

Abstract
To identify new structure-function correlations in the γ domain of streptokinase, mutants were generated by error-prone random mutagenesis of the γ domain and its adjoining region in the β domain followed by functional screening specifically for substrate plasminogen activation. Single-site mutants derived from various multipoint mutation clusters identified the importance of discrete residues in the γ domain that are important for substrate processing. Among the various residues, aspartate at position 328 was identified as critical for substrate human plasminogen activation through extensive mutagenesis of its side chain, namely D328R, D328H, D328N, and D328A. Other mutants found to be important in substrate plasminogen activation were, namely, R319H, N339S, K334A, K334E, and L335Q. When examined for their 1:1 interaction with human plasmin, these mutants were found to retain the native-like high affinity for plasmin and also to generate amidolytic activity with partner plasminogen in a manner similar to wild type streptokinase. Moreover, cofactor activities of the mutants precomplexed with plasmin against microplasminogen as the substrate as well as in silico modeling studies suggested that the region 315–340 of the γ domain interacts with the serine protease domain of the macromolecular substrate. Overall, our results identify the presence of a substrate specific exosite in the γ domain of streptokinase.
Keywords: Enzyme Catalysis, Enzyme Kinetics, Enzyme Mechanisms, Enzyme Turnover, Enzymes, Exosites
Introduction
Streptokinase (SK),3 secreted by several β-hemolytic group C (1, 2) strains of the genus Streptococcus, is a flexible multi-domain protein (3, 4). SK is often employed as a thrombolytic drug because it has the ability to activate human plasminogen (HPG), a zymogen, to its enzymatically active form, the serine protease, plasmin (HPN) (5, 6). However, unlike the physiological HPG activators such as tPA and urokinase, SK first forms a high affinity equimolar complex with HPG that results in the conformational activation of HPG to form SK·HPG* that specifically recruits substrate HPG and cleaves the Arg-561—Val-562 bond in substrate HPG molecules, thereby converting these into HPN (Pathway I, conformational activation pathway) (5, 6). The N-terminal Ile of SK has been demonstrated to play a critically important role in the activation of the “partner” after the formation of the 1:1 binary complex between SK and HPG (7–9). In addition, through several elegant studies on the mechanism of zymogen activation in the SK·HPG complex, it has been shown that HPN exhibits much higher affinity for SK compared with HPG, as a result of which an intermolecular exchange reaction takes place wherein the SK·HPN activates free substrate HPG molecules into HPN (10, 11). SK can also directly act as a co-factor for HPN, which upon complexation with HPN (12) (Pathway II, direct proteolytic activation pathway) virtually transforms the trypsin-like broad substrate specificity of HPN toward the cleavage of the scissile peptide bond, Arg-561—Val-562, in the incoming substrate HPG (12, 13).
The mechanism by which SK alters the substrate specificity of HPN/HPG and imparts it with a high catalytic turnover for substrate HPG processing has been a subject of detailed investigation, but some very fundamental mechanistic questions still remain unanswered (14–19). Mechanistic insights on the specificity switch engendered by SK upon binding onto the HPN active site are crucial in major part because of possibly enabling the engineering of altogether new target specificities into pre-existent protease active sites for biomedical applications (20, 21). The binding of SK to HPN results in the expression of substrate recognition “exosites” on the SK·HPG/HPN activator complex, which are located physically away from the active center of the enzyme (19). Binding studies employing fluorescently labeled active site of partner HPN of the SK·HPN complex have indicated that the docking of substrate HPG onto the activator complex does not perturb the active site of the enzyme per se; rather other epitopes in the SK·HPN complex exert their influence to change the specificity of the active site (19). The crystal structure of the binary complex of SK with the catalytic domain (SK·μPN)(17) presents a high resolution model for studying the interactions and functional properties of the SK·HPG activator system but is largely silent about definitive identification of the regions of the activator complex that are involved in enzyme-substrate HPG interaction. Nevertheless, it did point out the potential of the various surface-exposed flexible regions present in the three domains of SK to engage in both inducing the conformational activation in partner HPG and also imparting substrate specificity to the activator complex.
Till recently, most structure-function studies have shown the direct involvement of discrete regions in the N-terminal-located domains, namely the α and β domains in its mechanism of action particularly with regard to enzyme-substrate interactions (22–27), and have not assigned any specific role to the γ domain of SK in this step of catalysis, although its role in inducing partner activation has been elucidated (23, 28, 29). The crystal structure of SK·μPN complex supports the zymogen activation role of the γ domain, which being in close vicinity to the partner HPG molecule, had been proposed to be involved in its activation through a contact mechanism (17). Moreover, the role of the γ domain in zymogen activation, i.e. partner PG activation, also becomes evident from the fact that the single- and bi-domain bacterial PG activators, namely SAK and Streptococcus uberis plasminogen activator, respectively, which are devoid of the γ domain, seem to be adversely affected in terms of zymogen activation; SAK does not carry out zymogen activation at all, whereas Streptococcus uberis plasminogen activator exhibits significantly decreased rates of zymogen activation (30, 31). Comparisons of the catalytic efficiencies of plasminogen activation of SK with the other two functionally similar indirect activators, namely SAK (single domain) and Streptococcus uberis plasminogen activator (two domains), show that by and large, the three exhibit broadly similar catalytic constants, kcat, once complexed with a fully functional plasmin as the partner (30, 32, 33). However, some studies do indicate that both β and γ domains are involved in substrate activation (33–36). Other studies carried out with truncated SK derivatives suggest that the region constituting both β and γ domains are strongly involved in 1:1 binding with the partner and also in generating an activator complex (37–39). Although studies in the recent past have implicated specific lysine residues in both β and γ domains in enzyme-substrate interactions followed by identification of discrete structural epitopes/exosites in both the α and β domains (20, 21, 33, 35), any clear-cut involvement of an exosite in the γ domain solely with this function has not been demonstrated so far. Bi-domain construct of SK composed of α and β domains exhibits only ∼3% substrate activation ability after complexation with HPN compared with wtSK, which suggests the importance of γ domain in engendering the full-blown substrate activation characteristic of wt SK (40). Moreover, the observation that the γ domain binds closely to the catalytic active center of the SK·HPN complex suggests that it might indeed be a preferred area of substrate interaction also (17), and there is, therefore, a need to explore intensively whether this domain contains any site(s) playing a role specifically in substrate interaction under conditions that unambiguously rule out the involvement of any 1:1 interactions. Previously, alanine mutagenesis of the extensive charged side chains of the residues present in the coiled coil region of the γ domain of SK has revealed the overall functional relevance of this region in HPG activation in general (29), but to look for the possible presence of a discrete substrate-interacting exosite/epitope of the type demonstrated in the α and β domains (20, 21, 35, 41), in the present study we took recourse to a random mutagenesis approach.
The results, presented below, examine a series of random-mutant clones of the C-terminal region of SK (residues 210–414) for their functional importance in substrate HPG activation. Because this approach initially yielded several scores of functionally compromised mutants that carried multiple-site mutations, an analysis of their “deconvolved” forms, that is, all or most single-site mutants derived from the mutant-clusters with compromised activity, was then carried out to decipher the functional importance of residues present in the γ domain in substrate recognition and/or turnover. The mutants with specifically reduced activator activity after complexation with HPN (to rule out deficiencies in zymogen activation capability at the 1:1 level) were then fully characterized by steady-state kinetics as well as measurements of affinity with plasmin to specifically rule out apparent substrate interaction effects due to compromised capability to form functional activator complexes. Through this approach we could indeed unveil the existence of a substrate-specific locus spanning a reasonably large stretch of residues in the γ domain, which is quite distinct from another, adjoining locus in the same domain, which is likely to be involved only in zymogen activation.
EXPERIMENTAL PROCEDURES
Materials
Glu-plasminogen was either purchased from Roche Diagnostics or purified from human plasma by affinity chromatography (42). The T7 RNA polymerase promoter-based expression vector, pET 23(d), and Escherichia coli strain BL21 (DE3) were products of Novagen Inc. (Madison, WI). Thermostable DNA polymerase (Pfu Turbo TM) was obtained from Stratagene Inc. (La Jolla, CA). Oligonucleotide primers were supplied by Biobasic Inc.. Phenyl-agarose 6 XL was procured from Prometic Biosciences Ltd., and DEAE-SepharoseTM (fast-flow) was Amersham Biosciences. Urokinase, ϵ-aminocaproic acid, sodium cyanoborohydride, and l-lysine were purchased from Sigma. All other reagents used were of the highest analytical grade available.
