

Abstract
A characteristic of malignant cells is their capacity to invade their surrounding and to metastasize to distant organs. During these processes, proteolytic activities of tumor and stromal cells modify the extracellular matrix to produce a microenvironment suitable for their growth and migration. In recent years the family of ADAM proteases has been ascribed important roles in these processes. ADAM-9 is expressed in human melanoma at the tumor-stroma border where direct or indirect interactions between tumor cells and fibroblasts occur. To analyze the role of ADAM-9 for the interaction between melanoma cells and stromal fibroblasts, we produced the recombinant disintegrin-like and cysteine-rich domain of ADAM-9 (DC-9). Melanoma cells and human fibroblasts adhered to immobilized DC-9 in a Mn2+-dependent fashion suggesting an integrin-mediated process. Inhibition studies showed that adhesion of fibroblasts was mediated by several β1 integrin receptors independent of the RGD and ECD recognition motif. Furthermore, interaction of fibroblasts and high invasive melanoma cells with soluble recombinant DC-9 resulted in enhanced expression of MMP-1 and MMP-2. Silencing of ADAM-9 in melanoma cells significantly reduced cell adhesion to fibroblasts. Ablation of ADAM-9 in fibroblasts almost completely abolished these cellular interactions and melanoma cell invasion in vitro. In summary, these results suggest that ADAM-9 expression plays an important role in mediating cell-cell contacts between fibroblasts and melanoma cells and that these interactions contribute to proteolytic activities required during invasion of melanoma cells.
Keywords: ADAM ADAMTS, Cell Adhesion, Cell Surface Receptor, Cell-Cell Interaction, Fibroblast, Melanoma Cells
Introduction
Growth and progression of human melanoma is a process that requires a cascade of different cellular processes including cellular interactions and proteolytic cleavage of growth factors and extracellular matrix proteins. ADAMs (a disintegrin and metalloproteinase)2 with both their proteolytic and adhesive functions play a pivotal role in these processes (1). Most of the members of the ADAM family share a general phylogenetically well-conserved domain structure including a pro-, metalloprotease-, disintegrin-like-, cysteine rich-, EGF-like-, and cytoplasmic domain (reviewed by Refs. 2, 3). Apart from their proteolytic activity, ADAMs exert also adhesive functions, which are mediated for example in ADAM-15 by the RGD motif in the disintegrin-like domain (4). For other ADAMs, as ADAM-2 and ADAM-9, an electron capture detection motif (ECD) also located within the disintegrin-like domain has been reported to be responsible for this event (5, 6). In vitro studies have shown that ADAM-15 interacts with ανβ3 and α5β1 integrins (7), whereas ADAM-2 binds to α6β1 integrin and ADAM-9 to α6β1, ανβ5 and α3β1 integrins (6, 8, 9). The interactions of ADAM proteases with cellular receptors have been proven to be of major importance in cell adhesion and fusion processes as for instance during spermatogenesis (ADAM-1, -16, -20) and myo- and osteogenesis (ADAM-9, -12, -19) (1). We have recently shown that ADAM-9 is expressed in both human and mouse epidermis. In keratinocytes, the adhesive activity of ADAM-9 leads to modulation of MMP-9 expression and cell migration (10). Further, proteolysis via ADAM-9 in keratinocytes has been shown to be important for the constitutive shedding of collagen XVII (11). Altered collagen XVII shedding was also detected in skin of ADAM-9-deficient mice, resulting in increased keratinocyte migration and accelerated skin repair (11, 12).
Altered expression of certain ADAMs has been associated with a number of diseases including asthma, arthritis, atherosclerosis, and cancer (13). However, relatively sparse information is available on the functional role of ADAMs in malignancy. Expression of ADAM-9, -10, -12 is increased in breast cancer (13). ADAM-9 is also expressed in human melanoma where it is localized on melanoma cells and peritumoral stromal fibroblasts while this protein was not found in fibroblasts distant from the tumor site (14). In prostate cancer ADAM-9 plays a tumor promoting role which was attributed to its ability to cleave EGF and FGFR2IIIb thereby altering signaling (15). Furthermore, a soluble form of ADAM-9 is produced by activated stellate cells that bind via α6β4 integrin to tumor cells at the border of liver metastasis and promote in vitro invasion (16). Recently, ADAM-9 has also been implicated in pathological neovascularization likely by modulating expression of EphB4, Tie-2, Flk-1, CD40, VCAM, and VE-cadherin, which all have been identified as substrates of ADAM-9 (17).
Many studies over the last years were focused on the role of the proteolytic domain of ADAM-9. By contrast, less is known about the function of its adhesive domain for melanoma cell interactions. To elucidate the role of ADAM-9 as an adhesive receptor for human skin fibroblasts and melanoma cells, the recombinant disintegrin-like and cysteine-rich domains (DC-9) (10) were used as substrate for cell attachment experiments. Here we show that both fibroblasts and melanoma cells adhere to the disintegrin-like and cysteine-rich domains of ADAM-9. This interaction is mediated by β1-containing integrin receptors and leads to augmentation of proteolytic activities. More importantly, we provide evidence that ADAM-9 is directly involved in melanoma cell-fibroblast interactions which might affect the invasive behavior of melanoma cells.
