

Abstract
An αβ T-cell receptor (αβTCR)/hemagglutinin (HA) peptide/human leukocyte antigen (HLA)-DR1 complex was stabilized by flexibly linking the HA peptide with the human HA1.7 αβTCR, to increase the local concentration of the interacting proteins once the peptide has been loaded onto the major histocompatibility complex (MHC) molecule. The structure of the complex, determined by X-ray crystallography, has a binding mode similar to that of the human B7 αβTCR on a pMHCI molecule. Twelve of the 15 MHC residues contacted are at the same positions observed earlier in class I MHC/peptide/TCR complexes. One contact, to an MHC loop outside the peptide-binding site, is conserved and specific to pMHCII complexes. TCR gene usage in the response to HA/HLA-DR appears to conserve charged interactions between three lysines of the peptide and acidic residues on the TCR.
Keywords: antigen recognition/complex/MHC class II/T-cell receptor/X-ray crystallography
Introduction
Influenza virus A subtype H3N2 caused the Hong Kong flu pandemic in 1968 and is currently the influenza subtype with the greatest impact, being responsible for >400 000 deaths in the USA alone since its first appearance in 1968 (source: US Department of Health). Peripheral blood lymphocytes from healthy people or people previously exposed to influenza A virus H3N2 generate a potent human leukocyte antigen (HLA)-DR-restricted in vitro T-cell response to a peptide epitope of hemagglutinin (HA) from this subtype corresponding to amino acids 306–318 (HA306–318: PKYVKQNTLKLAT) (Lamb et al., 1982a,b; Krieger et al., 1991). The apparent dominance of this peptide in T-cell responses may result from its ability to bind to many DR alleles, HLA-DR1, DR2, DR4w4, DR5 and DR7 (Krieger et al., 1991; O’Sullivan et al., 1991), and to be recognized by some individual αβ T-cell receptors (αβTCRs) in the context of several different DR molecules (Krieger et al., 1991; Zeliszewski et al., 1994; Brawley and Concannon, 1996).
Mutational studies of the HA peptide (Jardetzky et al., 1990; O’Sullivan et al., 1991; Alexander et al., 1993) and the crystal structure of the HLA-DR1/HA complex (Stern et al., 1994) showed that HA is bound in an extended conformation deep in the binding groove of DR1, with the termini of HA extending out of the groove at either end. About a dozen hydrogen bonds between conserved DR1 residues and peptide main chain carbonyl and amide groups result in a binding of the HA peptide that is independent of its amino acid sequence. Burying of the HA peptide side chains of Y308 (P1) in a deep pocket and of Q311 (P4), T313 (P6), L314 (P7) and L316 (P9) in more shallow pockets of DR1 establishes peptide specificity. Binding studies using HA mutants suggest that the overall binding of HA to DR1, DR2, DR4w4, DR5 and DR7 is similar, with Tyr308, labeled the P1 position, always being the dominant anchor residue (Krieger et al., 1991; O’Sullivan et al., 1991). Side chains of HA residues in between these anchor residues point upward from the composite DR1/HA surface and thus are readily available for recognition by TCRs. In particular, Lys307 (P–1), Lys310 (P3), Asn312 (P5) and Lys315 (P8) have been identified as important residues for the activation of different HA-specific T cells (Krieger et al., 1991; De Magistris et al., 1992; Alexander et al., 1993; Ostrov et al., 1993; Snoke et al., 1993). Several acidic side chains in the Vα and Vβ domain of different αβTCRs have been predicted to interact, by making salt bridges, with the three positively charged lysines in the HA peptide (Ostrov et al., 1993; Wedderburn et al., 1995; Yassine-Diab et al., 1999). Attempts to model the overall orientation of TCR binding over the HA/DR1 surface using different experiments, however, have given inconsistent results (Wedderburn et al., 1995; Yassine-Diab et al., 1999).
In order to understand the binding of αβTCRs to MHC class II/peptide complexes in general and the recognition of the HA antigen in particular, we determined the X-ray structure of the human HA1.7 αβTCR in complex with DR1/HA at a resolution of 2.6 Å. HA1.7 is one of the first TCRs that was isolated and shown to bind to the HA peptide epitope HA306–318 from influenza A (Lamb et al., 1982a; Rothbard et al., 1988). To stabilize the αβTCR/peptide/MHCII complex for crystallization, the HA peptide was covalently attached to the Vβ chain of the TCR such that when the peptide was loaded into the MHCII-binding site, a very long-lived αβTCR/MHC complex resulted. Peptide-linked TCRs may provide a general reagent for preparing crystals of biologically interesting class II and class I TCR/pMHC complexes, and find uses in functional experiments. We find that HA1.7 binds to DR1/HA in an orientation with the TCR Vα domain mainly contacting the N-terminal half of HA and the DR1 β1 helix, and the TCR Vβ domain mainly contacting the C-terminal half of HA and the DR1 α1 helix, in an orientation very similar to that observed in the complex of a human MHCI molecule and the B7 αβTCR (Ding et al., 1998) and of the murine D10 αβTCR and an MHCII molecule (Reinherz et al., 1999). Contacts between the HA1.7 TCR and the HA peptide are dominated by salt bridges between the three lysines of HA and acidic residues of the TCR. Sequence comparisons among HA-specific TCRs indicate that these electrostatic interactions dominate in the selection of TCR genes in the HA-induced immune response.
Results
Stabilizing the αβTCR/peptide/class II MHC complex by protein engineering, expression and refolding
To overcome the tendency of the HA1.7 αβTCR ectodomains to dissociate into separate polypeptides and of the αβTCR/peptide/MHCII complexes to dissociate into αβTCR and pMHCII molecules before crystallizing, required both a controlled refolding reaction and protein engineering of the HA1.7 αβTCR. The HA1.7 αβTCR, with the cysteines that form the interchain disulfide bond deleted, could be expressed in bacteria, refolded in vitro and purified by gel filtration chromatography, but it was unstable, dissociating on native PAGE (data not shown). Making a disulfide-linked αβTCR heterodimer required screening redox conditions during the in vitro refolding reaction for a redox potential where the four intrachain disulfide bonds were stable, as assessed by mobility on SDS–PAGE, so that only the two cysteines near the C-termini participated in intermolecular disulfide bond formation (Pecorari et al., 1999) (data not shown). The disulfide-linked α-S-S-βTCR (Figure 1B, lane 4) is stable, migrating as a single band on native PAGE (Figure 1C, lane 1).

Fig. 1. Linking of the HA306–318 antigen peptide to the TCR β-chain leads to an SDS-stable αβTCR/pMHCII complex. (A) The HA peptide (magenta) was linked via an octapeptide linker (black) to the N-terminus of the Vβ-chain (blue) of the HA1.7 αβTCR (yellow, blue). HA of the resulting p-TCR, HA-HA1.7, was loaded on empty HLA-DR1 (red, green) with HLA-DM. (B) SDS–PAGE (8–16%) of the HA/HA1.7/DR1 complex. Pre-formed HA-HA1.7/DR1 complex was boiled and reduced, just boiled or loaded directly onto the SDS–polyacrylamide gel. Note that the non-boiled and non-reduced HA-HA1.7/DR1 complex does not dissociate on SDS–PAGE, in contrast to the unlinked complex between HA1.7 and DR1/HA. (C) Native gel band-shift assay shows a smear for the HA1.7 TCR/HA/DR1 complex and a distinct band for the HA-HA1.7-linked TCR/DR1 complex. (D) The structure of the p-TCR, HA-HA1.7/DR1 complex with the TCR at the top (yellow, blue) and DR1 at the bottom (red, green). The distance of 19 Å between the C-terminus of the HA peptide (magenta) and residue 3 of the TCR Vβ domain is indicated by an arrow. The figure was created with MOLSCRIPT (Kraulis, 1991) and Raster3D (Merritt and Murphy, 1994).
Mixing the disulfide-bonded HA1.7 αβTCR with the HA/HLA-DR1 complex generated a ternary complex that dissociated during gel filtration chromatography and migrated as a smear on native PAGE (Figure 1C, lane 3), suggesting that it was unstable. Crystallization screens resulted only in crystals of HA/HLA-DR1 and, under other conditions, precipitation of the HA1.7 αβTCR from the mixtures, but no crystals of the TCR/HA peptide/HLA-DR1 complex.
