. Media were conditioned for 24 h beginning 72 h after transfection. Equivalent volumes of conditioned media (CM) were resolved by SDS-PAGE. Immunoblot analyses reveal reduction in LR11 ectodomain shedding in ROCK2-depleted cells but no change in APPsα release. For analyses of total cell lysate (Lysate), 50 μg of protein was loaded per lane in SDS-PAGE. Immunoblotting indicated that the reduction in ROCK2 protein was accompanied by an increase in cellular LR11. Calnexin was used as a loading control. B, ROCK2 depletion produced significant changes in cell-associated ROCK2 and LR11 (60% decrease and 33% increase, respectively) and secreted LR11 ectodomain (44% decrease). C, semi-quantitative reverse transcription-PCR analyses demonstrate no change in LR11 mRNA, suggesting that the observed changes in protein levels are not likely to be due to changes in LR11 transcription. β-Actin was used as a control. Data are representative of three independent experiments.
Co-immunoprecipitations
For HEK293 cell co-immunoprecipitations (IP), cells were transiently transfected (when applicable) with indicated plasmids. After 48 h, cells were lysed as described above and 5% of the lysate was removed for later use as “lysates” in immunoblots. The remaining cell lysate was cleared with mouse immunoglobulin plus protein A-Sepharose beads (Invitrogen), incubated for 30 min at 4 °C, and IPs were then performed according to standard protocols. For human brain tissue, lysates for LR11 antisera IPs were pre-cleared with pre-immune sera plus beads. Additionally, control IPs were performed using beads alone and pre-immune sera to demonstrate specificity of LR11 antisera.
In Vitro and in Vivo Labeling with [32Pi]Orthophosphate
For in vitro labeling, HEK293 cells were transiently transfected with indicated plasmids, and IPs were performed as described above. IPs were washed four times in PIC+lysis buffer and washed once in kinase buffer (50 mm Tris-HCl, pH 7.4, 5 mm MgCl2, 1 mm EDTA, 1 mm EGTA). IPs were resuspended in kinase buffer plus cold 100 μm ATP and 1 μCi [32Pi]orthophosphate (NEX053002MC, PerkinElmer) with or without 10 μm Rho kinase inhibitor (555550, Calbiochem). IPs were incubated at 30 °C for 30 min, resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), exposed to x-ray film, and subjected to immunoblot. For in vivo labeling, HEK293 cells were transfected with indicated plasmids as described above. Forty-eight hours post-transfection, cells were washed twice with phosphate-free DMEM and incubated for 2 h in the same medium containing [32Pi]orthophosphate (1 mCi/ml). Metabolic labeling was stopped by washing cells twice with ice-cold PBS+PIC. Cells were lysed, and lysates were subjected to IP with monoclonal V5 antibody for 4 h at 4 °C, as described above. IPs were washed four times with PBS+PIC+Halt+lysis buffer and subjected to SDS-PAGE, autoradiography, and finally immunoblot. For calf-intestinal alkaline phosphatase (CIP) (New England Biolabs) treatment, immunoprecipitates were incubated with or without 5000 units of CIP for 60 min at 37 °C according to the manufacturer's instructions and then subjected to SDS-PAGE. Mock treated samples contained all elements except CIP. For drug inhibition of ROCK, 50 μm Rho kinase inhibitor or DMSO (mock) was added during 2 h [32Pi]orthophosphate metabolic labeling as described above.
Identification of LR11-interacting Proteins by Mass Spectrometry
HEK293 cells were transiently transfected with empty vector or plasmid expressing V5-tagged LR11 and harvested 48 h later. IP using monoclonal V5 antibody was as described above and immunoprecipitates were separated by SDS-PAGE followed by staining with Coomassie Blue G-250. Six gel pieces were excised per lane based on protein molecular mass (>250 kDa, 100–250 kDa, 75–100 kDa, 50–75 kDa, 25–50 kDa, <25 kDa). Twelve total gel slices were individually digested with trypsin, and the resulting peptides were analyzed independently by reverse-phase liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) as previously described (21). Briefly, peptide mixtures were loaded onto a C18 column (100 μm i.d., 10 cm long, 5 μm resin from Michrom Bioresources, Inc., Auburn, CA) and eluted over a 5–30% gradient (Buffer A: 0.4% acetic acid, 0.005% heptafluorobutyric acid, and 5% AcN; Buffer B: 0.4% acetic acid, 0.005% heptafluorobutyric acid, and 95% AcN). Eluates were monitored in a MS survey scan followed by nine data-dependent MS/MS scans on an LTQ-Orbitrap ion trap mass spectrometer (Thermo Finnigan, San Jose, CA). The LTQ was used to acquire MS/MS spectra (2 m/z isolation width, 35% collision energy, 5,000 AGC target, 150 ms maximum ion time). The Orbitrap was used to collect MS scans (300–1600 m/z, 1,000,000 AGC target, 750 ms maximum ion time, resolution 60,000). All data were converted from raw files to the .dta format using ExtractMS version 2.0 (Thermo Finnigan, San Jose, CA). The acquired MS/MS spectra were searched against a concatenated target decoy human reference data base of the National Center for Biotechnology Information (November 19, 2008 with 38,114 target entries) using the SEQUEST Sorcerer algorithm (version 3.11, SAGE-N) (22); Searching parameter including: partially tryptic restriction, parent ion mass tolerance (± 50 ppm), product ion tolerance (± 0.5 m/z), and dynamic modifications for oxidized Met (+15.9949 Da). The peptides were classified by charge state and tryptic state (fully and partial) and first filtered by mass accuracy (10 ppm for high-resolution MS), and then dynamically by increasing XCorr (minimal 1.0) and ΔCn values to reduce protein false discovery rate to less than 1%. All accepted proteins sharing peptides were grouped together, in which only the top protein with highest spectral counts was selected to represent the group. In total 2,462 peptides clustered in 305 protein groups in LR11 immunoprecipitates and 1662 peptides clustered in 238 groups for control immunoprecipitates were identified. All peptides identified in control and LR11 immunoprecipitates are provided in supplemental Tables S1 and S2, respectively.
Semi-quantitative Reverse-transcriptase-PCR
HEK293 cells were transfected with ROCK2 ON-TARGETplus SMARTpool or ON-TARGET Non-targeting Pool siRNA (Dharmacon) for 72 h, and total RNA was isolated with TRIzol (Sigma) according to the manufacturer's instructions. RNA was prepared and cDNA was generated as described (4) using Super-Script First-Strand Synthesis System for RT-PCR (Invitrogen). Subsequent PCR for LR11 and β-actin was performed exactly as described (4). PCR products were separated on a 2% agarose gel, stained with ethidium bromide, and visualized with a Fluorchem 8800 gel documentation system (Alpha Innotech Corp., San Leandro, CA).