EXPERIMENTAL PROCEDURES
Antibodies
For Western blot analysis goat polyclonal antibodies raised against human ADAM-9 were purchased from R&D Systems (Wiesbaden, Germany). Actin was detected with mouse monoclonal antibodies (MP Biomedicals, Irvine, CA). The blocking monoclonal mouse antibody 4B4 directed against the human β1-integrin chain was obtained from Coulter Corp. (Hialeah, FL); the antibodies against the human β3- and the α-integrin subunits from Chemicon (Beta1 Integrin Partners Kit; Hofheim, Germany). Antibodies directed against MMP-1 were a kind gift from P. Angel (German Cancer Research Center, Heidelberg).
Cells and Cell Culture
Primary human and murine dermal fibroblasts were obtained by outgrowth from skin explants as previously described (18). Cells were cultured in Dulbecco's modified medium (DMEM) supplemented with 10% FCS, 2 mm glutamine, 100 units/ml of penicillin, and 100 μg/ml streptomycin in 5% CO2 at 37 °C in a humidified atmosphere. Fibroblasts were passaged by trypsinization at a ratio of 1:2 every 5 days and used at passages 1–10. The human melanoma cell lines (19) were cultured in RPMI 1640 medium supplemented with 10% FCS, 2 mm glutamine, non-essential amino acids, 100 units/ml of penicillin, and 100 μg/ml streptomycin. MV3 cells were transiently transfected with double-stranded RNA encoding ADAM-9 sequences (20) using Lipofectamine according to manufacturer's instructions (Invitrogen, Darmstadt, Germany). After 6 h, medium was replaced and ADAM-9 expression analyzed after further 24 and 48 h of culture.
Cell Adhesion Assays
Semi-confluent mono-layer cultures of human primary fibroblasts or melanoma cells were detached with 0.05% EDTA. The cells were resuspended in HEPES buffer (1 mm Hepes, 0.5% BSA, 1 mm of each CaCl2 and MnCl2). Adhesion assays were performed as described before (10). Briefly, 96-well tissue culture plates were coated with recombinant DC-9 and His6 peptide (0.6 μm) at 4 °C overnight. Expression and purification of the disintegrin-cysteine rich domain of ADAM-9 has been described elsewhere (10). BSA coating and blocking of nonspecific binding sites were performed with heat-denatured BSA (1% BSA in Ca2+/Mg2+-free PBS) for 1 h at room temperature. After washing the wells, 2 × 104 cells/well were seeded and incubated for 1 h at 37 °C. For competition assays, antibodies (10 μg/ml) or peptides (0.6 μm) were added to the cell suspension before plating. Non-adherent cells were removed by washing with PBS. Adherent cells were fixed with 3% paraformaldehyde in PBS, pH 7.6, and stained with 0.5% crystal violet in 20% (v/v) methanol. The dye was released from the cells by addition of 0.1 m sodium citrate in 50% (v/v) ethanol. The absorbance (A) of the released dye solution was determined at 595 nm. Adhesion was either expressed directly as A595 nm or as percentage relative to untreated controls, which were set arbitrarily as 100%. Statistical analysis was performed with the ANOVA Dunnett's multiple comparison test or Student's t test.
Cell-Cell Adhesion Assays
Analysis of melanoma cell adhesion to fibroblasts was performed as described previously (21). Briefly, melanoma cells were labeled with 0.5 μm CellTrackerTM CMFDA (5-chloromethyl-fluorescein diacetate; Molecular Probes, Invitrogen) for 30 min at 37 °C. After extensive washes, 2 × 104 cells/well were seeded onto confluent monolayers of fibroblasts in 96-well plates for 30 min. After incubation, not adhered cells were removed and attached cells fixed with 4% paraformaldehyde for 10 min at room temperature. Bound cells were counted in 5 microscopic fields in three separate wells and averaged. Alternatively, cells were resuspended and their fluorescence measured in a Victor Multilabel Reader (Perkin Elmer).
In Vitro Invasion Assays
Melanoma cell invasion was assayed using transwell with polycarbonate filters (8-μm pore size, Corning Costar, Bodenheim, Germany). The filters were coated with 25 μl of Matrigel (Becton Dickinson, Heidelberg, Germany) and placed above the lower compartment, which contained either 200 μl of serum-free RPMI 1640 culture medium or fibroblast-conditioned medium. GFP-expressing B16F1 melanoma cells (22) mixed 2:1 with primary wild type or ADAM-9 knock-out fibroblasts were suspended in serum-free RPMI 1640 (1 × 105/ml) and seeded into the upper compartment of the chamber. The chambers were incubated at 37 °C for 24 to 48 h. Cells attached to the upper side of the filter were mechanically removed. The filters were fixed with 4% paraformaldehyde for 5 min and nuclei stained with 1 μg/ml DAPI. Invasion was determined by counting the GFP-positive cells that had migrated to the lower surface of the filter. For each filter, the number of cells in 5 randomly chosen microscope fields was determined and averaged. The experiment was performed twice each in quadruplicates.