To form a more stable αβTCR/HA/HLA-DR1 complex, we expressed HA1.7 Vβ with the HA peptide fused to the N-terminus by the octapeptide linker GGGGGSGG (Figure 1A). The termini of antigenic peptides extend out of the binding groove of class II MHC molecules (Stern and Wiley, 1994) and can be elongated by artificial sequences without interfering with function (Kozono et al., 1994). The HA peptide was loaded, as part of the HA/HA1.7 fusion protein, onto empty HLA-DR1 molecules with the help of the peptide exchange catalyst HLA-DM (Figure 1A). The bound half-life for the HA peptide on HLA-DR1 is very long, ∼800 h (Sato et al., 2000), so that attaching the HA peptide to the TCR β-chain establishes a stable link between the TCR and the MHCII. This link stabilizes the αβTCR/peptide/MHCII complex, presumably by increasing the local concentration of the interacting proteins and possibly by pre-orienting the complementarity determining regions (CDRs) of the TCR toward the peptide-binding site of the MHCII. The HA-HA1.7 αβTCR forms stable complexes with HLA-DR1 that resist dissociation by SDS when not boiled (Figure 1B, lane 3) and that migrate as a single band on native PAGE (Figure 1C, compare lanes 3 and 4). The stabilized complex crystallized readily.
The octapeptide linker that connects the C-terminus of the HA peptide with the N-terminus of Vβ was designed to be long enough (∼30 Å) to allow the TCR to bind in any orientation on the MHC peptide-binding site in a fan of angles ∼130° to either side of a horizontal line defined by the direction from the peptide N- to C-terminus. This would permit binding modes over two-thirds of the full 360° range, and a generous margin around the ‘diagonal’ range of between 46 and 80°, observed on all earlier αβTCR/peptide/MHC complexes (Reinherz et al., 1999).
Structure determination validates the protein engineering
Using X-ray diffraction from a single crystal, the three-dimensional structure of the HA1.7 αβTCR/HA/HLA-DR1 complex was determined by molecular replacement, first locating the HLA-DR1 (Stern et al., 1994), then the C, and then the V domains of the B7 αβTCR model (Ding et al., 1998). The final model was refined at 2.6 Å resolution (Rwork = 0.222, Rfree = 0.256; 20–2.6 Å resolution) and includes 811 of the 855 residues and 99 water molecules (see Materials and methods).
No electron density could be detected for any of the residues of the octapeptide linker or for the first two residues of Vβ, indicating that they are disordered and probably flexible. The octapeptide linker plus the first two amino acids of Vβ would be able to span a distance about twice that actually observed (19 Å) between the C-terminus of HA and residue 3 of Vβ (Figure 1D, arrow). The linker length, therefore, does not restrict the binding orientation of the TCR to the MHC. The presence of the linker does not cause even minor conformational changes at either Thr318 at the C-terminus of the HA peptide or the first observed residue (3) of Vβ, which both have the same structures as observed in the HA/HLA-DR1 and αβTCR/MHCI structures, respectively (Stern et al., 1994; Garboczi et al., 1996a; Ding et al., 1998).
Generality of the αβTCR/MHC binding mode
Twelve of the 15 MHCII residues contacted by the HA1.7 αβTCR (red in Figure 2A) are in the same positions on the MHC α-helices as residues previously observed to be contacted in complexes of αβTCRs with class I MHC molecules. Two of the remaining three contacts are to residues immediately adjacent to those contacted in αβTCR/pMHCI complexes (DR1α-62, -67). This level of overlap between positions recognized by the TCR on pMHCII and pMHCI molecules emphasizes the generality of the TCR/MHC binding mode.

Fig. 2. Binding of TCRs to MHC class II and class I molecules. Data from five αβTCR/peptide/MHC structures are compared: HA1.7/HA/DR1 (this study), D10/CA/I-Ak (Reinherz et al., 1999), A6/TAX/HLA-A2 (Garboczi et al., 1996a), B7/TAX/HLA-A2 (Ding et al., 1998) and 2C/DEV8/H-2Kb (Garcia et al., 1998). (A) Red MHC residues are contacted by the TCR in the class II and class I complexes named. Yellow underlined sequences correspond to the ‘top’ α-helices and blue underlined sequences to the ‘bottom’ α-helices of the peptide-binding grooves as shown in the ribbon diagrams. Residues of MHCII on the loop between β-strands 3 and 4 are underlined in magenta, and those that precede the N-terminal of the ‘top’ α-helix in green (also on the ribbon diagrams). (B) Orientations of the antigen-combining sites of TCRs with the peptides to define a horizontal axis (Vα is at top right, Vβ is at bottom left, the peptide N-terminus is at the left and the C-terminus is at the right). Positive and negative electrostatic surface potentials of the TCRs are indicated in blue and red, respectively. Figures were prepared with MOLSCRIPT (Kraulis, 1991), Raster3D (Merritt and Murphy, 1994) and GRASP (Nicholls et al., 1991).
The major structural difference between MHCII and MHCI molecules (green in Figure 2A) is the replacement of two turns of the α-helix in MHCI by an extended strand in MHCII. This unique MHCII feature is not contacted by either the human HA1.7 αβTCR studied here or the murine D10 αβTCR reported earlier (Reinherz et al., 1999) (Figure 2A). The two-turn α-helical segment of MHCI is contacted in all the reported αβTCR/pMHCI complexes (Garboczi et al., 1996a; Ding et al., 1998, 1999; Garcia et al., 1998; Degano et al., 2000) (but not by the same TCR residues).
A contact unique to pMHCII complexes is found to Lys39α on a loop between two of the β-strands of MHCII located outside the peptide-binding site (magenta in Figure 2A). This contact is observed on both the DR1 and I-Ak complexes (Figure 2A). In total, 7 of the 15 MHCII positions contacted by TCR in the HA1.7 complex are contacted in the D10 complex (Reinherz et al., 1999) (Figure 2A).
The general disposition of the HA1.7 αβTCR relative to a horizontal axis defined by the peptide is like that seen in MHCI complexes (Figure 2B). The binding mode is especially close to that observed in the human B7 αβTCR/Tax/HLA-A2 complex (Ding et al., 1998), as is that of D10 bound to I-Ak (Figure 2B).
Orientation of the TCR variable loops
As in pMHCI complexes, the variable CDRs, CDR1 and CDR3, of both the Vα and Vβ domains of the HA1.7 αβTCR contact both peptide and MHCII residues, but the CDR2 loops only contact the MHC molecule (Figure 3A). Both conserved and polymorphic MHCII residues are contacted (filled and open circles in Figure 3A). As discussed previously (Garboczi et al., 1996a), this ‘diagonal’ arrangement, first suggested from mutation data (Sun et al., 1995), contrasts with predictions from model building based on Fab structures that had suggested an ‘orthogonal’ binding mode, with CDR3 loops contacting the peptide, and CDR1 and CDR2 contacting the MHC molecule (Chothia et al., 1988; Davis and Bjorkman, 1988).

Fig. 3. Interaction of TCR HA1.7 with HLA-DR1 and HA peptide. (A) TCR contacts. Contacts of CDR residues (solid red lines) with HLA-DR1 (solid blue lines) and the HA peptide (solid black line) are indicated by dashed green and dashed red lines, respectively. Human MHC class II conserved (filled circles) and polymorphic residues (open circles) are shown. (B) Contacts between CDR2β and the conserved Lys39 of DR1α outside of the peptide-binding site (van der Waals contacts, dashed black lines; potential hydrogen bonds, dashed red lines). (C) MHC–peptide solvent-accessible surface buried by the TCR, colored by CDR type (see key). The total accessible surfaces buried on pMHC by the TCR are 1111 Å2 for HA1.7/DR1/HA, 1041 Å2 for D10/I-Ak/CA, 1031 Å2 for A6/A2/TAX, 918 Å2 for B7/A2/TAX and 1111 Å2 for 2C/H-2Kb/DEV8. The antigenic peptide is shown by a white line. Figures were prepared with MOLSCRIPT (Kraulis, 1991), Raster3D (Merritt and Murphy, 1994) and GRASP (Nicholls et al., 1991).
The range of TCR binding angles observed in pMHCI complexes, 45°, 54°, 56° and 70° (Table II of Reinherz et al., 1999), encompasses the HA1.7 angle of 70°. The D10 αβTCR binding angle to I-Ak of 80°, while close to the B7 αβTCR/pMHCI angle of 70°, suggested that MHCII recognition might be more ‘orthogonal’. The angle has been defined between the line formed by the peptide direction and a line between the centers of mass of the Vα and Vβ domains (Reinherz et al., 1999). Because the Vα and Vβ domains of different TCRs twist differently as they rise up from the MHC contact surface to the far end of the domains (e.g. Figure 1 of Ding et al., 1998), the angles reported depend not only on the angle of the contact but also on the VαVβ twists of the individual TCRs (Wilson, 1999).
Table II. Vβ gene usage by HA-specific TCRs.

The CDR2β of both the HA1.7 and D10 TCRs contact Lys39α of MHCII nearly identically. Both form salt-bridged hydrogen bonds from the carboxylate Glu56 of Vβ and a second hydrogen bond from the main chain carbonyl group of residue 55 to the ε-amino group of the lysine (Figure 3A and B). A third, water-mediated, hydrogen bond is observed in the HA1.7 structure from the main chain amide of residue 55 to the same ε-amino group (Figure 3B). Because Lys39α is conserved in all human MHCII molecules (Robinson et al., 2000) and 28 of the 46 functional human Vβ genes encode glutamate or aspartate at position 56 (Arden et al., 1995), this feature may occur in many human TCR/MHCII complexes. In 8 of the 16 Vβ genes that do not have aspartate or glutamate at position 56β, Met54β, which forms a van der Waals contact with the MHCII Lys39α ε-amino group (Figure 3B), is replaced by aspartate or glutamate, which could form an alternative hydrogen bond.