Design and Construction of Random Mutations of C-terminal Region of SK 210–414
The SK gene from Streptococcus equisimilis H46A was cloned earlier in pET 23d vector (43), and this construct was used to prepare random mutations. The cDNA in the open reading frame encoding for the C-terminal region of SK was randomly mutagenized using the GeneMorph® II Random Mutagenesis kit and strategy. The primers flanking the segment encoding residues 210–414 were used to set up PCR I as per the conditions of the Stratagene random mutagenesis kit. The PCR I product was used as megaprimer employing pET-23d-SK as template, and another PCR reaction was set up using Pfu Turbo DNA polymerase. The PCR product after DpnI digestion was transformed into BL21 (DE3) electro-competent cells. The transformants in BL21 (DE3) cells were obtained with a transformation efficiency of ∼105 and then pick-patched onto freshly prepared LB-Amp plates. The plasmid DNAs of the transformed clones of SK were subjected to automated sequencing on an Applied Biosystems DNA sequencer (model 3130 XL analyzer).
Casein-HPG Overlay Assay for Detection of SK Activity
All the “randomized” pET-23d-SK clones were first screened for their HPG activation ability using a plate-based caseinolytic assay. The primary cultures of different clones were spotted (∼2 μl) on LB-Amp agar plates + IPTG (final concentration of 50 mm) to induce the expression of SK/SK random mutants. The plate was then overlaid by casein and HPG in soft agar (44), incubated at 37 °C for 4 h, and thereafter observed for the generation of zones of clearance due to casein hydrolysis after HPG activation.
Expression and Purification of Different Mutant Constructs of pET-23d-SK Using Nickel-nitrilotriacetic acid Spin Columns
To purify several mutant proteins simultaneously, a mid-throughput strategy was devised. The SK mutants were expressed as proteins with hexa-His-tag extensions at their C termini, thus allowing their purification by Ni2+-immobilized metal affinity chromatography (45). Nickel-nitrilotriacetic acid spin columns (Qiagen) were used for purifying the mutants under native conditions. This process yielded the few μg of protein per mutant that was analyzed by SDS-PAGE gel electrophoresis, which showed nearly 95% purity.
Purification of SK/SK Random Mutants from E. coli BL21 (DE3) Strain
The various mutants of SK selected after three rounds of screening were purified at a larger scale using hydrophobic interaction chromatography. The selected clones were grown from their BL21 (DE3) glycerol stocks as inclusion bodies and purified as described (40). SK proteins eluted were generally more than 95% pure, as analyzed by SDS-PAGE. The amount of protein in each fraction was measured routinely using the Bradford method of protein estimation and confirmed by values at A280 nm.
Design and Construction of Various SK Single-site γ Domain Mutants
The substitution point mutations of the γ domain mutants of SK were prepared through the QuikChange® mutagenesis kit obtained from Stratagene Inc., which involves usage of two complementary primers having the desired mutation at the center. After the primers were extended by temperature cycling with the Pfu Turbo DNA polymerase, the procedure given by the manufacturer was followed to obtain the site-specific incorporation of desired mutations. The transformed clones obtained were selected by first screening mini-prep plasmid DNAs, and the various γ domain mutants of SK were confirmed by automated sequencing on an Applied Biosystems DNA sequencer (model 3130 XL analyzer). The plasmids carrying the desired mutations were transformed into chemically competent cells of E. coli BL21 (DE3) host strain for expression of the proteins.
Preparation of HPN
Plasmin (HPN), the active form of HPG, was prepared by digesting Glu-HPG with urokinase covalently immobilized on agarose beads using a ratio of 300 Plough units/mg of HPG in 50 mm Tris·Cl, pH 8.0, 25% glycerol, and 25 mm l-lysine at 22 °C for 10 h (43).
Preparation of μPG
Recombinant μPG was expressed and purified as described earlier (7).
Preparation of Active-site Fluorescent-labeled HPN
Nα-[(Acetylthio)acetyl]-d-Phe-Pro-Arg-CH2Cl (ATA-FPR-CK) was prepared by mixing 5 mg of d-Phe Pro-Arg-CH2Cl (FPR-CK), (procured from Bachem) in 50 mm sodium phosphate buffer, pH 7.0, with 0.5–1 volume of succinimidyl (acetylthio) acetate (SATA, Molecular Probes) in methanol to give final concentration of 3–6 mm FPR-CK and a 4–5-fold molar excess of SATA (46–49). The reaction mix was incubated for 30 min, and final product was then purified through gel filtration chromatography and monitored as described (49). The active site of HPN was then specifically inhibited by above-obtained ATA-FPR-CK in sodium phosphate buffer, pH 7.0, buffer, and its inhibition was measured by measuring the residual activity against chromogenic substrate (S2251). After complete inhibition of HPN, it was labeled with 5-(iodoacetamido) fluorescein, and fluorescein incorporation was determined to be 0.9–1.0 mol of fluorescein/mol of HPN as described (19).
Assays for Studying the Activation of HPG by wtSK and SK Mutants
A one-stage assay method was used to measure the kinetics of HPG activation by wtSK or its mutants (50). Purified wtSK or SK random mutants (0.5 nm) were added in a 100-μl reaction well of a 96-well plate containing HPG (1 μm) in assay buffer (50 mm Tris-Cl buffer, pH 7.5,100 mm NaCl) containing 0.5 mm chromogenic substrate, Chromozym®-PL (tosyl-Gly-l-Pro-l-Lys-pNA) obtained from Roche Diagnostics. The change in absorbance at 405 nm was then measured as a function of time (t) in a Molecular Devices Versamax microplate reader at 22 °C. The activator activities were obtained from the slopes of the activation progress curves as change in absorbance/t2 (50).
Assays for Determining the Steady-state Kinetic Constants for Amidolytic Activity of wtSK and SK Mutants
The kinetic parameters of amidolysis by wtSK and SK γ domain mutants complexed with HPN were determined by precomplexing equimolar ratios of SK and HPN (500 nm each) at 22 °C in 50 mm Tris·Cl, pH 7.5, containing 0.5% BSA. The amidolytic activity was measured by transferring an aliquot of this mixture to a 100-μl reaction well of a 96-well plate containing buffer (50 mm Tris·Cl) and varying the concentrations of the chromogenic substrates, namely Chromozym-PL (tosyl-Gly-l-Pro-l-Lys-pNA) and S-2444 (l-PyroGlu-Gly-l-Arg-pNA) (0.1–2.0 mm) to obtain a final concentration of 10 nm of the complex in the reaction. The reaction was continuously monitored spectrophotometrically at 405 nm for 10 min. The kinetic parameters were determined from Michaelis-Menten (V versus S) and inverse (1/v versus 1/s) Lineweaver-Burk plots by standard methods (50).
N-terminal Methionine Removal from wtSK/SK γ Domain Mutants
The N-terminal methionine was removed from wtSK and γ domain mutants using methionine aminopeptidase (pfu methionine aminopeptidase) as described (26). The extent of removal of the N-terminal methionine was determined by N-terminal sequence analysis on an Applied Biosystems sequencer, Model 491A, and also through quantitative amino acid composition analyses on a Waters Pico-tagTM system. By this methodology, generally 70–80% removal was observed.
Esterolytic Activation of Equimolar HPG·SK/SK γ Domain Mutant Complexes
To monitor the active site formation, 7 μm HPG was added to an assay cuvette containing 7.5 μm wtSK/mutant, 100 μm NPGB, and 10 mm sodium phosphate buffer, pH 7.5. The “burst” of p-nitrophenol release due to acylation of the active center was monitored at 410 nm as a function of time at 22 °C (5, 51, 52).
Fluorescence Studies
Fluorescence measurements were made with a Cary EclipseTM fluorescence spectrophotometer using acrylic cuvettes coated with polyethylene glycol 20,000. Fluorescence titrations were measured at the excitation and emission wavelength maxima for the 5-(iodoacetamido) fluorescein-labeled HPN at 513 and 525 nm, respectively. Fluorescence titrations were performed as described (19), and the fluorescence measurements were expressed as the fractional change in the initial fluorescence (Fobs − Fo)/Fo = ΔF/Fo. The titrations were analyzed by nonlinear least square fitting of the quadratic binding equation with maximum fluorescence change (ΔFmax/Fo), dissociation constant (KD), and stoichiometric factor (n) as the fitted parameters as described (19).
Determination of Steady-state Kinetic Constants for HPG/μPG Activator Activity of wtSK/SK γ Domain Mutants
The kinetics of HPG/μPG activation by HPN·wtSK/SK γ domain mutant complexes were measured by transferring suitable aliquots (usually, so as to attain 0.5 nm final concentration in an assay mix) of preformed HPN·wtSK/SK γ domain mutant complexes to the reaction wells containing varying concentrations of substrate HPG/μPG in assay buffer (50 mm Tris·Cl buffer, pH 7.5, and 100 mm NaCl) also containing 0.5 mm chromogenic substrate Chromozym®-PL. The generation of activator activity was monitored at 22 °C at 405 nm as before. The kinetic parameters for HPG activation were then calculated from Michaelis-Menten (V versus S) and inverse (1/v versus 1/s) Lineweaver-Burk plots (53).