RNA Isolation and RT-PCR
Total RNA was prepared using RNAzol (Wak-Chemie Medical GmbH, Bad Homburg, Germany). For RT-PCR (REDTaqTM ReadyMixTM PCR Reaction Mix, Sigma, Taufkirchen, Germany) 1 μg of RNA was reverse transcribed using oligo dT. The following primers were used for amplification of human ADAM-9: 5′-CCTCGGGGACCCTTCGTGT and 5′-ATCCCATAACTCGCATTCTCTAAA (10). Amplification of S26 was used for normalization (18). PCR reactions were performed for 32 cycles (within the linear range of amplification): denaturation (94 °C, 1 min), annealing (60 °C, 1 min) and extension (72 °C, 1 min). The products were analyzed on 2% agarose gels.
Zymographic Analysis
Serum-free conditioned media were analyzed by gelatin zymography as previously described (18). Briefly, 15–20 μl of supernatants (corresponding to 10 μg of cell lysate) were fractionated on 10% SDS-polyacrylamide gels containing 1 mg/ml gelatin (bovine, Sigma). Gels were washed in 2.5% Triton X-100 for 30 min before overnight incubation in metalloproteinase substrate buffer (50 mm Tris-HCl, pH 8.0, 5 mm CaCl2) and stained with Coomassie Blue R 250. Bands corresponding to gelatinase activities appeared white against a blue background.
Western Blot Analysis
Lysates were prepared by directly scraping off the cells on ice in RIPA buffer (50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1% Nonidet® P40, 0,5% Natriumdesoxycholat, 0,1% SDS) containing aprotinin (10 μg/ml), Pefabloc (0.25 mg/ml), and leupeptin (1 μg/ml). Protein concentration was determined using a commercial assay (MicroBCA, Perbio Science, Bonn, Germany). For detection of MMP-1 supernatants corresponding to 10 μg of cell lysates were fractionated on 10% SDS-polyacrylamide gels under reducing conditions and transferred onto Hybond-C SuperTM (GE Healthcare, München, Germany). The blots were incubated with the primary antibodies overnight at 4 °C. Bound primary antibodies were detected using a horseradish peroxidase-conjugated secondary antibody (1:2000; Dako, Hamburg, Germany) and visualized using ECL system (GE Healthcare).
RESULTS
Human Skin Fibroblasts Adhere To and Signal via the Disintegrin-like and Cysteine-rich Domains of ADAM-9
To analyze the role of ADAM-9 as an adhesive substrate for human fibroblasts we performed cell adhesion assays using the recombinant disintegrin-like and cysteine-rich domains of ADAM-9 (DC-9) as immobilized substrate (Fig. 1A). Adhesion of fibroblasts was maximal at a coating concentration of 50 μg/ml reaching about 60% of the adhesion level determined with serum, which was used as a positive control substrate. At all coating concentrations of DC-9 fibroblasts showed a spread morphology demonstrating an active process of substrate recognition and signaling. Furthermore, comparable adhesion to DC-9 was detected using human skin fibroblasts isolated from 8 individuals of different sex and age at passages between 4 and 7. This indicates that adhesion of fibroblasts to DC-9 is not dependent on the cell source (data not shown).
FIGURE 1.

Fibroblasts adhere to the adhesive domains of ADAM-9. A, microtiter plates were coated with increasing concentrations of recombinant His-tagged DC-9 protein as indicated. Coating with BSA (1% heat inactivated) or FCS were used as negative and positive control, respectively. Microphotographs were taken after fixation of the cells (scale bar 10 μm). B and C, microtiter plates were coated with His-tagged DC-9 protein (10 μg/ml) or his peptide (0.6 μm). Fibroblasts were incubated in assay buffer in the presence of 10 μg/ml inhibitory antibodies as indicated. IgG isotype control or 0.6 μm of each RGD, RAD (control), ECD or scrambled-ECD (scr.; control) peptide was added before plating. Adhesion is expressed as percentage of adhesion determined on FCS, which was arbitrarily set as 100%. In B, representative results of four independent experiments, each performed in triplicates; in C representative results of three independent experiments, each performed in triplicates.
Adhesion of human fibroblasts to recombinant DC-9 was dependent on the presence of Mn2+ ions, which is in line with an integrin-dependent adhesion process (data not shown). Human fibroblasts express several integrin receptors in vivo and in vitro and these include integrins of the β1 and β3 subgroups (23). To identify the integrin subunit mediating cell adhesion to DC-9, we incubated fibroblasts in the presence of different inhibitory anti-integrin antibodies before performing adhesion assays. Antibodies directed against the β1 but not the β3 integrin subunit efficiently inhibited adhesion (Fig. 1B). Furthermore, antibodies against the α3, α5, and α6 integrin chains reduced adhesion by 50, 30, and 60%, respectively, whereas inhibitory antibodies directed against the α1, α2, and α4 subunits had no significant effect when compared with the antibody-untreated control.