No interaction is observed between Lys68 of CDR4α and the DR1 molecule, although salt-bridged hydrogen bonds are observed between that conserved αβTCR residue and conserved acidic residues in both the D10/pMHCII complex and two of the three αβTCR/pMHCI complexes (not in B7 αβTCR/Tax/HLA-A2). The absence of this interaction in two of the five high resolution complexes (Figure 3C) provides no support for suggestions that this interaction between conserved residues might restrict the orientation of TCR on MHC (Wilson, 1999).
The solvent-accessible surface buried on pMHC by αβTCR is fairly constant, varying from 918 to 1111 Å2 (Figure 3C). In all cases reported, Vα (green, red and indigo in Figure 3C) buried more pMHC surface than Vβ (magenta, cyan and yellow in Figure 3C), ranging in ratio from ∼67/33 in A2–Tax–HLA-A2 to 54/46 in 2C/DEV8/H-2Kb. The HA1.7 αβTCR ratio is reversed, 45/54, with Vβ burying the majority of the pMHC surface that is covered.
The recognition of the HA306–318 peptide by the HA1.7 αβTCR is dominated by electrostatic interactions
While the HA peptide is buried deeply in the DR1 peptide-binding site (Figure 4A, bottom), a relatively flat surface of the TCR contacts the peptide (Figure 4A, top). Five side chains of the peptide, including three lysines, P–1, P3 and P8, an asparagine (P5) and a valine (P2), extend ‘upwards’ to contact the TCR (Figure 4A and B). The ‘flat’ TCR surface is electrostatically negatively charged (Figures 2B and 4A), so that the three peptide lysines make salt-bridged hydrogen bonds (red dashes in Figure 4B and C) with four acidic residues of the TCR. Seven of the 15 atomic contacts between the TCR and peptide involve the positively charged ε-amino groups of the peptide lysines. Two peptide residues are only contacted on main chain atoms (P6 and P7); both by Gly98β at the tip of the CDR3β loop (Figure 4B).

Fig. 4. Recognition of the HA peptide by TCR HA1.7. (A) Binding of the HA peptide (yellow) to the surface of the TCR HA1.7 (top) and in the groove of DR1 (bottom). HA1.7 and DR1 were moved apart and rotated around the long axis of the peptide by –20° and +20°, respectively, in order to allow a better view into the peptide-binding sites. Positive and negative electrostatic surface potentials of HA1.7 and DR1 are indicated in blue and red, respectively. (B) van der Waals contacts and potential hydrogen bonds between TCR HA1.7 and HA peptide are shown by black and red dashed lines, respectively. (C) Electrostatic interactions between the three lysines (P–1, P3 and P8) of HA with acidic residues of HA1.7 TCR. (D) HA and CA peptide residues that are contacted by TCR HA1.7 and D10, respectively, are shown in red. The number of peptide residues that are contacted by the different TCRs and the range over which they are distributed are indicated. (A–C) were prepared with MOLSCRIPT (Kraulis, 1991), Raster3D (Merritt and Murphy, 1994) and GRASP (Nicholls et al., 1991).
No difference is observed in the conformation of the bound HA peptide before and after TCR binding (compare Figure 4B with Figure 2B and C of Stern et al., 1994).
Although peptides bound by MHCII extend out of both ends of the peptide-binding site (Figures 3C and 4A), exposing a much longer peptide surface than those bound to MHCI (Figure 3C), TCR contacts span only nine residues in both the human and murine αβTCR/pMHCII complexes, only one more than in αβTCR/pMHCI complexes (Figure 4D). When the MHCII and MHCI structures are superimposed, the P8 residues in the respective bound peptides overlap, so that TCR recognition spans one more peptide residue toward the peptide N-terminus in TCR/MHCII complexes than TCR/MHCI complexes (Figure 4D). Because MHCII-bound peptides are in a more extended conformation than the kinked conformations found in MHCI-bound peptides, TCRs contact a span of 25 Å of peptide, P–1 to P8, in MHCII complexes, but only 20 Å, P1 to P8, in MHCI complexes (Figure 4D). Of the nine peptide residues spanned by TCR in MHCII complexes, only seven residues of the HA peptide and six of the CA peptide are contacted (Figure 4D). Of the eight peptide residues spanned in MHCI complexes, from five to seven are contacted (Figure 4D).
αβTCR orientation and N-terminal peptide residues on MHCII
CDR1α residues in HA1.7/pMHCII are positioned like those in pMHCI complexes and do not avoid the region above the N-terminus of the peptide as in the murine D10–MHCII structure. Compared with the D10–I-Ak binding mode, the CDR1α in HA1.7/HLA-DR1 is positioned ∼5 Å closer to the peptide P–1 residue than CDR1α in the D10–I-Ak complex. Therefore, avoidance of a ridge in the pMHCII surface formed by the peptide near P–1, as observed in D10–I-Ak, is not a general explanation for the angle of αβTCR contact on pMHCII (Reinherz et al., 1999), although HA1.7 and D10 have very close to the same angular disposition (Figure 2B).
TCR gene usage in the immune response to influenza HA/HLA-DR
The amino acid sequences of 23 Vα and 34 Vβ domains from human αβTCRs responding to the influenza virus HA306–318 peptide presented by HLA-DR1, DR2, DR4w4, DR5 or DR7 show substantial conservation of the acidic CDR residues that interact with the three lysines (P–1, P3 and P8) of the HA peptide (Hewitt et al., 1992; Ostrov et al., 1993; Snoke et al., 1993; Prevost-Blondel et al., 1995; Wedderburn et al., 1995; Brawley and Concannon, 1996; Yassine-Diab et al., 1999) (Table I). Among the Vβ sequences, residue 30 (Glu) of CDR1β, which is salt bridged with the P8-Lys of the HA peptide (Figure 4C), is highly conserved, being either glutamate or aspartate in 94% (32/34) of the HA/DR-specific αβTCRs (Table I). P8-Lys is also salt bridged with Asp28 of CDR1β and in contact with Ser96 of CDR3β (Figure 4B and C); residue 28 of CDR1β is aspartate in 59% (20/34) of the selected sequences, and when residue 28 is not an acidic residue, position 96 of CDR3β is acidic in 50% (7/14) of the sequences (Table I). In the 46 human Vβ genes, the frequency of acidic residues at these positions in CDRβ1 is not above that expected by chance (5–26%) so that the high frequencies of acidic residues in the Cβ genes selected by HA/HLA-DR1 are statistically very significant.
Table I. CDR sequences of HA-specific TCRs.

HA1.7 residues that contact HA/DR1 are shown in green, Basic (blue) and acidic (red) residues are indicated.
Among the Vα sequences, the acidic residues, Glu94α and Glu102α, that are salt bridged to P–1-Lys and P3-Lys (Figure 4C) are somewhat less conserved. CDR3α position 94 is acidic in 57% (13/23) of the αβTCRs (Table I). The absence of the negatively charged amino acid at residue 94 of CDR3α may, in some cases, be compensated for by the presence of a glutamate or aspartate in CDR1α at positions 26, 27 and 31, which occurs in 57% (8/14) of the sequences lacking an acidic residue at position 94 of CDR3α, and may be able to reach to P–1-Lys (Figure 3A). CDR3α position 102, which forms a salt bridge to P3-Lys (Figure 4C), is an acidic residue in only 26% (6/23) of the selected αβTCRs (Table I), but in cases where it is not acidic, nearby CDR2α positions 53, 54 or 55 are often (7/10) acidic. (However, in all αβTCR complexes studied crystallographically, CDR2 only contacts the MHC, not the peptide.)
Other TCR residues appear to have been selected to make conserved interactions with MHCII or DR subtypes. For example, Asp51 of CDR2β, which forms a salt bridge with Lys67α of DR1 (Figure 3A), is an acidic residue in 68% (23/34) of HA/DR-restricted αβTCRs (Table I).
There are also indications, in the conservation of TCR residues, of TCR positions that might be brought into contact with the HA peptide or MHCII by conformational changes of the CDRs or small overall rotations, sliding or tipping of the TCR over the surface of the pMHCII. For example, Lys48 of CDR2α would only need to move 2 Å to salt-bridge to Asp66β, which is 100% conserved in the DR subtypes, and acidic in DP and DQ; also, Pro96 and Phe97 of CDR3α, which contact Gln70β (Figure 3A), are missing in 61% (14/23) of the αβTCRs selected, but may be ‘replaced’ by nearby Lys/Arg (∼94α) in TCRs restricted to DR1*0103, DR5 and DR7 where Gln70β is replaced by aspartate (Yassine-Diab et al., 1999).