Aβ Measurements
HEK293 cells were transiently transfected with indicated plasmids, and media were conditioned for 48 h beginning 24 h after transfection. Conditioned media were collected at 72 h, and cells were harvested. Endogenous Aβ was detected using a sandwich enzyme-linked immunosorbent assay (ELISA) for human β amyloid 1–40 (Millipore Corp., Billerica, MA) following the manufacturer's instructions. Plates were read at 450 nm on a Spectra Max Plus plate reader (Molecular Devices, Sunnyvale, CA). Cell extracts and conditioned media samples were blotted for indicated proteins as described above. Experiments were performed in triplicate.
RESULTS
In Vivo Phosphorylation of LR11
Because of low endogenous expression of LR11 in cultured human cells, we overexpressed N-terminal V5-tagged LR11 to facilitate immunoprecipitation (IP). To determine whether LR11 is phosphorylated in vivo, HEK293 cells were transiently transfected with plasmid expressing N-terminal V5-tagged LR11or empty vector (pcDNA3.1) and incubated with [32Pi]orthophosphate for 2 h in phosphate-free medium to label the intracellular ATP pool and phosphoproteins. IP of LR11 using anti-V5 antibody followed by SDS-PAGE and autoradiography revealed covalent incorporation of phosphate into two closely spaced bands of ∼250 kDa (Fig. 1A). Immunoblot analysis for V5-LR11 revealed an identical pattern and electrophoretic mobility, consistent with the 32Pi-labeled bands being V5-LR11. That LR11 migrates in SDS-PAGE as a doublet has been previously attributed to differences in N-glycosylation (23). Lack of 32Pi-labeled and LR11-specific bands in controls confirms the specificity of the IP. Calf-intestinal alkaline phosphatase (CIP) treatment was employed to verify the incorporation of radioactive phosphate in V5-LR11. IPs prepared as in Fig. 1A were incubated with or without CIP prior to SDS-PAGE, and autoradiography revealed the loss of labeled bands in the CIP-treated sample (Fig. 1B). Hence, these results confirm that V5-LR11 is phosphorylated when expressed in HEK293 cells.
FIGURE 1.
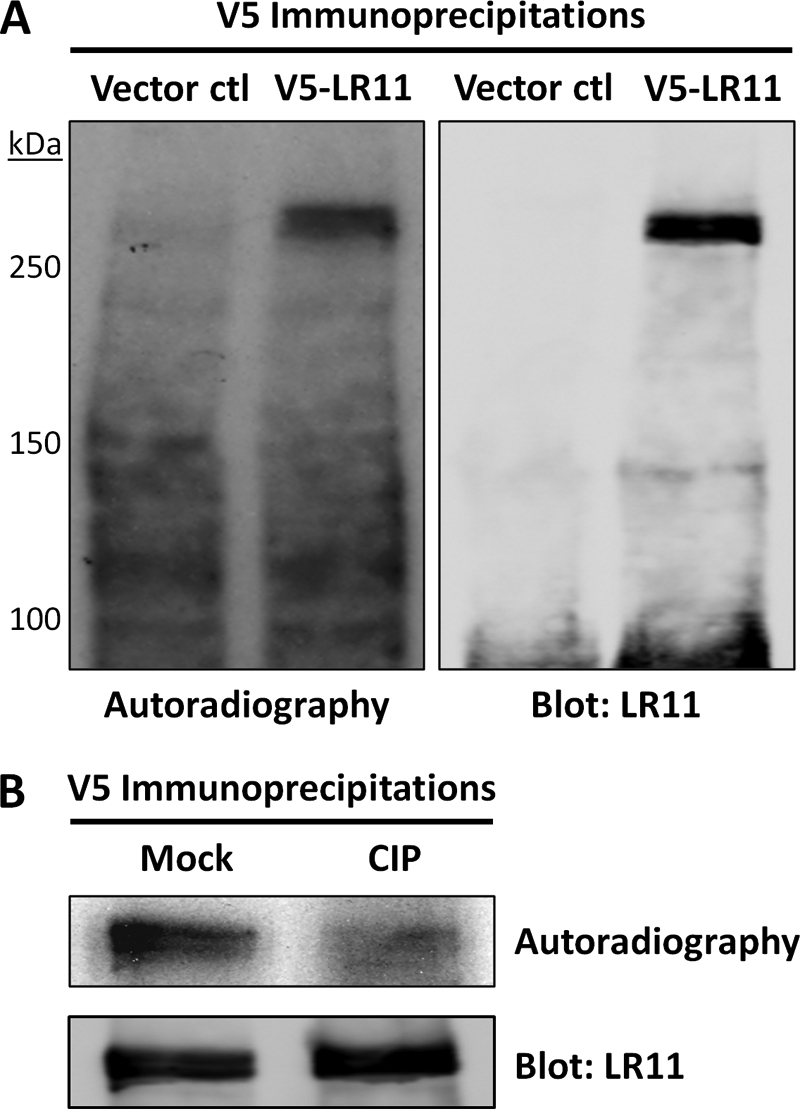
LR11 is phosphorylated in vivo. A, HEK293 cells transfected with empty vector (Vector ctl) or V5-LR11 were metabolically labeled with [32Pi]orthophosphate for 2 h, as described under “Experimental Procedures.” IPs were performed with V5 antibody and subjected to SDS-PAGE, autoradiography, and immunoblot. The incorporation of 32P into bands at ∼250 kDa that co-migrate with V5-LR11 immunoreactivity in subsequent immunoblots is consistent with V5-LR11 being a phosphoprotein. The absence of LR11 bands in Vector ctl lane indicates specificity of V5 IP. B, HEK293 cells expressing V5-LR11 were labeled as above and IPs were performed as described under “Experimental Procedures.” Before SDS-PAGE, immunoprecipitates were incubated with calf-intestinal alkaline phosphatase (CIP) or without (Mock) for 1 h at 37 °C. Autoradiography reveals loss of V5-LR11 phosphorylation in CIP-treated samples, and immunoblots indicate that V5-LR11 levels in the IPs are unchanged. Data shown are representative of two independent experiments.