This demonstrates that multiple β1-containing integrin receptors contribute to the adhesion of fibroblasts to DC-9. Interestingly, the ECD cyclic peptide did not affect adhesion to immobilized DC-9. This indicates that the ECD motif located in the disintegrin-like domain of ADAM-9 does not contribute to ADAM-9 mediated adhesion (Fig. 1B). The RGD sequence, albeit not present in ADAM-9, was used to exclude that binding through closely associated RGD dependent receptors e.g. integrins α5β1 and αvβ3, which are expressed by fibroblasts, might interfere with cell adhesion to DC-9. This peptide as well as the control peptides containing the RAD and ECD scrambled motifs had no effect on the adhesion of fibroblasts to DC-9. In addition, adhesion of fibroblasts to DC-9 either applied as immobilized or soluble substrate, induced synthesis of MMP-1 and MMP-2 as well as proMMP-2 activation (Fig. 2).
FIGURE 2.
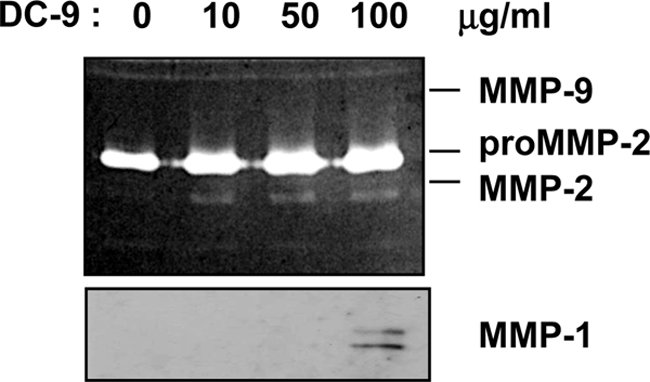
Adhesion of fibroblasts to DC-9 alters MMP-1 and -2 expression. Primary human fibroblasts were starved for 24 h in serum-free medium before incubation with DC-9 protein (10, 50, 100 μg/ml). After stimulation, supernatants were collected and analyzed by gelatin zymography. The 72 kDa inactive form and the 62/59 kDa active forms of MMP-2 are shown in the zymogram (upper panel). MMP-1 in supernatants was detected by Western blotting using rabbit-anti-MMP-1 antibodies. The antibodies detect two bands of 57 and 52 kDa.
Recombinant DC-9 Is an Adhesive and Signaling Molecule for Melanoma Cells
Melanoma cells in vivo and in vitro express ADAM-9 (14). Using melanoma cells of various invasive abilities we analyzed whether melanoma cells bind to the adhesive domains of ADAM-9 and whether the differences in adhesion correlate with their invasive potential. All melanoma cell lines adhered to DC-9 (Fig. 3). Adhesion to DC-9 was high for the low invasive cell lines IF6, Skmel 28, and WM 164, reaching at the coating concentration of 100 μg/ml almost the same adhesion level as determined on serum, which was used as positive control substrate. Interestingly, adhesion of all high invasive melanoma cell lines BLM, MV3, and VMM5 to DC-9 was significantly lower at all coating concentrations. MV3 cells showed the highest adhesion level at a coating concentration of 10 μg/ml. However, adhesion was significantly lower at the same substrate concentration when compared with low invasive Skmel 28 and WM164 cells. MMP-2 expression was slightly increased in the high invasive MV3 and BLM cells by treatment with soluble DC-9 as compared with low invasive melanoma cells (Fig. 3, right panel). In summary, these results indicate that adhesion to DC-9 and alterations of MMP-2 expression/activation do not directly correlate with the invasive capacity of the melanoma cells.
FIGURE 3.

Melanoma cells of different invasive potential adhere to DC-9. Melanoma cells were seeded on microtiter plates coated with 10 and 100 μg/ml of DC-9 protein for 1 h. Coating with BSA (1% heat-inactivated) and FCS were used as negative and positive controls, respectively. Adhesion is expressed as percentage of adhesion to FCS, which was set arbitrarily as 100%. For gelatin zymography melanoma cells were starved for 24 h in serum-free medium before stimulation with DC-9 protein (10, 50, 100 μg/ml). Then the supernatants were collected and analyzed by gelatin zymography. The 72 kDa inactive form and the 62/59 kDa active forms of MMP-2 are indicated in the zymogram. Significance of MV3 adhesion as compared with SKmel23 and WM164, *, p < 0,05.
ADAM-9 Mediates Fibroblast-Melanoma Cell Interactions
Adhesion of ADAMs to integrin receptors has been shown to result in cell-cell interactions as in the case of ADAM-15 (24) and ADAM-9. For ADAM-9 this leads to cellular fusion of monocytes and macrophages (25, 26). To determine whether ADAM-9 may function as a cell surface receptor mediating interactions of melanoma cells and fibroblasts, MV3 cells were labeled with the fluorochrome CMFDA and seeded on top of confluent fibroblast monolayers in the absence or presence of recombinant DC-9 (Fig. 4). While MV3 cells attached to fibroblasts in the absence of DC-9, this interaction was significantly reduced when DC-9 was added to the incubation buffer. Increasing concentrations of DC-9 up to 50 μg/ml had no further effect.