Kinetic Analysis of Ternary Protein-Protein Interactions by Surface Plasmon Resonance
A surface plasmon resonance-based biosensor was also used to measure the rate and equilibrium dissociation constants describing interactions between soluble analytes (HPG) and wtSK/SK γ domain mutants complexed with immobilized HPG, a situation simulating substrate binding to binary complex and hereafter referred to as ternary interaction, as described previously (35). Experiments were performed at 25 °C in HBS “running buffer” (30 mm Hepes, pH 7.4, 135 mm NaCl, and 1 mm EDTA) supplemented with 0.05% Tween 20 and 5 mm NPGB. The NPGB was included to prevent plasmin-mediated proteolysis (35). After subtracting the nonspecific response the association rate constants (kon values), the dissociation rate constants (koff values) and the equilibrium binding constant (KD) were calculated from sensorgrams by non-linear fitting of the association and dissociation curves according to a 1:1 binding model using the BIACORE 3000 evaluation software supplied by the manufacturer (19).
Molecular Modeling and Docking Studies for the Generation of μPN·SK·μPNsubstrate Ternary Complex
Cartesian coordinates of the SK β domain, SK·μPN complex, and ternary μPN·SAK·μPN complex were retrieved from the Protein Data Bank (PDB IDs 1C4P, 1BML and 1BUI, respectively) (17, 18, 54). Coordinates for 170 loop and 250 loop in the β domain of SK are missing in SK·μPN crystal structure due to high flexibility of the two disordered loops (17). However, coordinates for these two loops are intact in the isolated β domain crystal structure (PDB ID 1C4P) (18). The coordinates for the β domain in the SK were replaced with those of the isolated β domain, and the structure was energy-minimized for a total of 2000 steps with combination of steepest descent and conjugant gradient algorithms without any cut-off for calculation of non-bonded interactions in implicit solvent using FF03 force-field. Subsequently, rigid body docking of the energy-minimized structure of SK onto the μPNsubstrate·μPNpartner complex derived from the ternary complex coordinates of SAK·μPNpartner·μPNsubstrate (54) was performed using ZDOCK (55). A total of 100 docked conformations was generated. SAK, being structurally homologous to SK α domain (17), is known to share the same target sites on the PG surface (56, 57). Based on the structural equivalency of SK α domain in the SK·μPN complex with that of the SAK in the ternary complex of SAK, μPNpartner·μPNsubstrate, scoring of the modeled complex was done. Re-scoring of docked conformations was then performed based on their root mean square distance with respect to the structure of the SK·μPN complex (17). This step ensured that the relative orientation of SK and co-factor μPN was the same as that observed in the crystal structure (17), with the substrate μPN optimally positioned in the active site valley of the SK·μPN complex. Docked conformation with minimum root mean square distance was taken for further investigation.
RESULTS
Introduction and Screening of Random Mutations in the C-terminal Region of SK
The SK γ domain and adjoining β domain region, i.e. 210–414 residues was randomly mutagenized (see “Experimental Procedures” for details). To screen for mutants with apparently altered HPG activation activity, either increased or decreased due to an introduced random mutation(s) in SK, we selected a plate-based caseinolytic method (58) as an initial screen for the hundreds of clones generated in the experiments and then selected for clones with either bigger or smaller haloes compared with wtSK clones (58). Although the mutants in this study were cloned in pET-23(d), which is not a secretory plasmid, it has been shown earlier that SK deprived of any leader sequence is able to translocate slowly across the outer membranes of E. coli (59). The mutant library (about 1100 clones) was screened by the plate-based assay procedure, and the clones found “positive” after this initial round on the basis of weaker or smaller haloes compared with the wtSK were isolated and subjected to another round of plate-based testing by the caseinolytic assay screening. However, in the second round the clones were selected on casein plates containing trace amounts of HPN along with HPG (see “Experimental Procedures”). This was done so as not to “lose” the mutants defective in Pathway I (i.e. ability to open up the active site in partner HPG). Also, through the second round of caseinolytic assay, the reproducibility of the clones that were screened from the first round was confirmed. Approximately 400 clones with apparently impaired activator activity were selected from the screenings, and these were then purified individually after shake-flask culturing and checked for HPG activation activity.
Expression, Purification, and Characterization of SK Random Mutants
The mutant clones selected after two rounds of screening by caseinolytic plate-based assays were expressed and purified to near homogeneity through Ni+2 affinity chromatography (see “Experimental Procedures”). The cofactor activities of the purified random mutants were compared with that of wtSK against HPG substrate (see “Experimental Procedures”). Around 70 clones were selected and subjected to DNA sequencing that showed either no activity or activity that was significantly less compared with that of wtSK, which were then purified at a larger scale and studied for their cofactor activity and steady-state kinetics properties (Table 1). The results shown in Fig. 1A represent a few of the diverse combinations of mutants that exhibited overall reduced HPG activation compared with wt SK. Interestingly, nearly 40 clones that gave larger than wtSK haloes on the caseinolytic plate assay when sequenced invariably showed no mutation, suggesting that a larger halo occasionally did not necessarily mean a better HPG activator; rather, it was likely that the clone secreting the protein could be a fast growing variant (data not shown). The mutants that showed reproducibly low cofactor activities (Table 1) were also studied for their ability to induce an active site in the partner HPG (Pathway I) (12) using the active site acylating agent, NPGB (data not shown). NPGB has been previously used to titrate the active site formation in the SK·HPG complex (5, 51, 52). Neither SK nor HPG alone gave the burst observed at 410 nm but only when the 2 are mixed in equimolar proportions give a characteristic burst due to the formation of a “virgin” SK·HPG activator complex and consequent acylation of the active site. The residues altered in the mutants that failed in their ability to open the active site in the partner HPG molecule were located largely in the β7 sheet of the β domain. These results are in agreement with earlier studies (43) done in context of the charge reversal of selected residues in the β-domain, where it was observed that mutation of a few residues in and around the 250 loop region resulted in a significantly compromised Pathway I activation capability. Therefore, these random clusters of mutants obtained in the present study with mutations centered in this region were not investigated further. Esterolytic activity (NPGB burst) of all of the mutants bearing mutations in the γ domain, particularly the so-called coiled coil region, however, were positive and, thus, indicated an intact Pathway I in these mutants. All the mutants of the γ domain with altered co-factor activity and an intact esterolytic activity were further subjected to detailed structure-function analysis.
TABLE 1.
Random mutants of C-terminal SK (residues 210–414)
The first column represents selected random mutants (RM), and the second column represents the corresponding residues identified in the random mutants after DNA sequencing. The cofactor activities of the random mutants in complex with HPN (SK/SK*·HPN) were determined for substrate HPG (see “Experimental Procedures”) and were calculated as percent change in activity compared to wtSK, taken as 100%.
| Random mutant | Mutation | Specific activity (Pathway II) |
|---|---|---|
| % | ||
| wt SK | wt | 100 |
| RM 1 | P329L | 0.5 |
| R324G | ||
| RM 2 | G344D | 14 |
| RM 3 | N265Y | 3 |
| R330C | ||
| E355D | ||
| RM 4 | H226L | 10 |
| D328N | ||
| H381Q | ||
| RM 5 | D322R | 5 |
| D328H | ||
| RM 6 | K334E | 12 |
| RM 7 | R324G | 8 |
| RM 8 | D328G | 10 |
| RM 9 | F287S | 25 |
| N339S | ||
| D347V | ||
| RM 10 | D322G | 7.5 |
| R330H | ||
| R363L | ||
| T403R |
FIGURE 1.

Comparative analysis of HPG activation capability of mutants of the γ domain of SK obtained through random mutagenesis as well as through site-directed mutagenesis. A, the histogram depicts a comparison of HPG activator activity of the various clusters of random mutants, originally obtained by screening on activity plates, with wild type SK. Purified SK/various mutants of SK (1 nm each) were added individually to microtiter plate wells containing HPG (2 μm) and Chromozym®-PL, and the activator activities were measured spectrophotometrically at 405 nm as detailed under the “Experimental Procedures.” B, the histogram depicts the comparison of HPG activator activity of the various point mutants of the γ domain with that of wild type SK. SK/various mutants of SK (1 nm each) were added individually to microtiter plate wells containing HPG (2 μm) and Chromozym®-PL, and the activator activities were measured spectrophotometrically at 405 nm as detailed under “Experimental Procedures.”