Identification and Validation of LR11-interacting Proteins by Mass Spectrometry
To identify potential kinases acting on LR11 we tested for protein-binding partners using co-IP (Fig. 2A). HEK293 cells were transiently transfected with empty vector or plasmid expressing V5-LR11 and IPs were performed with V5 antibody. IPs were resolved by SDS-PAGE, each lane was cut into six pieces, and proteins were digested with trypsin. Peptides were independently analyzed by LC-MS/MS, and MS/MS spectra were collected and searched against a concatenated target-decoy database. Our LC-MS/MS data identified 2,462 peptides clustered in 305 protein groups in LR11 IPs and 1662 peptides clustered in 238 groups for control IPs. After V5-LR11, the most abundant protein in our V5-LR11 IPs was ROCK2, as quantified by spectral counts (Fig. 2, B and C). ROCK2 scored highest in total peptides (22), total spectral counts (24), and was in the top three candidates in percent protein coverage for potential LR11 interacting partners. Moreover, ROCK2 was the only kinase identified in LR11 IPs. Additional candidate LR11 interacting partners are listed in (Fig. 2C), but these interactions have not been biochemically validated using other methods. A list of all identified peptides and spectral counts in control and LR11 IPs is provided in supplemental Tables S1 and S2, respectively.
FIGURE 2.

Identification of LR11-ROCK2 interaction by LC-MS/MS. A, flowchart of experimental design to identify LR11-interacting partners. B, representative ROCK2 peptide spectrum (residues 1227–1239) from LR11 IP. C, list of the most abundant candidate LR11-binding partners. Each protein listed was absent from control IP samples. List of all spectra identified in control and V5-LR11 IPs is available in supplemental Table S1. TP, total peptides; TSC, total spectra counts; % Coverage, percent protein coverage.
ROCK2 is a serine/threonine kinase that is a downstream effector of the Rho family of small GTPases. The Rho-ROCK pathway is involved in multiple aspects of neuronal function including neurite outgrowth and retraction, and it has recently become an attractive target for drug development due to its role in spinal cord injuries, stroke, and Alzheimer disease (reviewed in Refs. 24, 25). ROCK exists as two isoforms, ROCK1 and ROCK2, but to date their functional differences remain largely uncharacterized. There is 65% similarity in their amino acid sequences overall and 92% identity in their kinase domains (residues 92–354 of ROCK2) (26). We found 21 peptides unique to ROCK2 in our LC-MS/MS analyses of LR11 IPs, but no unique peptides were identified for ROCK1. Notably, ROCK1 is predominantly expressed in non-neuronal tissues, whereas ROCK2 is preferentially expressed in brain (26). Like LR11, ROCK2 is highly expressed in pyramidal neurons of the hippocampus and Purkinje cells of the cerebellum (4, 27).
To validate the putative interaction between LR11 and ROCK2 identified by LC-MS/MS, we performed reciprocal co-IP experiments in HEK293 cells expressing V5-LR11 and endogenous ROCK2 (Fig. 3, A and B). V5 IPs were enriched with ROCK2 immunoreactivity while control IPs showed no detectable ROCK2 (Fig. 3A). Conversely, V5-LR11 was co-immunoprecipitated with endogenous ROCK2 (Fig. 3B). To confirm the physiological relevance and bona fide interactions between LR11 and ROCK2 in brain, we performed co-IP experiments using post-mortem human brain tissue from the Emory University Alzheimer Disease Research Center and NINDS Neuroscience Core Facilities Brain Bank. While low endogenous expression in HEK293 cells required LR11 overexpression, interaction between endogenous proteins was identified by co-IP from human frontal cortex (Fig. 3, C and D). ROCK2 was observed in IPs using LR11 anti-sera, and LR11 was found in IPs using antibodies to ROCK2. Control co-IPs using beads alone or pre-immune sera failed to enrich either LR11 or ROCK2. These findings were replicated in three independent postmortem cases and add strong support to the conclusion that LR11 and ROCK2 form a stable complex in human cells and tissues.
FIGURE 3.

Biochemical validation of LR11-ROCK2 interaction. A, HEK293 cells transfected with empty vector (Vector ctl) or V5-LR11 were harvested for IP with V5 antibody and proteins were resolved by SDS-PAGE. Immunoblotting revealed the presence of ROCK2 only in V5-LR11 IPs and not in control IPs. B, HEK293 cells were transfected with plasmid expressing V5-LR11 for IPs with mouse Ig plus protein A beads (Ig+beads) or ROCK2 antibody (ROCK2) and subjected to SDS-PAGE. Immunoblotting revealed LR11 only in ROCK2 IPs. Absence of ROCK2 in Ig+beads lane indicates specificity of ROCK2 IP. C and D, reciprocal co-IPs from post-mortem human frontal cortex. C, LR11 IPs using (left to right) protein A beads only (beads), pre-immune sera, or LR11 anti-sera (antisera). Immunoblotting revealed ROCK2 only in antisera IP while the absence of LR11 in control lanes (beads and pre-immune sera) indicate specificity of IP. D, ROCK2 IPs using (left to right) Ig+beads or ROCK2. Immunoblot reveals LR11 only in ROCK2 IP. Input lysate represents 5% by volume of IP. Blots in A and B are representative of three independent experiments. Findings in C and D were replicated in three independent postmortem cases.
Inhibition of Rho Kinase Reduces LR11 Phosphorylation in Vivo
The results of the LR11 phosphorylation and LR11 - ROCK2 interaction studies suggest that ROCK2 may phosphorylate LR11. To more directly test this hypothesis, we used pharmacological inhibitors of the kinase activity of ROCK2 and determined their effects on LR11 phosphorylation in cells. Several Rho kinase inhibitors have been used to study ROCK2 substrates, including myosin light chain (for review see Ref. 28). We chose RKI (dimethlyfausidil, Calbiochem), a reversible Rho kinase inhibitor that offers higher specificity than Y-27632, another widely used ROCK inhibitor (29, 30). Often to test whether a kinase can directly phosphorylate a potential substrate, full length recombinant proteins are generated and purified from bacteria and subjected to in vitro kinase assay. However, bacterial expression of large transmembrane proteins, like LR11, is quite challenging due to their hydrophobicity and structure, and previous attempts by our lab to generate full-length LR11 in bacteria have failed. To circumvent this issue, HEK293 cells were transiently transfected with plasmid directing expression of V5-LR11 and IPs were performed using the V5 antibody. IPs were washed in kinase buffer and resuspended in kinase buffer containing cold ATP, [32Pi]orthophosphate, and DMSO (mock) or 10 μm RKI. IPs were incubated at 30 °C for 30 min, and then subjected to SDS-PAGE, autoradiography, and immunoblot analyses. Autoradiography revealed covalent incorporation of phosphate into a band of ∼250 kDa in the mock-treated IP, but no band was observed in the RKI treated sample (Fig. 4A). Immunoblot analysis indicated the same relative intensities of LR11 and ROCK2 in all IP samples. Although the LC-MS/MS analysis described above identifies ROCK2 as the only kinase observed in V5-LR11 IPs, we cannot exclude the possibility that other kinases are present. The absence of phosphoproteins in the RKI treated sample suggests that V5-LR11 is a substrate for ROCK2, therefore we sought to test this hypothesis in vivo. HEK293 cells were transiently transfected with plasmid expressing V5-LR11, and cells were metabolically labeled with [32Pi]orthophosphate for 2 h in the presence or absence of 50 μm RKI. IP was performed using V5 antibody, followed by SDS-PAGE, autoradiography, immunoblot analysis, and quantification (normalized to the amount of LR11). The results revealed ∼42% reduction (p = 0.002) in LR11 phosphorylation in RKI treated samples compared with untreated controls (Fig. 4, B and C). Thus, LR11 phosphorylation is dependent upon ROCK2 activity, and because there was not complete loss of LR11 phosphorylation, other kinases may also be involved in LR11 phosphorylation in vivo and/or RKI inhibition was incomplete.