FIGURE 4.

The DC domain of ADAM-9 antagonise fibroblast-melanoma cell interaction. MV3 melanoma cells, labeled with the fluorochrome CMFDA, were preincubated in suspension with either 0.6 μm his peptide (Co) or 10 and 50 μg/ml of recombinant, His-tagged DC-9 protein before seeding on human primary fibroblasts. After 1 h, attached cells were counted in 5 microscopic fields of 3 separate wells. The bars represent the mean ± S.D. of cells attached per field. **, p < 0,006.
As fibroblasts as well as melanoma cells express ADAM-9 and thus both cell types may be involved in mediating cellular interactions, we used in a second approach murine dermal fibroblasts isolated from ADAM-9-deficient mice. In addition, to reduce expression of ADAM-9 also in melanoma cells we applied siRNA targeting sequences shown previously to be effective for silencing (27). Transient silencing of ADAM-9 for 48 h in MV3 melanoma cells resulted in an almost complete reduction of ADAM-9 mRNA. However, ADAM-9 protein was reduced by only about 50% (Fig. 5A), which might result from a slow turnover of the protein. Using these MV3 cells in cell-cell adhesion assays with murine wild-type fibroblasts, the number of melanoma cells attaching to fibroblasts was significantly lower (Fig. 5B) when compared with melanoma cells with normal ADAM-9 expression (+). In addition, cell-cell interaction between MV3 cells with reduced ADAM-9 expression (low) and ADAM-9 knock-out fibroblasts (Fb-) was further decreased when compared with the corresponding controls. The difference in attachment of MV3 cells with reduced ADAM-9 expression to wild-type fibroblasts compared with those attached to ADAM-9 knock-out fibroblasts was highly significant (***, p < 0.0004). Comparable results were obtained with B16F1 murine melanoma cells which express ADAM-9 (data not shown). The attachment to ADAM-9 knock-out fibroblasts was about 50% lower than the attachment to wild type fibroblasts (Fig. 5C).
FIGURE 5.

Silencing of ADAM-9 in MV3 melanoma cells results in reduced melanoma-fibroblast interaction. A, MV3 cells were transiently transfected with ADAM-9 siRNA (met) or control siRNA (scr). After 48 h, ADAM-9 mRNA levels were determined by RT-PCR. S26 levels were used for normalization. Protein expression was analyzed in cell lysates by Western blotting, and detection of actin was used as loading control. B, 48 h after transfection of MV3 cells (Mel) with ADAM-9 (low) or control siRNA (+), the MV3 cells were labeled with CMFDA and seeded on confluent fibroblast monolayers (Fb) derived from either wild type (+) or ADAM-9-deficient (−) mice. After 20 min, bound cells were quantified by fluorimetric analysis. Bars represent mean ± S.D. of the relative fluorescence units of three independent experiments. ****, p < 0.0001; ***, p < 0.0004. C, B16F1 murine melanoma cells were labeled with CMFDA and seeded on confluent fibroblast monolayers derived from either wild type (WT-Fb) or ADAM-9-deficient (KO-Fb) mice. Numbers of bound cells were quantified after 20 min. *, p < 0,006. D, invasion of GFP-transfected B16F1 melanoma cells was determined in transwell cultures with wild type (WT) or ADAM-9-deficient (KO) fibroblasts. Melanoma cells (mel; 5 × 105/ml) suspended in a 2:1 ratio with fibroblasts in 0.5-ml serum-free RPMI medium were filled in the upper compartment, while fibroblast-conditioned medium was used as a chemoattractant in the lower compartment. After 24 and 48 h, the numbers of GFP-positive melanoma cells that had migrated through the matrigel to the lower side of the filter were counted. Bars represent the mean ± S.D. of the cell numbers determined per filter in three independent experiments. ***, p < 0,0001.
To analyze whether ADAM-9-mediated cell-cell interactions affect the invasive behavior of melanoma cells, B16F1 melanoma cells expressing GFP were seeded in a 2:1 ratio with wild-type or ADAM-9-deficient fibroblasts on matrigel-coated transwells. The melanoma cells which have migrated through the matrigel were counted after 24 and 48 h, (Fig. 5D). After 24 h, the number of invaded melanoma cells was similar independent of the type of fibroblasts. However, after 48 h, the number of invaded melanoma cells was dramatically reduced in the presence of ADAM-9 knock-out fibroblasts when compared with the wild type control. This suggests that loss of ADAM-9-mediated cross-talk between fibroblasts and melanoma cells contributes significantly to the invasive behavior of melanoma cells in vitro.
DISCUSSION
Several lines of evidence point to a role for ADAM-9 in the development and spreading of a variety of tumors (28). ADAM-9 protein expression is increased in melanoma in vivo and melanoma cells in vitro (14). In addition, a high-throughput analysis of human melanomas revealed a correlation between ADAM-9 expression and melanoma progression (29). However, the understanding how ADAM-9 contributes to melanoma growth and invasion and by which function, proteolytic or adhesive, is still rudimentary.