Steady-state Kinetics of Screened Random Mutants for Substrate HPG Activation
The cofactor activity of the selected random mutants after complexation with equimolar HPN was then checked against a varied range of substrate HPG concentrations (see “Experimental Procedures” for details) to obtain the kinetic parameters for substrate HPG activation. Thus, the activity obtained for substrate activation by SK mutants after random mutagenesis is a true reflection of their differential substrate activation capability. The bioactivity data obtained with the mutants are summarized in Table 2, which clearly shows that several mutants were altered with respect to their rate of catalysis for HPG activation. For example, the mutant SK-P329L/R324G showed negligible activity with substrate HPG activity (Table 2), suggesting that either or both of these residues are crucial for substrate processivity. The kinetic constants determined with HPG as a substrate suggest that mutants with residues substituted in the γ domain, namely SK-K334E and SK-D322G/R330H/R363L, showed a significant decrease in catalytic activity, which was nearly an order of magnitude less compared with wtSK with no concomitant changes in apparent substrate affinity (Km). Many other mutants also showed no change in affinity with HPG as substrate but exhibited a differential effect in substrate turnover e.g. SK-G344D/E355D/R330C (a triple-site mutant), SK-D328E (a single-site random mutant), SK-D328N/H381Q and SK-D322R/D328H (double-site mutants), and SK-D328G/R324G/R330C and SK-F287S/N339S/D347V (triple-site mutants). These results with random mutants, which exhibited mainly altered kcat values for HPG, as substrate does not by its self establish a cause and effect correlation with the mutation sites as multiple residues were simultaneously mutated in a single cluster. Hence, we resorted to generating the corresponding single-site mutants to definitively identify the critical residues in the γ domain important for substrate interactions with SK·HPN enzyme complex.
TABLE 2.
Steady-state kinetic parameters for HPG activation by equimolar complex of HPN·SK/SK random mutant(s) (RM)
Fixed, catalytic amounts of the respective preformed activator complexes of each protein with HPN were added to the cuvette containing HPG as the substrate and Chromozym®-PL in 50 mm Tris·Cl buffer, pH 7.5, and the reactions were monitored at 405 nm at 22 °C. The kinetic parameters for substrate HPG activation were determined as described under “Experimental Procedures.” NO, not observable.
| Activator protein (SK/SK*·HPN) | km | kcat | kcat/km |
|---|---|---|---|
| μm | min−1 | min−1/μm | |
| wtSK | 0.4 ± 0.05 | 7.5 ± 0.5 | 19.0 |
| RM 1 | NO | NO | NO |
| RM 2 | 0.5 ± 0.1 | 1.0 ± 0.2 | 2.0 |
| RM 3 | 0.5 ± 0.1 | 0.22 ± 0.03 | 0.4 |
| RM 4 | 0.5 ± 0.05 | 0.75 ± 0.05 | 1.5 |
| RM 5 | 0.6 ± 0.1 | 0.37 ± 0.03 | 0.6 |
| RM 6 | 0.6 ± 0.2 | 0.9 ± 0.02 | 1.5 |
| RM 7 | 0.5 ± 0.1 | 0.5 ± 0.02 | 1.0 |
| RM 8 | 0.6 ± 0.1 | 0.8 ± 0.05 | 1.3 |
| RM 9 | 0.6 ± 0.1 | 1.9 ± 0.1 | 3.1 |
| RM 10 | 1.2 ± 0.1 | 0.56 ± 0.05 | 0.5 |
Construction, Purification, and Characterization of the Single-site Mutants of the Random Mutant Clusters
To deduce structure-function co-relations from the cluster of random mutants and to ascertain the functional importance of the residues in HPG activation among the series of γ domain mutants, we undertook to “deconvolve” the cluster of random mutants to their respective single-site substitution mutants. The mutants, as before, were expressed in E. coli BL21 (DE3) cells and harvested as inclusion bodies and then purified to near homogeneity (see “Experimental Procedures” for details). The far-UV CD spectra of the purified single-site mutants showed a close resemblance with that of the wtSK (data not shown), which was suggestive of the absence of any major structural impairment caused by the mutations. The mutants were first characterized for their ability to activate HPG by a continuous spectrophotometric assay (single-stage assay) that measures the overall HPG activator activity using catalytic amounts of SK and a large excess of HPG (60). Through this preliminary screening, deconvolved single-site forms that were wild type-like in terms of their functioning could be rapidly identified. The relative slopes of the progress curves obtained as a result of HPG activation (see Fig. 1B) is depicted in the form of bar diagrams. It compares the overall HPG activator activity of representative single-site mutants derived from bi- and multisite mutants with that of wtSK. The results clearly indicate that several of the single-site forms of the γ domain multisite “clustered” mutants that mostly comprised the residues of the coiled-coil region do exhibit varied extents of reduction in their HPG activation capabilities. Of all the single-site substitution mutants tested, we observed that SK-R319H, SK-D328G, SK-N339S, and SK-L335Q exhibited the most compromised plasminogen activator activity, roughly an order of magnitude less compared with that of wild type SK followed by SK-R330H and SK-K334E. However, some declustered mutants of SK γ domain, e.g. SK-P329L, SK-R324G, SK-D347V, SK-E355D, and SK-R363L, showed at most only a minor or no decrease in activity compared with wtSK. Overall, these results do suggest that several residues within the coiled-coil region are important in substrate HPG activation per se, whereas other residues placed relatively distal to the coiled-coil region seem not to be critical in HPG activation.
Interaction of the γ Domain Mutants of SK with Partner HPG and HPN
The single-site mutants of the γ domain of SK that exhibited significant reduction in their plasminogen activator capability, as monitored through single-stage assay, were then subjected to activity assays that would explore the underlying cause of their decline in activity, i.e. whether it was the amidolytic/zymogen activation step that operates through Pathway I wherein 1:1 complexation of HPG with SK and formation of the SK·HPG* activator complex occurs (7, 11, 21), was affected or a step subsequent to this, that is, in Pathway II, namely the interaction of substrate HPG with a fully mature mutant SK·HPN complex. To address this, all the above studied single-site γ domain mutants, namely SK-D328A, SK-K334E, and SK-G344D were investigated using the active site acylating agent, NPGB. All three mutants showed a native-like burst with NPGB (Fig. 2), indicating an intact Pathway I in these mutants.
FIGURE 2.

Active-site titration of HPG on complexing with wtSK/SK mutant using the active site acylating agent, NPGB. The figure shows progress curves of NPGB hydrolysis as followed by color generation because of p-nitrophenol liberation by methionine aminopeptidase-treated wtSK (▼), methionine aminopeptidase-treated SK-D328G mutant (■), SK-K334E (●), and SK-G344D (▵) (see “Experimental Procedures” for details).
In the recent past elegant equilibrium binding studies with active site-labeled fluorescent HPN derivatives have revealed that SK has a much higher order of affinity with HPN as compared with HPG (9, 19, 61, 62). From these studies it became evident that formation of a stable, high affinity SK·HPN complex is a prerequisite for its interaction with substrate HPG. Therefore, it is important to exclude the probable dissociation of mutant SK·HPN complexes (even when the apparent SK-HPG interaction is native-like in terms of the NPGB burst and amidolytic activity) under the conditions of activator assays due to a possible lowered affinity between the mutant(s) and HPN, which are generally carried out in the low nanomolar to picomolar range of concentration, which tends to overlap the KD for SK and HPN (19). Accordingly, the various γ domain SK mutants were examined for their capability of forming high affinity complexes with HPN by sensitive spectroscopic titration procedures developed for the interaction between the two proteins in the picomolar range (9, 19, 61, 62) in which titrations of SK/SK mutants are performed with 5-(iodoacetamido) fluorescein-labeled HPN. Given that these assays are able to detect low picomolar range of SK-HPN dissociation constants, one can safely assume that if the binary activator complexes of any or all the mutant(s) and HPN were relatively unstable due to their compromised affinity with HPN as compared with the wt SK·HPN complex at the concentrations (0.5–1 nm) at which the activator assays of the mutants and wtSK were conducted, then this procedure would detect if this was indeed the underlying cause of the observed highly reduced substrate HPG activity rather than a selective inactivation with respect to enzyme-substrate interactions per se.
However, analysis of the titrations of SK/SK γ domain mutants with fluorescein-labeled HPN (see Fig. 3) indicated that the various mutants indeed bound with similar high affinity with HPN as the wtSK. The dissociation constants obtained for the SK/SK γ domain mutants were indistinguishable from that of wtSK, which is in the range of 10 ± 2 pm. Overall, these results strongly indicate that the Pathway II is native-like in the mutants of the γ domain and that the significantly lowered HPG activator activity of SK-D328A, SK-K334E, and SK-G344D did not arise from an altered affinity for HPN during the formation of the mature SK·HPN activator complex.
FIGURE 3.