FIGURE 4.

Inhibition of Rho kinase reduces LR11 phosphorylation in vivo. A, LR11 IPs from HEK293 cells transfected with V5-LR11 were incubated with kinase buffer-containing cold ATP, [32Pi]orthophosphate, and DMSO (Mock) or 10 μm Rho kinase inhibitor (RKI) for 30 min at 30 °C, as described under “Experimental Procedures.” Autoradiography indicated that RKI treatment inhibits LR11 phosphorylation in vitro. Immunoblotting identified V5-LR11 and ROCK2 in V5-LR11 IPs. B, HEK293 cells transfected with V5-LR11 were metabolically labeled with [32Pi]orthophosphate for 2 h in the presence of 50 μm RKI (RKI) or DMSO (Mock). LR11 was IPed with V5 antibody and subjected to SDS-PAGE. Autoradiography revealed that RKI reduced LR11 phosphorylation in vivo. Immunoblot data indicated that relatively equivalent amounts of LR11 were present in each IP. C, intensity of phosphoproteins at 250 kDa was quantified and normalized to the amount of LR11 in the IPs. Data shown in A and B are representative of three independent experiments.
ROCK2 Influences LR11 Ectodomain Shedding
Protein-protein interactions are physiologically relevant when modification of one interacting protein occurs in parallel with a change in function of its partner. LR11 ectodomain shedding is stimulated by receptor-ligand interaction and blockade of LR11 shedding affects cell proliferation (23). Therefore, we investigated effects of ROCK2 depletion on LR11 ectodomain cleavage using siRNA techniques. HEK293 cells were transfected with ROCK2 targeted or control (non-targeting) siRNA smart pools and cells were collected after 96 h to assess the efficiency of knockdown. As seen in Fig. 5, A and B, ROCK2 was reduced to ∼41% of controls. The incomplete depletion of ROCK2 can be at least partially explained by the transfection efficiency of HEK293 cells, which in our hands is ∼60% under these conditions.
To assess any changes in LR11 ectodomain shedding resulting from ROCK2 knockdown, media were conditioned for 24 h beginning 72 h after transfection with ROCK2 siRNA smart pools. Immunoblotting was performed on conditioned media and corresponding cell lysates to determine the levels of shed ectodomain and cell associated LR11, respectively. LR11 ectodomain shedding decreased to ∼56% of controls (p = 0.0006) while cellular LR11 increased ∼33% (p = 0.002) in cells depleted of ROCK2 (Fig. 5, A and B). In contrast, no changes in APP secreted products were observed. Because APP ectodomain release is mediated primarily by metalloprotease cleavage of APP at the cell surface, we infer that general ectodomain shedding of receptors is not affected by the depletion of ROCK2, and there is specificity to the reduced ectodomain cleavage of LR11 upon ROCK2 knockdown.
The increase in cell-associated LR11 protein might be predicted in cells in which LR11 ectodomain shedding was decreased but other explanations are possible. To determine if changes in ROCK2 expression alter LR11 gene expression, total RNA was harvested in parallel from control and ROCK2 siRNA-transfected cells, and LR11 mRNA levels were determined using semi-quantitative reverse transcription-PCR (RT-PCR). LR11 mRNA levels were unchanged between ROCK2 knockdown and control samples (Fig. 5C). Because no change in the level of LR11 mRNA was observed it is more likely that the increase in protein level seen in ROCK2-depleted cells results from an increase in the protein stability or half-life. This interpretation is also consistent with the decrease in LR11 ectodomain shedding in ROCK2-depleted cells.
Mutagenesis of LR11 Ser-2206 Reduces LR11 Ectodomain Shedding
Based on the results of the Rho kinase drug inhibition and ROCK2 siRNA knockdown experiments we hypothesized that ROCK2 phosphorylation of LR11 enhances LR11 shedding. To investigate this hypothesis, we sought to identify LR11 phosphorylation sites by mass spectrometry. HEK293 cells were transiently transfected with plasmid directing expression of V5-LR11 and IPs were performed with V5 antibody. IPs were resolved by SDS-PAGE and a single gel piece containing proteins ≥ 250 kDa was excised for trypsin digestion. Additionally, parallel V5-LR11 IPs were in-solution digested with trypsin. To enrich for LR11 phosphopeptides, we employed immobilized metal-affinity chromatography (IMAC) incorporating Fe3+ ion followed by LC-MS/MS as previously described (31). Calcium phosphate precipitation (CPP) was also performed as an alternative to enrich for phosphopeptides (32). These strategies failed to identify LR11 phosphopeptides, however complete sequence coverage of LR11 was not achieved. Specifically, peptides containing amino acids 2180–2213 (Fig. 6A), in the LR11 cytoplasmic tail, were not observed. Notably, C-terminal peptides are under-represented in complex proteomic samples and only recently have C terminus-centric techniques been reported (33). To circumvent these issues, the Group-based Prediction System (GPS) version 2.1 software, with a modified version of Group-based Phosphorylation Scoring algorithm, was used to predict potential Rho kinase phosphorylation sites in the LR11 cytoplasmic tail (34, 35). GPS calculated that serine (Ser)-2167 and Ser-2206 were the most likely candidates for Rho kinase activity. Therefore, LR11 mutants S2167A and S2206A were generated by site-directed mutagenesis, substituting alanine for Ser-2167 and Ser-2206, respectively. To evaluate the effects of these mutations on LR11 ectodomain shedding, HEK293 cells were transiently transfected with empty vector, V5-LR11, V5-LR11S2167A, or V5-LR11S2206A, and media were conditioned for 24 h beginning 24 h after transfection. Conditioned media samples and corresponding cell lysates were subjected to immunoblot analyses to determine the levels of shed ectodomain and cell associated LR11, respectively. Mutagenesis of Ser-2167 had no observable impact on LR11 ectodomain shedding, but mutation of Ser-2206 resulted in a ∼46% reduction (p = 0.008) of shedding as compared with wild-type LR11 (Fig. 6, B and C). Although the LR11S2206A phenotype mimicked the reduction of LR11 shedding that was observed under ROCK2 knockdown conditions (Fig. 5, A and B), LR11S2206A did not display an increase of cell associated LR11. These results suggest that Ser-2206, and possibly phosphorylation of Ser-2206, is necessary for LR11 shedding but may not impact LR11 half-life or stability. To address whether mutagenesis of Ser-2206 affected LR11 interaction with or phosphorylation by ROCK2, HEK293 cells were transiently transfected with empty vector, V5-LR11, or V5-LR11S2206A, and IPs were performed using V5 antibody. V5 IPs from empty vector, V5-LR11, and V5-LR11S2206A-transfected cells were incubated with kinase buffer containing cold ATP and [32Pi]orthophosphate for 30 min at 30 °C and subjected to SDS-PAGE, autoradiography, and immunoblot analyses. Autoradiography revealed decreased phosphorylation of V5-LR11S2206A, compared with V5-LR11, and immunoblotting indicated that comparable amounts of LR11 and ROCK2 were present in the IPs (Fig. 6D). These data support the hypothesis that ROCK2 phosphorylates LR11 Ser-2206 and increases LR11 ectodomain shedding. Moreover, these results suggest a means of uncoupling the regulation of domain shedding from that of LR11 protein half-life that may provide important insights in future studies of LR11 biology.