In this report we have addressed the adhesive role of ADAM-9 for cellular processes involved in melanoma progression. Using the recombinant disintegrin-like and cysteine-rich domains of ADAM-9 as immobilized substrates we show that human primary dermal fibroblasts strongly adhere to this substrate and that adhesion is primarily mediated by β1 integrin receptors (see Fig. 1B). Keratinocytes also interact with ADAM-9 via a β1 integrin receptor (10) indicating a narrow specificity of receptor recognition. Interestingly, in both studies adhesion was not altered in the presence of peptides containing the ECD motif, which is located within the disintegrin-like domain of ADAM-9 (30). These results are in good agreement with a crystal structure study of the vascular apoptosis-inducing protein-1 (VAP-1), a snake venom homologue of mammalian ADAMs. In this study the high variable region in the cysteine-rich domain was shown to be responsible for substrate interaction, whereas the disintegrin loop containing the ECD motif turned out to be masked and inaccessible for protein binding (31–34).
Our results demonstrate that various integrins, including α3β1, α5β1, and α6β1, are involved in DC-9 mediated adhesion of fibroblasts. This suggests, as already observed for other cell types which recognize the soluble ADAM-9 domain via both α6β4 and α2β1 integrins (16), that several integrin receptors are able to contribute to the interaction with the adhesive domains of ADAM-9. Adhesion to the DC-9 was also detected using different melanoma cell lines. Adhesion was dependent on the presence of Mn2+ but not of Ca2+ ions indicating an integrin-mediated process (data not shown). The number of adhering cells tended to be higher for the low invasive melanoma cells than for the high invasive ones. This difference may be attributed to different integrin expression profiles on the cell surface of differently invasive cells. In vivo studies revealed that expression of integrin α6β1 and/or α6β4 is more common in nevi than in malignant melanomas, whereas most lesions of malignant melanoma seem to contain cells expressing α2β1, α3β1, and αvβ3 integrins (35, 36). Thus it is tempting to speculate that interaction of melanoma cells with ADAM-9 depends on the expression of specific integrin receptors which in turn might play a role for the invasiveness of these cells.
Interestingly, adhesion of both fibroblasts and melanoma cells to recombinant ADAM-9 domains was paralleled by alterations of cellular activities. Fibroblast displayed increased spreading and secretion of MMP-1 and -2 when plated on DC-9. Incubation of melanoma cells with increasing concentrations of soluble DC-9 also resulted in slightly increased levels of MMP-2 expression and activation, however, no effect was seen in low invasive melanoma cells or in control cells treated with the histidine peptide. Induction of MMPs can be stimulated by growth factors and cytokines but also by engagement of integrin receptors (18, 36). We have previously shown that increased expression and activation of MMP-2 and MMP-14 in MV3 and BLM melanoma cells occur upon α2β1 interaction with collagen (18, 37). However, these changes are only observed in the presence of fibrillar but not monomeric collagen type I suggesting that integrin clustering by a more complex substrate may be required for this response. Similarly, while we could detect adhesion to ADAM-9 already at low substrate levels, a prominent increase in proteolytic activities occurred only in the presence of high amounts. These high levels may be a prerequisite for integrin clustering and hence for inhanced expression of MMP-1 and -2 as detected in fibroblasts and high invasive melanoma cells.
In vivo expression of both MMP-2 and MMP-1 has been detected in peritumoral fibroblasts. MMP-2 is also found in tumor cells invading the underlying tissue in human melanoma (38–40). In line with these observations our data suggest that ADAM-9 mediated cell interaction might provide an additional mechanism to increase proteolytic activities in the tumor microenvironment thus contributing to melanoma cell invasion.
Importantly, we show that ADAM-9 mediates direct interactions between melanoma cells and fibroblasts as incubation of fibroblasts and melanoma cells with soluble DC-9 resulted in significant reduction of cell-cell adhesion. This finding is stressed by the observation that a strong reduction of cell-cell adhesion occurs when ADAM-9 was ablated in either melanoma cells or fibroblasts. Adhesion of fibroblasts to DC-9 was almost completely abolished in the presence of anti-β1 integrin antibodies, suggesting a heterotypic adhesion process mediated by the interaction of ADAM-9 with integrin receptors. This notion is supported by the finding that ADAM-9 depletion in both cell types resulted in a further reduction of cell-cell adhesion between fibroblasts and melanoma cells, indicating that homotypic interactions between ADAM molecules are not significantly contributing to this adhesion process. Adhesion is not completely abrogated, which we think is most likely due to the incomplete silencing of ADAM-9 protein in melanoma cells.
Similar types of cellular interactions have been shown for ADAM-15 between T-lymphocytes and epithelial cells (24). In these cells interactions led to secretion of pro-inflammatory factors thus suggesting a putative role of ADAM-15 in eliciting inflammatory responses. In this report we show that interactions through ADAM-9 also elicit cellular responses such as secretion of proteolytic enzymes. As ablation of ADAM-9 in fibroblasts resulted in strong inhibition of melanoma cell invasion in vitro, we speculate that these interactions leading to altered secretion of proteases are involved in the invasion process. In line with this hypothesis is the work from Stuelten and co-authors (41) showing that transient interactions between tumor cells and normal fibroblasts are necessary to modify the local microenvironment and increase tumorigenicity of cancer cells in vivo.