Discussion
Engineering stable TCR/pMHC complexes
The determination of X-ray structures of assemblies of αβTCR ectodomains with peptide/MHC ectodomain complexes, which mimic the intercellular contact between T cell and antigen-presenting cell membranes, has required overcoming the relative instability of both the αβTCR heterodimers (in the absence of their transmembrane anchors and associated CD3 and ζ-chains) and the weak association constants of the αβTCR with pMHC (1–100 µM) (Davis et al., 1998; Baker et al., 1999; Ding et al., 1999).
Here, we stabilized the αβTCR ectodomain heterodimer by including a native interchain disulfide bond, which we formed during in vitro refolding by carefully regulating the redox potential to ensure that the four intrachain disulfide bonds were formed first and stabilized by the folded protein domains (Pecorari et al., 1999). This strategy excluded those cysteines that formed intrachain disulfides from interfering with the relatively exposed cysteines near the C-termini of the α- and β-chains that form the interchain disulfide bond. We subsequently stabilized the complex of HA1.7 αβTCR with HA/HLA-DR1 by linking the HA peptide to the TCR β-chain N-terminus, establishing, via a flexible linkage, a stable interaction between the αβTCR and the MHC once the peptide was loaded onto the MHC molecule (Figure 1A and B). This protein engineering stabilized the αβTCR/peptide/MHCII complex by increasing the local concentration of the two proteins relative to each other and possibly by pre-orienting the TCR CDRs toward the peptide-binding site of the MHCII molecule. Peptides have been fused via flexible linkers to the N-terminus of the MHCII β-chain (Kozono et al., 1994) to facilitate peptide loading, which stabilizes MHCII molecules (Stern and Wiley, 1992).
Earlier studies of αβTCR/peptide/MHC complexes required other methods to create or select complexes stable enough to crystallize. To determine the structure of the human B7 αβTCR complex with Tax/HLA-A2, a large excess (20 mg/l) of folded Tax/HLA-A2 needed to be added during the in vitro refolding of the B7 αβTCR to prevent dissociation of the α- from the β-chains (Ding et al., 1998). The murine 2C αβTCR/pMHCI structure was determined only after screening many expressed αβTCR and peptide–MHC ectodomains for pairs that would crystallize (Garcia et al., 1997). In that case, locating a crystalline pair where the TCR had not been induced specifically by the pMHC, its role in biology is uncertain (Garcia et al., 1998; Degano et al., 2000). The murine D10 αβTCR/pMHCII was only determined after the αβTCR was replaced by a single chain construct of Vα–Vβ TCR domains, omitting the TCR C domains (Reinherz et al., 1999). Two TCR/pMHCI complexes required no special stabilization, N15 αβTCR/VSV8/H-2Kb and A6 αβTCR/Tax/HLA-A2, but the former, which used leucine zippers to assemble the αβTCR, diffracted to only 6 Å resolution (Teng et al., 1998) and the A6 αβTCR of the latter dissociated during non-denaturing electrophoresis (native PAGE) into α- and β-chains when not bound to the Tax/HLA-A2 ligand (Garboczi et al., 1996a,b).
Peptide-linked TCRs as reagents for crystallography and functional studies
We suggest that linking the antigenic peptide to the Vβ domain of the TCR is a general strategy for stabilizing TCR/peptide/class II MHC complexes that should allow the crystallization and structure determination of more biologically interesting complexes in the future. The strategy may work on pMHCI complexes, if the peptide can be engineered to exit the binding groove, as observed with a decamer peptide bound to HLA-A2 (Collins et al., 1994). Peptide-linked TCRs may also be useful in functional studies to localize and quantitate the level of specific MHC expression on cells.
Disordered linker
Three observations from the structure determination provide evidence that the TCR binding mode was not influenced by the presence of the linker peptide used to stabilize the αβTCR/pMHCII complex. First, the entire eight-residue linker and the first two residues of the Vβ domain are completely missing in the electron density map, indicating that they are disordered and probably flexible and, therefore, probably could not alter the structure of the TCR/pMHCII binding mode. Secondly, the atoms at both ends of the disordered segment, the C-terminus of the HA peptide and the N-terminus of the residue Vβ-3, are in exactly the same location as those atoms in the structure of HA bound to DR1 without the linker (Stern et al., 1994) and in other αβTCR structures (e.g. Garboczi et al., 1996a; Ding et al., 1998), indicating that the linker has not distorted the residues to which it was attached (residues Vβ-1 and -2 were also disordered in other αβTCR structures). Thirdly, the distance between the atoms connected by the linker is only 19 Å, whereas the linker could extend about twice that length, so linker length has not constrained the binding mode.
αβTCR binding mode
When the three pMHCI and two pMHCII complexes with αβTCR whose structures are known to high resolution are superimposed on their MHC peptide-binding domains, the αβTCRs all cluster within an envelope of similar orientations (Figure 5). The long CDR3 loops of Vα and Vβ from all the TCRs extend down over the center of the bound peptides and the CDR2 loops contact the prominent α-helices of the MHC molecules that lie adjacent to the bound peptides (Figure 5A). The TCRs are tipped, twisted and rotated differently relative to the MHC molecules, but all within a narrow range of 35° about a vertical axis on Figure 5A (or perpendicular to Figure 2B) and only a few degrees about an axis perpendicular to Figure 5A. The positions on the MHC α-helices contacted by the TCRs are very similar both within MHC classes and between pMHCI and pMHCII complexes (red in Figure 2A), emphasizing the similarity of the recognition events.

Fig. 5. Stereographic diagram of the α-carbon paths of five αβTCR/pMHC complexes. (A) Complexes were superimposed on their peptide-binding domains (center). The similar orientations of the TCRs (top) on the pMHC (bottom) are evident when viewed in stereo. (B) As in (A), but rotated by 90° about the vertical axis. TCRs (top) all fit at an angle between high points on the MHC surfaces, when viewed in stereo. Arrows show the difference locations of the CD4 and CD8 binding sites. Complexes shown are as in Figure 2.
There is greater variation in the positions of the Vβ domains than the Vα domains, as though the different binding angles pivot around Vα. For example, as previously noted, the Vα domains of the A6 and B7 αβTCRs contact Tax/HLA-A2 very similarly (green, red and indigo in Figure 3C; Figure 1A and B in Ding et al., 1998), but Vβ of A6 makes almost no contact, while the Vβ in the B7 αβTCR/pMHCI complex has tipped down to make contact (cyan in Figure 3C; Figure 3C in Ding et al., 1998).
The solvent-accessible surface areas buried by αβTCRs in each complex are in a narrow range of ∼1000 ± 100 Å. In four of the complexes, Vα dominates slightly, burying between 55 and 66% of the total, but, in the structure determined here, Vβ dominates slightly, burying 55% of the total.
In every case, the TCR presents a relatively flat surface that binds on an angle to the MHC peptide-binding site, avoiding the peaks or the α-helical borders of the MHC peptide-binding groove (visible in stereo in Figure 5B).
Antigenic peptide recognition
Overall, a very similar interaction is observed between the HA1.7 TCR and the HA peptide bound to HLA-DR1, as was previously observed between other TCRs and peptides on both MHCI and MHCII. The relatively flat surface of the TCR (although A6 and B7 TCRs had a deep pocket in the center of the flat surface) spans nine peptide residues in pMHCII complexes and eight in pMHCI complexes (Figure 4D). The HA1.7 TCR does not appear to have made any special accommodation to contact the longer peptides bound to MHCII, as the footprints of TCRs are similar on pMHCI and pMHCII (Figure 3C). Perhaps because the TCR footprints all extend beyond the peptide N-termini on MHCI, they readily contact the extra exposed peptide residues found on pMHCII.
Some details of peptide recognition are also conserved. The very same CDR1β residue Glu30 hydrogen-bonds to P8-Tyr in the A6/pMHCI complex as hydrogen-bonds to P8-Lys in the HA/DR1 complex studied here (Figure 4C) (Garcia et al., 1996; Ding et al., 1999). Similarly, the CDR3α and CDR3β loops surround the central P5 tyrosine in the TCR/Tax–MHCI complexes and the central P5 asparagine in the HA/MHCII complex studied here (Figure 4B and C) (Garcia et al., 1996; Ding et al., 1998, 1999).
Although peptides bound to MHCII are longer than the nine-residue span contacted by the HA1.7 and D10 αβTCRs, and peptide residues are observed extending out of the peptide-binding site on both ends in those two TCR/pMHCII complexes (Figure 3C), the structures provide no evidence for contacts to the distal peptide residues (Carson et al., 1997). Effects attributed to such distal contacts would require either a different TCR binding mode from those observed on pMHCI and pMHCII to date, or a change in the conformation of bound peptides, peeling away from contacts on the MHCII to form contacts with the TCR. The absence of contacts beyond P8 in TCR/pMHCII (or TCR/MHCI complexes) (Figures 3C and 4D) provides no support for the suggestion that TCR residues might bind into partially vacant P9 pockets in some peptide–I-Ag7 complexes (Corper et al., 2000).