FIGURE 6.

Mutagenesis of LR11 Ser-2206 reduces LR11 ectodomain shedding. A, primary sequence of the cytoplasmic tail of human LR11 indicating in silico predicted sites of ROCK2 phosphorylation and the two serines targeted for mutagenesis. Serine residues at amino acid 2167 and 2206 were substituted for alanines by site-directed mutagenesis, as described under “Experimental Procedures.” B, HEK293 cells were transfected with empty vector (Vector ctl), V5-LR11, V5-LR11S2167A, or V5-LR11S2206A and harvested after 48 h. Media were conditioned for 24 h beginning 24 h after transfection. Equivalent volumes of conditioned media (Media) were loaded for SDS-PAGE. Immunoblot analyses indicate ∼46% reduction in LR11 ectodomain shedding in V5-LR11S2206A samples but no change in V5-LR11S2167A shedding. For cell lysate (Lysate) analyses, 50 μg of protein was loaded per lane for SDS-PAGE. Immunoblot reveals similar levels of cellular V5-LR11, V5-LR11S2167A, and V5-LR11S2206A. For Media and Lysate, a nonspecific immunoreactive band and calnexin, respectively, were used as loading controls. C, intensity of immunoblot bands were quantified and show significant reduction in LR11 ectodomain shedding in LR11S2206A samples. D, HEK293 cells transfected with empty vector (Vector ctl), V5-LR11 (LR11wt), or V5-LR11S2206A were harvested for IP with V5 antibody. IPs were incubated with kinase buffer containing cold ATP and [32Pi]orthophosphate for 30 min at 30 °C. Autoradiography indicates greater intensity of labeled bands in V5-LR11 immunoprecipitates compared with V5-LR11S2206A, suggesting that Ser-2206 is a potential ROCK2 phosphorylation site in vitro. Immunoblot reveals ROCK2 in V5-LR11 and LR11S2206A immunoprecipitates, indicating that mutagenesis of LR11 Ser-2206 does not impact LR11-ROCK2 co-IP. Further, immunoblot shows equivalent LR11 enrichment in V5-LR11 and V5-LR11S2206A IPs. Input lysate represents 5% by volume of IP. Data are representative of three independent experiments. TM, transmembrane domain; wt, wild-type.
Mutagenesis of LR11 Ser-2206 impacts LR11-mediated Aβ Reduction
LR11 interacts with APP and when overexpressed, LR11 consistently reduces Aβ secretion (4, 11, 36). However, which of the many LR11 domains are important for these effects in human cells remains unclear. To assess whether mutagenesis of LR11 Ser-2206 influences LR11 mediated Aβ reduction, HEK293 cells were transiently transfected with empty vector, V5-LR11, or V5-LR11S2206A, and media were conditioned for 48 h beginning 24 h after transfection. Endogenous full length APP and α-secretase cleaved secreted APP (APPsα) were analyzed by immunoblotting cell extract and conditioned media, respectively, while secreted Aβ1–40 was detected by sandwich ELISA. There were no differences in the levels of full length, cell-associated APP or APPsα in the conditioned media after expression of V5-LR11 or V5-LR11S2206A, as compared with empty vector control (Fig. 7A). However, Aβ1–40 levels were reduced by ∼44% (p < 0.0001) in conditioned media from cells transfected with V5-LR11 and reduced by only ∼28% (p = 0.0004 compared with empty vector, p = 0.0043 compared with V5-LR11) in V5-LR11S2206A samples (Fig. 7B). Because LR11-mediated reduction of Aβ secretion was significantly hampered by mutagenesis of Ser-2206, we hypothesize that phosphorylation of LR11, most likely by ROCK2, enhances LR11 modulation of APP processing in a non-amyloidogenic manner.
FIGURE 7.

LR11 Ser-2206 is necessary for LR11-mediated Aβ reduction. A, HEK293 cells were transfected with empty vector (Vector ctl), V5-LR11, orV5-LR11S2206A and harvested after 72 h. Media were conditioned for 48 h beginning 24 h after transfection. Equivalent volumes of conditioned media (Media) and 50 μg of protein for cell lysates (Lysate) were loaded for SDS-PAGE. Immunoblot analyses indicate similar levels of secreted APPsα and full-length APP among all samples. Calnexin was used as a loading control. B, endogenous secreted Aβ1–40 was detected by sandwich ELISA. Expression of V5-LR11 reduced secreted Aβ1–40 by 44% while expression of LR11S2206A reduced secreted Aβ1–40 by only 28% compared with empty vector control. Data shown are representative of three independent experiments. wt, wild-type.