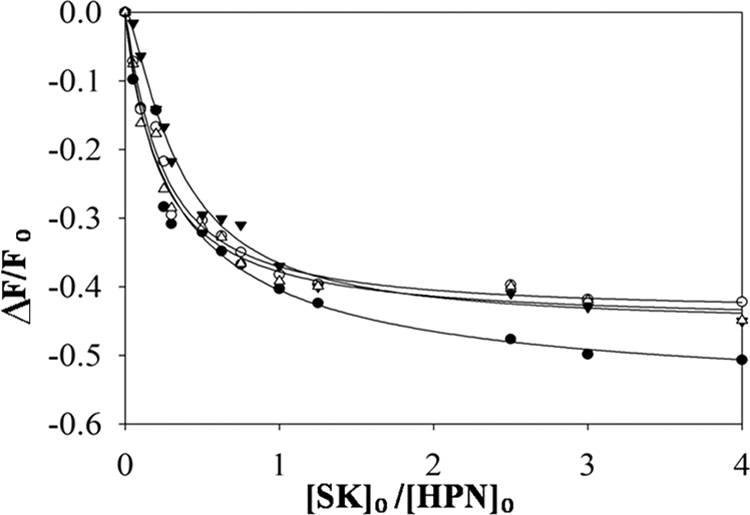
Fluorescence titrations of 5-(iodoacetamido) fluorescein-labeled HPN with wild type SK and γ domain mutants of SK. The fractional change in fluorescence (ΔF/Fo) is plotted versus the ratio of varied concentrations of SK/SK mutant with labeled HPN, prepared by inactivation with ATA-FPR-CH2Cl and labeling with thiol reactive probe 5-(iodoacetamido) fluorescein. The lines represent the least squares fits of the binding equation for the fluorescence titrations performed and analyzed as described under “Experimental Procedures.” The fitted curves depicting change in fluorescence as a function of ratio of SK/HPN concentrations of wtSK (○), SK-D328G (●), SK-K334E (▵), and SK-G344D (▿) are shown.
Steady-state Kinetic Parameters for Amidolytic Activation of Small Chromogenic Peptides by HPN·SK/SK-γ Domain Mutant Complexes
To characterize the covalent specificity of the active site of HPN in complexation with different γ domain mutants of SK, the amidolytic activation of two small chromogenic peptide substrates, namely Chromozym-PL and S-2444, with distinctly different peptide sequences attached to the scissile ester bond by HPN·SK/SK γ domain mutant complexes was performed and compared with that of amidolytic activation by HPN alone. These peptides would, thus, probe the S-site specificities of the plasmin and SK/mutant SK-plasmin complexes. The results (Table 3) suggest that the Km values of HPN complexes of different γ domain mutants, namely SK-D328A, SK-K334E, and SK-G344D were close to that of wt SK, and the turnover rates (kcat values) were also similar. From the comparison of the amidolytic constants of wtSK/SK mutant complexes with HPN with amidolytic parameters of HPN alone, it is evident that there is an increase in the Km for the amidolytic substrate over that of free HPN, which can be accounted for in overall reduced accessibility of the HPN active site by small molecular weight substrates due to steric hindrance brought about by SK binding in the vicinity of the HPN active site (17, 52, 63). These results suggest that the primary characteristics of the HPN active site after complexation with the γ domain mutants remained unaltered as compared with that of wt SK in terms of its ability to catalyze the hydrolysis of the small peptide substrates.
TABLE 3.
Steady-state kinetic parameters for amidase activity of equimolar complexes of SK or the various γ domain mutants and HPN
For the determination of the amidolytic parameters, wt SK/SK mutants were precomplexed with HPN in an equimolar ratio, and an aliquot of this mixture was assayed for amidolysis at varying concentrations of two chromogenic substrates with different peptide sequences, namely chromozym-PL(tosyl-Gly-l-Pro-l-Lys-pNA) and S-2444 (l-PyroGlu-Gly-l-Arg-pNA) as described under “Experimental Procedures.” The data represent the mean of three independent determinations.
| Activator specie | Chromozym-PL |
S-2444 |
||||
|---|---|---|---|---|---|---|
| Km | kcat | kcat/Km | Km | kcat | kcat/Km | |
| mm | min−1 | min−1/mm | mm | min−1 | min−1/mm | |
| HPN | 0.2 ± 0.06 | 32 ± 2 | 160 | 1.0 ± 0.1 | 8.0 ± 0.4 | 8.0 |
| wtSK·HPN | 0.5 ± 0.05 | 30 ± 2 | 60 | 3.1 ± 0.2 | 6.0 ± 0.4 | 1.9 |
| SK-D328A·HPN | 0.6 ± 0.03 | 29 ± 1 | 48 | 3.2 ± 0.4 | 6.5 ± 0.2 | 2.0 |
| SK-K334E·HPN | 0.5 ± 0.05 | 30 ± 1.5 | 60 | 2.8 ± 0.2 | 6.0 ± 0.3 | 2.1 |
| SK-G344D·HPN | 0.6 ± 0.04 | 31 ± 3 | 51 | 3.5 ± 0.3 | 6.2 ± 0.1 | 1.7 |
Steady-state Kinetics of γ Domain Mutants for HPG Activation
To further evaluate the mutants in context of activation of substrate HPG specifically, we determined the steady-state kinetic constants of the various γ domain mutants. The cofactor activity of the selected single-site mutants after complexation with equimolar HPN was checked against a concentration range of substrate HPG (see “Experimental Procedures” for details). The data obtained are summarized in Table 4 and clearly show that several mutants were found to be strongly altered in their rate of catalysis. The single-site mutants of SK-P329L, R324G, viz. SK-P329L and SK-R324G, showed nearly similar activity as wtSK for substrate HPG, suggesting that only if both of these residues were simultaneously mutated would loss of substrate processivity result, as the single-site mutants of this cluster were unaltered in HPG activation. SK-K334E, which exhibited nearly 10% activity as compared with wt SK when converted to its single-site alanine counterpart, exhibited only a 2-fold drop in catalytic activity, with an essentially unaltered substrate affinity. Another random mutant (SK-D322G, R330H, R363L), which showed a 2–3-fold increase in km for substrate HPG and a significant decrease in processing of full-length HPG when declustered to the respective single-site mutants, namely SK-D322G, SK-R330H, or SK-R363L, and characterized for substrate HPG activation, showed that mutants SK-D322G and SK-R363L exhibited wt SK-like activity, and only the mutant SK-R330H from this three-site cluster exhibited an order of drop in kcat (with no significant changes in km for substrate HPG). This suggests that of all the three residues mutated in the random cluster, only arginine at position 330 is crucial, as mutation at this very site resulted in a strong phenotypic effect related to substrate processing. Similarly, the “random” two-site cluster (SK-R330C, E355D), which exhibited decreased catalytic activity, when resolved into its single-site mutant counterparts exhibited similar result as described above, where the mutant SK-E355D had wt SK properties, but SK-R330C was found to be the underlying cause of the reduced catalytic activity of this cluster. Another random cluster, namely SK-D322G, R330H, R363L, and T403R, which exhibited nearly 10% kcat as compared with wtSK upon resolution to its point mutants, revealed that the single-site mutants SK-D322G, SK-R363L, or SK-T403R had nearly wtSK-like properties, with low catalytic activity of this cluster remaining associated with only SK-R330H, which showed a kcat that was 10-fold less as compared with wt SK. Another double-site random mutant (SK-D322R/D328H), with around 20-fold reduction in catalytic turnover, when further resolved to single-site mutants revealed that SK-D328H exhibited a similar 20-fold reduction in kcat, whereas the mutant SK-D322R exhibited unchanged kinetic parameters compared with wtSK. Similarly, the random double mutant (SK-D328N/H381Q), with the kcat compared with wtSK, when subsequently resolved into its two single-site mutants, viz. SK-D328N and SK-H381Q, further reinforced the importance of a negatively charged aspartate at position 328, as asparagine at this site resulted in only 5-fold reduced catalytic activity, whereas the mutation H381Q had no alteration in HPG activation. Overall, the results of the single-site-based simplification of the initial random mutant isolates suggest that there clearly exists a region in the γ domain that is important in substrate plasminogen. Residues downstream to the coiled-coil region seem not to be crucial in substrate plasminogen activation as the steady-state kinetic parameters of mutants in this region are wt SK-like in terms of substrate HPG activation by the preformed SK·HPN complex.
TABLE 4.