TCR gene usage
Because HA1.7 TCR recognition of the HA peptide is dominated by the formation of salt-bridged hydrogen bonds between three peptide lysines, P–1, P3 and P8, and four acidic TCR residues, Glu94α, Glu102α, Asp28β and Glu30β (Figure 4C), the acidic positions in sequences of other TCRs selected by HA/DR ligands are recognized readily (Table I). The importance of the positively charged lysines on the HA peptide and negative counter charges in a few TCR sequences was recognized earlier in mutational studies (Alexander et al., 1993; Ostrov et al., 1993; Wedderburn et al., 1995; Brawley and Concannon, 1996; Yassine-Diab et al., 1999). The structure reveals that two of the lysines, P3 and P8, are almost completely buried by formation of the TCR interface, which also completely buries E30β. Reversing the charges of either of those two peptide lysines by substitution with glutamic acid results in null ligands, with neither agonist nor antagonist activity, an indication that the substitution abrogates TCR binding to the clones examined (Alexander et al., 1993). It may be significant that the most completely buried TCR residue, Glu30β, which interacts with the P8-Lys, is the most conserved acidic residue, found in 32 of 34 HA/DR selected Vβ sequences. Such a buried salt bridge should contribute substantial free energy to the stabilization of the complex, as buried salt bridges in protein structures are strongly stabilizing (Anderson et al., 1990; Tissot et al., 1996).
Although the overall strategy for recognizing HA peptide by charged interactions is apparently very conserved, the TCR sequences suggest that the detailed mode of binding may involve small tips and rotations, and the specific contacts may differ in different TCRs.
The structural and energetic requirement for a charged interaction between P8-Lys and an acidic residue at position 30 in CDR1β seems to dominate Vβ gene selection. Among the 46 functional Vβ genes in the genome, only 12 have a negatively charged residue at this position (Table II). Eight of these 12 Vβ genes have been selected by the HA-specific TCRs reported.
In addition to Glu30β, Asp28β also makes a charged interaction with P8-Lys (Figure 4C). BV3S1 is the only Vβ gene segment in humans with negatively charged residues at both of these positions (#1 in Table II). Twenty of the 34 HA/DR-specific TCRs sequenced used the BV3S1 gene segment, indicating a strong selection for these two charges in response to the HA peptide.
Among the HA/DR-specific Vα sequences, AV1S2A1 (4×) and AV1S3A2 (6×), which are very similar to each other, were selected most often (10/23) (Table I). Those 10 TCRs do not have any negatively charged residues in CDR1α or CDR2α, as expected from their lack of contact with the peptide lysines (although those regions of other Vαs that contain charges may contact lysines at P–1 and P3, as discussed in Results). The pronounced preference for certain V gene segments in response to an antigenic peptide has also been reported in other cases (Acha-Orbea et al., 1988; Urban et al., 1988; Jorgensen et al., 1992a). At least in one case, recognition of charged peptide residues is achieved by the CDR3s and hence does not have an obvious impact on V gene selection (Jorgensen et al., 1992a).
V gene selection also appears to be influenced by charged interactions with MHC residues. Position 67 of MHCII α-chains is a lysine in DRα and DQα (asparagine in DPα). In the structure presented here, Lys67α of DR1 forms a salt bridge to Asp51 of CDR2β (Figure 3A). An acidic residue is found in only 8 of the 46 functional Vβ genes (17%), but in 68% (23/34) of the TCRs that recognize HA/DR (Table II). Similarly, Lys39 of DR1α, which is conserved among all human MHCIIs, is contacted by residues 54 and 56 of CDR2β (Figure 3A and B). Of the 46 functional human Vβ genes, 78% have acidic residues at positions 54, 56 or both; the frequency is 91% for HA/DR-specific TCRs (Table II), suggesting a selection at these positions for MHCII binding.
The HA/DR1-specific Vα gene sequences also suggest some selection by the MHCII molecule (Table I). CDR1α residues Val28 and Tyr31 and CDR2α residues Ser51 and Ala/Gly52, which contact MHCII, are found in half or more of the HA/DR-specific TCRs (Figure 3A).
Signaling and the αβTCR binding mode
Substitutions in the HA peptide at all of the positions contacted by the HA1.7 TCR (P–1, P2, P3, P5, P7 and P8) create antagonist ligands (Alexander et al., 1993; Ostrov et al., 1993; Wedderburn et al., 1995), while the substitutions at residues not contacted (P1, P4 and P6) did not; an indication that a large part of the TCR/pMHC interface is important for specific recognition
A difference in the location of the CD4 and CD8 co-receptor binding loops on class II and class I MHC molecules is evident in the five known αβTCR/pMHC structures (arrows in Figure 5B), as a result of the different location of the class II β2 and class I α3 domains (Brown et al., 1993). Whether this difference is related to any presumptive contacts between co-receptor and TCR is unknown (Vignali et al., 1996; Vignali and Vignali, 1999).
The likelihood of a conserved binding mode between MHC molecules and TCRs has been suggested from theories (Jerne, 1971), mutational studies (Jorgensen et al., 1992b; Sun et al., 1995; Ignatowicz et al., 1996; Sant’Angelo et al., 1996) and the crystal structure of αβTCR/pMHC complexes (e.g. Garboczi et al., 1996a). The conserved orientation of Vα and Vβ domains (very approximately over the N- and C-terminal regions of the bound peptides, respectively) and the narrow range of ‘diagonal’ angular orientations, ∼35°, observed in six αβTCR/pMHC complexes confirm these suggestions (Garboczi et al., 1996a; Ding et al., 1998; Garcia et al., 1998; Teng et al., 1998; Reinherz et al., 1999). The approximately conserved binding mode could provide the structural basis for the assembly of an oligomer to initiate a signal in the T cell (e.g. a dimer; Brown et al., 1993). The variation in the geometry of TCR binding, although within limits, may represent a challenge to simple geometrical assemblies, requiring the ability to adopt a number of closely related oligomeric shapes (J.Kappler, personal communication). It remains possible that the addition of the CD3 chains and/or the possible presence of two αβTCRs within one pre-formed cell surface TCR (Exley et al., 1995; San Jose et al., 1998; Fernandez-Miguel et al., 1999) will restrain the geometry of TCR/MHC interactions or permit all the observed variations to stabilize an ‘active, signaling’ conformation of a cell surface TCR.
Materials and methods
Protein expression and purification
Empty HLA-DR1 and HLA-DM were overexpressed from stable Schneider cell lines and purified from the cell culture supernatant by immunoaffinity chromatography as described (Sloan et al., 1995; Dessen et al., 1997; Mosyak et al., 1998).
The genes coding for the α- and β-chain ectodomains of TCR HA1.7 (Lamb et al., 1982a) have been modified by PCR and inserted into the bacterial expression plasmid pLM1 (Sodeoka et al., 1993) under the control of the T7 polymerase promotor as described previously (Garboczi et al., 1996b).
The α-chain construct encodes residues Q1–K214 of HA1.7α, including C211, which forms an intermolecular disulfide bond with C248 of the β-chain. The β-chain construct encodes a fusion protein, termed HA/HA1.7β, comprising the influenza virus HA peptide antigen HA306–318 (PKYVKQNTLKLAT) linked via an octapeptide linker (GGSGGGGG) to residues D1–T251 of HA1.7β. The unpaired Cys191 was replaced by serine.
The p-TCR HA/HA1.7 was overproduced and refolded as described (Garboczi et al., 1996b), but with the following modifications. Both chains were expressed separately as inclusion bodies from BL21(DE3) (Studier et al., 1990) grown in rich medium (20 g/l tryptone, 10 g/l yeast extract, 5 g/l NaCl, 20 ml/l glycerol, 50 mM K2HPO4, 10 mM MgCl2, 10 g/l glucose, 100 mg/l ampicillin) at 37°C. Bacterial cultures were induced at an OD600 of 1.0 by the addition of 1 mM isopropyl-β-d-thio galactopyranoside (IPTG), and harvested 3 h later. Cells were washed with 50 mM Tris–HCl pH 8.0 and lysed by resuspending the cell pellets in 50 mM Tris–HCl pH 8.0, 1 mM MgCl2, 0.4 mg/ml DNase I, 0.4 mg/ml RNase A, 1 mg/ml lysozyme and brief sonication. Insoluble material was sedimented by centrifugation, and inclusion bodies were washed five times with 20 mM Tris–HCl pH 8.0, 23% (w/v) sucrose, 0.5% (v/v) Triton X-100, 1 mM EDTA and once with 20 mM Tris–HCl pH 8.0, 1 mM EDTA. Inclusion body pellets were resolubilized in 9.5 M urea, 20 mM Tris–HCl pH 8.0, 2 mM EDTA, 5 mM dithiothreitol (DTT). The p-TCR HA/HA1.7 was refolded by rapid dilution (a total of three injections at 0, 12 and 24 h) of both unfolded chains into ice-cold refolding buffer [1 M arginine, 100 mM Tris–HCl pH 8.2, 2 mM EDTA, 0.3 mM oxidized glutathione, 0.3 mM reduced glutathione, 60 mg/l phenylmethylsulfonyl fluoride (PMSF), 2 mg/ml leupeptin, 2 mM benzamidine, 2 mg/l pepstatin A, two protease cocktail tablets (Complete™)] at a final protein concentration of 110 mg/l HA1.7α and 90 mg/l HA/HA1.7β, and stored on ice for another 24 h after the last injection.