DISCUSSION
LR11 is a multifunctional low-density lipoprotein receptor that influences AD susceptibility. While LR11 has multiple cell biological activities that may be relevant to AD pathogenesis, we hypothesize that LR11 is an endosomal chaperone that increases APP traffic in a non-amyloidogenic pathway and helps retard amyloid accumulation (3, 4, 6, 11–13). Therefore, it is important to better understand the functional motifs of LR11 and the proteins that regulate LR11 activity. This study provides direct demonstration that LR11 is phosphorylated in vivo and presents compelling evidence that LR11 interacts with the serine/threonine kinase, ROCK2, in human brain. Moreover, we observed a reduction in LR11 phosphorylation in vivo following drug inhibition of ROCK2, suggesting that LR11 is a substrate of ROCK2. Targeted knockdown of ROCK2 with siRNA resulted in decreased LR11 ectodomain shedding, thus implicating ROCK2 in the mechanism(s) that promote shedding of LR11. Site-directed mutagenesis of potential ROCK2 phosphorylation sites in the LR11 cytoplasmic tail revealed that Ser-2206 is necessary for LR11 shedding as well as LR11 mediated Aβ reduction. Based on these observations, we conclude that phosphorylation of LR11 by ROCK2 exerts physiologically relevant influences on LR11 regulation of APP processing.
The current study suggests that ROCK2 is stably complexed with and phosphorylates LR11. ROCK2 is a cytosolic serine/threonine kinase that translocates to membranes following stimulation of RhoA (37, 38). If ROCK2 binds directly to LR11, the most plausible site of interaction and phosphorylation is LR11's cytoplasmic tail. Investigation of the 54 amino acid long intracellular tail of LR11 reveals 12 potential phosphorylation sites, including 9 serine, 2 threonine, and 1 tyrosine residues, however it is also possible that the LR11 ectodomain, like APP, harbors sites of phosphorylation (39). GPS version 2.1 software predicted two potential ROCK2 phosphorylation sites in the LR11 intracellular tail at Ser-2167 and Ser-2206 (34, 35). The consensus amino acid sequence for Rho kinase phosphorylation is considered to be R/XXS/T or RXS/T (40–52), and while LR11 Ser-2167 falls within a consensus motif, Ser-2206 does not (Fig. 6A). Notably, several Rho kinase substrates are phosphorylated at non-consensus motifs, including Calponin, Tau, and the ERM family (ezrin/radixin/moesin) (40, 43, 48).
How does ROCK2 modulate LR11 shedding? Ectodomain shedding is influenced by intracellular protein-protein interactions between the substrate and signaling molecules (reviewed in Ref. 53), and it is hypothesized that kinases induce conformational change in the substrate to facilitate cleavage. Furthermore, metalloproteases, like TACE (TNFα-converting enzyme/ADAM 17), are activated by phosphorylation of their intracellular tail (54). Therefore, ROCK2 may promote LR11 shedding by phosphorylation of LR11 cytoplasmic tail, possibly at Ser-2206, and/or by activating the sheddase(s) responsible for LR11 cleavage. The extracellular portion of LR11 includes a cluster of LDL receptor type A repeats, which binds ApoE with high affinity (55), and the VPS10p homology domain that binds known ligands, including the neuropeptide head activator (HA) (23). Phorbol esters and HA stimulate release of the LR11 ectodomain possibly mediated by TACE, but the functional implications of LR11 shedding are unclear (23). It is hypothesized that shed LR11 ectodomain may complex with ligands to facilitate the availability of extracellular signaling factors. Following ectodomain release, the remaining intracellular tail of LR11 undergoes intramembranous cleavage by the γ-secretase complex (56), allowing nuclear translocation of the LR11 intracellular domain, where it may activate gene transcription (57).
Previous reports have described a link between ROCK and APP processing. Drug inhibition of ROCK with Y-27632 lowered brain levels of pathogenic Aβ in a transgenic mouse model of AD and reduced levels of Aβ in neuronal cell lines (58). Although Y-27632 is selective for ROCKs, it does not distinguish between ROCK1 and ROCK2. In cell models, overexpression of a constitutively active ROCK1 mutant reduced α-secretase cleavage of APP, whereas a dominant-negative ROCK1 mutant elevated levels of secreted APPα (59). We observed no affect on endogenous secreted APPα following targeted knockdown of ROCK2 with siRNA (Fig. 5A), but experiments directly testing effects of ROCK2 on amyloidogenic processing of APP have not been reported. Although the precise molecular mechanisms underlying ROCK regulation of Aβ production are not fully understood, this pathway remains an exciting avenue for rational design of AD therapeutics.
Interestingly, ROCK2 depletion by siRNA reduces LR11 shedding and simultaneously increases the level of cellular LR11. The latter finding is of considerable therapeutic interest given that increasing expression of LR11 results in a reduction in Aβ (4, 11). Greater protein stability or half-life of LR11 within the cell could allow for enhanced chaperoning of APP to non-amyloidogenic compartments. Depleting cells of ROCK2 would potentially increase endocytosis of full-length LR11 or possibly redirect traffic of LR11 to an alternative intracellular compartment. Either scenario could increase LR11 traffic to endosomes, where we hypothesize LR11 exerts its effects on APP processing. Although the contribution of LR11 shedding to amyloidogenic processing of APP is unknown, mutagenesis of LR11 Ser-2206 decreased shedding of LR11 ectodomain and attenuated LR11-mediated Aβ reduction. These results suggest an association between LR11 shedding and its modulation of APP processing. Future studies using site-specific LR11 mutants that ablate interaction with ROCK2 should allow us to directly test models of LR11-ROCK2 interactions and their impact on LR11 phosphorylation, shedding, and APP processing.
Supplementary Material
Acknowledgments
We thank members of the Lah/Levey, Peng, and Mao Laboratories for constructive discussion regarding this manuscript, Marla Gearing, and the National Institutes of Health through the Emory NINDS Neuroscience Core Facilities (NS055077). Acknowledgment is made to the donors of ADR, a program of the American Health Assistance Foundation, for support of this research.
This work was supported, in whole or in part, by National Institutes of Health NINDS Training and Translational Research in Neurology Grant T32 NS007480-07, and Grants NIA P01AG1449 and R01GM067226. This work was also supported by Alzheimer Disease Research Center Grant AG025688, Training Grant (F32AG032848-02, to N. T. S.), and a Research Scholar Grant RSG-09-181-01 from the American Cancer Society.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Tables S1 and S2.
- LDLR
- low-density lipoprotein receptor
- AD
- Alzheimer disease
- Aβ
- amyloid-β peptide
- APP
- amyloid-β precursor protein
- HEK
- human embryonic kidney
- IP
- immunoprecipitation
- LC-MS/MS
- liquid chromatography coupled with tandem mass spectrometry
- LR11
- low-density lipoprotein receptor with 11 class A ligand binding repeats
- ROCK
- Rho-associated coiled-coil containing protein kinase
- RKI
- Rho kinase inhibitor.