Steady-state kinetic parameters for HPG activation by equimolar complexes of SK or the various deconvolved random mutants and HPN
The kinetic parameters for substrate HPG activation were determined at 22 °C with the chromogenic substrate in 50 mm Tris-Cl buffer, pH 7.5, 100 mm NaCl as described under “Experimental Procedures.” The data represent the mean of three independent determinations.
| Activator species | Km | kcat | kcat/Km |
|---|---|---|---|
| μm | min−1 | min−1μm−1 | |
| wt SK | 0.4 ± 0.05 | 7.5 ± 0.3 | 19.00 |
| R319H | 0.5 ± 0.02 | 0.17 ± 0.03 | 0.34 |
| R324G | 0.5 ± 0.01 | 7.0 ± 0.2 | 14.0 |
| D328A | 0.4 ± 0.03 | 1.7 ± 0.03 | 4.275 |
| D328E | 0.6 ± 0.04 | 3.2 ± 0.2 | 5.28 |
| D328G | 0.5 ± 0.01 | 0.75 ± 0.04 | 1.5 |
| D328N | 0.4 ± 0.03 | 2.2 ± 0.2 | 5.5 |
| D328R | 0.6 ± 0.02 | 0.84 ± 0.03 | 1.4 |
| P329L | 0.4 ± 0.03 | 7.0 ± 0.3 | 17.5 |
| R330H | 0.6 ± 0.06 | 0.68 ± 0.04 | 1.13 |
| K334A | 0.4 ± 0.02 | 3.42 ± 0.4 | 8.55 |
| K334E | 0.5 ± 0.06 | 0.9 ± 0.02 | 1.8 |
| L335Q | 0.7 ± 0.07 | 0.15 ± 0.01 | 0.2 |
| E355D | 0.5 ± 0.08 | 7.5 ± 0.2 | 15 |
| R363L | 0.5 ± 0.06 | 7.6 ± 0.4 | 15.2 |
| H381Q | 0.4 ± 0.04 | 7.7 ± 0.2 | 19.25 |
Probing the Importance of Residues in the γ Domain of Streptokinase in Substrate HPG Activation
To further discern the relative importance of the residues in the γ domain, we carried out side-chain substitutions of some of its key residues. Among the various residues of the γ domain, aspartate at position 328 seems to be important in plasminogen activation as SK-D328G showed a nearly 10-fold drop in catalytic activity compared with wtSK, with no discernable alteration in the substrate affinity. We then investigated the importance of residue 328 through extensive mutagenesis of its side chains. From the results obtained (Table 4), it is evident that charge-neutralization mutants, namely SK-D328A, and SK-D328N, showed decreased activation of HPG to levels about 5-fold lower compared with wtSK (Fig. 2). Interestingly, charge-reversal mutation at this position, namely SK-D328R, significantly reduced the catalytic activity of the mutant to nearly that of wild type, whereas the mutant in which the negative charge was retained in the form of glutamate, namely SK-D328E, the activity drop was just 2-fold compared with wtSK. These observations clearly suggest the importance of negative charge (as well as, possibly, side-chain length) at this position in the catalytic processing of substrate HPG. Similarly, the role of lysine at position 334 was also probed through mutagenesis. The results presented in Table 3 suggest that the alanine substitution mutant SK-K334A showed only a 2-fold decline in catalytic activity, whereas charge-reversal mutation to glutamate, i.e. SK-K334E, resulted in an order of magnitude drop in catalytic activity. Overall, these results suggest that a constellation of charges rather than a critical side chain, present in the coiled coil region of SK, probably plays an important role in interaction with, and activation of substrate HPG, but this site has no overt role in promoting substrate affinity to the SK·HPN activator complex.
Next, we wanted to evaluate the mutants of the γ domain in the context of the “target” site in the macromolecular substrate so as to evaluate the role of kringle-mediated interactions, if any, during the interaction of SK·HPN with HPG, i.e. whether the substrate-specific site in SK γ domain was “aimed” at the catalytic domain or kringle domains of the substrate. For this purpose, the activity of the mutants was checked using the isolated catalytic domain devoid of the five kringles (μPG). Thus, the cofactor activity of the selected mutants after complexation with equimolar HPN was measured spectrophotometrically against a varied range of substrate μPG concentrations. Interestingly, the three single-point mutants of the coiled coil region, namely SK-D328G, SK-K334E, and SK-G344D, exhibited a similar pattern of decline in the kcat as they exhibited with the full-length substrate HPG while not showing any apparent change in affinity for substrate μPG compared with wtSK (see Table 5). Therefore, the present results identify the broad locale to be the serine protease domain (or components therein, such as the scissile peptide bond region) in substrate HPG that might interact with the γ domain of SK as the γ domain mutants are catalytically impaired in a quantitatively similar pattern with both HPG as well as its truncated derivative μPG in relative terms with respect to wtSK.
TABLE 5.
Steady-state kinetic parameters for microplasminogen activation by equimolar complexes of SK/SK γ domain mutants and HPN
The kinetic parameters for substrate μPG activation were determined at 22 °C with the chromogenic substrate in 50 mm Tris-Cl buffer, pH 7.5, 100 mm NaCl as described under “Experimental Procedures.” The data represent the mean of three independent determinations.
| Activator species | Km | kcat | kcat/Km |
|---|---|---|---|
| μm | min−1 | min−1μm−1 | |
| wtSK | 1.2 ± 0.1 | 0.13 | 0.1 |
| SK-D328G | 1.2 ± 0.4 | 0.01 | 0.008 |
| SK-K334E | 1.4 ± 0.3 | 0.04 | 0.02 |
| SK-G344D | 1.2 ± 0.2 | 0.02 | 0.016 |
Real-time Surface Plasmon Resonance Studies for Studying the Ternary-mode Interactions of γ Domain Mutants with Full-length Substrate HPG
With a view to further substantiate the results of steady-state kinetics, that the mutations in the γ domain of SK do not alter the substrate affinity of the activator complex for substrate HPG (as opposed to strong effects on kcat), we employed the surface-plasmon resonance approach to characterize the affinity parameters of some of these mutants. Because various mutations studied in the γ domain of SK clearly suggested their interaction with substrate HPG, we undertook the surface plasmon resonance study with selected mutants, namely SK-D328G, SK-K334E, and SK-G344D, for determining the kinetic and equilibrium affinity parameters. Substrate HPG at varying concentrations was “docked” onto the preformed binary complex of immobilized HPN and wtSK/γ domain mutants on the sensor surface by procedures described earlier, and the respective rates were determined in real time (35, 40). The data, summarized in Table 6, suggest that the equilibrium dissociation constant (KD) values for the ternary interaction of substrate HPG and the three mutants, namely SK-D328G SK-K334E and SK-G344D, were closely similar to that observed in the case of ternary interaction of substrate HPG with wtSK (∼0.15 μm). It is noteworthy that despite the low catalytic activity of the single-point mutants of the γ domain, the examined association and dissociation rate constants of ternary interactions were found to be closely similar in all cases to those observed in case of the ternary interaction of substrate HPG with wtSK. These observations, which essentially support the results of the steady-state kinetics using activity measurements, clearly confirm the contention that discrete epitope in the γ domain of SK is important for endowing the activator complex with the ability to activate substrate HPG in terms of turnover but do not provide additional substrate-enzyme affinity during the catalytic step(s).
TABLE 6.
Association (kon) and dissociation (koff) rate constants and apparent equilibrium dissociation (KD) constants were measured by surface plasmon resonance for the interaction of substrate plasminogen with wtSK/SK γ domain mutants precomplexed with immobilized HPN
Kinetic constants for the interaction of substrate HPG with wtSK/γ domain mutants·HPN (binary complexes) were determined by global fitting to a 1:1 binding model using the BIAcore 3000 evaluation software as described under “Experimental Procedures.” A stable binary complex between wtSK/SK γ domain mutants and HPN immobilized on to the SA chip was first made, and the binding of varying concentrations of substrate HPG (0.05–1 μm) was then monitored in real time.