Optimization of the redox conditions to the glutathione concentrations used above was essential for the successful refolding of HA/HA1.7 and the formation of the intermolecular disulfide bond (Pecorari et al., 1999).
The refolding reaction was dialyzed (molecular weight cut-off 6000–8000) against 10 mM Tris–HCl pH 8.0 and loaded onto a Q Sepharose Fast Flow anion exchange column (Pharmacia Biotech). Disulfide-linked p-TCR α/β heterodimer was separated from non-linked dimer (∼5% of total protein) by elution with a linear gradient of 0–400 mM NaCl in 10 mM Tris–HCl pH 8.0. The linked dimer was purified further by size exclusion chromatography (Superdex 200, Pharmacia Biotech) in 20 mM Tris–HCl pH 8.0, 150 mM NaCl and anion exchange chromatography (MonoQ, Pharmacia Biotech) in 20 mM Tris–HCl pH 8.0 with a linear gradient from 0 to 400 mM NaCl. The final yield was 10 mg of α/β heterodimer per liter of refolding reaction.
Assembly of the HA-HA1.7/DR1 complex
The whole HA-HA1.7/DR1 complex was assembled by loading the HA peptide that is part of the p-TCR HA-HA1.7 onto empty DR1 with the help of the peptide exchange catalyst HLA-DM. HA-HA1.7, DR1 and DM were mixed in a stoichiometric ratio of 1:1:0.3 in 20 mM Tris–HCl pH 8.0, concentrated to an overall protein concentration of ∼1 mg/ml, adjusted to pH 6 and incubated for 12 h at 37°C. The complex was separated from DM and uncomplexed HA-HA1.7 and DR1 by size exclusion chromatography (Superdex 200, Pharmacia Biotech) in 20 mM Tris–HCl pH 8.0, 150 mM NaCl. The complex could be purified further by anion exchange chromatography (MonoQ, Pharmacia Biotech) in 20 mM Tris–HCl pH 8.0 using a linear gradient from 0 to 400 mM NaCl without falling apart. The pure complex was concentrated to 10 mg/ml, buffer exchanged to 10 mM HEPES pH 7.0, 50 mM NaCl and stored at –80°C.
Crystallization, structure determination and refinement
Small crystals of HA-HA1.7/DR1 were initially grown overnight at 18°C by vapor diffusion in hanging drops combining 1 µl of protein (10 mg/ml) with 1 µl of well solution (14% PEG 8000, 1 M NaCl, 100 mM HEPES pH 7.0). Single, large crystals were obtained by streak seeding sitting drops of 1 µl of protein (10 mg/ml) and 1 µl of well solution (11% PEG 8000, 1 M NaCl, 100 mM HEPES pH 7.0) after 12 h pre-equilibration. Crystals are monoclinic, space group C2, with a = 142.9 Å, b = 73.4 Å, c = 122.4 Å, β = 108.2° and one complex molecule per asymmetric unit. For data collection, crystals were first transferred into a cryo-protecting solution of 20% glycerol, 16% PEG 8000, 1 M NaCl, 100 mM HEPES pH 7.0 and then flash cooled at 100 K in liquid nitrogen. X-ray diffraction data were collected from a single crystal to 2.6 Å resolution (Table III) at the BIOCARS station 14-BM-C at the APS at Argonne National Laboratory using 1 Å wavelength X-rays and a Quantum4 CCD device detector. Oscillation images were processed with DENZO and data reduction was carried out with SCALEPACK (Otwinowski, 1993).
Table III. Crystallographic data.
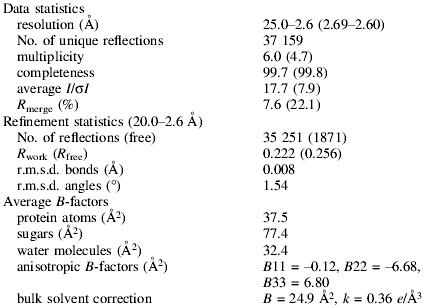
Rmerge = (Σhkl|I − <I>|)/(Σhkl<I>), ∀hkl ∈ (independent Miller indices).
Rfree = (Σh|Fo − Fc|)/(ΣhFo), ∀h ∈ (free set, 5% of reflections).
Rwork = (Σh|Fo − Fc|)/(ΣhFo), ∀h ∈ (working set).
The structure of the complex was determined by molecular replacement using the program AMoRe (Navaza and Saludjian, 1997). The refined coordinates of a monomer of HLA-DR1 without the HA peptide (Stern et al., 1994) and the CαCβ and VαVβ domains of the B7 αβTCR from the B7/TAX/HLA-A2 complex (Ding et al., 1998) were used as search models. First, the position of the α- and β-chains of DR1 was determined as a rigid body (R = 50.7, correlation coefficient = 31.5). With the location of DR1 fixed, the positions of the Cα and Cβ domains (R = 45.7, correlation coefficient = 43.9) and then of the Vα and Vβ domains were found (R = 43.9, correlation coefficient = 48.6). Rigid-body refinement of the individual domains of this model with CNS (Brünger et al., 1998) yielded Rfree = 43.0% (Rwork = 41.1%) using all the data between 20 and 3.5 Å resolution. At the beginning of the refinement and for calculations of electron density maps, all non-conserved residues were changed to alanines, and the HA peptide and the CDR loops of Vα and Vβ were omitted from the model.
Model building and correction were performed in O (Jones et al., 1991) using σA-weighted (2mFo – DFc) and (mFo – DFc) electron density maps (Read, 1986). Positional refinement was performed in CNS using a maximum likelihood (ML) target. A bulk solvent correction and anisotropic B-factor tensor were applied throughout the refinement. Toward the end, individual B-factors were refined and water molecules were added.
The final model includes 811 of 855 residues and 99 water molecules. Weak or no electron density was observed for residues 105–113 of DR1β, residues 130–132 of the HA1.7 Cα domain, the last 10 residues of the Cα as well as the Cβ domain, for 0–3 residues at the N- or C-termini of the different chains and for the octapeptide linker between the HA peptide and the HA1.7β chain (Table III).
Protein database code
Coordinates have been deposited in the PDB under entry code 1FYT.
Acknowledgments
Acknowledgements
We thank K.Mahan and A.Haykov for excellent technical assistance, B.Harris for help with crystallization, Dr S.Liemann and the staff at APS at Argonne National Laboratory beamline BIOCARS 14-BM-C for help with data collection, Dr Dennis Zaller (Merck) for the generous gifts of insect cell lines expressing both HLA-DR1 and DM, Dr Kai Wucherpfennig for useful discussions, and members of the Harrison/Wiley groups for assistance. The research was supported by the National Institutes of Health and the Howard Hughes Medical Institute (HHMI). J.H. was supported by fellowships from the Schweizerischer Nationalfonds and the Deutsche Forschungsgemeinschaft, and A.C. by a fellowship from the Irvington Institute for Immunological Research. D.C.W. is an investigator of the HHMI.