REFERENCES
- 1. Motoi Y., Aizawa T., Haga S., Nakamura S., Namba Y., Ikeda K. (1999) Brain Res. 833, 209–215 [DOI] [PubMed] [Google Scholar]
- 2. Jacobsen L., Madsen P., Moestrup S. K., Lund A. H., Tommerup N., Nykjaer A., Sottrup-Jensen L., Gliemann J., Petersen C. M. (1996) J. Biol. Chem. 271, 31379–31383 [DOI] [PubMed] [Google Scholar]
- 3. Scherzer C. R., Offe K., Gearing M., Rees H. D., Fang G., Heilman C. J., Schaller C., Bujo H., Levey A. I., Lah J. J. (2004) Arch. Neurol. 61, 1200–1205 [DOI] [PubMed] [Google Scholar]
- 4. Offe K., Dodson S. E., Shoemaker J. T., Fritz J. J., Gearing M., Levey A. I., Lah J. J. (2006) J. Neurosci. 26, 1596–1603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sager K. L., Wuu J., Leurgans S. E., Rees H. D., Gearing M., Mufson E. J., Levey A. I., Lah J. J. (2007) Ann. Neurol. 62, 640–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rogaeva E., Meng Y., Lee J. H., Gu Y., Kawarai T., Zou F., Katayama T., Baldwin C. T., Cheng R., Hasegawa H., Chen F., Shibata N., Lunetta K. L., Pardossi-Piquard R., Bohm C., Wakutani Y., Cupples L. A., Cuenco K. T., Green R. C., Pinessi L., Rainero I., Sorbi S., Bruni A., Duara R., Friedland R. P., Inzelberg R., Hampe W., Bujo H., Song Y. Q., Andersen O. M., Willnow T. E., Graff-Radford N., Petersen R. C., Dickson D., Der S. D., Fraser P. E., Schmitt-Ulms G., Younkin S., Mayeux R., Farrer L. A., St George-Hyslop P. (2007) Nat. Genet. 39, 168–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sisodia S. S. (1992) Proc. Natl. Acad. Sci. U.S.A. 89, 6075–6079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Koo E. H., Squazzo S. L. (1994) J. Biol. Chem. 269, 17386–17389 [PubMed] [Google Scholar]
- 9. Hartmann T., Bieger S. C., Brühl B., Tienari P. J., Ida N., Allsop D., Roberts G. W., Masters C. L., Dotti C. G., Unsicker K., Beyreuther K. (1997) Nat. Med. 3, 1016–1020 [DOI] [PubMed] [Google Scholar]
- 10. Xu H., Sweeney D., Wang R., Thinakaran G., Lo A. C., Sisodia S. S., Greengard P., Gandy S. (1997) Proc. Natl. Acad. Sci. U.S.A. 94, 3748–3752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Andersen O. M., Reiche J., Schmidt V., Gotthardt M., Spoelgen R., Behlke J., von Arnim C. A., Breiderhoff T., Jansen P., Wu X., Bales K. R., Cappai R., Masters C. L., Gliemann J., Mufson E. J., Hyman B. T., Paul S. M., Nykjaer A., Willnow T. E. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 13461–13466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dodson S. E., Andersen O. M., Karmali V., Fritz J. J., Cheng D., Peng J., Levey A. I., Willnow T. E., Lah J. J. (2008) J. Neurosci. 28, 12877–12886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rohe M., Carlo A. S., Breyhan H., Sporbert A., Militz D., Schmidt V., Wozny C., Harmeier A., Erdmann B., Bales K. R., Wolf S., Kempermann G., Paul S. M., Schmitz D., Bayer T. A., Willnow T. E., Andersen O. M. (2008) J. Biol. Chem. 283, 14826–14834 [DOI] [PubMed] [Google Scholar]
- 14. Jacobsen L., Madsen P., Nielsen M. S., Geraerts W. P., Gliemann J., Smit A. B., Petersen C. M. (2002) FEBS Lett. 511, 155–158 [DOI] [PubMed] [Google Scholar]
- 15. Jacobsen L., Madsen P., Jacobsen C., Nielsen M. S., Gliemann J., Petersen C. M. (2001) J. Biol. Chem. 276, 22788–22796 [DOI] [PubMed] [Google Scholar]
- 16. Nielsen M. S., Madsen P., Christensen E. I., Nykjaer A., Gliemann J., Kasper D., Pohlmann R., Petersen C. M. (2001) EMBO J. 20, 2180–2190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Puertollano R., Aguilar R. C., Gorshkova I., Crouch R. J., Bonifacino J. S. (2001) Science 292, 1712–1716 [DOI] [PubMed] [Google Scholar]
- 18. Zhu Y., Doray B., Poussu A., Lehto V. P., Kornfeld S. (2001) Science 292, 1716–1718 [DOI] [PubMed] [Google Scholar]
- 19. Nielsen M. S., Gustafsen C., Madsen P., Nyengaard J. R., Hermey G., Bakke O., Mari M., Schu P., Pohlmann R., Dennes A., Petersen C. M. (2007) Mol. Cell Biol. 27, 6842–6851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cramer J. F., Gustafsen C., Behrens M. A., Oliveira C. L., Pedersen J. S., Madsen P., Petersen C. M., Thirup S. S. Traffic 11, 259–273 [DOI] [PubMed] [Google Scholar]
- 21. Xu P., Duong D. M., Peng J. (2010) J. Proteome Res. 8, 3944–3950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Eng J., McCormack A. L., Yates J. R., 3rd. (1994) J. Am. Soc. Mass Spectrom. 5, 976–989 [DOI] [PubMed] [Google Scholar]
- 23. Hampe W., Riedel I. B., Lintzel J., Bader C. O., Franke I., Schaller H. C. (2000) J. Cell Sci. 113, 4475–4485 [DOI] [PubMed] [Google Scholar]
- 24. Kubo T., Yamaguchi A., Iwata N., Yamashita T. (2008) Ther. Clin. Risk Manag. 4, 605–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schmandke A., Strittmatter S. M. (2007) Neuroscientist 13, 454–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nakagawa O., Fujisawa K., Ishizaki T., Saito Y., Nakao K., Narumiya S. (1996) FEBS Lett. 392, 189–193 [DOI] [PubMed] [Google Scholar]
- 27. Hashimoto R., Nakamura Y., Kosako H., Amano M., Kaibuchi K., Inagaki M., Takeda M. (1999) Biochem. Biophys. Res. Commun. 263, 575–579 [DOI] [PubMed] [Google Scholar]