| Ligand | Ligate HPG |
||
|---|---|---|---|
| Kon | koff | Kd | |
| m−1s−1 × 105 | s−1 × 10−1 | m × 10−6 | |
| wtSK | 12 ± 1.5 | 1.8 ± 0.2 | 0.15 ± 0.03 |
| SK-D328G | 14 ± 2.0 | 1.6 ± 0.2 | 0.11 ± 0.01 |
| SK-K334E | 10 ± 2.0 | 1.5 ± 0.3 | 0.15 ± 0.02 |
| SK-G344D | 12 ± 3.0 | 1.7 ± 0.3 | 0.14 ± 0.01 |
DISCUSSION
The results described above strongly indicate the presence of a substrate-interacting region in the γ domain of SK. The region spanning approximately residues 314–347 in SK is a catalytically important locus that primarily contributes toward processing of substrate HPG by the SK·HPN complex. This has been clearly elucidated by results of kinetic and equilibrium binding studies. The present study also reconciles the apparent contradictions in the literature regarding this region of SK. Site-specific mutagenesis of the “coiled coil” region by Wu et al. (29) had first demonstrated its importance in virgin enzyme induction, and before the present study, the importance of the C-terminal domain of SK (residues 285–414) in the catalytic activation of HPG had been largely understood to be limited to its role in its interaction with partner plasmin(ogen) (28, 29). Application of sensitive kinetic and fluorescent approaches developed later (7, 19, 53), however, have enabled us to resolve much more clearly whether a given locus or mutation is important in 1:1 zymogen activation through the so-called Pathway I or specifically in the turnover of substrate HPG by the fully activated SK·plasmin complex (Pathway II) (see Fig. 1 in Aneja et al. (21)). As a result, in the present study the significance of the “upstream” section of the coiled-coil region of SK in substrate HPG activation has been unambiguously demonstrated. Interestingly, computer modeling and docking studies based on the available crystal coordinates for μPG, SK β domain, μPN·SAK·μPN complex, and the SK·μPN complex to obtain a ternary model of μPNpartner·SK·μPNsubstrate (17, 18, 54, 64) wherein the catalytic domain of the substrate was docked in silico into the SK·μPN enzyme complex (see “Experimental Procedures” and Fig. 4) essentially confirmed our mutagenesis and steady-state kinetics results, as the modeled structure also strongly suggested the same discrete regions (residues 314–346) in the γ domain as optimally positioned for activator enzyme-substrate μPG interactions. Similar to earlier models of the μPpartner·SK·μPNsubstrate ternary complex proposed (17, 65), the ternary complex model presented in this study was generated utilizing the PDB coordinates of the available crystal structures of the μPN·SK binary complex and μPNpartner·SAK·μPNsubstrate ternary complex. The current ternary model also envisions the distinct positioning of the activation loop of substrate μPN in the active site valley of the enzyme complex as reported by the previously modeled ternary complex of μPpartner·SK·μPNsubstrate (17, 65). The ternary modeling studies by Bode and coworkers (65) assign a distinct function to each of the domains of SK, where the γ domain of SK has been shown to participate in the binding activation of the complex with partner HPG. Taking into account the present understanding of μPpartner·SK·μPNsubstrate ternary complex as well as the results of our steady-state kinetic studies, there clearly seem to be two functionally distinct loci, one encompassing residues roughly 345–380 of the SK γ domain, which faces toward the partner μPN and might be involved in formation and/or activation of the 1:1 activator complex, and another encompassing the region from residues roughly 320–340, which seems to be interacting with substrate μPG in which, through mutagenesis, critical residues have been shown to be functionally important in enzyme-substrate interactions. From the thermal factors of the SK-μPN structure (17), it also appears that the peptide backbone bearing the constellation of charged residues in the coiled coil region of the γ domain of SK, especially Asp322, Arg324, Asp328, Asp325, Arg330, Asp331, Lys332, and Lys334 is highly flexible and can potentially participate in an extended network of salt bridges and hydrogen bonds within the coiled-coil region or with partner and/or substrate HPG. In particular, residues Asp328 and Lys334 of the coiled coil region seem to closely interact with residues of the methionine loop and the 186 loop of substrate μPG, whereas the side chain of neighboring Lys332, which is shown not to be critical in substrate HPG activation (data not shown) seems to face the partner μPN (see Fig. 4). A priori, a salt bridge between Arg330 of SK and Glu714 of the methionine loop of substrate μPG seems to provide a rigidifying anchor to facilitate optimal interaction with substrate. Previously, the importance of charged residues (lysines 256, 257, 332, and 334) in SK-HPG interactions has been demonstrated through substitution mutagenesis (33) even though at that time it was not clear that these residues actually constituted discrete epitopes for substrate-specific interactions. The present study, as also recent studies in the case of other exosites in SK (17–20), has convincingly shown that this region is indeed catalytically important through its interaction with the substrate catalytic domain and is not important in zymogen activation per se.
FIGURE 4.

Putative ternary interaction model of partner and substrate μPG interaction(s) with the SK γ domain. The ternary interaction model of μPN·SK·μPN complex generated by ZDOCK is shown. Close-up of interactions of SK γ domain (pink) is shown. Catalytically important residues (yellow; ball and stick forms) can be observed to be interacting with substrate μPN (blue), particularly residues of the methionine loop and 186 loop (red; ball and stick), and the region of SK γ domain is visualized to be interacting with partner μPN (shown in green). Molecular modeling was performed as described in “Experimental Procedures.”
Based upon the observations reported in this study and our earlier findings (20, 21, 35, 41), it can be envisioned that the substrate HPG interaction sites are not centered at one or two epitopes in SK, but rather, these seem to be strategically “scattered” over all the three domains of SK. Site-specific mutational studies in SK (16, 22, 37, 38, 40) have also broadly defined the roles for all the three domains of SK in both activator complex formation as also in substrate HPG activation. Thus, the present work along with previously gleaned understanding on SK exosites opens a new and interesting paradigm for the catalytic mechanism of action of this medically important co-factor protein (20, 21, 35, 41). Earlier, the importance of exosites, especially in providing substrate specificity via facilitated docking of the latter, is well demonstrated in blood coagulation proteinases, especially prothrombinases (66–68). In the case of SK too, it seems reasonable to assume that the different exosites in SK probably modify the substrate specificity by providing additional docking sites for an enhanced presentation of the scissile peptide bond in substrate HPG onto the enzyme active site through their precise interactions onto cognate “receptor” sites in the catalytic domain of substrate HPG. However, it appears that the exosite in the γ domain discussed in the present study does not alter at least the primary covalent specificity (particularly in terms of the S site) of the HPN active site as indicated by the results of the amidolytic assays (Table 3) using two different chromogenic peptidic substrates. Thus, even though the results suggest but do not unambiguously prove (as the S′ site specificity was not probed) that the primary active site of HPN is probably unaltered in the γ domain mutant complexes with HPN, what is clearly established by our results is that the exosite in the γ domain does constitute an important component in the surface-located epitopes on SK that mediate the interaction of the SK-HPN complex with its macromolecular substrate. Unpublished results from our laboratory indeed suggest that all these exosites very likely modulate the release of the product after scission of the Arg-560—Val-561 peptide bond.4 Studies in our laboratory are currently aimed at exploring the cooperativity, if any, that exists among the various exosites of SK. It is expected that such an understanding would greatly aid the future de novo design of more efficacious thrombolytic molecules.
Acknowledgments
We thank Paramjit Kaur, Deepak Bhatt, and Sharanjot Kaur for expert technical support and Dr. Subita Basu of the Blood Bank, Government Medical College, Chandigarh for human plasma.
This study was largely supported with intramural funds from the Council of Scientific and Industrial Research, India.