In conclusion, we have shown that ADAM-9 is an adhesive protein, which induces cell signaling leading to increased proteolytic activities. Most importantly, it mediates fibroblast-melanoma cell interactions and seems to be important for melanoma invasion. Further studies will be needed to elucidate the mechanisms by which ADAM-9 ablation interferes with melanoma cell invasion and whether this results not only in impaired invasion but also in alterations of tumor growth in vivo.
Acknowledgments
We thank Julia Steiger and Claudia Coerper-Ochsmann for excellent technical assistance, Dr. Carl Blobel (Hospital for Special Surgery New York) for kindly providing the ADAM-9-deficient mice, and Dr. Beate Eckes for helpful comments on this manuscript.
This work was supported by the Melanoma Research Network of the Deutsche Krebshilfe (Melanoma Verbund, to P. Z. and C. M.).
- ADAM
- a disintegrin and metalloproteinase
- DC
- disintegrin-like and cysteine-rich domain
- MMP
- matrix metalloproteinase
- CMFDA
- 5-chloromethylfluorescein diacetate
- FGFR2iiib
- fibroblast growth factor receptor 2(iiib)
- EphB4
- ephrin-type B receptor 4
- VCAM-1
- vascular cell adhesion molecule-1
- VE-cadherin
- vascular endothelial cadherin.
REFERENCES
- 1. Edwards D. R., Handsley M. M., Pennington C. J. (2008) Mol. Aspects Med. 29, 258–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bergers G., Coussens L. M. (2000) Curr. Opin. Genet. Dev. 10, 120–127 [DOI] [PubMed] [Google Scholar]
- 3. Schlöndorff J., Blobel C. P. (1999) J. Cell Sci. 112, 3603–3617 [DOI] [PubMed] [Google Scholar]
- 4. Zhang X. P., Kamata T., Yokoyama K., Puzon-McLaughlin W., Takada Y. (1998) J. Biol. Chem. 273, 7345–7350 [DOI] [PubMed] [Google Scholar]
- 5. Chen M. S., Tung K. S., Coonrod S. A., Takahashi Y., Bigler D., Chang A., Yamashita Y., Kincade P. W., Herr J. C., White J. M. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 11830–11835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nath D., Slocombe P. M., Webster A., Stephens P. E., Docherty A. J., Murphy G. (2000) J. Cell Sci. 113, 2319–2328 [DOI] [PubMed] [Google Scholar]
- 7. Nath D., Slocombe P. M., Stephens P. E., Warn A., Hutchinson G. R., Yamada K. M., Docherty A. J., Murphy G. (1999) J. Cell Sci. 112, 579–587 [DOI] [PubMed] [Google Scholar]
- 8. Zhou M., Graham R., Russell G., Croucher P. I. (2001) Biochem. Biophys. Res. Commun. 280, 574–580 [DOI] [PubMed] [Google Scholar]
- 9. Almeida E. A., Huovila A. P., Sutherland A. E., Stephens L. E., Calarco P. G., Shaw L. M., Mercurio A. M., Sonnenberg A., Primakoff P., Myles D. G., White J. M. (1995) Cell 81, 1095–1104 [DOI] [PubMed] [Google Scholar]
- 10. Zigrino P., Steiger J., Fox J. W., Löffek S., Schild A., Nischt R., Mauch C. (2007) J. Biol. Chem. 282, 30785–30793 [DOI] [PubMed] [Google Scholar]
- 11. Franzke C. W., Tasanen K., Schäcke H., Zhou Z., Tryggvason K., Mauch C., Zigrino P., Sunnarborg S., Lee D. C., Fahrenholz F., Bruckner-Tuderman L. (2002) EMBO J. 21, 5026–5035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mauch C., Zamek J., Abety A. N., Grimberg G., Fox J. W., Zigrino P. (2010) J. Invest Dermatol. 130, 2120–2130 [DOI] [PubMed] [Google Scholar]
- 13. O'Shea C., McKie N., Buggy Y., Duggan C., Hill A. D., McDermott E., O'Higgins N., Duffy M. J. (2003) Int. J. Cancer 105, 754–761 [DOI] [PubMed] [Google Scholar]
- 14. Zigrino P., Mauch C., Fox J. W., Nischt R. (2005) Int. J. Cancer 116, 853–859 [DOI] [PubMed] [Google Scholar]
- 15. Peduto L., Reuter V. E., Shaffer D. R., Scher H. I., Blobel C. P. (2005) Cancer Res. 65, 9312–9319 [DOI] [PubMed] [Google Scholar]
- 16. Mazzocca A., Coppari R., De Franco R., Cho J. Y., Libermann T. A., Pinzani M., Toker A. (2005) Cancer Res. 65, 4728–4738 [DOI] [PubMed] [Google Scholar]