References
- Acha-Orbea H., Mitchell,D.J., Timmermann,L., Wraith,D.C., Tausch,G.S., Waldor,M.K., Zamvil,S.S., McDevitt,H.O. and Steinman,L. (1988) Limited heterogeneity of T cell receptors from lymphocytes mediating autoimmune encephalomyelitis allows specific immune intervention. Cell, 54, 263–273. [DOI] [PubMed] [Google Scholar]
- Alexander J. et al. (1993) Functional consequences of engagement of the T cell receptor by low affinity ligands. J. Immunol., 150, 1–7. [PubMed] [Google Scholar]
- Anderson D.E., Becktel,W.J. and Dahlquist,F.W. (1990) pH-induced denaturation of proteins: a single salt bridge contributes 3–5 kcal/mol to the free energy of folding of T4 lysozyme. Biochemistry, 29, 2403–2408. [DOI] [PubMed] [Google Scholar]
- Arden B., Clark,S.P., Kabelitz,D. and Mak,T.W. (1995) Human T-cell receptor variable gene segment families. Immunogenetics, 42, 455–500. [DOI] [PubMed] [Google Scholar]
- Baker B.M., Ding,Y.-H., Garboczi,D.N., Biddison,W.E. and Wiley,D.C. (1999) Structural, biochemical and biophysical studies of HLA-A2/altered peptide ligands binding to viral-peptide-specific human T-cell receptors. Cold Spring Harb. Symp. Quant. Biol., 64, 235–241. [DOI] [PubMed] [Google Scholar]
- Brawley J.V. and Concannon,P. (1996) Modulation of promiscuous T cell receptor recognition by mutagenesis of CDR2 residues. J. Exp. Med., 183, 2043–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown J.H., Jardetzky,T.S., Gorga,J.C., Stern,L.J., Urban,R.G., Strominger,J.L. and Wiley,D.C. (1993) Three-dimensional structure of the human class II histocompatibility antigen HLA-DR1. Nature, 364, 33–39. [DOI] [PubMed] [Google Scholar]
- Brünger A.T. et al. (1998) Crystallography and NMR system—a new software suite for macromolecular structure determination. Acta Crystallogr. D, 54, 905–921. [DOI] [PubMed] [Google Scholar]
- Carson R.T., Vignali,K.M., Woodland,D.L. and Vignali,D.A. (1997) T cell receptor recognition of MHC class II-bound peptide flanking residues enhances immunogenicity and results in altered TCR V region usage. Immunity, 7, 387–399. [DOI] [PubMed] [Google Scholar]
- Chothia C., Boswell,D.R. and Lesk,A.M. (1988) The outline structure of the T-cell αβ receptor. EMBO J., 7, 3745–3755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins E.J., Garboczi,D.N. and Wiley,D.C. (1994) Three-dimensional structure of a peptide extending from one end of a class I MHC binding site. Nature, 371, 626–629. [DOI] [PubMed] [Google Scholar]
- Corper A.L., Stratmann,T., Apostolopoulos,V., Scott,C.A., Garcia,K.C., Kang,A.S., Wilson,I.A. and Teyton,L. (2000) A structural framework for deciphering the link between I-Ag7 and autoimmune diabetes. Science, 288, 505–511. [DOI] [PubMed] [Google Scholar]
- Davis M.M. and Bjorkman,P.J. (1988) T-cell antigen receptor genes and T-cell recognition. Nature, 334, 395–402. [DOI] [PubMed] [Google Scholar]
- Davis M.M., Boniface,J.J., Reich,Z., Lyons,D., Hampl,J., Arden,B. and Chien,Y.H. (1998) Ligand recognition by α-β T cell receptors. Annu. Rev. Immunol., 16, 523–544. [DOI] [PubMed] [Google Scholar]
- Degano M., Garcia,K.C., Apostolopoulos,V., Rudolph,M.G., Teyton,L. and Wilson,I.A. (2000) A functional hot spot for antigen recognition in a superagonist TCR/MHC complex. Immunity, 12, 251–261. [DOI] [PubMed] [Google Scholar]
- De Magistris M.T., Alexander,J., Coggeshall,M., Altman,A., Gaeta,F.C., Grey,H.M. and Sette,A. (1992) Antigen analog–major histocompatibility complexes act as antagonists of the T cell receptor. Cell, 68, 625–634. [DOI] [PubMed] [Google Scholar]
- Dessen A., Lawrence,C.M., Cupo,S., Zaller,D.M. and Wiley,D.C. (1997) X-ray crystal structure of HLA-DR4 (DRA*0101, DRB1*0401) complexed with a peptide from human collagen II. Immunity, 7, 473–481. [DOI] [PubMed] [Google Scholar]
- Ding Y.H., Smith,K.J., Garboczi,D.N., Utz,U., Biddison,W.E. and Wiley,D.C. (1998) Two human T cell receptors bind in a similar diagonal mode to the HLA-A2/Tax peptide complex using different TCR amino acids. Immunity, 8, 403–411. [DOI] [PubMed] [Google Scholar]
- Ding Y.H., Baker,B.M., Garboczi,D.N., Biddison,W.E. and Wiley,D.C. (1999) Four A6-TCR/peptide/HLA-A2 structures that generate very different T cell signals are nearly identical. Immunity, 11, 45–56. [DOI] [PubMed] [Google Scholar]
- Exley M., Wileman,T., Mueller,B. and Terhorst,C. (1995) Evidence for multivalent structure of T-cell antigen receptor complex. Mol. Immunol., 32, 829–839. [DOI] [PubMed] [Google Scholar]
- Fernandez-Miguel G., Alarcon,B., Iglesias,A., Bluethmann,H., Alvarez-Mon,M., Sanz,E. and de la Hera,A. (1999) Multivalent structure of an αβT cell receptor. Proc. Natl Acad. Sci. USA, 96, 1547–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garboczi D.N., Ghosh,P., Utz,U., Fan,Q.R., Biddison,W.E. and Wiley,D.C. (1996a) Structure of the complex between human T-cell receptor, viral peptide and HLA-A2. Nature, 384, 134–141. [DOI] [PubMed] [Google Scholar]
- Garboczi D.N., Utz,U., Ghosh,P., Seth,A., Kim,J., VanTienhoven,E.A., Biddison,W.E. and Wiley,D.C. (1996b) Assembly, specific binding and crystallization of a human TCR-αβ with an antigenic Tax peptide from human T lymphotropic virus type 1 and the class I MHC molecule HLA-A2. J. Immunol., 157, 5403–5410. [PubMed] [Google Scholar]
- Garcia K.C., Degano,M., Stanfield,R.L., Brunmark,A., Jackson,M.R., Peterson,P.A., Teyton,L. and Wilson,I.A. (1996) An αβ T cell receptor structure at 2.5 Å and its orientation in the TCR/MHC complex. Science, 274, 209–219. [DOI] [PubMed] [Google Scholar]
- Garcia K.C. et al. (1997) αβ T cell receptor interactions with syngeneic and allogeneic ligands: affinity measurements and crystallization. Proc. Natl Acad. Sci. USA, 94, 13838–13843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia K.C., Degano,M., Pease,L.R., Huang,M., Peterson,P.A., Teyton,L. and Wilson,I.A. (1998) Structural basis of plasticity in T cell receptor recognition of a self peptide-MHC antigen. Science, 279, 1166–1172. [DOI] [PubMed] [Google Scholar]
- Hewitt C.R., Lamb,J.R., Hayball,J., Hill,M., Owen,M.J. and O’Hehir,R.E. (1992) Major histocompatibility complex independent clonal T cell anergy by direct interaction of Staphylococcus aureus enterotoxin B with the T cell antigen receptor. J. Exp. Med., 175, 1493–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignatowicz L., Kappler,J. and Marrack,P. (1996) The repertoire of T cells shaped by a single MHC/peptide ligand. Cell, 84, 521–529. [DOI] [PubMed] [Google Scholar]
- Jardetzky T.S., Gorga,J.C., Busch,R., Rothbard,J., Strominger,J.L. and Wiley,D.C. (1990) Peptide binding to HLA-DR1: a peptide with most residues substituted to alanine retains MHC binding. EMBO J., 9, 1797–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerne N.K. (1971) The somatic generation of immune recognition. Eur. J. Immunol., 1, 1–9. [DOI] [PubMed] [Google Scholar]
- Jones T.A., Zou,J.Y., Cowan,S.W. and Kjeldgaard,M. (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A, 47, 110–119. [DOI] [PubMed] [Google Scholar]
- Jorgensen J.L., Esser,U., Fazekas de St. Groth,B., Reay,P.A. and Davis,M.M. (1992a) Mapping T-cell receptor–peptide contacts by variant peptide immunization of single-chain transgenics. Nature, 355, 224–230. [DOI] [PubMed] [Google Scholar]
- Jorgensen J.L., Reay,P.A., Ehrich,E.W. and Davis,M.M. (1992b) Molecular components of T-cell recognition. Annu. Rev. Immunol., 10, 835–873. [DOI] [PubMed] [Google Scholar]
- Kozono H., White,J., Clements,J., Marrack,P. and Kappler,J. (1994) Production of soluble MHC class II proteins with covalently bound single peptides. Nature, 369, 151–154. [DOI] [PubMed] [Google Scholar]
- Kraulis P.J. (1991) MOLSCRIPT—a program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr., 24, 946–950. [Google Scholar]
- Krieger J.I. et al. (1991) Single amino acid changes in DR and antigen define residues critical for peptide–MHC binding and T cell recognition. J. Immunol., 146, 2331–2340. [PubMed] [Google Scholar]