S. Kumar and G. Sahni, unpublished observations.
- SK
- streptokinase
- ATA-FPR-CK
- Nα-[(acetylthio)acetyl]-d-Phe-Pro-Arg-CH2Cl
- μPG
- microplasminogen
- μPN
- microplasmin
- HPG
- human plasminogen
- HPN
- human plasmin
- kcat
- rate of catalysis at substrate saturation
- NPGB
- p-nitrophenyl p-guanidinobenzoate
- pNA
- 4-nitroanilide
- SAK
- staphylokinase.
REFERENCES
- 1. Jackson K. W., Tang J. (1982) Biochemistry 21, 6620–6625 [DOI] [PubMed] [Google Scholar]
- 2. Huang T. T., Malke H., Ferretti J. J. (1989) Mol. Microbiol. 3, 197–205 [DOI] [PubMed] [Google Scholar]
- 3. Radek J. T., Castellino F. J. (1989) J. Biol. Chem. 264, 9915–9922 [PubMed] [Google Scholar]
- 4. Damaschun G., Damaschun H., Gast K., Gerlach D., Misselwitz R., Welfle H., Zirwer D. (1992) Eur. Biophys. J. 20, 355–361 [DOI] [PubMed] [Google Scholar]
- 5. McClintock D. K., Bell P. H. (1971) Biochem. Biophys. Res. Commun. 43, 694–702 [DOI] [PubMed] [Google Scholar]
- 6. Bajaj A. P., Castellino F. J. (1977) J. Biol. Chem. 252, 492–498 [PubMed] [Google Scholar]
- 7. Wang S., Reed G. L., Hedstrom L. (1999) Biochemistry 38, 5232–5240 [DOI] [PubMed] [Google Scholar]
- 8. Wang S., Reed G. L., Hedstrom L. (2000) Eur. J. Biochem. 267, 3994–4001 [DOI] [PubMed] [Google Scholar]
- 9. Boxrud P. D., Verhamme I. M., Fay W. P., Bock P. E. (2001) J. Biol. Chem. 276, 26084–26089 [DOI] [PubMed] [Google Scholar]
- 10. Boxrud P. D., Verhamme I. M., Bock P. E. (2004) J. Biol. Chem. 279, 36633–36641 [DOI] [PubMed] [Google Scholar]
- 11. Boxrud P. D., Bock P. E. (2004) J. Biol. Chem. 279, 36642–36649 [DOI] [PubMed] [Google Scholar]
- 12. Reddy K. N., Markus G. (1972) J. Biol. Chem. 247, 1683–1691 [PubMed] [Google Scholar]
- 13. Schick L. A., Castellino F. J. (1973) Biochemistry 12, 4315–4321 [DOI] [PubMed] [Google Scholar]
- 14. Robbins K. C., Summaria L., Wohl R. C., Bell W. R. (1983) Thromb. Haemost. 50, 787–791 [PubMed] [Google Scholar]
- 15. Summaria L., Wohl R. C., Boreisha I. G., Robbins K. C. (1982) Biochemistry 21, 2056–2059 [DOI] [PubMed] [Google Scholar]
- 16. Nihalani D., Raghava G. P., Sahni G. (1997) Protein Sci. 6, 1284–1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang X., Lin X., Loy J. A., Tang J., Zhang X. C. (1998) Science 281, 1662–1665 [DOI] [PubMed] [Google Scholar]
- 18. Wang X., Tang J., Hunter B., Zhang X. C. (1999) FEBS Lett. 459, 85–89 [DOI] [PubMed] [Google Scholar]
- 19. Boxrud P. D., Fay W. P., Bock P. E. (2000) J. Biol. Chem. 275, 14579–14589 [DOI] [PubMed] [Google Scholar]
- 20. Yadav S., Datt M., Singh B., Sahni G. (2008) Biochim. Biophys. Acta 1784, 1310–1318 [DOI] [PubMed] [Google Scholar]
- 21. Aneja R., Datt M., Singh B., Kumar S., Sahni G. (2009) J. Biol. Chem. 284, 32642–32650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nihalani D., Sahni G. (1995) Biochem. Biophys. Res. Commun. 217, 1245–1254 [DOI] [PubMed] [Google Scholar]
- 23. Parrado J., Conejero-Lara F., Smith R. A., Marshall J. M., Ponting C. P., Dobson C. M. (1996) Protein Sci. 5, 693–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fay W. P., Bokka L. V. (1998) Thromb. Haemost. 79, 985–991 [PubMed] [Google Scholar]
- 25. Reed G. L., Houng A. K., Liu L., Parhami-Seren B., Matsueda L. H., Wang S., Hedstrom L. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 8879–8883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wakeham N., Terzyan S., Zhai P., Loy J. A., Tang J., Zhang X. C. (2002) Protein Eng. 15, 753–761 [DOI] [PubMed] [Google Scholar]
- 27. Mundada L. V., Prorok M., DeFord M. E., Figuera M., Castellino F. J., Fay W. P. (2003) J. Biol. Chem. 278, 24421–24427 [DOI] [PubMed] [Google Scholar]
- 28. Young K. C., Shi G. Y., Chang Y. F., Chang B. I., Chang L. C., Lai M. D., Chuang W. J., Wu H. L. (1995) J. Biol. Chem. 270, 29601–29606 [DOI] [PubMed] [Google Scholar]
- 29. Wu D. H., Shi G. Y., Chuang W. J., Hsu J. M., Young K. C., Chang C. W., Wu H. L. (2001) J. Biol. Chem. 276, 15025–15033 [DOI] [PubMed] [Google Scholar]
- 30. Sazonova I. Y., Houng A. K., Chowdhry S. A., Robinson B. R., Hedstrom L., Reed G. L. (2001) J. Biol. Chem. 276, 12609–12613 [DOI] [PubMed] [Google Scholar]
- 31. Collen D., Schlott B., Engelborghs Y., Van Hoef B., Hartmann M., Lijnen H. R., Behnke D. (1993) J. Biol. Chem. 268, 8284–8289 [PubMed] [Google Scholar]
- 32. Collen D., Van Hoef B., Schlott B., Hartmann M., Gührs K. H., Lijnen H. R. (1993) Eur. J. Biochem. 216, 307–314 [DOI] [PubMed] [Google Scholar]
- 33. Lin L. F., Oeun S., Houng A., Reed G. L. (1996) Biochemistry 35, 16879–16885 [DOI] [PubMed] [Google Scholar]
- 34. Nihalani D., Kumar R., Rajagopal K., Sahni G. (1998) Protein Sci. 7, 637–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dhar J., Pande A. H., Sundram V., Nanda J. S., Mande S. C., Sahni G. (2002) J. Biol. Chem. 277, 13257–13267 [DOI] [PubMed] [Google Scholar]
- 36. Dahiya M., Rajamohan G., Dikshit K. L. (2005) FEBS Lett. 579, 1565–1572 [DOI] [PubMed] [Google Scholar]
- 37. Reed G. L., Lin L. F., Parhami-Seren B., Kussie P. (1995) Biochemistry 34, 10266–10271 [DOI] [PubMed] [Google Scholar]
- 38. Rodríguez P., Fuentes P., Barro M., Alvarez J. G., Muñoz E., Collen D., Lijnen H. R. (1995) Eur. J. Biochem. 229, 83–90 [DOI] [PubMed] [Google Scholar]
- 39. Kim D. M., Lee S. J., Yoon S. K., Byun S. M. (2002) Biochem. Biophys. Res. Commun. 290, 585–588 [DOI] [PubMed] [Google Scholar]
- 40. Sundram V., Nanda J. S., Rajagopal K., Dhar J., Chaudhary A., Sahni G. (2003) J. Biol. Chem. 278, 30569–30577 [DOI] [PubMed] [Google Scholar]
- 41. Tharp A. C., Laha M., Panizzi P., Thompson M. W., Fuentes-Prior P., Bock P. E. (2009) J. Biol. Chem. 284, 19511–19521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Deutsch D. G., Mertz E. T. (1970) Science 170, 1095–1096 [DOI] [PubMed] [Google Scholar]
- 43. Chaudhary A., Vasudha S., Rajagopal K., Komath S. S., Garg N., Yadav M., Mande S. C., Sahni G. (1999) Protein Sci. 8, 2791–2805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Malke H., Gerlach D., Köhler W., Ferretti J. J. (1984) Mol. Gen. Genet 196, 360–363 [DOI] [PubMed] [Google Scholar]
- 45. Porath J., Carlsson J., Olsson I., Belfrage G. (1975) Nature 258, 598–599 [DOI] [PubMed] [Google Scholar]
- 46. Bock P. E. (1992) J. Biol. Chem. 267, 14974–14981 [PubMed] [Google Scholar]
- 47. Bock P. E. (1992) J. Biol. Chem. 267, 14963–14973 [PubMed] [Google Scholar]
- 48. Bock P. E. (1993) Methods Enzymol. 222, 478–503 [DOI] [PubMed] [Google Scholar]
- 49. Bock P. E. (1988) Biochemistry 27, 6633–6639 [DOI] [PubMed] [Google Scholar]
- 50. Wohl R. C., Summaria L., Robbins K. C. (1980) J. Biol. Chem. 255, 2005–2013 [PubMed] [Google Scholar]
- 51. Chase T., Jr., Shaw E. (1969) Biochemistry 8, 2212–2224 [DOI] [PubMed] [Google Scholar]
- 52. Wohl R. C. (1984) Biochemistry 23, 3799–3804 [DOI] [PubMed] [Google Scholar]
- 53. Radek J. T., Davidson D. J., Castellino F. J. (1993) Methods Enzymol. 223, 145–155 [DOI] [PubMed] [Google Scholar]
- 54. Parry M. A., Fernandez-Catalan C., Bergner A., Huber R., Hopfner K. P., Schlott B., Gührs K. H., Bode W. (1998) Nat. Struct. Biol. 5, 917–923 [DOI] [PubMed] [Google Scholar]
- 55. Chen R., Li L., Weng Z. (2003) Proteins 52, 80–87 [DOI] [PubMed] [Google Scholar]
- 56. Dawson K. M., Marshall J. M., Raper R. H., Gilbert R. J., Ponting C. P. (1994) Biochemistry 33, 12042–12047 [DOI] [PubMed] [Google Scholar]
- 57. Jespers L., Van Herzeele N., Lijnen H. R., Van Hoef B., De Maeyer M., Collen D., Lasters I. (1998) Biochemistry 37, 6380–6386 [DOI] [PubMed] [Google Scholar]
- 58. Malke H., Ferretti J. J. (1984) Proc. Natl. Acad. Sci. U.S.A. 81, 3557–3561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Pratap J., Kaur J., RajaMohan G., Singh D., Dikshit K. L. (1996) Biochem. Biophys. Res. Commun. 227, 303–310 [DOI] [PubMed] [Google Scholar]
- 60. De Renzo E. C., Siiteri P. K., Hutchings B. L., Bell P. H. (1967) J. Biol. Chem. 242, 533–542 [PubMed] [Google Scholar]
- 61. Boxrud P. D., Bock P. E. (2000) Biochemistry 39, 13974–13981 [DOI] [PubMed] [Google Scholar]
- 62. Bock P. E., Day D. E., Verhamme I. M., Bernardo M. M., Olson S. T., Shore J. D. (1996) J. Biol. Chem. 271, 1072–1080 [DOI] [PubMed] [Google Scholar]
- 63. Robbins K. C., Summaria L., Wohl R. C. (1981) Methods Enzymol. 80, 379–387 [DOI] [PubMed] [Google Scholar]
- 64. Wang X., Terzyan S., Tang J., Loy J. A., Lin X., Zhang X. C. (2000) J. Mol. Biol. 295, 903–914 [DOI] [PubMed] [Google Scholar]
- 65. Parry M. A., Zhang X. C., Bode I. (2000) Trends Biochem. Sci. 25, 53–59 [DOI] [PubMed] [Google Scholar]
- 66. Bock P. E., Panizzi P., Verhamme I. M. (2007) J. Thromb. Haemost. 5, 81–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Boskovic D. S., Krishnaswamy S. (2000) J. Biol. Chem. 275, 38561–38570 [DOI] [PubMed] [Google Scholar]
- 68. Huntington J. A. (2005) J. Thromb. Haemost. 3, 1861–1872 [DOI] [PubMed] [Google Scholar]