- 17. Guaiquil V., Swendeman S., Yoshida T., Chavala S., Campochiaro P. A., Blobel C. P. (2009) Mol. Cell. Biol. 29, 2694–2703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zigrino P., Drescher C., Mauch C. (2001) Eur. J. Cell Biol. 80, 68–77 [DOI] [PubMed] [Google Scholar]
- 19. Löffek S., Zigrino P., Steiger J., Kurschat P., Smola H., Mauch C. (2006) Eur. J. Cell Biol. 85, 1167–1177 [DOI] [PubMed] [Google Scholar]
- 20. Asai M., Hattori C., Szabó B., Sasagawa N., Maruyama K., Tanuma S., Ishiura S. (2003) Biochem. Biophys. Res. Commun. 301, 231–235 [DOI] [PubMed] [Google Scholar]
- 21. Yoshida M., Westlin W. F., Wang N., Ingber D. E., Rosenzweig A., Resnick N., Gimbrone M. A., Jr. (1996) J. Cell Biol. 133, 445–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zigrino P., Kuhn I., Bäuerle T., Zamek J., Fox J. W., Neumann S., Licht A., Schorpp-Kistner M., Angel P., Mauch C. (2009) J. Invest Dermatol 129, 2686–2693 [DOI] [PubMed] [Google Scholar]
- 23. Margadant C., Sonnenberg A. (2010) EMBO Rep. 11, 97–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Charrier L., Yan Y., Nguyen H. T., Dalmasso G., Laboisse C. L., Gewirtz A. T., Sitaraman S. V., Merlin D. (2007) J. Biol. Chem. 282, 16948–16958 [DOI] [PubMed] [Google Scholar]
- 25. Namba K., Nishio M., Mori K., Miyamoto N., Tsurudome M., Ito M., Kawano M., Uchida A., Ito Y. (2001) Cell. Immunol. 213, 104–113 [DOI] [PubMed] [Google Scholar]
- 26. Puissegur M. P., Lay G., Gilleron M., Botella L., Nigou J., Marrakchi H., Mari B., Duteyrat J. L., Guerardel Y., Kremer L., Barbry P., Puzo G., Altare F. (2007) J. Immunol. 178, 3161–3169 [DOI] [PubMed] [Google Scholar]
- 27. Fischer O. M., Hart S., Gschwind A., Prenzel N., Ullrich A. (2004) Mol. Cell. Biol. 24, 5172–5183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Peduto L. (2009) Curr. Pharm. Des. 15, 2282–2287 [DOI] [PubMed] [Google Scholar]
- 29. Alonso S. R., Tracey L., Ortiz P., Pérez-Gómez B., Palacios J., Pollán M., Linares J., Serrano S., Sáez-Castillo A. I., Sánchez L., Pajares R., Sánchez-Aguilera A., Artiga M. J., Piris M. A., Rodríguez-Peralto J. L. (2007) Cancer Res. 67, 3450–3460 [DOI] [PubMed] [Google Scholar]
- 30. Blobel C. P., White J. M. (1992) Curr. Opin Cell Biol. 4, 760–765 [DOI] [PubMed] [Google Scholar]
- 31. Takeda S., Igarashi T., Mori H., Araki S. (2006) EMBO J. 25, 2388–2396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Serrano S. M., Jia L. G., Wang D., Shannon J. D., Fox J. W. (2005) Biochem. J. 391, 69–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jia L. G., Wang X. M., Shannon J. D., Bjarnason J. B., Fox J. W. (2000) Arch. Biochem. Biophys. 373, 281–286 [DOI] [PubMed] [Google Scholar]
- 34. Kamiguti A. S., Gallagher P., Marcinkiewicz C., Theakston R. D., Zuzel M., Fox J. W. (2003) FEBS Lett. 549, 129–134 [DOI] [PubMed] [Google Scholar]
- 35. Heino J. (1996) Int. J. Cancer 65, 717–722 [DOI] [PubMed] [Google Scholar]
- 36. Klein C. E., Dressel D., Steinmayer T., Mauch C., Eckes B., Krieg T., Bankert R. B., Weber L. (1991) J. Cell Biol. 115, 1427–1436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kurschat P., Zigrino P., Nischt R., Breitkopf K., Steurer P., Klein C. E., Krieg T., Mauch C. (1999) J. Biol. Chem. 274, 21056–21062 [DOI] [PubMed] [Google Scholar]
- 38. Kurschat P., Wickenhauser C., Groth W., Krieg T., Mauch C. (2002) J. Pathol. 197, 179–187 [DOI] [PubMed] [Google Scholar]
- 39. Hofmann U. B., Westphal J. R., Waas E. T., Zendman A. J., Cornelissen I. M., Ruiter D. J., van Muijen G. N. (1999) Br. J. Cancer 81, 774–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wandel E., Grasshoff A., Mittag M., Haustein U. F., Saalbach A. (2000) Exp. Dermatol. 9, 34–41 [DOI] [PubMed] [Google Scholar]
- 41. Stuelten C. H., Busch J. I., Tang B., Flanders K. C., Oshima A., Sutton E., Karpova T. S., Roberts A. B., Wakefield L. M., Niederhuber J. E. (2010) PLoS One 5, e9832 [DOI] [PMC free article] [PubMed] [Google Scholar]