- Lamb J.R., Eckels,D.D., Lake,P., Woody,J.N. and Green,N. (1982a) Human T-cell clones recognize chemically synthesized peptides of influenza haemagglutinin. Nature, 300, 66–69. [DOI] [PubMed] [Google Scholar]
- Lamb J.R., Eckels,D.D., Phelan,M., Lake,P. and Woody,J.N. (1982b) Antigen-specific human T lymphocyte clones: viral antigen specificity of influenza virus-immune clones. J. Immunol., 128, 1428–1432. [PubMed] [Google Scholar]
- Merritt E.A. and Murphy,M.E.P. (1994) Raster3d version 2.0—a program for photorealistic molecular graphics. Acta Crystallogr. D, 50, 869–873. [DOI] [PubMed] [Google Scholar]
- Mosyak L., Zaller,D.M. and Wiley,D.C. (1998) The structure of HLA-DM, the peptide exchange catalyst that loads antigen onto class II MHC molecules during antigen presentation. Immunity, 9, 377–383. [DOI] [PubMed] [Google Scholar]
- Navaza J. and Saludjian,P. (1997) AMoRe: an automated molecular replacement program package. Methods Enzymol., 276, 581–594. [DOI] [PubMed] [Google Scholar]
- Nicholls A., Sharp,K.A. and Honig,B. (1991) Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins, 11, 281–296. [DOI] [PubMed] [Google Scholar]
- Ostrov D., Krieger,J., Sidney,J., Sette,A. and Concannon,P. (1993) T cell receptor antagonism mediated by interaction between T cell receptor junctional residues and peptide antigen analogues. J. Immunol., 150, 4277–4283. [PubMed] [Google Scholar]
- O’Sullivan D. et al. (1991) On the interaction of promiscuous antigenic peptides with different DR alleles. Identification of common structural motifs. J. Immunol., 147, 2663–2669. [PubMed] [Google Scholar]
- Otwinowski Z. (1993) Data collection and processing. In Sawyer,L., Isaacs,N. and Bailey,S. (eds), Proceedings of the CCP4 Study Weekend. SERC Daresbury Laboratory, Warrington, UK, pp. 56–62. [Google Scholar]
- Pecorari F., Tissot,A.C. and Pluckthun,A. (1999) Folding, heterodimeric association and specific peptide recognition of a murine αβ T-cell receptor expressed in Escherichia coli. J. Mol. Biol., 285, 1831–1843. [DOI] [PubMed] [Google Scholar]
- Prevost-Blondel A., Chassin,D., Zeliszewski,D., Dorval,I., Sterkers,G., Pannetier,C. and Guillet,J.G. (1995) Preferential usage of the T-cell receptor by influenza virus hemagglutinin-specific human CD4+ T lymphocytes: in vitro life span of clonotypic T cells. J. Virol., 69, 8046–8050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read R.J. (1986) Improved Fourier coefficients for maps using phases from partial structures with errors. Acta Crystallogr. A, 42, 140–149. [Google Scholar]
- Reinherz E.L. et al. (1999) The crystal structure of a T cell receptor in complex with peptide and MHC class II. Science, 286, 1913–1921. [DOI] [PubMed] [Google Scholar]
- Robinson J., Malik,A., Parham,P., Bodmer,J.G. and Marsh,S.G.E. (2000) IMGT/HLA database—a sequence database for the human major histocompatibility complex. Tissue Antigens, 55, 280–287. [DOI] [PubMed] [Google Scholar]
- Rothbard J.B., Lechler,R.I., Howland,K., Bal,V., Eckels,D.D., Sekaly,R., Long,E.O., Taylor,W.R. and Lamb,J.R. (1988) Structural model of HLA-DR1 restricted T cell antigen recognition. Cell, 52, 515–523. [DOI] [PubMed] [Google Scholar]
- San Jose E., Sahuquillo,A.G., Bragado,R. and Alarcon,B. (1998) Assembly of the TCR/CD3 complex: CD3 ε/δ and CD3 ε/γ dimers associate indistinctly with both TCR α and TCR β chains. Evidence for a double TCR heterodimer model. Eur. J. Immunol., 28, 12–21. [DOI] [PubMed] [Google Scholar]
- Sant’Angelo D.B., Waterbury,G., Preston-Hurlburt,P., Yoon,S.T., Medzhitov,R., Hong,S.C. and Janeway,C.A.,Jr (1996) The specificity and orientation of a TCR to its peptide–MHC class II ligands. Immunity, 4, 367–376. [DOI] [PubMed] [Google Scholar]
- Sato A.K., Zarutskie,J.A., Rushe,M.M., Lomakin,A., Natarajan,S.K., Sadegh-Nasseri,S., Benedek,G.B. and Stern,L.J. (2000) Determinants of the peptide-induced conformational change in the human class II major histocompatibility complex protein HLA-DR1. J. Biol. Chem., 275, 2165–2173. [DOI] [PubMed] [Google Scholar]
- Sloan V.S., Cameron,P., Porter,G., Gammon,M., Amaya,M., Mellins,E. and Zaller,D.M. (1995) Mediation by HLA-DM of dissociation of peptides from HLA-DR. Nature, 375, 802–806. [DOI] [PubMed] [Google Scholar]
- Snoke K., Alexander,J., Franco,A., Smith,L., Brawley,J.V., Concannon,P., Grey,H.M., Sette,A. and Wentworth,P. (1993) The inhibition of different T cell lines specific for the same antigen with TCR antagonist peptides. J. Immunol., 151, 6815–6821. [PubMed] [Google Scholar]
- Sodeoka M., Larson,C.J., Chen,L., Leclair,K.P. and Verdine,G.L. (1993) A multifunctional plasmid for protein expression by Ecpcr—overproduction of the P50 subunit of NF-κ-B. Bioorg. Med. Chem. Lett., 3, 1089–1094. [Google Scholar]
- Stern L.J. and Wiley,D.C. (1992) The human class II MHC protein HLA-DR1 assembles as empty αβ heterodimers in the absence of antigenic peptide. Cell, 68, 465–477. [DOI] [PubMed] [Google Scholar]
- Stern L.J. and Wiley,D.C. (1994) Antigenic peptide binding by class I and class II histocompatibility proteins. Structure, 2, 245–251. [DOI] [PubMed] [Google Scholar]
- Stern L.J., Brown,J.H., Jardetzky,T.S., Gorga,J.C., Urban,R.G., Strominger,J.L. and Wiley,D.C. (1994) Crystal structure of the human class II MHC protein HLA-DR1 complexed with an influenza virus peptide. Nature, 368, 215–221. [DOI] [PubMed] [Google Scholar]
- Studier F.W., Rosenberg,A.H., Dunn,J.J. and Dubendorff,J.W. (1990) Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol., 185, 60–89. [DOI] [PubMed] [Google Scholar]
- Sun R., Shepherd,S.E., Geier,S.S., Thomson,C.T., Sheil,J.M. and Nathenson,S.G. (1995) Evidence that the antigen receptors of cytotoxic T lymphocytes interact with a common recognition pattern on the H-2Kb molecule. Immunity, 3, 573–582. [DOI] [PubMed] [Google Scholar]
- Teng M.K. et al. (1998) Identification of a common docking topology with substantial variation among different TCR/peptide–MHC complexes. Curr. Biol., 8, 409–412. [DOI] [PubMed] [Google Scholar]
- Tissot A.C., Vuilleumier,S. and Fersht,A.R. (1996) Importance of two buried salt bridges in the stability and folding pathway of barnase. Biochemistry, 35, 6786–6794. [DOI] [PubMed] [Google Scholar]
- Urban J.L., Kumar,V., Kono,D.H., Gomez,C., Horvath,S.J., Clayton,J., Ando,D.G., Sercarz,E.E. and Hood,L. (1988) Restricted use of T cell receptor V genes in murine autoimmune encephalomyelitis raises possibilities for antibody therapy. Cell, 54, 577–592. [DOI] [PubMed] [Google Scholar]
- Vignali D.A. and Vignali,K.M. (1999) Profound enhancement of T cell activation mediated by the interaction between the TCR and the D3 domain of CD4. J. Immunol., 162, 1431–1439. [PubMed] [Google Scholar]
- Vignali D.A., Carson,R.T., Chang,B., Mittler,R.S. and Strominger,J.L. (1996) The two membrane proximal domains of CD4 interact with the T cell receptor. J. Exp. Med., 183, 2097–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wedderburn L.R., Searle,S.J.M., Rees,A.R., Lamb,J.R. and Owen,M.J. (1995) Mapping T cell recognition—the identification of a T cell receptor residue critical to the specific interaction with an influenza hemagglutinin peptide. Eur. J. Immunol., 25, 1654–1662. [DOI] [PubMed] [Google Scholar]
- Wilson I.A. (1999) Perspectives: protein structure. Class-conscious TCR? Science, 286, 1867–1868. [DOI] [PubMed] [Google Scholar]
- Yassine-Diab B., Carmichael,P., L’Faqihi,F.E., Lombardi,G., Deacock,S., de Preval,C., Coppin,H. and Lechler,R.I. (1999) Biased T-cell receptor usage is associated with allelic variation in the MHC class II peptide binding groove. Immunogenetics, 49, 532–540. [DOI] [PubMed] [Google Scholar]
- Zeliszewski D., Gaudebout,P., Golvano,J.J., Dorval,I., Prevost,A., Borras-Cuesta,F. and Sterkers,G. (1994) Molecular basis for degenerate T-cell recognition of one peptide in the context of several DR molecules. Hum. Immunol., 41, 28–33. [DOI] [PubMed] [Google Scholar]