- 28. Riento K., Ridley A. J. (2003) Nat. Rev. Mol. Cell Biol. 4, 446–456 [DOI] [PubMed] [Google Scholar]
- 29. Ikenoya M., Hidaka H., Hosoya T., Suzuki M., Yamamoto N., Sasaki Y. (2002) J. Neurochem. 81, 9–16 [DOI] [PubMed] [Google Scholar]
- 30. Sasaki Y., Suzuki M., Hidaka H. (2002) Pharmacol. Ther. 93, 225–232 [DOI] [PubMed] [Google Scholar]
- 31. Herskowitz J. H., Seyfried N. T., Duong D. M., Xia Q., Rees H., Gearing M., Peng J., Lah J., Levey A. (2010) J. Proteome Res. 9, 6368–6379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Xia Q., Cheng D., Duong D. M., Gearing M., Lah J. J., Levey A. I., Peng J. (2008) J. Proteome Res. 7, 2845–2851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schilling O., Barre O., Huesgen P. F., Overall C. M. (2010) Nat. Methods 7, 508–511 [DOI] [PubMed] [Google Scholar]
- 34. Xue Y., Zhou F., Zhu M., Ahmed K., Chen G., Yao X. (2005) Nucleic Acids Res. 33, W184–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhou F. F., Xue Y., Chen G. L., Yao X. (2004) Biochem. Biophys. Res. Commun. 325, 1443–1448 [DOI] [PubMed] [Google Scholar]
- 36. Spoelgen R., von Arnim C. A., Thomas A. V., Peltan I. D., Koker M., Deng A., Irizarry M. C., Andersen O. M., Willnow T. E., Hyman B. T. (2006) J. Neurosci. 26, 418–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Leung T., Manser E., Tan L., Lim L. (1995) J. Biol. Chem. 270, 29051–29054 [DOI] [PubMed] [Google Scholar]
- 38. Matsui T., Amano M., Yamamoto T., Chihara K., Nakafuku M., Ito M., Nakano T., Okawa K., Iwamatsu A., Kaibuchi K. (1996) EMBO J. 15, 2208–2216 [PMC free article] [PubMed] [Google Scholar]
- 39. Hung A. Y., Selkoe D. J. (1994) EMBO J. 13, 534–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Amano M., Kaneko T., Maeda A., Nakayama M., Ito M., Yamauchi T., Goto H., Fukata Y., Oshiro N., Shinohara A., Iwamatsu A., Kaibuchi K. (2003) J. Neurochem. 87, 780–790 [DOI] [PubMed] [Google Scholar]
- 41. Kawano Y., Fukata Y., Oshiro N., Amano M., Nakamura T., Ito M., Matsumura F., Inagaki M., Kaibuchi K. (1999) J. Cell Biol. 147, 1023–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Goto H., Kosako H., Tanabe K., Yanagida M., Sakurai M., Amano M., Kaibuchi K., Inagaki M. (1998) J. Biol. Chem. 273, 11728–11736 [DOI] [PubMed] [Google Scholar]
- 43. Kaneko T., Amano M., Maeda A., Goto H., Takahashi K., Ito M., Kaibuchi K. (2000) Biochem. Biophys. Res. Commun. 273, 110–116 [DOI] [PubMed] [Google Scholar]
- 44. Ohashi K., Nagata K., Maekawa M., Ishizaki T., Narumiya S., Mizuno K. (2000) J. Biol. Chem. 275, 3577–3582 [DOI] [PubMed] [Google Scholar]
- 45. Fukata Y., Oshiro N., Kinoshita N., Kawano Y., Matsuoka Y., Bennett V., Matsuura Y., Kaibuchi K. (1999) J. Cell Biol. 145, 347–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kosako H., Amano M., Yanagida M., Tanabe K., Nishi Y., Kaibuchi K., Inagaki M. (1997) J. Biol. Chem. 272, 10333–10336 [DOI] [PubMed] [Google Scholar]
- 47. Arimura N., Inagaki N., Chihara K., Ménager C., Nakamura N., Amano M., Iwamatsu A., Goshima Y., Kaibuchi K. (2000) J. Biol. Chem. 275, 23973–23980 [DOI] [PubMed] [Google Scholar]
- 48. Matsui T., Maeda M., Doi Y., Yonemura S., Amano M., Kaibuchi K., Tsukita S. (1998) J. Cell Biol. 140, 647–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Amano M., Ito M., Kimura K., Fukata Y., Chihara K., Nakano T., Matsuura Y., Kaibuchi K. (1996) J. Biol. Chem. 271, 20246–20249 [DOI] [PubMed] [Google Scholar]
- 50. Sasaki Y. (2003) J. Pharmacol. Sci. 93, 35–40 [DOI] [PubMed] [Google Scholar]
- 51. Nagumo H., Ikenoya M., Sakurada K., Furuya K., Ikuhara T., Hiraoka H., Sasaki Y. (2001) Biochem. Biophys. Res. Commun. 280, 605–609 [DOI] [PubMed] [Google Scholar]
- 52. Kang J. H., Jiang Y., Toita R., Oishi J., Kawamura K., Han A., Mori T., Niidome T., Ishida M., Tatematsu K., Tanizawa K., Katayama Y. (2007) Biochimie 89, 39–47 [DOI] [PubMed] [Google Scholar]
- 53. Hayashida K., Bartlett A. H., Chen Y., Park P. W. Anat. Rec. 293, 925–937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Díaz-Rodríguez E., Montero J. C., Esparís-Ogando A., Yuste L., Pandiella A. (2002) Mol. Biol. Cell 13, 2031–2044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Taira K., Bujo H., Hirayama S., Yamazaki H., Kanaki T., Takahashi K., Ishii I., Miida T., Schneider W. J., Saito Y. (2001) Arterioscler. Thromb. Vasc. Biol. 21, 1501–1506 [DOI] [PubMed] [Google Scholar]
- 56. Nyborg A. C., Ladd T. B., Zwizinski C. W., Lah J. J., Golde T. E. (2006) Mol. Neurodegener. 1, 3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Böhm C., Seibel N. M., Henkel B., Steiner H., Haass C., Hampe W. (2006) J. Biol. Chem. 281, 14547–14553 [DOI] [PubMed] [Google Scholar]
- 58. Zhou Y., Su Y., Li B., Liu F., Ryder J. W., Wu X., Gonzalez-DeWhitt P. A., Gelfanova V., Hale J. E., May P. C., Paul S. M., Ni B. (2003) Science 302, 1215–1217 [DOI] [PubMed] [Google Scholar]
- 59. Pedrini S., Carter T. L., Prendergast G., Petanceska S., Ehrlich M. E., Gandy S. (2005) PLoS Med. 2, e18 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.